
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complexes trans-Pt(BODIPY)X(PEt3)2: excitation energy-dependent fluorescence and phosphorescence emissions, oxygen sensing and photocatalysis†
Peter
Irmler
and
Rainer F.
Winter
*
Fachbereich Chemie der Universität Konstanz, Universitätsstraße 10, D-78464 Konstanz, Germany. E-mail: rainer.winter@uni-konstanz.de
First published on 3rd June 2016
Abstract
We report on five new complexes with the general formula trans-Pt(BODIPY)X(PEt3)2 (Pt–X), where the platinum(II) ion is σ-bonded to a 4,4-difluoro-4-bora-3a,4a-diaza-s-indacen-8-yl (BODIPY) and an anionic ligand X− (X− = Cl−, I−, NO2−, NCS−, CH3−). All five complexes were characterized by multinuclear NMR, electronic absorption and luminescence spectroscopy and by X-ray diffraction analysis. Four of these complexes show efficient intersystem crossing (ISC) from an excited singlet state to a BODIPY-centred T1 state and exhibit dual fluorescence and phosphorescence emission from the BODIPY ligand. In Pt–I, the fluorescence is almost completely quenched, whereas the phosphorescence quantum yield reaches a value of 40%. The rate of ISC and the ratio of phosphorescence to fluorescence emissions depend on the excitation wavelength (i.e. on which specific transition is excited). The performance of these complexes as one-component oxygen sensors and their photocatalytic activities were tested by Stern–Volmer quenching experiments and by monitoring the oxidation of 1,5-dihydroxynaphthalene with 1O2 generated from the long-lived triplet state of the sensitizer by triplet–triplet annihilation with 3O2. Exceptionally high 1O2 generation quantum yields of up to near unity were obtained.
Introduction
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) dyes have been known for three decades for their excellent performances as fluorophores, their versatility and their chemical and photochemical robustness.1–10 A particularly favourable asset of the BODIPY family of dyes is their modular construction from readily available building blocks, thus allowing for easy implementation of desirable properties or functionalities, e.g. for substrate binding, with important implications in the analytical sciences, or fine-tuning of the absorption and emission wavelengths.3,11,12 Phosphorescence from BODIPY dyes has, however, only rarely been observed and usually relies on the heavy atom effect of bromine or iodine substituents.13–16 In particular, there are only a handful of phosphorescent metal–organic BODIPY derivatives, and until very recently, the phosphorescence quantum yields of such compounds did not exceed the rather modest value of 3.5%.17 In these complexes, the BODIPY dye(s) are either appended to a 2,2′-bipyridine ligand as in Ru–BDP or Ir–BDP or bonded to Pt(N^C^N) entities with cyclometalating bis(benzimidazol-2-yl)phenyl-derived ligands as in Pt2–BDP, Fig. 1.3,14,17–19 Much higher quantum yields of up to 31% for the PEt3 derivative Pt–Br (Fig. 1) were achieved in complexes trans-Pt(BODIPY)Br(PR3)2 (R = Ph, Et) featuring a σ-bonded 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenyl dye, which connects to the platinum(II) ion via its meso position.20 In the latter complexes, the Pt coordination centre acts as a remote heavy metal ion, as the HOMO and the LUMO are heavily biased to the BODIPY ligand and receive only very minor contributions from the coordination centre. As a consequence, the relevant excitation is adequately described as a BODIPY-based π → π* transition with essentially no charge-transfer contributions from the {PtBr(PR3)2} fragment. Long-lived excited triplet states of BODIPY dyes are of great interest for applications such as chemical sensing,16,21,22 triplet–triplet annhilation-based upconversion17–19,23 and photodynamic therapy.13,15,24,25 For photodynamic therapy the ability of triplet emitters to transform triplet oxygen (3O2) to singlet oxygen (1O2) in a triplet–triplet annihilation process is of pivotal relevance. Its cell toxicity makes the highly reactive 1O2 molecule a powerful weapon against cancer cells.26–29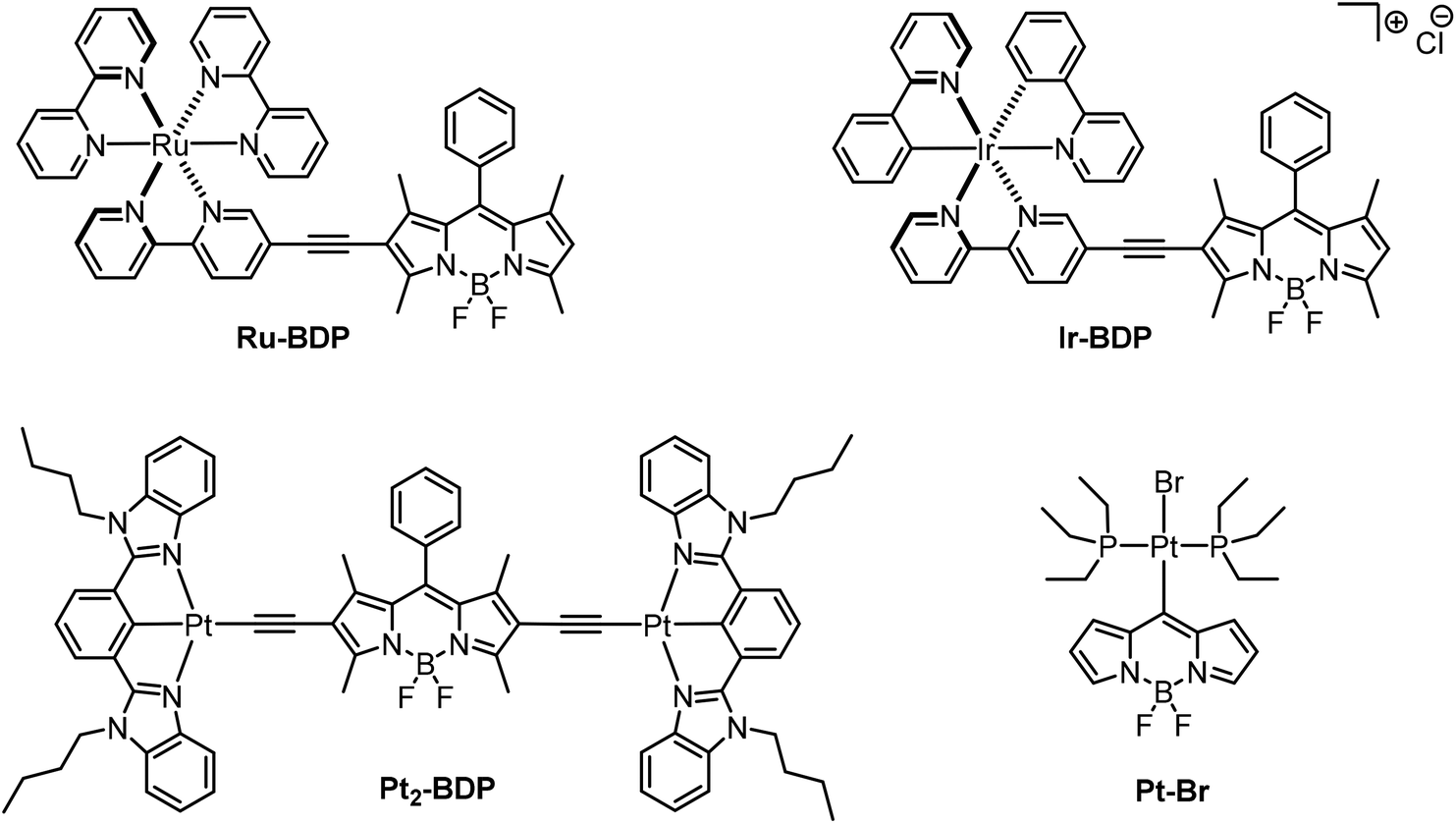 | ||
| Fig. 1 Molecular structures of room-temperature phosphorescent BODIPY complexes Ru–BDP, Ir–BDP, Pt2–BDP and Pt–Br. | ||
The different structural and electronic influences of a transition metal coligand entity on the photophysical properties of complexes, particularly the phosphorescence quantum yield ΦPh, are not trivial, though. Decisive factors are the rate constant of the intersystem crossing (kISC), the ratio of the radiative and non-radiative decay rates, and the thermal accessibility of excited d-states, which typically provide non-radiative deactivation pathways.30 In the case of square-planar Pt(II) complexes, the relative positioning of the dz2 orbital with respect to the emissive T1 state is often of crucial relevance.31 This energy separation largely depends on the ligand-field splitting. Thus, by introducing strong-field ligands, the dz2 orbital can be pushed to higher energy, increasing the energy barrier for non-radiative decay via excited d-states.30–33 In complexes of the type trans-Pt(Dye)X(PR3)2 (X− = Br−, Cl−, I− or CN−), where Dye represents a σ-bonded thioxanthonyl or a BODIPY attached via its meso position, the ligand-field splitting can be modulated by the PR3 ligand and the anionic ligand X−.34 Our previous study has already shown that PEt3 ligands endow the BODIPY complexes with superior photophysical properties when compared to their PPh3 counterparts.20 Here we report our results on five new BODIPY complexes trans-Pt(BODIPY)X(PEt3)2 with anionic ligands X− that cover a wider range of the spectrochemical series and differ with respect to their trans-influence35 and the results of our investigations into the performance of some representatives as one-component triplet sensors and sensitizers for the photocatalytic oxidation of 1,5-dihydroxynaphthalene (DHN) with molecular oxygen.
