
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic mono and dicarbonyl pincer complexes of rhodium and iridium to assess the donor properties of PCcarbeneP ligands†
Joel D.
Smith
a,
Jessamyn R.
Logan
a,
Lauren E.
Doyle
a,
Richard J.
Burford
a,
Shun
Sugawara
b,
Chiho
Ohnita
b,
Yohsuke
Yamamoto
b,
Warren E.
Piers
*a,
Denis M.
Spasyuk
a and
Javier
Borau-Garcia
a
aDepartment of Chemistry, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: wpiers@ucalgary.ca
bDepartment of Chemistry, Graduate School of Science, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, 739-8526, Japan
First published on 20th July 2016
Abstract
The donor properties of five different PCcarbeneP ligands are assessed by evaluation of the CO stretching frequencies in iridium(I) and rhodium(I) carbonyl cations. The ligands feature dialkyl phosphine units (R = iPr or tBu) linked to the central benzylic carbon by either an ortho-phenylene bridge, or a 2,3-benzo[b]thiophene linker; in the former, substituent patterns on the phenyl linker are varied. The carbonyl complexes are synthesized from the (PCcarbeneP)M–Cl starting materials via abstraction of the chlorides in the presence of CO gas. In addition to the expected mono carbonyl cations, products with two carbonyl ligands are produced, and for the rhodium example, a novel product in which the second carbonyl ligand adds reversibly across the Rh![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to give an η2 ketene moiety was characterized. The IR data for the complexes shows the 2,3-benzo[b]thiophene linked system to be the poorest overall donor, while the phenyl bridged ligands incorporating electron donating dialkyl amino groups para to the anchoring carbene are very strongly donating pincer arrays.
C bond to give an η2 ketene moiety was characterized. The IR data for the complexes shows the 2,3-benzo[b]thiophene linked system to be the poorest overall donor, while the phenyl bridged ligands incorporating electron donating dialkyl amino groups para to the anchoring carbene are very strongly donating pincer arrays.
Introduction
Pincer ligands are tridentate frameworks that provide a multitude of permutations for supporting complexes of main group elements, transition metals and lanthanides.1 They are often classified according to the three donor atoms directly involved in chelating the central element; for example, the archetypical ligand I2 is a “PNP” pincer donor system. For late transition metals,3,4 flanking phosphorus-based donors comprises a common motif, and when the anchoring element of the framework is also a strong donor, the complexes that result are capable of a variety of bond activations for catalytic applications.5–7
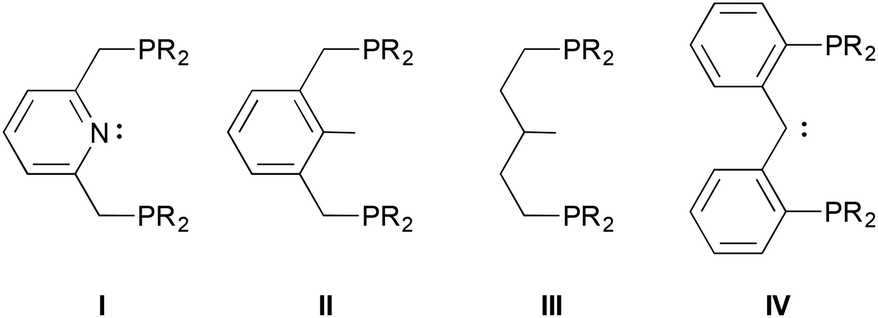
Although the diphenylphosphino version of ligand IV was initially reported in the 1990s,15 the dialkylphosphino examples we recently introduced16 have been explored more intensively in the past few years,22–30 in part because of this opportunity for ligand cooperativity involving the carbene moiety31,32 and also because of the electron richness of the donor ensemble. Indeed, we initially envisioned these ligands as being capable of supporting E–H bond activations (E = N, O) because of their strong donor character.33,34 However, lacking concrete data that suggest this expectation is rooted in reality, we have endeavoured to garner a measure of their donor character by preparing some carbonyl compounds to establish their place within the pantheon of other electron rich pincer ligands. Herein, we describe the synthesis of cationic mono and dicarbonyl complexes starting from the collection of rhodium and iridium(I) PCcarbeneP chlorido compounds shown in Scheme 1. The trends in the IR stretching frequencies of these complexes are used to assess the relative donor ability of the various PCcarbeneP donor arrays.
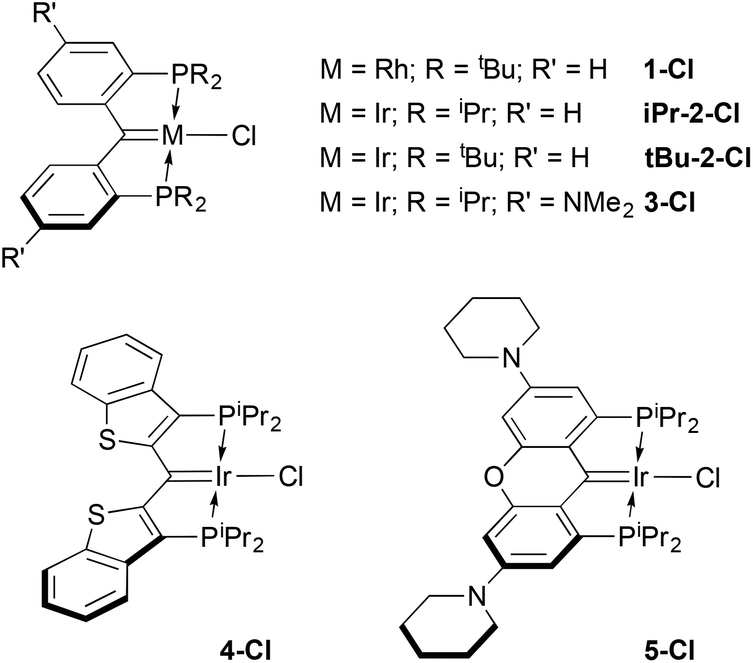 | ||
| Scheme 1 Ensemble of PCcarbeneP metal chloro compounds evaluated in this study. | ||
Results and discussion
Details concerning the syntheses of the ligands and their rhodium(I) (1-Cl18) or iridium(I) chloro carbene complexes (iPr-2-Cl,16tBu-2-Cl,233-Cl,35 and 4-Cl25) have been previously published. The xanthene-based ligand variation seen in 5-Cl is, however, a new ligand/complex. Related to the NMe2 substituted ligand in 3-Cl, the oxygen atom linker is designed to planarize and rigidify the ligand framework, improving stability,35 while retaining the electron donating dialkylamino groups. This ligand was prepared in two steps (54% yield) from the known 1,8-dibromo-3,6-di(piperidin-1-yl)-9H-xanthen-9-one36 by B(C6F5)3 catalyzed reductive silation of the ketone unit,37–39 followed by dilithiation with tert-butyl lithium and quenching with ClP(iPr)2. The resulting proligand 1,1′-(1,8-bis(diisopropylphosphino)-9H-xanthene-3,6-diyl)dipiperidine was attached to iridium using [Ir(COE)2(μ-Cl)]2via a double C–H activation protocol,16,40 yielding the new complex 5-Cl as an analytically pure dark brown solid in 75% isolated yield. The carbene carbon resonates at 183.0 ppm (2JCP = 3.7 Hz) and X-ray quality crystals were obtained via slow evaporation of a pentane solution of the complex. A depiction of the structure is shown in Fig. S17 in the ESI;† the IrC distance of 1.921(6) Å is typical for these complexes16 and reflective of double bond character.