Results and discussion
Synthesis and NMR spectroscopy
All complexes were synthesized starting from cis-Pt(η2-C2H4)(PEt3)2, which is obtained by heating cis-Pt(Et)2(PEt3)2 in C6D6 for 45 min to 114 °C.36 Oxidative addition of 8-bromo-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (Br–BODIPY, see Fig. 2) to the reactive Pt0-species is fast at room temperature (r.t.). The resulting complex cis-Pt(BODIPY)Br(PEt3)2 is then transformed by AgOTf to trans-Pt(BODIPY)(OTf)(PEt3)2. Subsequent treatment with NaX (X− = Cl−, NO2−, NCS−) resulted in the replacement of the weakly coordinated OTf− by the respective counter ion and provided complexes Pt–Cl, Pt–NO2 and Pt–NCS (see Fig. 2) in moderate to good yields. Our attempts to introduce a methyl ligand by transmetalation using the Grignard reagent MeMgI failed and the complex trans-Pt(BODIPY)I(PEt3)2 (Pt–I) was formed instead. The use of MgMe2 as a transmetalating agent was likewise unsuccessful. Reaction of Pt–OTf with MeLi finally afforded Pt–CH3 (Fig. 2).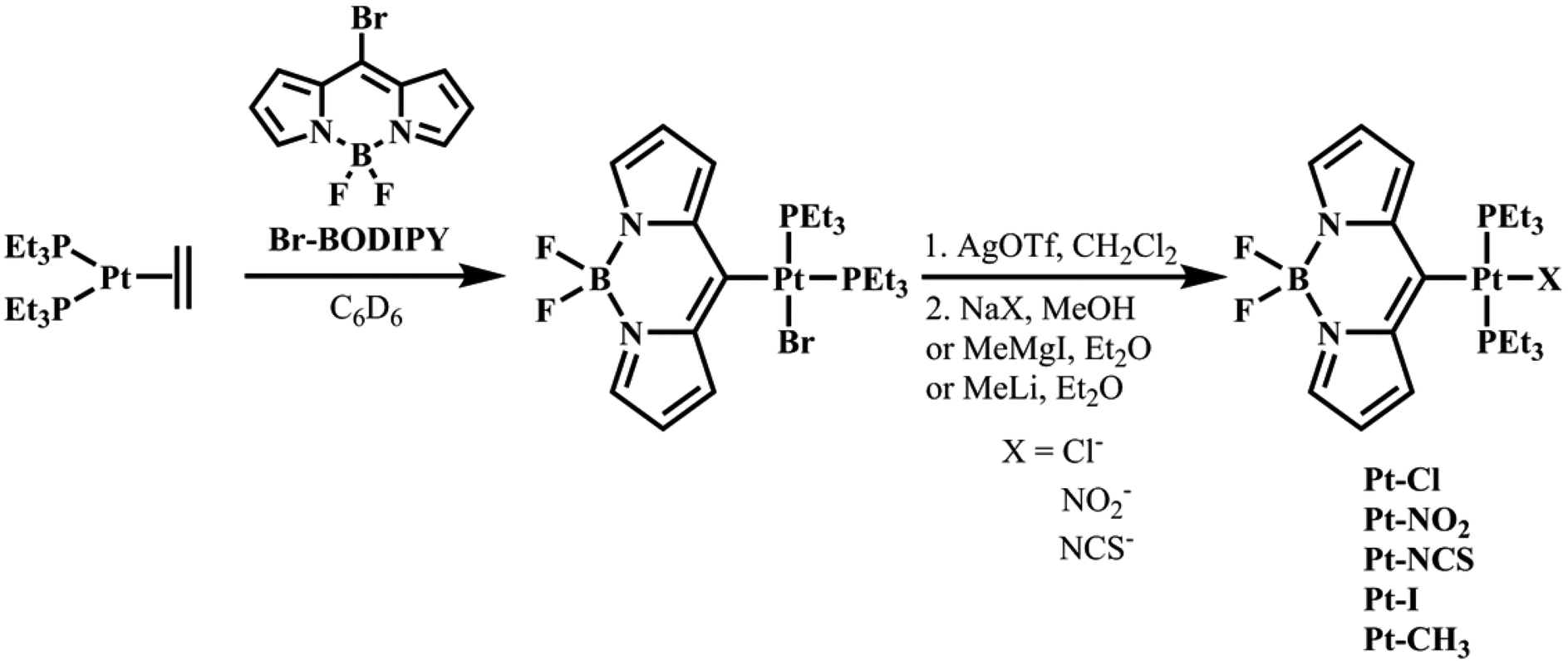 | ||
| Fig. 2 Synthesis of the complexes Pt–X. | ||
195Pt NMR spectra of the trans-complexes show a triplet with a coupling constant JPtP in the range of 2692 Hz to 2450 Hz. Correspondingly, the 31P NMR spectra give a singlet for the two trans-disposed P donors, which is flanked by the 195Pt satellite doublet with the same JPtP coupling constant. The formation of a direct Pt–C σ-bond is confirmed by the observation of platinum satellites in the 13C NMR spectra, which range from 492 Hz to 409 Hz for JPtC and from 25 Hz to 17 Hz for 2JPtC and 3JPtC couplings, respectively. Some couplings could, however, not be detected due to a low signal-to-noise ratio. The NMR spectra can be found in the ESI, Fig. S1–S23.†
Single crystal X-ray diffraction
Single crystals suitable for X-ray diffraction analysis were obtained for all five Pt complexes. Fig. 3 displays the ORTEP representations of their molecular structures. Relevant bond lengths and angles can be taken from Table 1, and Table S1 of the ESI† summarizes the crystal and refinement data. Pt–Cl, Pt–NCS, Pt–NO2 and Pt–CH3 crystallize in the monoclinic space groups P21/c, P21/n, P21/c, or P21, respectively. The single crystal of Pt–I complies with the symmetry operations of the Cmc21 space group in the orthorhombic crystal system. The unit cells of Pt–Cl and Pt–CH3 contain two independent molecules with different sets of bond lengths and angles as given in Table 1.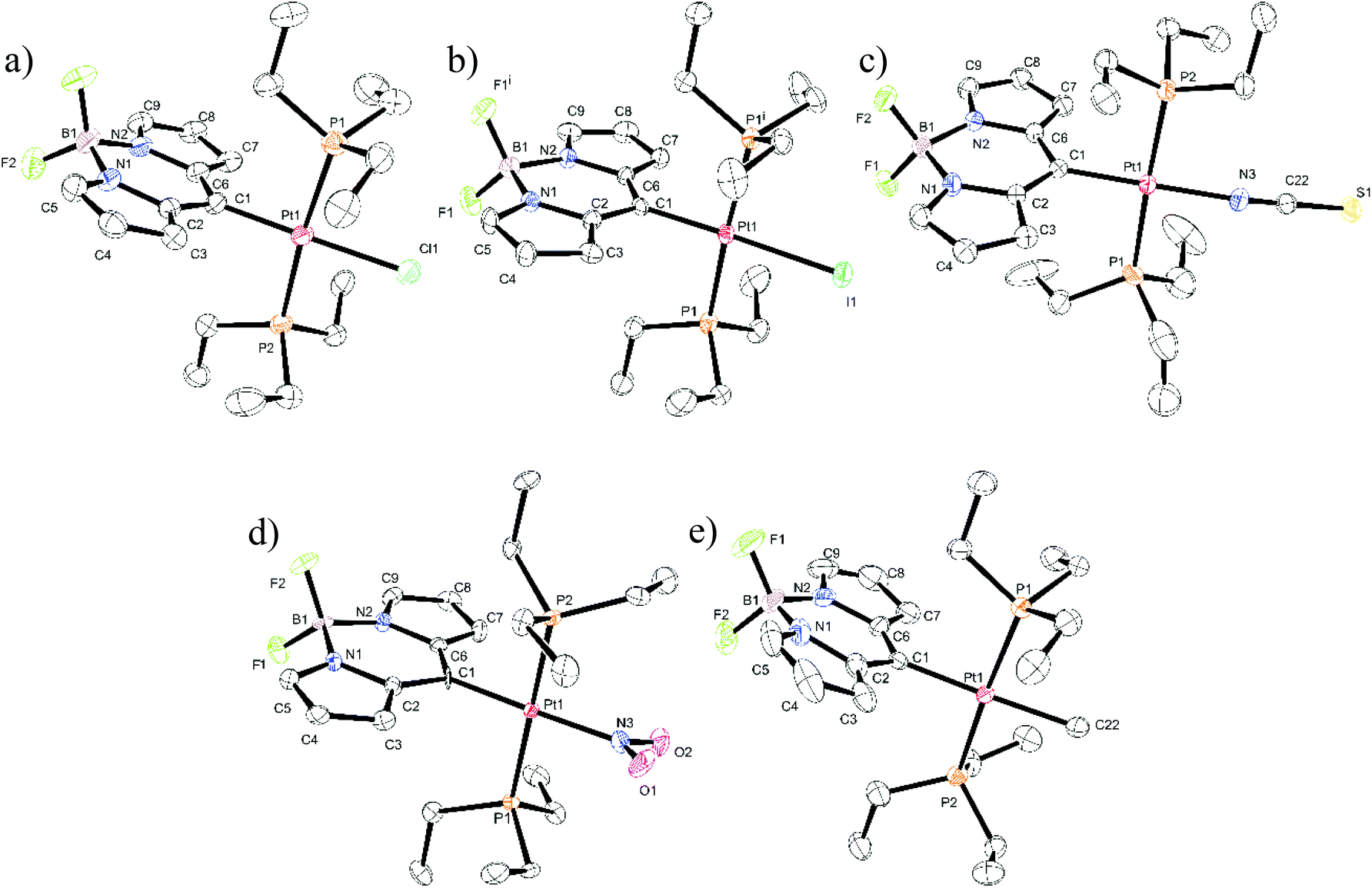 | ||
| Fig. 3 ORTEP representations of the molecular structures of (a) Pt–Cl, (b) Pt–I, (c) Pt–NCS, (d) Pt–NO2, and (e) Pt–CH3. For Pt–Cl and Pt–CH3 only one of the independent molecules per unit cell are shown. The ellipsoids are drawn at a 40% probability level. Hydrogen atoms are omitted for reasons of clarity. Atom C1 of Pt–NO2 remained isotropic and could not be refined further. | ||
| Pt–Br | Pt–Cl | Pt–I | Pt–NCS | Pt–NO2 | Pt–CH3 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Molecule 1 | Molecule 2 | Molecule 1 | Molecule 2 | Molecule 1 | Molecule 2 | ||||
| a X represents the donor atom of the anionic ligand in trans-position to the dye at the Pt ion. b The molecule has a mirror plane which is defined by the plane of the dye's inner heterocycle. c Atom C1 could not be refined anisotropically. | |||||||||
| Bond lengths/Å | |||||||||
| C1—Pt1 | 1.984(10) | 1.964(11) | 1.976(7) | 1.971(8) | 1.994(10) | 1.984(4) | 1.956(9)c | 2.039(11) | 2.053(12) |
| X—Pt1a | 2.4973(12) | 2.5118(12) | 2.3750(19) | 2.385(2) | 2.6689(8) | 2.048(4) | 2.019(8) | 2.127(12) | 2.137(11) |
| P1—Pt1 | 2.315(3) | 2.320(3) | 2.319(2) | 2.316(2) | 2.3206(15) | 2.3135(13) | 2.329(2) | 2.299(3) | 2.304(3) |
| P2—Pt1 | 2.327(3) | 2.324(3) | 2.309(2) | 2.310(2) | 2.3206(15)b | 2.3249(11) | 2.334(2) | 2.290(3) | 2.285(3) |
| Bond angles/° | |||||||||
| C1–Pt1–P1 | 93.7(3) | 92.9(3) | 91.2(2) | 91.6(2) | 91.37(7) | 91.44(12) | 92.3(3) c | 91.3(3) | 90.7(3) |
| C1–Pt1–P2 | 91.3(3) | 90.5(3) | 94.0(2) | 93.0(2) | 91.37(7)b | 90.79(12) | 89.3(3) c | 91.9(3) | 92.7(3) |
| P1–Pt1–Xa | 87.73(8) | 88.41(8) | 87.13(7) | 87.68(8) | 88.71(7) | 87.28(12) | 88.6(2) | 86.3(3) | 86.7(3) |
| P2–Pt1–Xa | 87.28(8) | 88.16(8) | 87.61(7) | 87.76(8) | 88.71(7)b | 90.62(11) | 90.0(2) | 90.5(3) | 89.9(3) |
| P1–Pt1–P2 | 175.01(11) | 176.02(11) | 174.74(8) | 175.00(7) | 176.8(3) | 177.17(5) | 173.83(8) | 176.05(12) | 176.57(12) |
| C1–Pt1–Xa | 178.6(3) | 178.6(3) | 178.3(2) | 179.0(2) | 176.03(8) | 175.77(17) | 178.2(4)c | 177.6(4) | 177.2(5) |
In the present series of complexes the length of the C1–Pt σ-bond provides a measure for the trans-influence and consequently for the σ-donor strength of the anionic ligand X−,35 which increases in the order Pt–NO2 < Pt–Cl ≈ Pt–Br < Pt–I ≈ Pt–NCS < Pt–CH3. This ordering complies with that of a related series of platinum complexes with a σ-bonded perylene or perylene monoimide dye.37 For Pt–CH3 the difference between the Pt–C bond lengths to the methyl (2.127(12) or 2.137(11) Å) and the BODIPY ligands (2.039(11) or 2.053(12) Å for the two independent molecules of the unit cell) reflects the difference of the covalent radii of a sp3 and a sp2 carbon atom. Similar differences have e.g. been observed for trans-Pt(CH3)(Ph)(PPh3)2d(Pt−CH3) = 2.226(4) Å, d(Pt−Ph = 2.058(4) Å).38 The Pt–Me bond of Pt–CH3 is expectedly longer than in complexes trans-Pt(CH3)Cl(PR3)2 owing to the opposite placement of two σ-carbyl ligands, which both exert a strong σ-trans-influence (cf. 2.08(1) Å for R = Ph or 2.069(8) Å for R = C6H4F-4).39,40
With deviations of 2.8° to 6.2° for the angle P1–Pt–P2 and 1.0° to 4.2° for bond angle C1–Pt–X (X = donor atom of the anionic ligand) and a maximum deviation of 4.0° for cis-angles X–Pt–P and C1–Pt–P from the ideal values and a coplanarity of all donor atoms with the Pt(II) ion the coordination centre exhibits a close to ideal square planar coordination geometry. This is also indicated by the summations of bond angles at the Pt(II) ion, which range from 359.94° to 360.20°. The P1–Pt–P2 angle opens to the side of the sterically demanding BODIPY ligand.