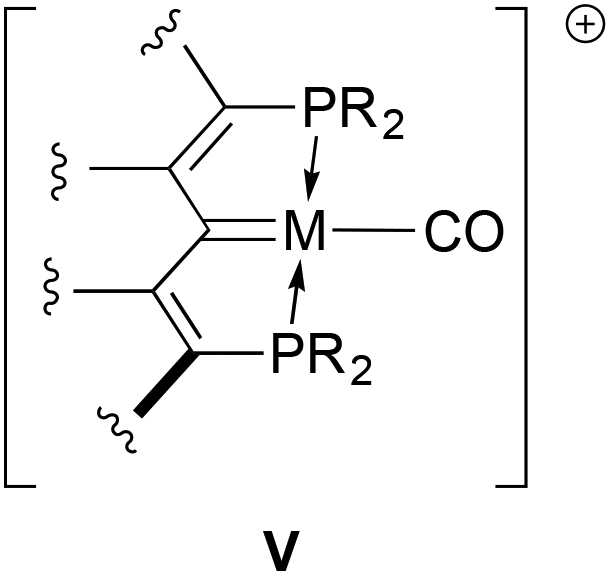
It was envisioned that a compilation of the CO stretching frequencies of carbonyl derivatives of these rhodium and iridium complexes could be used to assess the relative donor abilities of these ligands. Cationic M(I) carbonyl complexes of the form depicted in V were therefore targeted. Examples of several of these complexes containing other pincer ligands are available in the literature,41–49 allowing for comparison with our PCcarbeneP systems; accordingly, protocols towards such compounds starting from the chloro compounds in Scheme 1 were developed. While carbonyl compounds akin to V were obtained for all examples, the chemistry is somewhat more complex in that dicarbonyl cations and products arising from reactions of carbon monoxide with the MC unit of the ligand were also characterized.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1-Cl and sodium tetrakis-3,5-bis-trifluoromethylphenyl borate (Na[B(ArF)4]) was suspended in dichloromethane at −78 °C and exposed to one atmosphere of CO. Upon warming, a rapid colour change from the green colour of 1-Cl to deep purple was followed by a slow lightening to pale yellow over the course of ≈30 minutes. Monitoring the reaction by 31P NMR spectroscopy showed that an initial product characterized by a doublet at 96.3 ppm (1JRhP = 140.6 Hz) converted to the final product (79.0 ppm, 1JRhP = 106.8 Hz).
:1 mixture of 1-Cl and sodium tetrakis-3,5-bis-trifluoromethylphenyl borate (Na[B(ArF)4]) was suspended in dichloromethane at −78 °C and exposed to one atmosphere of CO. Upon warming, a rapid colour change from the green colour of 1-Cl to deep purple was followed by a slow lightening to pale yellow over the course of ≈30 minutes. Monitoring the reaction by 31P NMR spectroscopy showed that an initial product characterized by a doublet at 96.3 ppm (1JRhP = 140.6 Hz) converted to the final product (79.0 ppm, 1JRhP = 106.8 Hz).
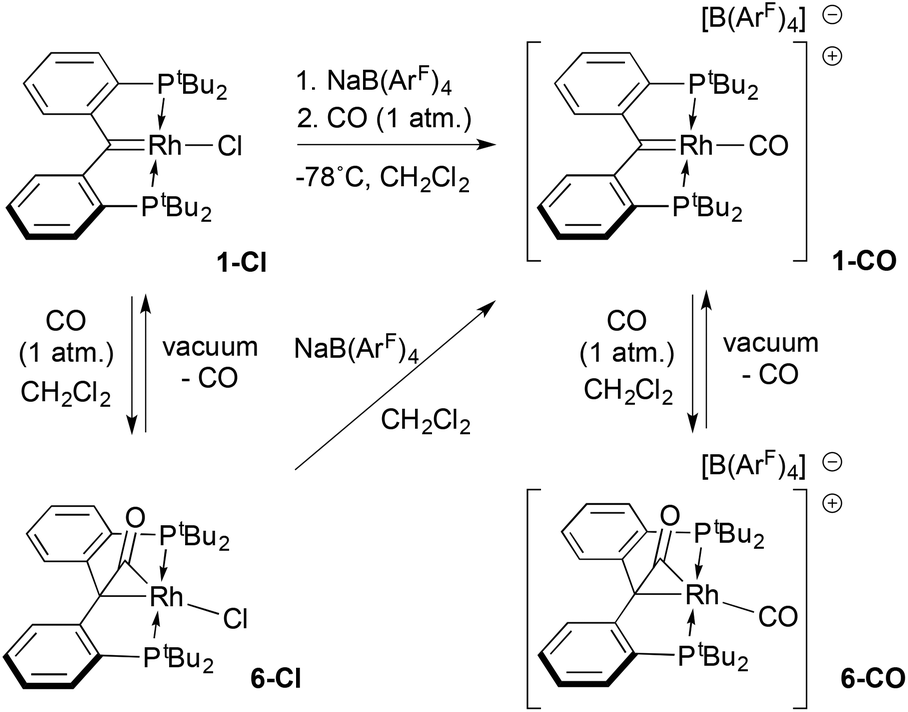 | ||
| Scheme 2 CO chemistry of compound 1-Cl. | ||
The magnitude of the RhP coupling constants in these species imply that the purple intermediate is a Rh(I) complex, while the final product is a Rh(III) derivative.50 Spectroscopic and structural data identify the latter species as the pale yellow cationic carbonyl complex 6-CO (Scheme 2) in which a second CO molecule has added to the RhC moiety of the ligand to form an η2 ketene ligand. An analogous complex, 6-Cl, can be prepared by treating 1-Cl with one atmosphere of CO in the absence of Na[B(ArF)4]. In the proton NMR spectra of both 6-CO and 6-Cl, the pattern of two virtual triplet resonances for the tert-butyl groups on phosphorus reflect the Cs symmetry of these molecules as opposed to the C2v symmetry of the starting complex 1-Cl or the cationic monocarbonyl target 1-CO. For 6-CO, the 13C NMR spectrum exhibits two doublets of triplets at 195.3 ppm (1JRhC = 22.1 Hz; 2JPC = 5.3 Hz), and 190.1 ppm (1JRhC = 66 Hz; 2JPC = 12.4 Hz) for the carbons of the ketene carbonyl moiety and the terminal carbonyl, respectively. This assignment is based on an observed carbon shift of 212.3 ppm (1JRhC = 26.5 Hz; 2JPC = 5.8 Hz) for the ketene CO carbon in 6-Cl. In 6-CO, the resonance for the former carbene carbon of the PCcarbeneP was shifted significantly upfield, appearing as a doublet of triplets (1JRhC = 6.7 Hz; 2JPC = 3.5 Hz) at 43.2 ppm. In the IR spectrum, two νCO stretches at 2050 and 1860 cm−1 were recorded; a band at 1787 cm−1 was observed for 6-Cl, indicating a CO formulation for the ketene ligand.
While addition of CO to the RhC bond was reversible (see below), release of CO was slow enough at atmospheric pressure to permit growth of crystals of both 6-Cl and 6-CO from dichloromethane solutions layered with pentane. The structure of the cation of 6-CO is shown in Fig. 1, along with selected metrical parameters; the structure of 6-Cl can be found in Fig. S18 in the ESI.† In addition to the terminal CO ligand, the structure confirms the addition of a CO to the RhC double bond; the Rh1–C1 distance of 2.105(3) Å is significantly elongated from the 1.900(3) Å Rh–C1 distance found in 1-Cl.18 The C1–C30 bond length of 1.442(5) Å is consistent with a Csp3–Csp2 single bond, while the C30–O1 length of 1.171(4) is similar to the C–O bond length of 1.162 Å in CO2.51
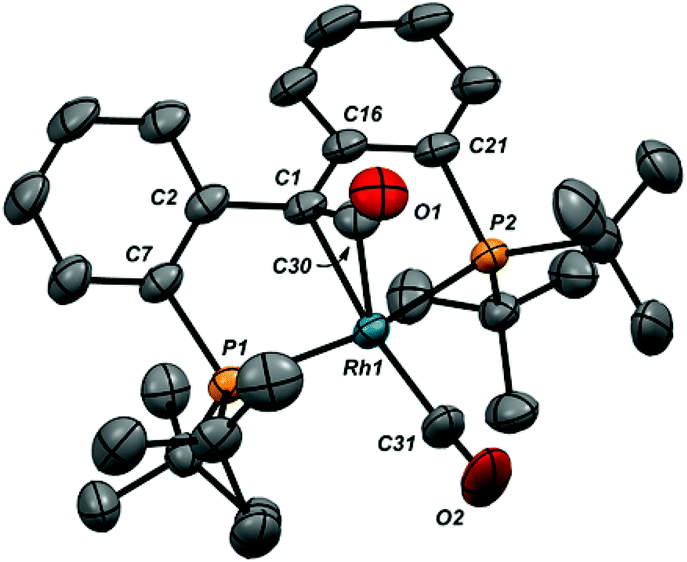 | ||
| Fig. 1 X-ray structure of the cation in 6-CO (thermal ellipsoids drawn to 50% probability level). Hydrogen atoms and the B(ArF)4 counteranion are omitted for clarity. Selected bond distances (Å): Rh1–C1, 2.105(3); Rh1–C30, 1.992(4); Rh1–C31, 1.929(4); Rh1–P1, 2.3537(8); Rh1–P2, 2.3541(9); C1–C30, 1.442(5); C30–O1, 1.171(5); C31–O2, 1.124(5). Selected bond angles (°): P1–Rh1–P2, 161.87(3); C1–Rh1–C31, 160.22(18); C1–Rh1–C30, 41.10(14); C1–C30–Rh1, 73.6(2); C1–C30–O1, 149.6(4); O1–C30–Rh1, 136.5(3); C30–C1–Rh1, 65.26(18); C2–C1–C16, 121.7(3). | ||
Exposure of compounds 6-Cl or 6-CO to high vacuum resulted in loss of CO by reversal of the CO addition to the RhC moiety; in the former case, this regenerated carbene chloride 1-Cl, while in the latter this allowed for isolation of the targeted cationic carbonyl complex of the PCcarbeneP ligand as a purple solid. NMR spectroscopic data for samples of 1-CO isolated in this way indicate that this was the intermediate observed when the reaction of 1-Cl/Na[B(ArF)4] mixtures with CO was monitored spectroscopically. The νCO stretching frequency for the carbonyl ligand was measured to be 2026 cm−1, 24 wavenumbers below that recorded for the terminal CO ligand in 6-CO. As indicated in Scheme 1, analytically pure samples of 1-CO were best prepared from isolated 6-Cl and Na[B(ArF)4]; this observation suggests that 6-Cl is an intermediate on the way to formation of 1-CO, which gets converted in a slower step to 6-CO when excess carbon monoxide is present.