The various steric and electronic influences of a PtL3 fragment for tipping the scale towards either κN or κS coordination of a thiocyanate ligand are textbook examples for the phenomenon of coordination isomerism.41,42N coordination in spite of the soft character of the {Pt(BODIPY)(PEt3)2} fragment is here favoured by the strong trans-influence of the opposite carbyl ligand, the light donor atom, and by steric effects. Thus, N coordination maintains a near coincidence of the NCS− axis with the C1–Pt–N vector Pt–N3–C22 = 162.2(4)°, S1–C22–N3 = 179.7(4)°, thus avoiding unfavourable steric interactions with the cis-disposed PEt3 ligands (Fig. 3c). N coordination of the NCS− ligand has likewise been observed in the related perylene complex of Espinet and coworkers.37
Packing diagrams of individual molecules in the crystal lattice are shown in Fig. S24–S28 of the ESI.† All structures exhibit several short intermolecular contacts. Most prevalent are hydrogen bonding interactions H⋯F–B between pyrrolic or methyl protons and the BF2− fluorine atoms. These latter contacts are in the range of 2.330 to 2.539 Å, which is by 0.330 to 0.131 Å shorter than the sum of the van der Waals radii. Most notably, H⋯F contacts to methyl hydrogens of the PEt3 ligands are frequently shorter than those to the hydrogen atoms attached to the heterocycles. These hydrogen bonds are sometimes augmented by C–H⋯π interactions between methyl protons and a pyrrolic carbon atom ranging from 2.634 to 2.757 Å. In several cases, additional contacts exist between pyrrolic or methyl protons and heteroatoms of the anionic ligand X−, most importantly to the oxygen atoms of the nitrite ligand of Pt–NO2 (2.378 to 2.487 Å with the shorter contacts again to PEt3 methyl protons), the S atom of the κN-thiocyanate ligand in Pt–NCS (2.842 and 2.921 Å) or, very weakly, to the I− ligand in Pt–I (3.127 Å). The latter complex exhibits an interesting brick-wall packing in the ac plane, where individual molecules associate weakly along the c axis via C–H⋯I interactions and, more strongly so, along the a axis by CH⋯π interactions between the pyrrolic carbon atom C9 and a PEt3 methyl proton of neighbouring molecules positioned above and below (C–H⋯C = 2.684 Å, see Fig. S25b of the ESI†). The structural relevance of CH⋯π interactions has recently been highlighted.43
UV-vis spectroscopy, TD-DFT calculations and luminescence properties
The UV-Vis absorption spectra of complexes Pt–Cl to Pt–CH3 are shown in Fig. 4. They are dominated by the sharp, vibrationally structured band of the attached BODIPY dye with extinction coefficients ε of 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 to 57400 M−1 cm−1. Peaking at a narrow range of 461 to 472 nm (Table 2), the position is almost invariant to the identity of the ligand X−. At higher energies in the near UV another weaker, asymmetric absorption is observed at λ = 370 to 300 nm with a maximum extinction coefficient of ca. 11000 M−1 cm−1. In some cases that feature is resolved into two distinct bands which are separated by 20 to 30 nm. Time-dependent DFT (TD-DFT) calculations carried out on geometry optimized structures accordingly predict two separate absorptions in this energy range. The comparison of experimental and calculated TD-DFT data in Table 2 shows that our calculations reproduce the general absorption features well but overestimate the energy of the prominent BODIPY-based π → π* transition by ca. 4200 cm−1. The TD-DFT data reveal that the intense band at the lowest energy arises from the HOMO → LUMO transition. As it is evident from the graphical depictions of the relevant orbitals of Pt–NO2 and Pt–I in Fig. 5 and the compilation in Tables 2 and 3, the latter is adequately described as a π → π* transition of the BODIPY ligand with only very small contributions of the {PtX(PEt3)2} fragment. This also explains the negligible influence of the X− ligand on the transition energies. The absorption near 320 nm originates from two energetically close-lying transitions (HOMO−5 → LUMO, HOMO−6 → LUMO for Pt–I, HOMO−6 → LUMO, HOMO−7 → LUMO/HOMO−8 → LUMO for Pt–NO2, Table 2). One has distinct Pt(PEt3)2 → BODIPY charge-transfer (CT) character, while the second one involves another π → π* transition within the dye ligand. As we will see later, the more significant metal contribution to the higher energy transition has important implications on the intersystem crossing rate constants kISC from the different excited states.
600 to 57400 M−1 cm−1. Peaking at a narrow range of 461 to 472 nm (Table 2), the position is almost invariant to the identity of the ligand X−. At higher energies in the near UV another weaker, asymmetric absorption is observed at λ = 370 to 300 nm with a maximum extinction coefficient of ca. 11000 M−1 cm−1. In some cases that feature is resolved into two distinct bands which are separated by 20 to 30 nm. Time-dependent DFT (TD-DFT) calculations carried out on geometry optimized structures accordingly predict two separate absorptions in this energy range. The comparison of experimental and calculated TD-DFT data in Table 2 shows that our calculations reproduce the general absorption features well but overestimate the energy of the prominent BODIPY-based π → π* transition by ca. 4200 cm−1. The TD-DFT data reveal that the intense band at the lowest energy arises from the HOMO → LUMO transition. As it is evident from the graphical depictions of the relevant orbitals of Pt–NO2 and Pt–I in Fig. 5 and the compilation in Tables 2 and 3, the latter is adequately described as a π → π* transition of the BODIPY ligand with only very small contributions of the {PtX(PEt3)2} fragment. This also explains the negligible influence of the X− ligand on the transition energies. The absorption near 320 nm originates from two energetically close-lying transitions (HOMO−5 → LUMO, HOMO−6 → LUMO for Pt–I, HOMO−6 → LUMO, HOMO−7 → LUMO/HOMO−8 → LUMO for Pt–NO2, Table 2). One has distinct Pt(PEt3)2 → BODIPY charge-transfer (CT) character, while the second one involves another π → π* transition within the dye ligand. As we will see later, the more significant metal contribution to the higher energy transition has important implications on the intersystem crossing rate constants kISC from the different excited states.