Ion pair 1-CO was also characterized crystallographically; the structure of the cation, along with selected metrical parameters, is given in Fig. 2. The structure represents a fairly typical Rh(I) square planar complex, and the Rh1–C1 distance of 1.944(10) Å, while slightly elongated from that in 1-Cl, is still clearly indicative of restoration of the RhC bond.48 Addition of an excess of CO to solutions of 1-CO regenerates 6-CO quantitatively over the course of roughly 30 minutes. The addition of carbon monoxide to a metal carbene with C–C bond formation to give a ketene is a model reaction for a potentially important C–C bond forming mechanism in the Fischer–Tropsch process.52,53 While several model systems exist in the literature, examples that exhibit facile reversibility are rare.54,55
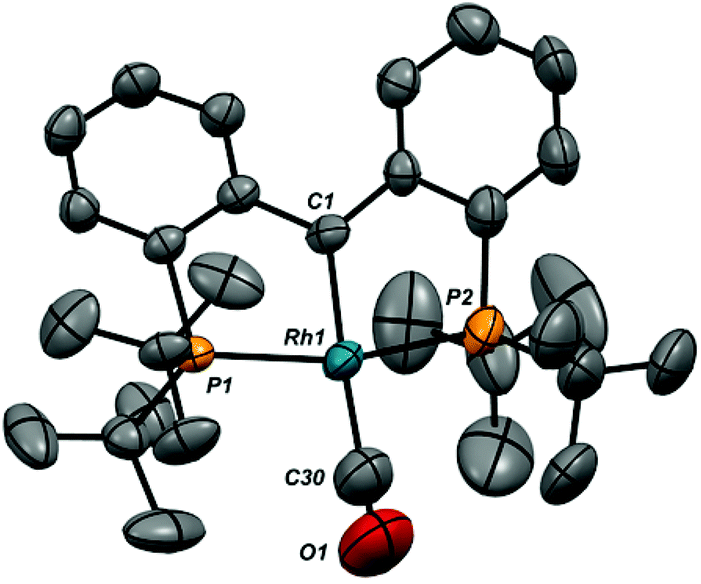 | ||
| Fig. 2 X-ray structure of the cation in 1-CO (thermal ellipsoids drawn to 50% probability level). Hydrogen atoms and the B(ArF)4 counteranion are omitted for clarity. Selected bond distances (Å): Rh1–C1, 1.944(10); Rh1–C30, 1.940(19); Rh1–P1, 2.311(3); Rh1–P2, 2.324(3); C30–O1, 1.15(2). Selected bond angles (°): P1–Rh1–P2, 164.03(11); C1–Rh1–C30, 165.3(7); C1–Rh1–P1, 83.5(3); C1–Rh1–P2, 81.5(3); P1–Rh1–C30, 95.5(6); P2–Rh1–C30, 100.4(6). | ||
Analogous reactions using the iridium complexes 2–5-Cl, lead to somewhat different outcomes compared to that seen for the rhodium carbene chloride 1-Cl (Scheme 3). Equimolar suspensions of the iridium starting material and the chloride abstracting agent Na[B(ArF)4] in CH2Cl2 did not visibly react until an atmosphere of CO was admitted into the vessel, whereupon a rapid color change from the generally dark green colors of the carbene chlorides to red or purple solutions were observed. Concentration of these solutions by removal of the solvent in vacuo, while keeping the vessel cooled below room temperature, allowed for isolation of the dicarbonyl cations N-(CO)2 shown in Scheme 3 in moderate to excellent yields (62–99%) rather than the expected monocarbonyl cations. In only one case, that of 4-Cl, was a second product observed in these reactions. This minor species was formed in 15–20% yield but could be minimized to less than 10% by using 1.5 equivalents of Na[B(ArF)4] and gently heating the 4-Cl/Na[B(ArF)4] mixture prior to addition of CO. We speculate that this minor species, which is manifested by a signal at 29.6 ppm in the 31P NMR spectrum, is analogous to the rhodium ketene carbonyl cation 6-CO described above. This is supported by its apparent Cs symmetry in the 1H NMR spectrum, but attempts to prepare this species cleanly met with failure. Furthermore, reactions of any of the iridium carbene chlorides N-Cl (N = 1–5) with CO in the absence of Na[B(ArF)4] were much more complex than the reaction of 1-Cl with CO to form 6-CO (Scheme 2). It thus appears that addition of CO to the IrC bond in these complexes is not as facile as addition to the RhC bonds in 1-Cl or 1-CO; rather, more conventional carbonyl complexes such as the observed dicarbonyl cations are the preferred products of CO addition to the iridium monocarbonyl cations 2–5-CO and the carbene chlorides 2–5-Cl.