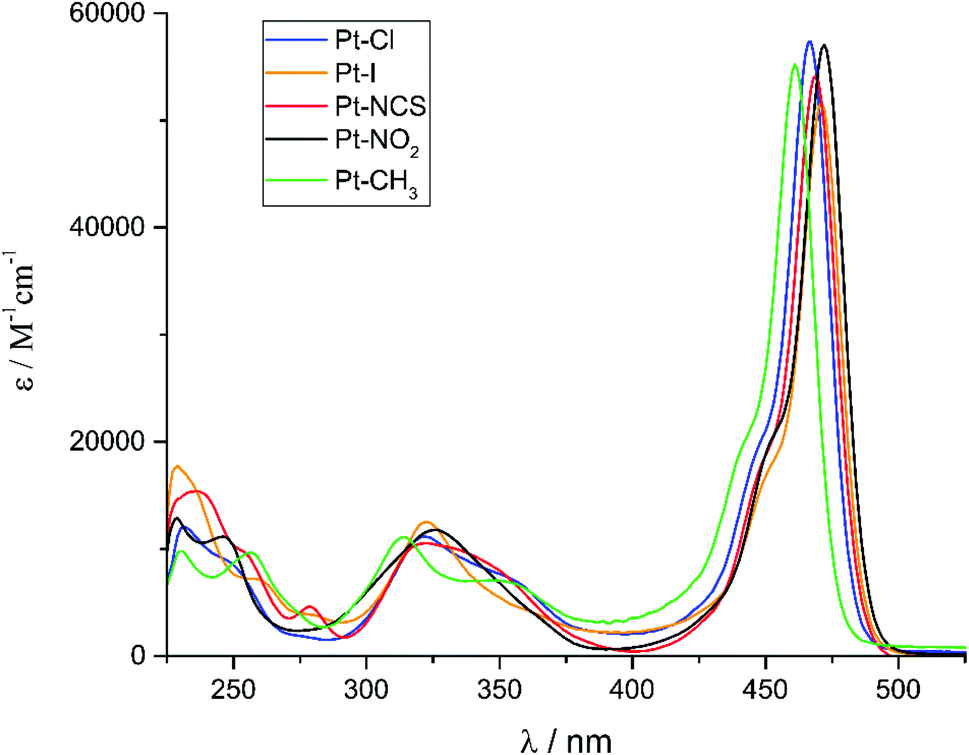 | ||
| Fig. 4 Electronic absorption spectra of Pt–Cl, Pt–I, Pt–NCS, Pt–NO2 and Pt–CH3 in a ca. 10−5 M CH2Cl2 solution at 298 K. | ||
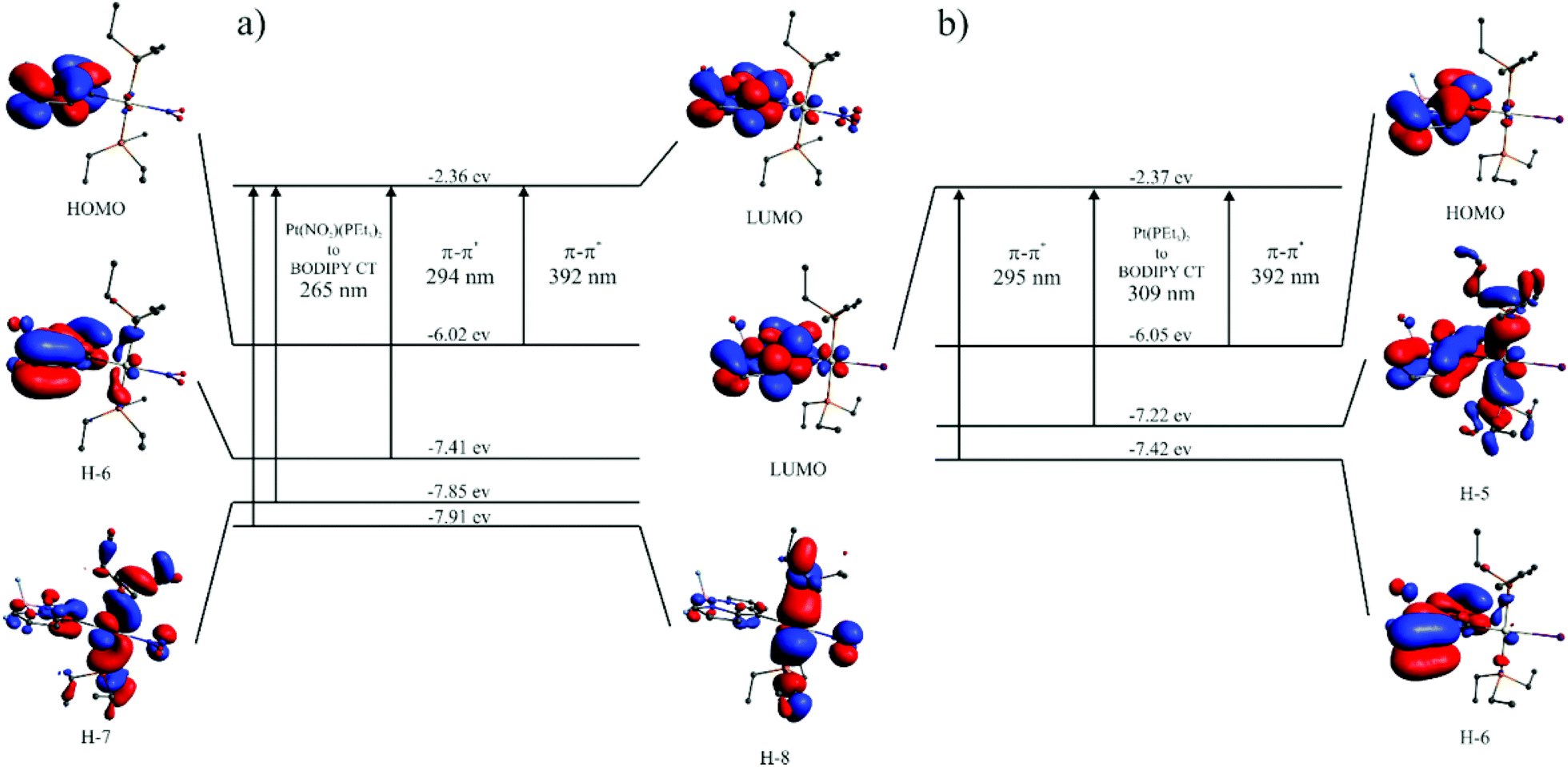 | ||
| Fig. 5 Energies and graphical representations of the relevant molecular orbitals along with calculated electronic transitions of (a) Pt–NO2 and (b) Pt–I. | ||
| Absorption data | TD-DFT data | ||||
|---|---|---|---|---|---|
| λ max [nm] (ε × 10−3 [M−1 cm−1]) | λ [nm] | Major contributions [%] | f | Assignment | |
| a Oscillator strength. b n.c. = not calculated. | |||||
| Pt–Cl | 321 (11.1), 340 (7.4), 467 (57.4) | 293 | H−6 → LUMO (93) | 0.16 | π → π* (BODIPY) |
| 310 | H−5 → LUMO (81) | 0.10 | Pt(PEt3)2 → BODIPY CT | ||
| 390 | HOMO → LUMO (97) | 0.39 | π → π* (BODIPY) | ||
| Pt–I | 322 (12.5), 352 (4.3) 471 (52.6) | 295 | H−6 → LUMO (90) | 0.17 | π → π* (BODIPY) |
| 309 | H−5 → LUMO (81) | 0.07 | Pt(PEt3)2 → BODIPY CT | ||
| 392 | HOMO → LUMO (97) | 0.39 | π → π* (BODIPY) | ||
| Pt–NCS | 320 (10.5), 337 (9.2), 469 (54.2) | 294 | H−6 → LUMO (93) | 0.18 | π → π* (BODIPY) |
| 310 | H−5 → LUMO (88) | 0.07 | Pt(PEt3)2 → BODIPY CT | ||
| 393 | HOMO → LUMO (95) | 0.37 | π → π* (BODIPY) | ||
| Pt–NO2 | 325 (11.7), 472 (57.0) | 265 | H−8 → LUMO (39) | 0.08 | Pt(NO2)(PEt3)2 → BODIPY CT |
| H−7 → LUMO (56) | Pt(NO2)(PEt3)2 → BODIPY CT | ||||
| 294 | H−6 → LUMO (90) | 0.17 | π → π* (BODIPY) | ||
| 392 | HOMO → LUMO (97) | 0.40 | π → π* (BODIPY) | ||
|
Pt–CH3b |
314 (11.1), 346 (7.1) 461 (55.2) | n.c. | |||
a
| Pt | BODIPY | PEt3 |
X−b |
||
|---|---|---|---|---|---|
| a Percent contributions of the given fragments. b X− represents the anionic ligand in trans-position to the dye at the Pt ion. c Spin density contribution of the respective fragment to the spin density surface. | |||||
| Pt–Cl | LUMO | 4 | 94 | 2 | 0 |
| HOMO | 1 | 98 | 1 | 0 | |
| H−5 | 16 | 43 | 40 | 1 | |
| H−6 | 2 | 95 | 3 | 0 | |
| Spin densityc | 0.012 | 1.975 | 0.017 | −0.004 | |
| Pt–I | LUMO | 3 | 94 | 2 | 0 |
| HOMO | 1 | 98 | 1 | 0 | |
| H−5 | 14 | 47 | 39 | 0 | |
| H−6 | 3 | 92 | 5 | 1 | |
| Spin densityc | 0.009 | 1.978 | 0.019 | −0.006 | |
| Pt–NCS | LUMO | 3 | 94 | 2 | 1 |
| HOMO | 1 | 98 | 1 | 0 | |
| H−5 | 18 | 41 | 41 | 0 | |
| H−6 | 2 | 93 | 5 | 0 | |
| Spin densityc | 0.003 | 1.978 | 0.020 | 0.000 | |
| Pt–NO2 | LUMO | 3 | 93 | 2 | 2 |
| HOMO | 1 | 98 | 1 | 0 | |
| H−6 | 3 | 90 | 7 | 0 | |
| H−7 | 40 | 16 | 37 | 7 | |
| H−8 | 50 | 6 | 31 | 13 | |
| Spin densityc | −0.003 | 1.982 | 0.020 | 0.001 | |
Table S15 of the ESI† compares the calculated structure parameters of complexes Pt–Cl, Pt–I, Pt–NCS, and Pt–NO2 to the experimental data from X-ray crystal diffraction and to those calculated for the T1 state. Calculated bond parameters for the S0 state retrace experimentally observed bond lengths and angles well. The only structural difference between the T1 and the S0 states is a slight elongation of the Pt–C1 bond by 2–3 pm while all other bond lengths and bond angles remain essentially unaffected.
Like the previously reported complex Pt–Br20 all complexes exhibit dual fluorescence at λ ≈ 480 nm and phosphorescence at λ ≈ 640 nm when excited into their lowest energy absorption band. Emission spectra of the complexes and of Br–BODIPY are compared in Fig. 6 while relevant photophysical data are collected in Table 4. The small Stokes shifts of <500 cm−1 and luminescence decay rates in the subnanosecond range are typical assets of BODIPY-based fluorescence emissions. The congruence of electronic absorption and excitation spectra as documented in Fig. S29–S33 of the ESI† and the blue shift of the fluorescence peaks compared to that of the Br–BODIPY precursor (λFl = 517 nm) demonstrate, that both emissions originate from the complexes and not from impurities or unreacted Br–BODIPY. That blue shift has been traced to a preferential lifting of the BODIPY LUMO owing to a slightly larger contribution of the strongly electron-donating {Pt(PEt3)2X} moiety44 to the receptor orbital.20 The long lifetimes of 162 to 439 μs at r.t. and the large Stokes shifts of ca. 5600 cm−1 characterize the low-energy emission band as phosphorescence (Table 4). From the comparison of emission spectra in Fig. 6 and the data in Table 4 it becomes immediately apparent that larger phosphorescence quantum yields ΦPh go along with a decrease of those of the fluorescence emission ΦFl and vice versa. No luminescence data could be obtained for Pt–CH3, as this complex decomposed when irradiated in the fluorescence spectrometer. Fig. S34 of the ESI† illustrates that the decomposition product still constitutes a BODIPY dye. The distinct red-shift of the fluorescence peak and its similar position to that of the Br–BODIPY precursor suggest that the BODIPY-ligand is detached from the Pt atom during photochemical degradation. Facile Pt-BODIPY bond breaking in this complex is likely caused by the strong σ-trans-influence of the methyl ligand and the concomitant weakening of the Pt-C(BODIPY) σ-bond, which is even amplified in the excited T1 state (Tables 1 and S15 of the ESI†).
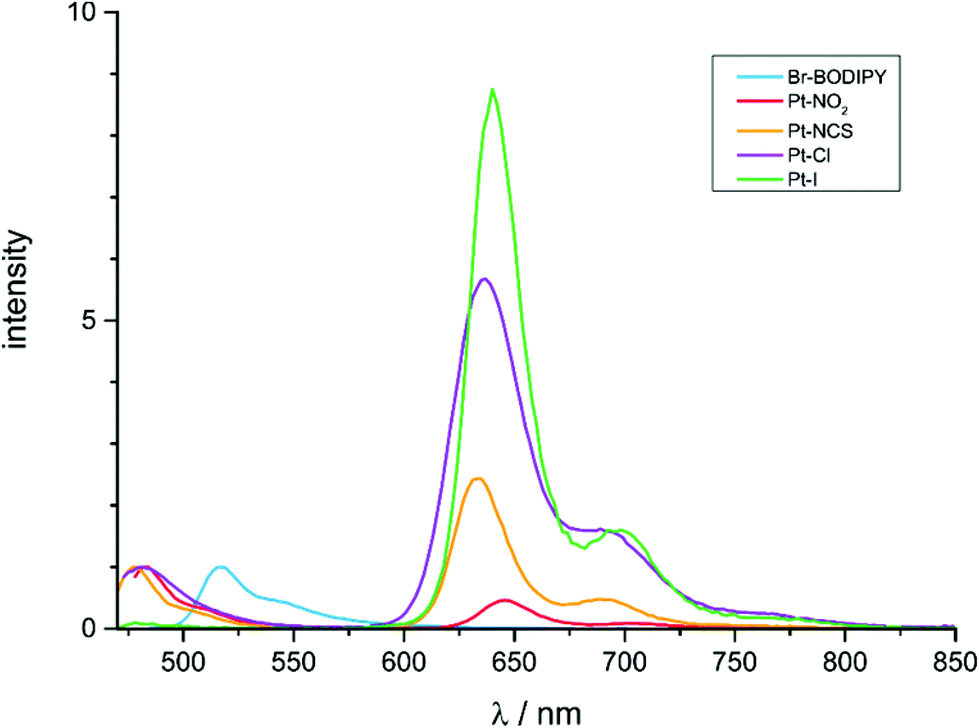 | ||
| Fig. 6 Emission spectra of Pt–Cl, Pt–I, Pt–NO2Pt–NCS and Br–BODIPY in degassed CH2Cl2 solutions at concentrations of ca. 10−6 M upon irradiation into the lowest energy absorption band of the complexes. | ||
| λ max,Fl [nm] (Stokes shift [cm−1]) | λ max,Ph [nm] (Stokes shift [cm−1]) | Φ Fl,exc467 (ΦFl,exc322)a | Φ Ph,exc467 (ΦPh,exc322)a | τ Fl [ns] | τ Ph [μs] | |
|---|---|---|---|---|---|---|
| a Fluorescence and phosphorescence quantum yields measured at an excitation wavelength of 467 nm or 322 nm, respectively. b Not determined. c Measured in toluene solution at r.t. d Measured in a toluene glass at 77 K. | ||||||
| Pt–Br 20 | 479 (491) | 637(5669) | 0.011 | 0.312 | n.d.b | 162 |
| Pt–Cl | 478 (493) | 633 (5615) | 0.016 (0.005) | 0.349 (0.356) | 0.174 | 277 |
| 479 (491)c | 631 (5520)c | 243c | ||||
| 478d | 626d | 450d | ||||
| Pt–I | 481 (441) | 641 (5631) | 0.002 (0.000) | 0.364 (0.397) | 0.484 | 297 |
| Pt–NO2 | 483 (483) | 645 (5683) | 0.115 (0.052) | 0.166 (0.209) | 0.470 | 439 |
| Pt–NCS | 480 (489) | 637 (5623) | 0.048 (0.024) | 0.244 (0.323) | 1.027 | 313 |
The ratio of phosphorescence to fluorescence intensities increases in the order Pt–NO2 < Pt–NCS < Pt–Cl < Pt–I; Pt–Br occupies a position intermediate between Pt–NCS and Pt–Cl. This ordering parallels an increasing trans-influence of the ligand X−,35 but shows no clear correlation to its positioning within the spectroelectrochemical series. This indicates that thermal population of excited d-states is most probably not the dominant pathway for radiationless decay of the excited states, although the documented complexities of such processes still warrant caution.30
As the already very weak fluorescence of Pt–I was found to vanish altogether on excitation into the high-energy absorption band at 322 nm, the intensities of the phosphorescence and fluorescence emissions were monitored at different excitation wavelengths. Fig. 7a and b illustrate that, on irradiation into the higher energy absorption band(s), the phosphorescence quantum yield ΦPh of Pt–NO2 further increases at the expense of that of the fluorescence emission (ΦFl). The notion that the ratio of phosphorescence and fluorescence emission intensities may depend on the excitation wavelength has been perspectively proposed by Chou et al.33 and was experimentally demonstrated soon after.45–47 This phenomenon relies on the different involvement of a heavy atom in the different excited states. In particular, a larger degree of charge-transfer between a metal/coligand entity and the emissive ligand (metal-to-ligand or ligand-to-metal charge-transfer) provides a more direct pathway for ISC, and hence a larger rate constant kISC, than the remote heavy-metal effect alone.45,46,48 The efficiencies of the ISC from a higher-lying Sn state (Sn → Tm → T1) and from the S1 state (Sn → S1 → T1) may thus drastically differ if the initially populated states differ in character.
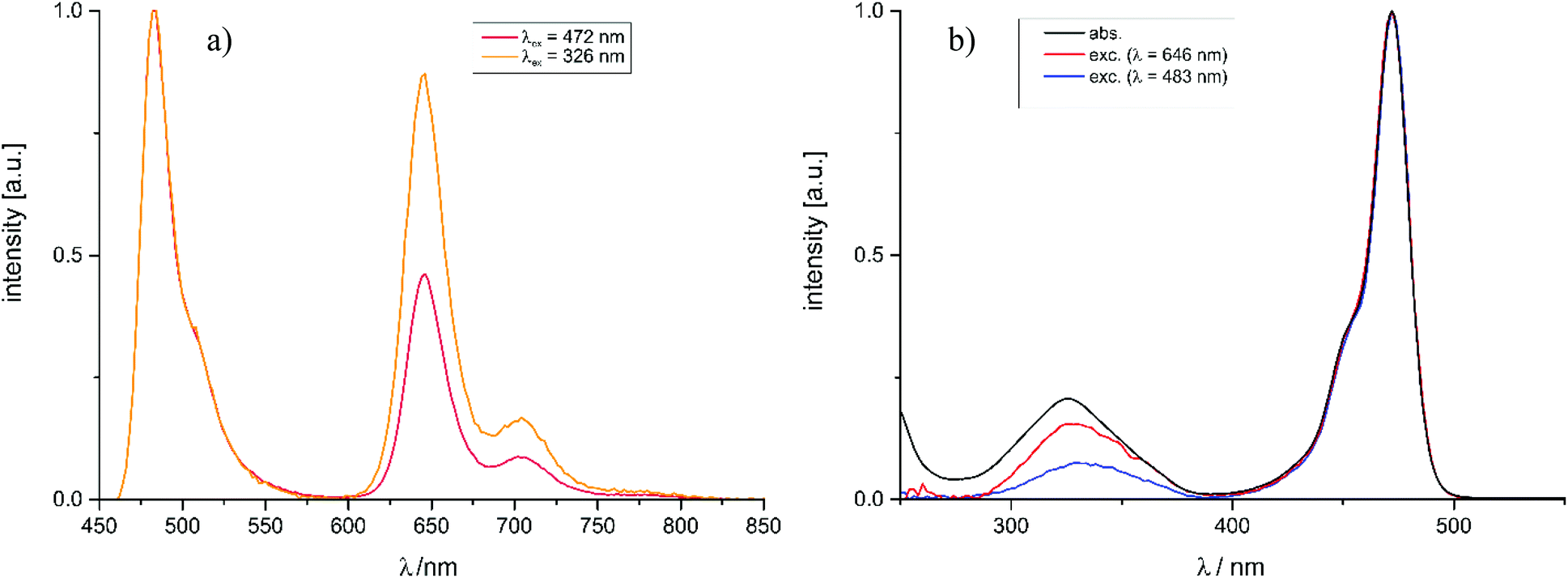 | ||
| Fig. 7 (a) Emission spectra of Pt–NO2 on excitation at λ = 326 nm and λ = 472 nm, respectively. (b) Absorption and excitation spectra of Pt–NO2. The excitation spectra were recorded for the fluorescence band at 483 nm and the phosphorescence band at 646 nm. Measurements were performed on degassed CH2Cl2 solutions at concentrations of ca. 10−6 M. | ||
For the BODIPY-centred excited S1 state, which is initially populated by irradiation into the prominent HOMO → LUMO π → π* absorption band, the coordination centre merely acts as a remote heavy metal atom, and the efficiency of ISC relies on the close proximity of the Pt ion to the dye (note that kISC in that case relates to r−6 where r is the distance of the heavy metal atom to the midpoint of the dye).47 This is readily inferred from the spin density surfaces for the excited triplet states of Pt–Cl, Pt–I, Pt–NCS, and Pt–NO2 in Fig. 8. Complying with the compositions of the HOMO and the LUMO, almost the entire spin density resides at the BODIPY ligand with only very modest contributions of 0.3% to 1.2% from the Pt ion. As it was already discussed, the higher energy absorption band, populating (a) higher Sn state(s), has more significant contributions from Pt(PEt3)2 → BODIPY charge-transfer (ML → L′CT, Fig. 5 and