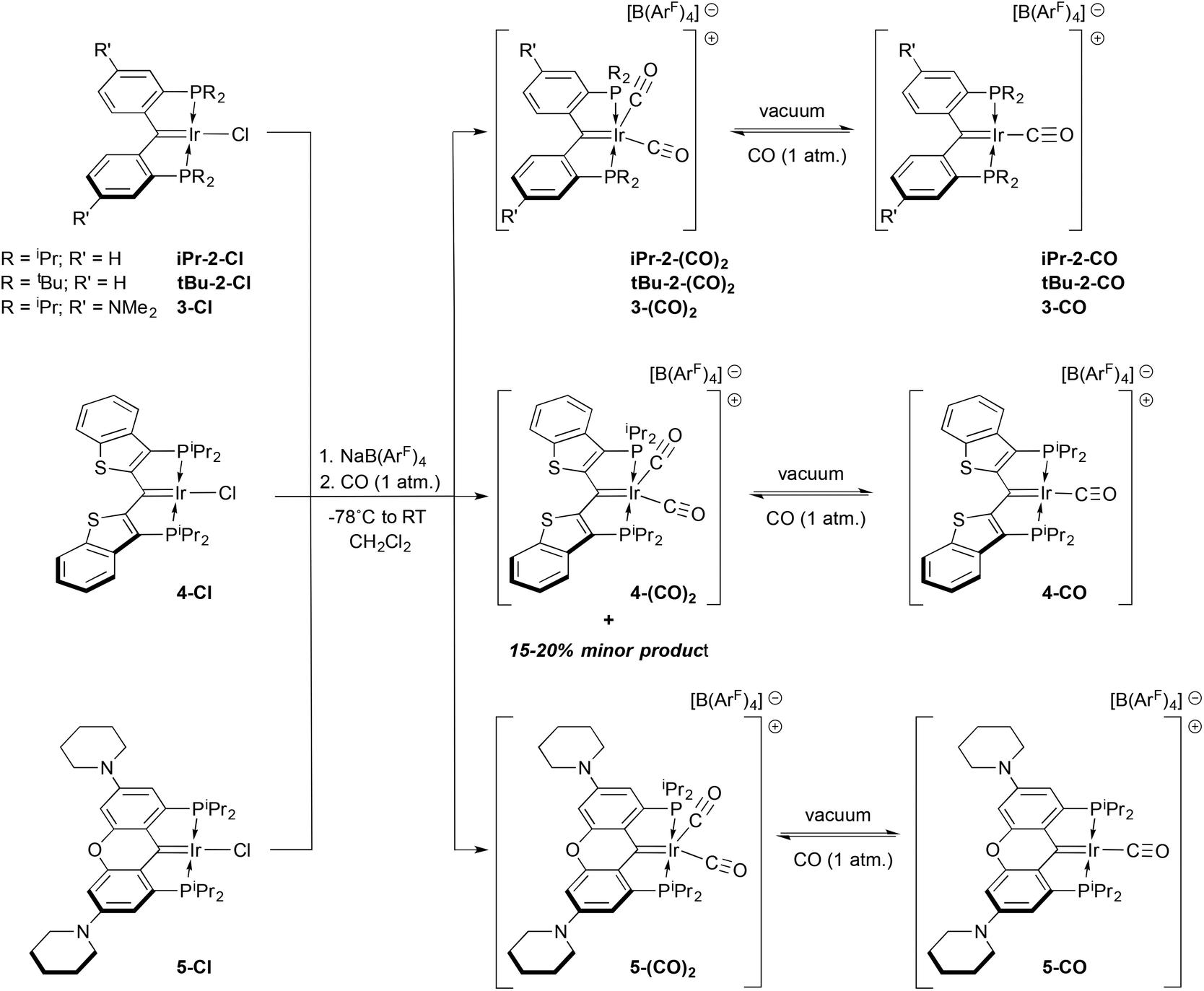 | ||
| Scheme 3 Synthesis iridium dicarbonyl cations and monocarbonyl cations. | ||
The monocarbonyl cations could be prepared via removal of the solvent in vacuo from solutions of the dicarbonyl complexes with heating briefly to high temperatures using a heat gun. In some instances, this would need to be repeated by addition of more CH2Cl2 solvent and further heating under dynamic vacuum to ensure sufficient conversion to the monocarbonyl products N-CO. Alternatively, in some cases methods that limited the time of exposure of mixtures of N-Cl and Na[B(ArF)4] to excess CO were the preferred routes to the monocarbonyl cations. The interconversion of these species was monitored via31P NMR spectroscopy, since the color changes were not distinct enough to allow for visual confirmation of complete reaction. Because of this tendency towards dicarbonyl complexes, it was not always possible to obtain samples of monocarbonyl cations that were analytically pure. Nonetheless, purities of >95% were possible in all cases and the spectroscopic data obtained for each of the monocarbonyl cations reported here is completely consistent with their formulation as depicted in Scheme 3.
All dicarbonyl and monocarbonyl cations were characterized by IR and multinuclear NMR spectroscopies, see Table 1 for selected spectroscopic data. The IR data will be discussed below. In the 31P NMR spectra, the dicarbonyl compounds exhibited resonances upfield by 7–15 ppm in comparison to those observed for the monocarbonyl cations. Characteristic low field resonances for the carbene carbon were observed and a similar trend towards more upfield-shifted resonances for the dicarbonyl derivatives in comparison to the monocarbonyl analogues was also noted here. Less variation in the chemical shift for the carbonyl carbons was seen, but again the observed shifts for the dicarbonyl species were upfield of the range seen for the monocarbonyl compounds. In the former complexes, the two carbonyl ligands are likely made equivalent due to a fast exchange process that could not be frozen out at lower temperatures. In light of the lability of the second CO ligand, it is reasonable to propose a dissociative mechanism for this exchange, but fluxionality through a pseudo rotation is also feasible. The mechanistic nature of this exchange process was not probed in further detail.
| Compound | 31P (ppm) |
13CIrC (ppm) |
13CCO (ppm) | ν CO (cm−1) |
|---|---|---|---|---|
| a IR spectra acquired on thin films deposited on salt plates. b Sample contaminated with 10–15% of 4-(CO)2. c Number in parentheses is the average value of the two stretching frequencies. | ||||
| 1-CO | 96.3 | 294.8 | 200.5 | 2026 |
| 2-iPr-(CO)2 | 61.0 | 233.9 | 178.2 | 2050, 2002 (2026)c |
| 2-tBu-(CO)2 | 81.0 | 231.8 | 183.1 | 2040, 2002 (2021)c |
| 3-(CO)2 | 58.2 | 242.4 | 179.7 | 2014, 1971 (1993)c |
| 4-(CO)2 | 47.1 | 200.1 | 182.1 | 2055, 2014 (2035)c |
| 5-(CO)2 | 62.9 | 207.4 | 178.2 | 2014, 1973 (1994)c |
| 2-iPr-CO | 68.6 | 267.8 | 195.1 | 2004 |
| 2-tBu-CO | 82.0 | 264.2 | 198.7 | 2005 |
| 3-CO | 72.2 | 260.4 | 195.9 | 1963 |
| 4-CO | 59.6 | 232.2 | 188.6 | 2048b |
| 5-CO | 78.6 | 214.9 | 196.4 | 1974 |
The X-ray structures of compounds 2-iPr-(CO)2, 4-(CO)2 and 3-CO were determined via single crystal X-ray diffraction; the structures of the cations of these ion pairs are shown in Fig. 3–5, respectively, along with selected metrical data. In the dicarbonyl cations 2-iPr-(CO)2 and 4-(CO)2, the iridium center is closer to a square pyramidal geometry than trigonal bipyramidal, as defined by their low τ values56 (0.22 for 2-iPr-(CO)2 and 0.19 for 4-(CO)2). Nonetheless, the equitorial carbonyl ligand in these structures dips below the basal plane; for 2-iPr-(CO)2 the C1–Ir1–C27 angle is 146.3(4)° while for 4-(CO)2 the C1–Ir1–C19 angle is 149.8(3)°. The Ir–Ccarbene distances in the complexes (2-iPr-(CO)2, 1.967(8) Å; 4-(CO)2, 1.996(6) Å) are somewhat elongated from the range of 1.899(7)–1.947(4) Å found in neutral, four-coordinate (PCcarbeneP)IrX complexes.16,22,23,26,35 The Ir–Ccarbonyl bond lengths are shorter than the Ir–C1 distances, with the bonds to the apical carbonyl carbons shorter than those to the basal carbonyl carbons by 0.038 Å in 2-iPr-(CO)2 and 0.024 Å in 4-(CO)2. The structure of the four coordinate monocarbonyl cation 3-CO is a distorted square planar geometry with a rather long Ir1–C1 distance of 2.038(9) Å, outside the range previously observed.