Tables 2 and 3). As is illustrated in Scheme 1, the faster kISC,n from the higher-lying ML → L′CT excited state provides an even more competitive pathway for population of the phosphorescent T1 state than ISC from S1. Excitation into (a) higher Sn state(s) thus decreases the fluorescence quantum yield ΦFl while further boosting ΦPh. The highest phosphorescence quantum yields are found for the simple halogenido complexes. The values of ΦPh of 36.4% or 39.7% for Pt–I on excitation at 467 or at 322 nm, respectively, are, to the best of our knowledge, the highest phosphorescence quantum yields of any BODIPY derivative, even surpassing those of Pt–Br.20
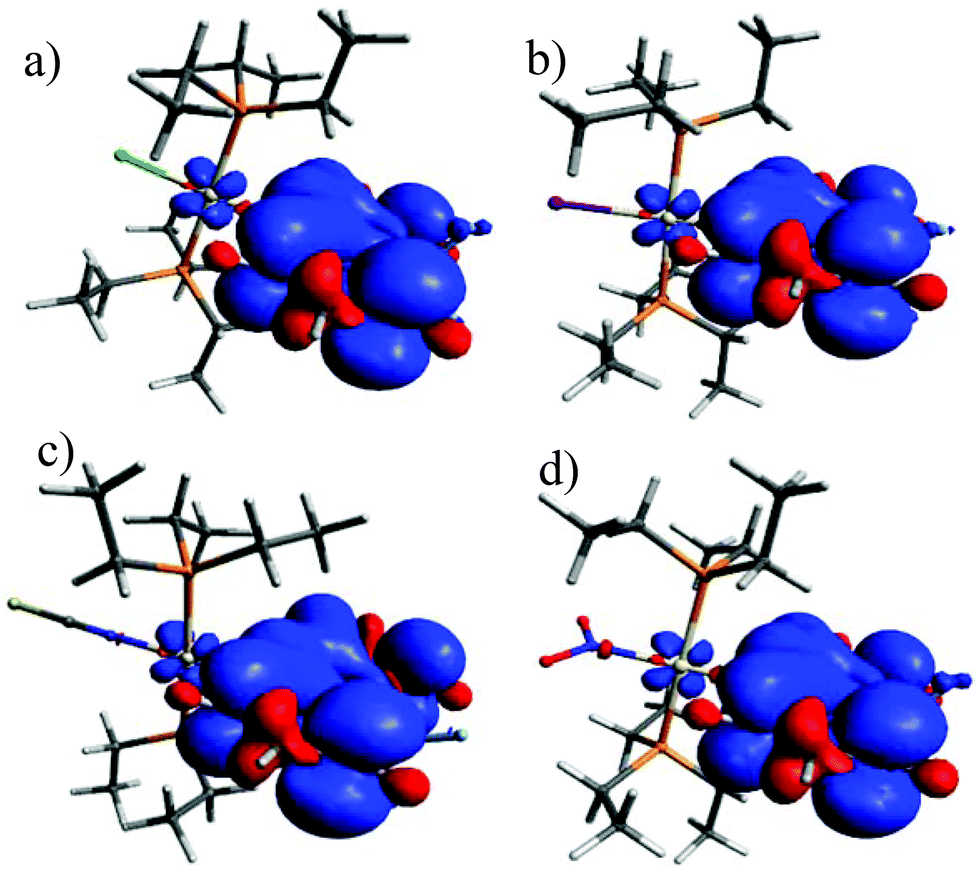 | ||
| Fig. 8 Spin density surfaces of the T1 state of (a) Pt–Cl, (b) Pt–I, (c) Pt–NCS, and (d) Pt–NO2. | ||
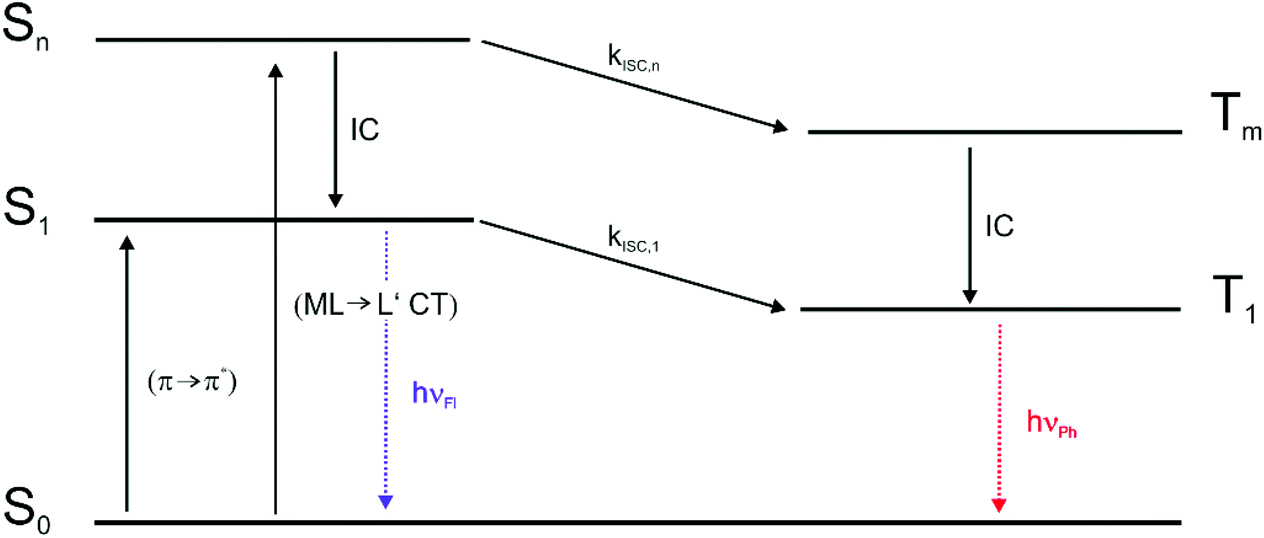 | ||
| Scheme 1 Jablonski diagram for the relevant optical processes in the complexes trans-Pt(BODIPY)X(PEt3)2. | ||
Emission quenching by 3O2 and 1O2 generation
The very long lifetimes of the excited triplet states of up to 439 μs make these compounds interesting candidates for applications such as triplet molecule sensing and photocatalysis. Their capabilities to act as one-component sensors for triplet molecules were tested by Stern–Volmer quenching experiments using 3O2 as the quencher. Fig. 9 and as Fig. S35 and S37 of the ESI† illustrate the results of such experiments for Pt–I, Pt–Cl and Pt–NO2. The Stern–Volmer equation is given as I0/I = 1 + KSV[O2], where I0 is the luminescence intensity under exclusion of oxygen, I is the luminescence intensity at a specific oxygen concentration, and KSV is the Stern–Volmer quenching constant, which is a measure for the sensitivity of the sensor. Fig. 10 displays plots of (I0/I) − 1 and (τ0/τ) − 1 as a function of the partial oxygen pressure (p(O2)). The quenching constants of KSV = 2380 ± 170 bar−1 for Pt–Cl and KSV = 2580 ± 70 bar−1 for Pt–I are identical within the experimental error limits. As expected from the longer triplet state lifetime, Pt–NO2 has an even larger KSV of 2810 ± 110 bar−1. Quenching constants evaluated by the ratios of lifetimes are somewhat smaller but still reach values of close to 2000 to 2200 bar−1. All complexes show high sensitivities for small partial oxygen pressures. Above p(O2) = 0.1 bar the plots start to deviate from linearity which relates to the low intensity of the residual signal. Our results render complexes Pt–Cl, Pt–I and Pt–NO2 particularly efficient oxygen sensors when compared to other successful platinum-based systems.49–54 We note here that a lower lifetime of the phosphorescence emission and hence lesser sensitivity towards O2 quenching as it was observed for Pt–Br allows for O2 detection in solution up to atmospheric concentration levels of the surrounding gas phase.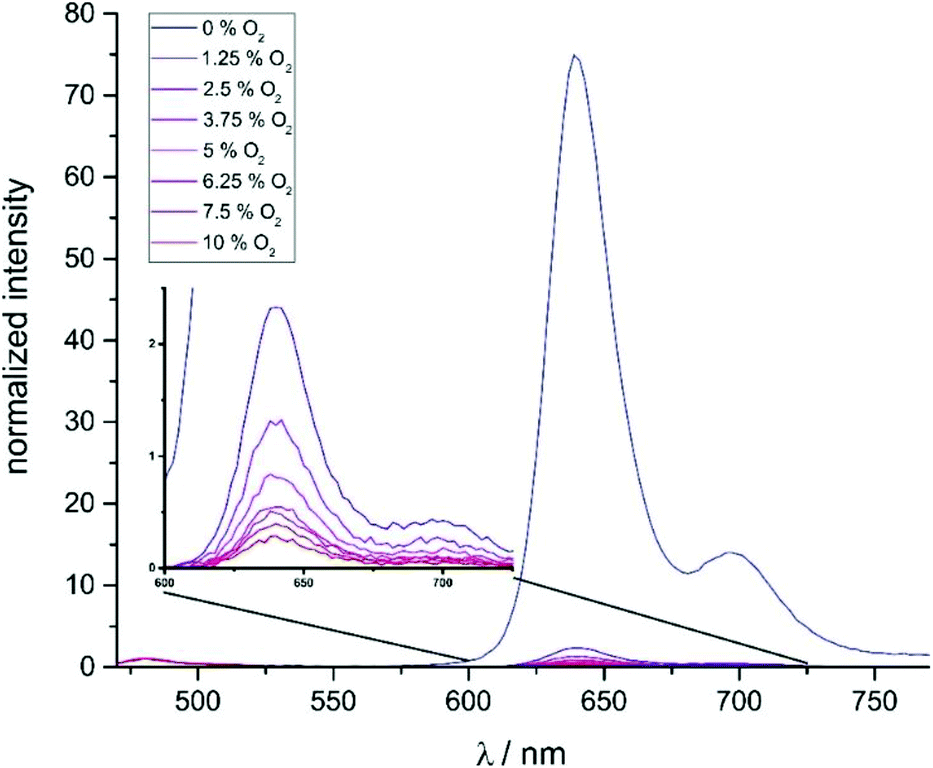 | ||
| Fig. 9 Stacked luminescence spectra of Pt–I in CH2Cl2 solution at different oxygen concentration levels. | ||
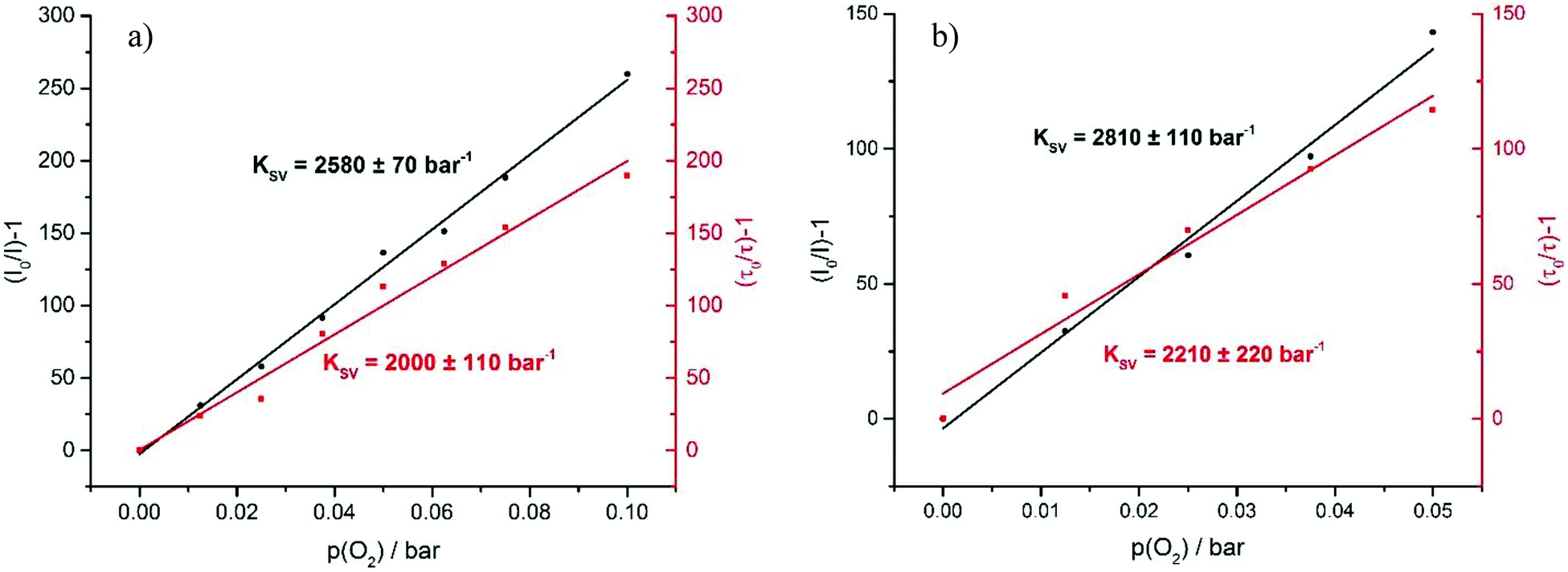 | ||
| Fig. 10 Stern–Volmer plot (a) of Pt–I and (b) of Pt–NO2. | ||
The feasibility of using these complexes as sensitizers for 1O2 generation from 3O2 by triplet–triplet annihilation in productive chemical reactions55,56 such as the oxidation of 1,5-dihydroxynaphthalene (DHN) was investigated using the complexes Pt–Cl and Pt–I as catalysts. The catalytic cycle of the photocatalytic system consisting of the sensitizer, aereal O2 and DHN is shown in Scheme 2. On the basis of this mechanism, the rate-law of DHN consumption can be written as νi = kr [O2][DHN]. At the initial stage of the reaction oxygen concentration can be treated as constant. The previous equation can therefore be simplified to νi = kobs·[DHN] using a pseudo first-order rate constant kobs. Rewriting this formula as ln(Ct/C0) = −kobs·t, where Ct denotes the concentration of DHN at a certain reaction time t while C0 is the initial concentration of DHN, allows for determining kobs from the slope of a plot of ln(Ct/C0) vs. reaction time t. The associated values of νi and the number of photons absorbed by the sensitizer provide the 1O2 generation quantum yield (ΦΔ) by using the relative method with methylene blue (MB) as a reference sensitizer.57 Details of these experiments are provided