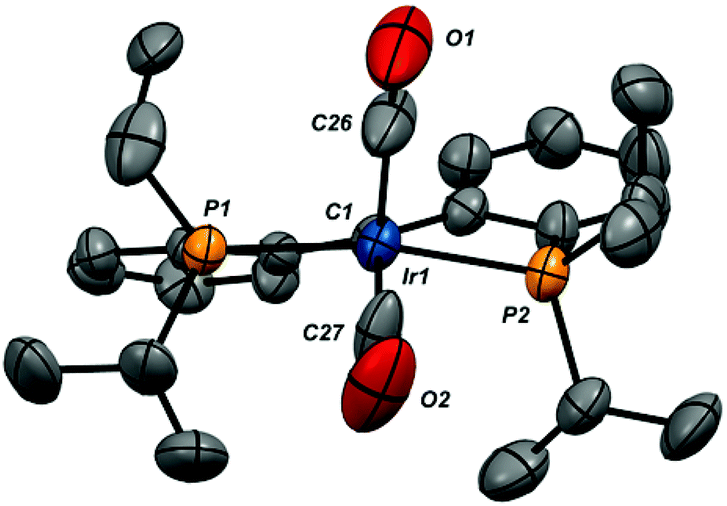 | ||
| Fig. 3 X-ray structure of the cation in iPr-2-(CO)2 (thermal ellipsoids drawn to 50% probability level). Hydrogen atoms and the B(ArF)4 counteranion are omitted for clarity. Selected bond distances (Å): Ir1–C1, 1.967(8); Ir1–C26, 1.894(10); Ir1–C27, 1.932(9); Ir1–P1, 2.3325(18); Ir1–P2, 2.328(2); C26–O1, 1.128(11); C27–O2, 1.121(10). Selected bond angles (°): P1–Ir1–P2, 159.59(8); C1–Ir1–C26, 117.8(4); C1–Ir1–C27, 146.3(4); C26–Ir1–C27, 95.8(5); C1–Ir1–P1, 82.1(2); C1–Ir1–P2, 81.5(2); P1–Ir1–C26, 98.7(3); P2–Ir1–C26, 99.6(3); P1–Ir1–C27, 93.9(3); P2–Ir1–C27, 93.1(3). | ||
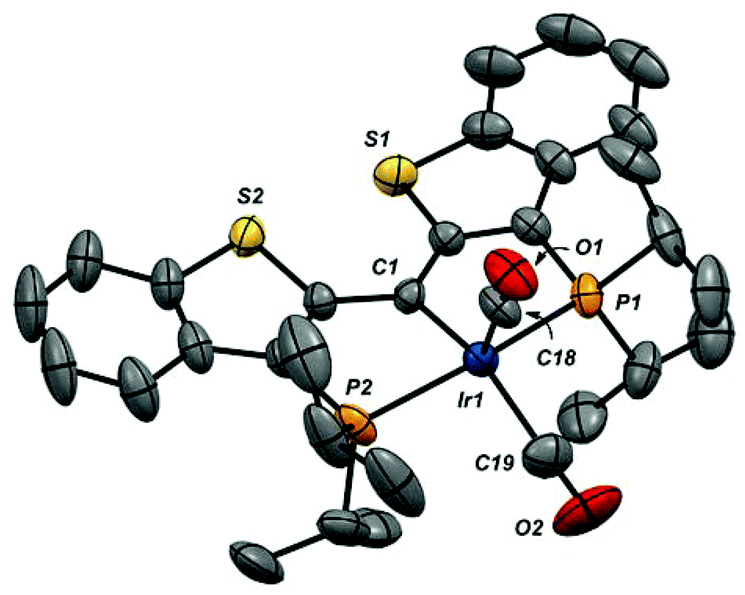 | ||
| Fig. 4 X-ray structure of the cation in 4-(CO)2 (thermal ellipsoids drawn to 50% probability level). Hydrogen atoms and the B(ArF)4 counteranion are omitted for clarity. Selected bond distances (Å): Ir1–C1, 1.996(6); Ir1–C18, 1.914(7); Ir1–C19, 1.938(8); Ir1–P1, 2.3396(17); Ir1–P2, 2.3401(18); C18–O1, 1.121(8); C19–O2, 1.115(10). Selected bond angles (°): P1–Ir1–P2, 160.98(6); C1–Ir1–C18, 115.4(3); C1–Ir1–C19, 149.8(3); C18–Ir1–C19, 94.8(3); C1–Ir1–P1, 82.53(17); C1–Ir1–P2, 82.44(18); P1–Ir1–C18, 98.4(2); P2–Ir1–C18, 98.6(2); P1–Ir1–C19, 93.0(3); P2–Ir1–C19, 94.2(3). | ||
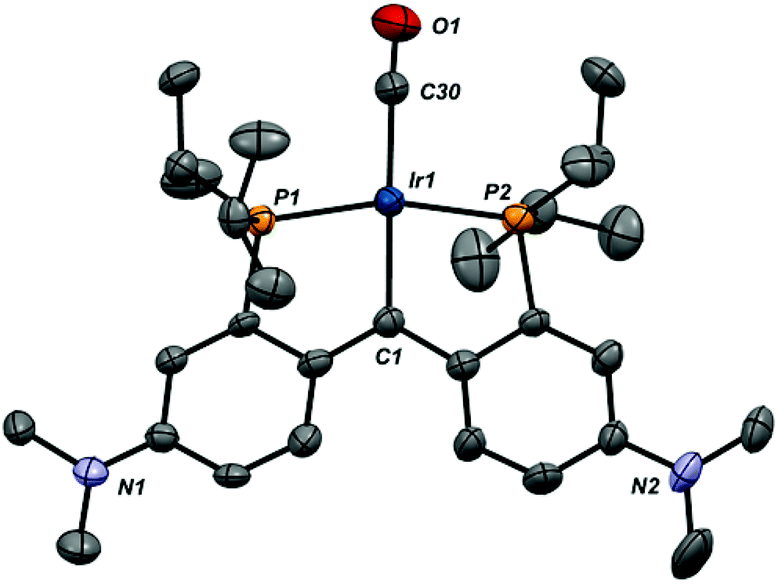 | ||
| Fig. 5 X-ray structure of the cation in 3-CO (thermal ellipsoids drawn to 50% probability level). Hydrogen atoms and the B(ArF)4 counteranion are omitted for clarity. Selected bond distances (Å): Ir1–C1, 2.038(9); Ir1–C30, 1.915(10); Ir1–P1, 2.285(2); Ir1–P2, 2.292(2); C30–O1, 1.117(12). Selected bond angles (°): P1–Ir1–P2, 164.25(8); C1–Ir1–C30, 178.0(4); C1–Ir1–P1, 82.5(2); C1–Ir1–P2, 81.7(2); P1–Ir1–C30, 98.0(3); P2–Ir1–C30, 97.7(3). | ||
The comparative donor properties of the various ligands employed here can be evaluated by the IR data collected in Table 1. In general, the observed stretching frequencies for the cationic species here are relatively high in comparison to typical neutral iridium(I) carbonyl complexes, with CO stretching frequencies spanning a range of 1963–2048 cm−1 in the monocarbonyl derivatives. In comparison, the value for a related Vaska's complex analogue IrCl(CO)(PMe2Ph)2,57 which also features dialkylarylphosphine donors, exhibits a νCO of 1938 cm−1 (1960 in a nujol mull, 1950 in THF58). The dicarbonyl compounds, as expected, give rise to two νCO bands, the low energy band in the range 1971–2014 cm−1, the high energy stretch appearing between 2014 and 2055 cm−1. The average value is given in italics in Table 1, and is in most cases larger than the stretch for the analogous monocarbonyl derivative by about 20 wavenumbers; nevertheless, the trends within each family of compounds with respect to ligand variation are consistent. That the stretching frequencies of these cationic compounds are larger than comparable neutral Ir(I) compounds is consistent with the expectation that π backbonding will be attenuated in cationic compounds.
Based on these data, the ligand featuring a 2,3-benzo[b]thiophene linker in compounds 4 is the poorest overall donor. The parent system, with the simple C6H4 linker is somewhat more donating, but incorporating N,N-dimethylamino groups in the positions para to the carbene carbon increases the donor properties of the PCcarbeneP ligand significantly. The installation of the rigidifying oxygen atom bridge in the xanthene-based ligand was motivated by a desire to increase the stability of the ligand system,35 similar to that found for the a 2,3-benzo[b]thiophene linked ligand, while retaining the stronger donating character of the N,N-dimethylamino adorned ligand in compounds 3. The IR data shows that the oxygen atom makes this ligand only slightly less donating, and preliminary reaction chemistry indicates that the ligand framework featured in compounds 5 is not prone to the C–C bond cleavage processes observed in the parent, unbridged systems.35
These dialkylamino substituted ligands are comparable or better overall donors than other common pincer systems in the literature. The IR data for several related monocarbonyl complexes containing other pincer ligands is given in Table S1 in the ESI.† The PNP ligands of type I (see Introduction), where the R groups on phosphorus are tert-butyl41–43 or iso-propyl,42 give mono carbonyl cations with comparable stretches of 1962–1970 cm−1 to those observed for compounds 3-CO and 5-CO. These PNP contain more electron-rich tris-alkylphosphine donor arms in comparison to the dialkylaryl donors found in the PCcarbeneP ligands discussed here. A related PNP system containing phosphinato donor arms is significantly less donating, with a nCO stretch of 2010 cm−1 found for its monocarbonyl cation.44 Other systems, including SNS,42 NNS,45 and NNN46,47 pincer complexes are all less donating than the ligands in compounds 3 and 5 by this criterion.
Conclusions
Assessment of the global donor properties of a given ligand set is important for rationalizing trends in reactivity, catalytic behaviour and structural properties within related families of compounds. The use of IR stretching frequencies of carbonyl ligands in families of compounds is a common way of evaluating the electronic effects of ligand variation. For relatively new ligand families such as the PCcarbeneP ligands discussed here, it is therefore important to establish relative donor properties so as to inform future ligand designs and to rationalize observed reaction trends.A comparison with available literature data shows that these aryl-linked ligands presented here exhibit better or comparable electron donating ability than common donor arrays. Logical trends in overall electron donating ability are observed based on the expectation that NMe2 groups placed para to the carbene carbon donor should increase electron density at this center by resonance. This suggests that the electronic properties of the ligands can be tuned in a rather sensitive way by varying groups in this position. Linking substituents ortho to the carbene carbon also have an effect; these features may be required to maintain ligand integrity,35 but also offer a further ligand design handle for tuning the overall properties of the ligand. Interestingly, the ligand featuring a 2,3-benzo[b]thiophene linker is significantly less donating overall, providing another platform for applications in which less basic ligands would be beneficial.