in the Experimental section. Fig. 11a depicts the changes of the absorption spectra of the reaction mixture with irradiation time t using Pt–Cl as a sensitizer, while Fig. 11b compares plots of ln(Ct/C0) as a function of t for Pt–Cl, Pt–I and the MB standard. The rate constants kobs, the rates νi of DHN consumption, and quantum yields for the generation of 1O2 (ΦΔ) in the photooxidation of DHN are summarized in Table 5. Both platinum complexes obey a linear relation between ln(Ct/C0) and the irradiation time t from which νi was determined. This precludes side reactions and proves that the sensitizers are stable under these conditions. Control experiments in the absence of light showed that none of the sensitizers promotes oxidation of DHN to Juglone under dark conditions (see Fig. S38 and S39 in the ESI†). Both complexes show a significantly higher rate νi of DHN consumption than MB. Most remarkably, Pt–I and Pt–Cl have exceptionally high quantum efficiencies ΦΔ of 0.95 (Pt–I) or even near unity (Pt–Cl) and clearly outperform the MB standard (ΦΔ of 0.57).58–60 Contributing factors are the high ISC efficiencies and the long lifetimes of the triplet state (τPh = 277 μs for Pt–Cl, τPh = 297 μs for Pt–I).
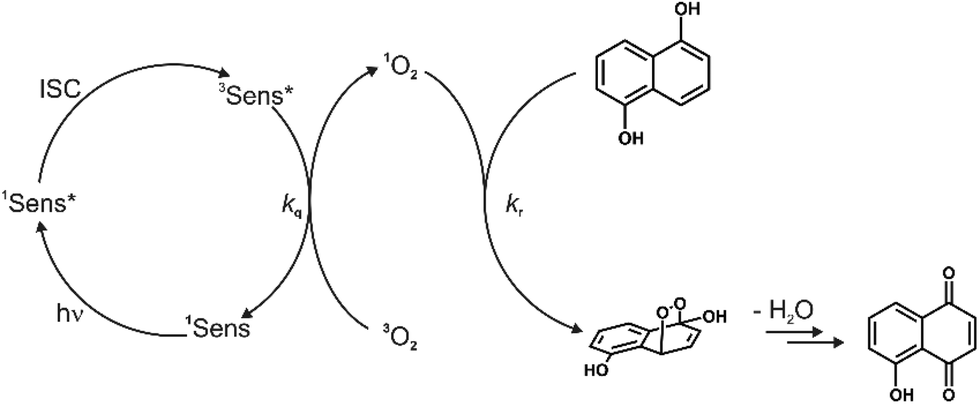 | ||
| Scheme 2 Mechanism for the photooxidation of 1,5-dihydroxynaphthalene (DHN) by 1O2 catalyzed by a sensitizer, producing Juglone after the elimination of a water molecule. | ||
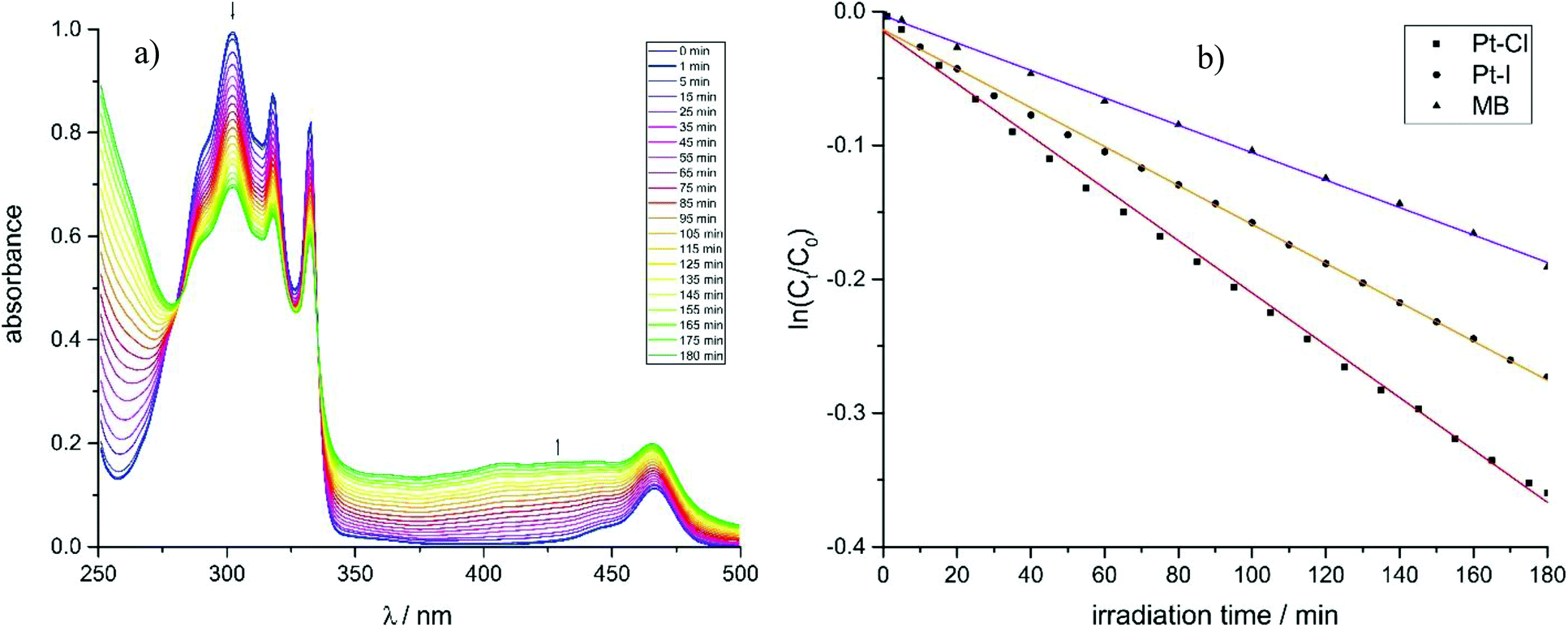 | ||
| Fig. 11 (a) Spectral change in the UV-Vis region for the photooxidation of DHN using Pt–Cl as the sensitizer. (b) Plots of ln(Ct/C0) vs. irradiation time for the photooxidation using complexes Pt–Cl, Pt–I and MB. | ||
|
k
obsa [min−1] |
ν
ib [×10−6 M min−1] |
I |
Φ
Δd |
Yielde [%] | TOFf [s−1] |
K
SVg [bar−1] |
|
|---|---|---|---|---|---|---|---|
| a Pseudo first-order rate constant for DHN consumption. b Rate of DHN consumption. c Relative value of the number of photons absorbed by the sensitizer (I = 1 for the standard sensitizer MB). d Corrected 1O2 generation quantum yield using the value of MB (ΦΔ = 0.57)58–60 as a reference. e Yield of Juglone after a reaction time of 180 min. f Turnover frequency. g In CH2Cl2 solution. | |||||||
| Pt–Cl | 0.00195 | 0.236 | 1.033 | 1.00 | 35 | 0.0019 | 2380 ± 170 |
| Pt–I | 0.00145 | 0.176 | 0.810 | 0.95 | 25 | 0.0014 | 2580 ± 70 |
| MB | 0.00102 | 0.130 | 1.000 | 0.57 | 14 | 0.0008 | |
Summary and conclusions
We report on the synthesis and the spectroscopic and photophysical properties of five new complexes trans-Pt(BODIPY)X(PEt3)2 (Pt–X, X− = Cl−, I−, NO2−, NCS−, CH3−). All contain a σ-bonded BODIPY ligand that binds to the platinum ion via its meso position. With the exception of Pt–CH3, all complexes show dual fluorescence and phosphorescence emissions from the attached BODIPY dye at wavelengths that are largely invariant to the nature of the ligand X−. Phosphorescence quantum yields and Pt–C(BODIPY) bond lengths increase in the order Pt–NO2 < Pt–NCS < Pt–Cl < Pt–I in parallel with the σ-trans-influence of the ligand X−.Most importantly, the ratio of phosphorescence to fluorescence intensities of each complex depends on the excitation wavelength. This is a direct consequence of the different natures of the initially populated excited states (BODIPY-based π → π* or a higher excited state with appreciable Pt(PEt3)2 → BODIPY π* ML → L′ charge-transfer character), which results in different rate constants kISC. Thus, the higher-energy MLCT absorption offers a more direct pathway for Pt-triggered ISC than just the heavy atom effect. Our present results provide experimental manifestation of the concept of excitation energy-dependent emission properties as recently discussed and observed by Chou and his coworkers.33,45–47
Additional studies into phosphorescence quenching by 3O2 have yielded exceptionally large Stern–Volmer quenching constants of ca. 2000 bar−1 and demonstrated that these complexes are excellent one-component sensors for triplet molecules. Moreover, they constitute highly efficient sensitizers for photocatalytic reactions involving 1O2 as the reactant, combining exceptionally high quantum efficiencies near unity for 1O2 generation with good photostabilities. These treats will be further explored in our future work.