Experimental section
General considerations
For a description of general procedures and the synthesis of the xanthene-based ligand and complex 5-Cl can be found in the ESI.†C), 200.5 (dt, 1JCRh = 50.8 Hz, 2JCP = 11.0 Hz, Rh–CO), 164.1 (dvt, JCP = 16.2 Hz, 2JCRh = 2.3 Hz, ArC) 162.2 (q, 1JCB = 49.8 Hz, C–B, B(ArF)4, Ar), 143.1 (dvt, JCP = 13.8 Hz, 2JCRh = 2.9 Hz, ArC), 136.7 (vt, JCP = 2.6 Hz, ArCH), 136.0 (s, ArCH), 135.2 (s, CH-ortho, B(ArF)4, Ar), 133.3 (s, ArCH), 132.5 (vt, JCP = 6.6 Hz, ArCH), 129.3 (qq, 2JCF = 31.5 Hz, 4JCF = 2.7 Hz, C–CF3, B(ArF)4), 124.9 (q, 1JCF = 272.4 Hz, CF3, B(ArF)4), 117.9 (sept, 3JCF = 4.0, CH-para, B(ArF)4, Ar), 37.3 (vt, JCP = 7.9 Hz, C(CH3)3), 30.7 (vt, JCP = 2.8 Hz, C(CH3)3). 31P{1H} NMR (203 MHz, CD2Cl2): δ 96.3 (d, JPRh = 140.6 Hz). 19F{1H} NMR (471 MHz, CD2Cl2): δ −63.8 (s). 11B{1H} NMR (161 MHz, CD2Cl2): δ −6.0 (s). IR (AgCl plates) cm−1: νCO 2026 (m). Elemental Anal. Calcd (%) for C58H48BF24OP2Rh: C 51.40, H 3.90; Found: C 51.38, H 3.94.
C), 178.1 (t, 2JCP = 4.3 Hz, Ir–CO), 166.4 (vt, JCP = 17.1 Hz, Ar), 162.2 (q, 1JCB = 49.8 Hz, C–B, B(ArF)4, Ar), 135.2 (s, CH-ortho, B(ArF)4, Ar), 133.9 (vt, JCP = 1.7 Hz, ArCH), 133.6 (s, ArCH), 133.4 (vt, JCP = 25.9 Hz, ArC), 129.8 (vt, JCP = 4.0 Hz, ArCH), 129.3 (qq, 2JCF = 30.2 Hz, 4JCF = 2.9 Hz, B(ArF)4, C–CF3, Ar), 125.5 (vt, JCP = 7.2 Hz, ArCH), 125.0 (q, 1JCF = 272.2 Hz, B(ArF)4, CF3), 117.9 (sept, 3JCF = 4.3 Hz, CH-para, B(ArF)4, Ar), 25.9 (vt, JCP = 15.7 Hz, –CH(CH3)2), 18.5 (s, –CH(CH3)2), 17.4 (s, –CH(CH3)2). 31P{1H} NMR (243 MHz, CD2Cl2): δ 61.9 (s). 19F{1H} NMR (376 MHz, CD2Cl2): δ −63.4 (s). 11B{1H} NMR (128 MHz, CD2Cl2): δ −7.1 (s). IR (NaCl plates) cm−1: νCO 2050 (s), νCO 2002 (s). Elemental Anal. Calcd (%) for C59H48BF24IrOP2: C 46.93, H 3.20; Found: C 46.92, H 3.48.
C), 195.1 (t, 2JCP = 7.5 Hz, Ir–CO), 167.2 (vt, JCP = 15.5 Hz, ArC), 162.2 (q, 1JCB = 49.8 Hz, C–B, B(ArF)4, Ar), 141.7 (vt, JCP = 20.4 Hz, ArC), 135.7 (m, ArCH), 135.6 (s, ArCH), 135.3 (s, CH-ortho, B(ArF)4, Ar), 135.1 (s, ArCH), 130.9 (vt, JCP = 6.3 Hz, ArCH), 129.4 (qq, 2JCF = 31.8 Hz, 4JCF = 2.4 Hz B(ArF)4, C–CF3, Ar), 125.0 (q, 1JCF = 272.6 Hz, B(ArF)4, CF3), 117.9 (m, CH-para, B(ArF)4, Ar), 25.6 (vt, JCP = 15.3 Hz, –CH(CH3)2), 19.8 (s, –CH(CH3)2), 18.9 (s, –CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2): δ 68.0 (s). 19F{1H} NMR (471 MHz, CD2Cl2): δ −63.8 (s). 11B{1H} NMR (161 MHz, CD2Cl2): δ −6.0 (s). IR (NaCl plates) cm−1: νCO 2004 (s). Elemental Anal. Calcd (%) for C58H48BF24IrOP2: C 47.01, H 3.26; Found: C 47.05, H 3.19.
C), 183.1 (t, 2JCP = 3.4 Hz, Ir–CO), 166.6 (vt, JCP = 16.1 Hz, ArC), 162.4 (q, 1JCB = 49.9 Hz, C–B, B(ArF)4, Ar), 135.4 (m, CH-ortho, B(ArF)4, Ar), 135.3 (s, ArCH), 134.5 (vt, JCP = 22.6 Hz, ArC), 133.2 (s, ArCH), 129.5 (qq, 2JCF = 31.3 Hz, 4JCF = 2.9 Hz, B(ArF)4, C–CF3, Ar), 129.3 (vt, JCP = 3.4 Hz, ArCH), 126.2 (vt, JCP = 7.0 Hz, ArCH), 125.2 (q, 1JCF = 272.4 Hz, CF3, B(ArF)4) 118.1 (sept, 3JCF = 4.4 Hz, CH-para, B(ArF)4, Ar), 39.3 (vt, JCP = 10.6 Hz, C(CH3)3), 30.9 (vt, JCP = 2.2 Hz, C(CH3)3). 31P{1H} NMR (243 MHz, CD2Cl2): δ 81.4 (s). 19F{1H} NMR (376 MHz, CD2Cl2): δ −62.8 (s). 11B{1H} NMR (128 MHz, CD2Cl2): δ −6.6 (s). IR (NaCl plates) cm−1: νCO 2044 (s), νCO 2002 (s). Elemental Anal. Calcd (%) for C63H56BF24O2P2Ir: C 48.32, H 3.60; Found: C 48.32, H 3.80.
C), 198.7 (t, 2JCP = 7.2 Hz, Ir–CO), 168.0 (vt, JCP = 15.3 Hz, ArC), 162.2 (q, 1JCB = 49.9 Hz, C–B, B(ArF)4, Ar), 141.7 (vt, JCP = 18.0 Hz, ArC), 136.9 (s, ArCH), 135.2 (m, CH-ortho, B(ArF)4, Ar), 135.1 (vt, JCP = 3.2 Hz, ArCH), 134.5 (s, ArCH), 131.8 (vt, JCP = 6.1 Hz, ArCH), 129.2 (qq, 2JCF = 31.5 Hz, 4JCF = 2.8 Hz, B(ArF)4, C–CF3, Ar), 125.0 (q, 1JCF = 272.8 Hz, CF3, B(ArF)4), 117.9 (sept, 3JCF = 4.0 Hz, CH-para, B(ArF)4, Ar), 37.7 (vt, JCP = 10.6 Hz, C(CH3)3), 30.9 (vt, JCP = 2.6 Hz, C(CH3)3). 31P{1H} NMR (243 MHz, CD2Cl2) δ 82.0. 19F{1H} NMR (471 MHz, CD2Cl2) δ −63.8. 11B{1H} NMR (161 MHz, CD2Cl2) δ −6.1. IR (AgCl plates) cm−1: νCO 2008 (s). HRMS (ESI+) for C30H44IrOP2; calculated (M+): 675.2491 (100%); Found (M+): 675.2473 (100%).
C), 179.7 (t, 2JCP = 4.9 Hz, Ir–CO), 162.2 (q, 1JCB = 49.9 Hz, C–B, B(ArF)4, Ar), 152.5 (vt, JCP = 4.5 Hz, ArC), 151.8 (vt, JCP = 15.8 Hz, ArC), 143.8 (vt, JCP = 23.5 Hz, ArC), 135.2 (s, CH-ortho, B(ArF)4, Ar), 129.3 (qq, 2JCF = 31.5 Hz, 4JCF = 2.7 Hz, C–CF3, B(ArF)4), 128.9 (m, ArCH), 125.0 (q, 1JCF = 272.6 Hz, CF3, B(ArF)4), 117.9 (sept, 3JCF = 4.0, CH-para, B(ArF)4, Ar), 115.4 (s, ArCH), 113.8 (s, ArCH), 40.7 (s, –N(CH3)2), 27.4 (vt, JCP = 15.7 Hz, –CH(CH3)2), 18.8 (vt, JCP = 2.0 Hz, –CH(CH3)2), 17.7 (s, –CH(CH3)2). 31P{1H} NMR (243 MHz, CD2Cl2) δ 58.2 (s). 19F{1H} NMR (376 MHz, CD2Cl2) δ −62.91 (s).11B{1H} NMR (128 MHz, CD2Cl2) δ −6.60 (s). IR (NaCl plates) cm−1: νCO 2014 (s), 1971 (s). Elemental Anal. Calcd (%) for C63H58BF24IrN2O2P2: C 47.41; H 3.66; N 1.76. Found: C 47.08; H 3.34; N 1.76.
C), 195.9 (t, 2JCP = 7.6 Hz, Ir–CO), 162.1 (q, 1JCB = 49.8 Hz, C–B, B(ArF)4, Ar) 155.7 (vt, JCP = 4.0 Hz, ArC), 151.2 (vt, JCP = 19.1 Hz, ArC), 150.0 (vt, JCP = 15.7 Hz, ArC), 135.2 (s, CH-ortho, B(ArF)4, Ar), 134.0 (vt, JCP = 6.6 Hz, ArCH), 129.3 (qq, 2JCF = 31.5 Hz, 4JCF = 2.7 Hz, C–CF3, B(ArF)4), 124.9 (q, 1JCF = 272.4 Hz, CF3, B(ArF)4), 117.9 (sept, 3JCF = 4.0, CH-para, B(ArF)4, Ar), 115.3 (s, ArCH), 115.2 (s, ArCH), 41.1 (s, –N(CH3)2), 26.4 (vt, JCP = 14.9 Hz, –CH(CH3)2), 19.5 (vt, JCP = 2.0 Hz, –CH(CH3)2), 19.0 (s, –CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2) δ 72.2 (s). 19F{1H} NMR (471 MHz, CD2Cl2) δ −63.84 (s). 11B{1H} NMR (161 MHz, CD2Cl2) δ −6.04 (s). IR (NaCl plates) cm−1: νCO 1963 (s). Elemental Anal. Calcd (%) for C62H58BF24IrN2OP2: C 47.49; H 3.73; N 1.79. Found: C 47.44; H 3.94; N 1.95.
Ir), 175.6 (vt, JCP = 20.8 Hz, ArC), 162.1 (q, 1JCB = 49.7 Hz, C–B B(ArF)4, Ar), 147.5 (vt, JCP = 5.0 Hz, ArC), 138.8 (vt, JCP = 3.4 Hz, ArC), 138.3 (vt, JCP = 23.9 Hz, ArC), 135.2 (s, CH-ortho, B(ArF)4, Ar) 129.2 (qq, 2JCF = 31.5 Hz, 4JCF = 3.0 Hz, C–CF3 B(ArF)4), 128.1 (s, ArCH), 127.9 (s, ArCH), 126.1 (s, ArCH), 125.0 (q, 1JCF = 272.3 Hz, CF3 B(ArF)4), 123.5 (s, ArCH), 117.9 (sept, 3JCF = 4.0 Hz, CH-para, B(ArF)4, Ar), 27.9 (vt, JCP = 15.4 Hz, –CH(CH3)2), 20.3 (s, –CH(CH3)2), 18.5 (s, –CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2) δ 47.1 (s). 19F NMR (471 MHz, CD2Cl2) δ −63.8 (s). 11B{1H} NMR (161 MHz, CD2Cl2) δ −6.0 (s). IR (NaCl) cm−1: 2014 (s), 2055 (s). Elemental Anal. Calcd (%) for C63H48BF24IrO2P2S2: C 46.65; H 2.98. Found: C 46.52; H 2.99.
Ir), 188.6 (t, 2JCP = 7.8 Hz, Ir–CO), 173.1 (vt, JCP = 19.2 Hz, ArC), 162.1 (q, 1JCB = 49.7 Hz, C–B B(ArF)4, Ar), 161.4 (vt, JCP = 16.4 Hz, ArC), 153.9 (vt, JCP = 4.2 Hz, ArC), 139.4 (vt, JCP = 1.7 Hz, ArC), 135.2 (s, CH-ortho B(ArF)4), 132.8 (s, ArCH), 129.2 (qq, 2JCF = 31.5 Hz, 4JCF = 3.0 Hz, C–CF3 B(ArF)4), 129.2 (s, ArCH), 127.2 (s, ArCH), 127.0 (s, ArCH), 125.0 (q, 1JCF = 272.3 Hz, CF3 B(ArF)4), 117.9 (sept, 3JCF = 4.0 Hz, CH-para, B(ArF)4, Ar), 27.4 (vt, JCP = 15.4 Hz, –CH(CH3)2), 20.8 (vt, JCP = 2.7 Hz, –CH(CH3)2), 20.7 (s, –CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2) δ 59.6 (s). 19F{1H} NMR (471 MHz, CD2Cl2) δ −63.8 (s). 11B{1H} NMR (161 MHz, CD2Cl2) δ −6.0 (s). IR (NaCl) cm−1: νCO 2048 (s). HRMS (APCI) for C30H36IrOP2S2; calculated (M+): 731.1312 (100%); found (M+): 731.1331 (100%).
C) 178.2 (t, 2JCP = 5.7 Hz, Ir–CO), 162.3 (q, 1JCB = 49.8 Hz, C–B, B(ArF)4, Ar), 156.2 (vt, 3JCP = 5.1 Hz, C–N(CH2)5, ArC), 150.8 (vt, JCP = 9.5 Hz, C–O, ArC), 143.9 (vt, JCP = 23.5 Hz, C–P(iPr)2, ArC), 136.4 (vt, JCP = 18.3 Hz, ArC), 135.3 (s, CH-ortho, B(ArF)4, Ar), 129.4 (qq, 2JCF = 31.4 Hz, 4JCF = 2.7 Hz, C–CF3, B(ArF)4), 125.1 (q, 1JCF = 272.5 Hz, CF3, B(ArF)4), 118.0 (sept, 3JCF = 3.8 Hz, CH-para, B(ArF)4, Ar), 116.2 (s, ArCH), 101.8 (s, ArCH), 49.5 (s, –N(CH2)5), 27.9 (vt, JCP = 15.6 Hz, –CH(CH3)2), 25.7 (s, –N(CH2)5), 24.5 (s, –N(CH2)5), 18.9 (s, –CH(CH3)2), 17.6 (s, –CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2) δ 65.8 (s). 19F{1H} NMR (376 MHz, CD2Cl2) δ −62.9 (s). 11B{1H} NMR (128 MHz, CD2Cl2) δ −6.57 (s). IR (NaCl plates) cm−1: νCO 2014 (s), 1973 (s). Repeated attempts to obtain satisfactory elemental analyses met with failure for this sample.
C), 196.4 (t, 2JCP = 6.6 Hz, Ir–CO), 162.2 (q, 1JCB = 49.7 Hz, C–B, B(ArF)4, Ar), 157.8 (vt, JCP = 4.5 Hz, C–N(CH2)5, ArC), 154.5 (vt, JCP = 8.9 Hz, C–O, ArC), 149.5 (vt, JCP = 19.7 Hz, C–P(iPr)2, ArC), 136.9 (vt, JCP = 19.4 Hz, ArC), 135.3 (s, CH-ortho, B(ArF)4, Ar), 129.4 (qq, 2JCF = 31.5 Hz, 4JCF = 2.6 Hz, C–CF3, B(ArF)4), 125.1 (q, 1JCF = 273.0 Hz, CF3, B(ArF)4), 117.9 (sept, 3JCF = 4.1 Hz, CH-para, B(ArF)4, Ar), 116.2 (s, ArCH), 100.0 (s, ArCH), 49.6 (s, –N(CH2)5), 27.7 (vt, JCP = 14.4 Hz, –CH(CH3)2), 25.9 (s, –N(CH2)5), 24.4 (s, –N(CH2)5), 19.9 (vt, 2J = 2.4 Hz, –CH(CH3)2), 19.4 (s, –CH(CH3)2). 31P{1H} NMR (203 MHz, CD2Cl2) δ 78.6 (s). 19F{1H} NMR (376 MHz, CD2Cl2) δ −62.9 (s). 11B{1H} NMR (128 MHz, CD2Cl2) δ −6.58 (s). IR (NaCl plates) cm−1: νCO 1974 (s). Elemental Anal. Calcd (%) for C68H64BF24IrN2O2P2: C 49.14; H 3.88; N 1.69. Found: C 49.04; H 4.19; N 1.61.
Acknowledgements
Funding for this work was provided by NSERC of Canada in the form of a Discovery Grant and an Accelerator Supplement to W. E. P. W. E. P. also thanks the Canada Research Chair secretariat for a Tier I CRC (2013–2020) and the Alexander von Humboldt Stiflung for a Research Award. L. E. D. thanks NSERC for a CGSD scholarship and Alberta Innovates Technology Futures for a Graduate Student Scholarship. Y. Y. acknowledges a Grant-in-Aid for Scientific Research on Innovative Areas “Stimuli-responsive Chemical Species for the Creation of Functional Molecules (No. 2408)” (JSPS KAKENHI Grant Number JP24109002).Notes and references
- G. van Koten and D. Milstein, Topics in Organometallic Chemistry: Organometallic Pincer Chemistry, Springer, Heidelberg, 2013, vol. 40 Search PubMed.
- D. Hermann, M. Gandelman, H. Rozenberg, L. J. W. Shimon and D. Milstein, Organometallics, 2002, 21, 812–818 CrossRef CAS.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed.
- W. Leis, H. A. Mayer and W. C. Kaska, Coord. Chem. Rev., 2008, 252, 1787–1797 CrossRef CAS.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- D. Gelman and R. Romm, PC(sp 3)P Transition Metal Pincer Complexes: Properties and Catalytic Applications, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Berlin, Heidelberg, 2013, vol. 40, pp. 289–317 Search PubMed.
- C. Crocker, H. D. Empsall, R. J. Errington, E. M. Hyde, W. S. McDonald, R. Markham, M. C. Norton, B. L. Shaw and B. Weeks, J. Chem. Soc., Dalton Trans., 1982, 1217–1224 RSC.
- H. M. Lee, J. Y. Zeng, C.-H. Hu and M.-T. Lee, Inorg. Chem., 2004, 43, 6822–6829 CrossRef CAS PubMed.
- W. Weng, S. Parkin and O. V. Ozerov, Organometallics, 2006, 25, 5345–5354 CrossRef CAS.
- G. Zhu, X. Li, G. Xu, L. Wang and H. Sun, Dalton Trans., 2014, 43, 8595–8598 RSC.
- W. Lesueur, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1997, 36, 3354–3362 CrossRef CAS PubMed.
- R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949–2952 CrossRef CAS.
- J. Arras, H. Speth, H. A. Mayer and L. Wesemann, Organometallics, 2015, 34, 3629–3636 CrossRef CAS.
- J. R. Logan, W. E. Piers, J. Borau-Garcia and D. M. Spasyuk, Organometallics, 2016, 35, 1279–1286 CrossRef CAS.
- D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed.
- C. C. Comanescu, M. Vyushkova and V. M. Iluc, Chem. Sci., 2015, 6, 4570–4579 RSC.
- P. Cui and V. M. Iluc, Chem. Sci., 2015, 6, 7343–7354 RSC.
- R. J. Burford, W. E. Piers and M. Parvez, Eur. J. Inorg. Chem., 2013, 3826–3830 CrossRef CAS.
- R. J. Burford, W. E. Piers, D. H. Ess and M. Parvez, J. Am. Chem. Soc., 2014, 136, 3256–3263 CrossRef CAS PubMed.
- J. Borau-Garcia, D. V. Gutsulyak, R. J. Burford and W. E. Piers, Dalton Trans., 2015, 44, 12082–12085 RSC.
- L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187–2190 CrossRef CAS PubMed.
- L. E. Doyle, W. E. Piers, J. Borau-Garcia, M. J. Sgro and D. M. Spasyuk, Chem. Sci., 2016, 7, 921–931 RSC.
- C. C. Comanescu and V. M. Iluc, Organometallics, 2014, 33, 6059–6064 CrossRef CAS.
- C. C. Comanescu and V. M. Iluc, Inorg. Chem., 2014, 53, 8517–8528 CrossRef CAS PubMed.
- C. C. Comanescu and V. M. Iluc, Organometallics, 2015, 34, 4684–4692 CrossRef CAS.
- P. Cui, C. C. Comanescu and V. M. Iluc, Chem. Commun., 2015, 51, 6206–6209 RSC.
- J. Weismann and V. H. Gessner, Chem. Commun., 2015, 51, 14909–14912 RSC.
- K.-S. Feichtner, S. Englert and V. H. Gessner, Chem. – Eur. J., 2016, 22, 506–510 CrossRef CAS PubMed.
- J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed.
- D. Y. Wang, Y. Choliy, M. C. Haibach, J. F. Hartwig, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2016, 138, 149–163 CrossRef CAS PubMed.
- J. D. Smith, J. Borau-Garcia, W. E. Piers and D. Spasyuk, Can. J. Chem., 2016, 94, 293–296 CrossRef CAS.
- S. Sugawara, M. Abe, Y. Fujiwara, M. Wakioka, F. Ozawa and Y. Yamamoto, Eur. J. Inorg. Chem., 2015, 2015, 534–541 CrossRef CAS.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed.
- V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS PubMed.
- H. Werner, Angew. Chem., Int. Ed., 2010, 49, 4714–4728 CrossRef CAS PubMed.
- S. M. Kloek, D. M. Heinekey and K. I. Goldberg, Organometallics, 2006, 25, 3007–3011 CrossRef CAS.
- Y. Klerman, E. Ben-Ari, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Dalton Trans., 2008, 3226–3234 RSC.
- E. Ben-Ari, R. Cohen, M. Gandelman, L. J. W. Shimon, J. M. L. Martin and D. Milstein, Organometallics, 2006, 25, 3190–3210 CrossRef CAS.
- J. Campos, S. Kundu, D. R. Pahls, M. Brookhart, E. Carmona and T. R. Cundari, J. Am. Chem. Soc., 2013, 135, 1217–1220 CrossRef CAS PubMed.
- T. Schaub, U. Radius, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2008, 27, 1892–1901 CrossRef CAS.
- J. Díez, M. P. Gamasa, J. Gimeno and P. Paredes, Organometallics, 2005, 24, 1799–1802 CrossRef.
- D. Cuervo, J. Díez, M. P. Gamasa, J. Gimeno and P. Paredes, Eur. J. Inorg. Chem., 2006, 599–608 CrossRef CAS.
- D. A. Valyaev, O. A. Filippov, N. Lugan, G. Lavigne and N. A. Ustynyuk, Angew. Chem., Int. Ed., 2015, 54, 6315–6319 CrossRef CAS PubMed.
- P. D. Newman, K. J. Cavell and B. M. Kariuki, Dalton Trans., 2012, 41, 12395–12407 RSC.
- R. J. Goodfellow, Group VIII Transition Metals, in Multinuclear NMR, ed. J. Mason, Springer, US, 1987, pp. 521–561 Search PubMed.
- G. Glockler, J. Phys. Chem., 1958, 62, 1049–1054 CrossRef CAS.
- Z. Zhang, Y. Zhang and J. Wang, ACS Catal., 2011, 1, 1621–1630 CrossRef CAS.
- W. A. Herrmann and J. Plank, Angew. Chem., Int. Ed. Engl., 1978, 17, 525–526 CrossRef.
- A. Miyashita, H. Shitara and H. Nohira, Organometallics, 1985, 4, 1463–1464 CrossRef CAS.
- D. B. Grotjahn, G. A. Bikzhanova, L. S. B. Collins, T. Concolino, K.-C. Lam and A. L. Rheingold, J. Am. Chem. Soc., 2000, 122, 5222–5223 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. J. Deeming and B. L. Shaw, J. Chem. Soc. A, 1968, 1887–1889 RSC.
- M. Rahim and K. J. Ahmed, Inorg. Chem., 1994, 33, 3003–3004 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and spectroscopic details and crystallographic data for 1-CO, 3-CO, 4-(CO)2, 5-Cl, 6-Cl, and 6-CO. CCDC 1485351–1485357. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02615j |
| This journal is © The Royal Society of Chemistry 2016 |