Experimental section
Materials and general methods
DHN was bought form Acros Organics and purified by sublimation (p = 4 × 10−3 mbar, 160° C oil bath). cis-Pt(BODIPY)Br(PEt3)2 was prepared as described elsewhere.20 All manipulations where conducted under air except for reactions involving MeMgI, MgMe2 and MeLi, which were performed under N2 atmosphere by standard Schlenk techniques. Solvents for the reactions under inert gas atmosphere were distilled over adequate drying agents and stored under N2 atmosphere. All other solvents were used as received from the suppliers.NMR experiments were carried out on a Bruker Avance III DRX 400 or a Bruker Avance DRX 600 spectrometer. 1H and 13C NMR spectra were referenced to the solvent signal, while 31P and 195Pt NMR spectra were referenced using the Absolute Reference tool in the MestReNova software. NMR data are given as follows: chemical shift (δ in ppm), multiplicity (br, broad; d, doublet; dd, doublet of doublets; m, multiplet; s, singlet; t, triplet), integration, coupling constant (Hz). Unequivocal signal assignments were achieved by 2D NMR experiments. The numbering of the nuclei follows that of the crystal structures in Fig. 3. Combustion analysis was conducted with an Elementar vario MICRO cube CHN-analyzer from Heraeus.
X-ray diffraction analysis of single crystals was performed at 100 K on a STOE IPDS-II diffractometer equipped with a graphite-monochromated radiation source (λ = 0.71073 Å) and an image plate detection system. A crystal mounted on a fine glass fiber with silicon grease was employed. If not indicated otherwise, the selection, integration, and averaging procedure of the measured reflex intensities, the determination of the unit cell dimensions and a least-squares fit of the 2θ values as well as data reduction, LP-correction, and space group determination were performed using the X-Area software package delivered with the diffractometer. A semiempirical absorption correction was performed.61 All structures were solved by the heavy-atom methods (SHELXS-97, SHELXS-2013, or SHELXS-2014).62,63 Structure solutions were completed with difference Fourier syntheses and full-matrix last-squares refinements using SHELXL-97, SHELXS-2013, or SHELXS-2014,63 minimizing ω(Fo2 − Fc2)2. The weighted R factor (wR2) and the goodness of the fit GOF are based on F2. All non-hydrogen atoms were refined with anisotropic displacement parameters, while hydrogen atoms were treated in a riding model. Molecular structures in this work are plotted with ORTEP 3264,65 or Mercury.66 CIF files of Pt–Cl, Pt–I, Pt–NCS, Pt–NO2, and Pt–CH3 have been deposited at the Cambridge Structure Data Base as CCDC 1474955 (Pt–Cl), 1474956 (Pt–NCS), 1474957 (Pt–I), 1474958 (Pt–NO2), and 1474959 (Pt–CH3).
UV-Vis absorption spectra were recorded on a TIDAS fiberoptic diode array spectrometer MCS from j&m in HELLMA quartz cuvettes with 1 cm optical path length at room temperature.
Computational details
The ground sate electronic structures were calculated by density functional theory (DFT) methods using the Gaussian 0967 program packages. Quantum chemical studies were performed without any symmetry constraints. Open shell systems were calculated by the unrestricted Kohn-Sham approach (UKS).68 Geometry optimization followed by vibrational analysis was made either in vacuum or in solvent media. The quasirelativistic Wood-Boring small-core pseudopotentials (MWB)69,70 and the corresponding optimized set of basis functions71 for Pt and the 6-31G(d) polarized double-ζ basis set72 for the remaining atoms were employed together with the Perdew, Burke and Ernzerhof exchange and correlation functional (PBE1PBE).73–75 Solvent effects were accounted for by the Polarizable Conductor Continuum Model (PCM)76–78 with standard parameters for dichloromethane. Absorption spectra and orbital energies were calculated using time-dependent DFT (TD-DFT)79 with the same functional/basis set combination as mentioned above. For easier comparison with the experiment, the obtained absorption and emission energies were converted into wavelengths and broadened by a Gaussian distribution (full width at half-maximum = 3000 cm−1) using the programm GaussSum.80 Molecular orbitals were visualized with the GaussView programm81 or with Avogadro.82Luminescence spectroscopy and quenching experiments
All luminescence spectra and excited state lifetimes were recorded for ca. 10−6 M solutions in CH2Cl2 or toluene with a PicoQuant FluoTime 300 spectrometer at room temperature, if not stated otherwise. Luminescence experiments under inert gas atmosphere and defined O2 concentrations were conducted in a quartz cuvette modified with an angle valve from Normag. Defined O2 concentrations were adjusted by completely degassing the sample and subsequent injection of adequate volumes of air and nitrogen by syringe. Quantum yields were measured using a Hamamatsu Absolute PL Quantum Yield Measurement System C9920-02 equipped with an integrating sphere.1O2 generation from Pt–X sensitizers
For the photoreactions involving 1O2 generation, a CH2Cl2/MeOH (9/1) solution containing DHN (1.2 × 10−4 M) and a sensitizer (1.7 mol% with respect to DHN) was irradiated in a quartz cell of 1 cm path length using the Xenon lamp of a PicoQuant FluoTime 300 spectrometer (λex(Pt–Cl, Pt–I) = 460 ± 5 nm, If(460 ± 5 nm) = 1.3 mW; λex(MB) = 655 ± 5 nm, If(655 ± 5 nm) = 580 μW). UV-Vis absorption spectra were recorded at intervals of 5–20 min on a Varian Cary 50 spectrometer. The consumption of DHN was monitored by the decrease of the absorption at 301 nm (ε = 7664 M−1 cm−1),57 while Juglone production was monitored by an increase of the absorption at 427 nm (ε = 3811 M−1 cm−1).57 The yield of Juglone was calculated from the concentration of Juglone and the initial concentration of DHN. The singlet oxygen quantum yield (ΦΔ) was determined using eqn (1),57,83| ΦΔ = ΦΔ,std(νi{·}Istd/νi,std{·}I) | (1) |
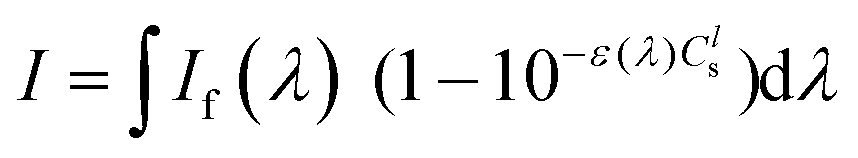 | (2) |
trans-Chloro-(4,4-difluoro-4-bora-3a,4a-diaza-s-indacen-8-yl)-bis(triethylphosphine)-platinum(II) (Pt–Cl)
40 mg (57 μmol, 1 eq.) of cis-Pt(BODIPY)Br(PEt3)2 and 19.5 mg (76 μmol, 1.4 eq.) of AgOTf were dissolved in 0.8 ml of CD2Cl2. The solution was heated to reflux for 5 min which led to the formation of trans-Pt(BODIPY)(OTf)(PEt3)2. The heterogeneous mixture was filtered and the filtrate was added to a solution of 66.6 mg (114 μmol, 2 eq.) of NaCl in 2.6 ml of MeOH. The orange suspension was stirred for 45 min. The solvents were then removed in vacuo and the solid was extracted with CH2Cl2. The product was purified by column chromatography (silica, CH2Cl2, Rf = 0.72). After washing with n-pentane (2 × 0.6 ml) the analytically pure product was obtained. Yield: 32%. Single crystals were obtained by slow evaporation of a CH2Cl2 solution. 1H NMR (400 MHz, CD2Cl2): δ 7.65 (br s, 2H, H5, H9), 7.46 (d, 2H, 3JHH = 3.68 Hz, H3, H7), 6.46 (dd, 2H, 3JHH = 3.68 Hz, 3JHH = 2.04 Hz, H4, H8), 1.74 (m, 12H, P–CH2–), 1.06 (dt, 18H, 3JHH = 7.66 Hz, 3JPH = 15.58 Hz, P–CH2–CH3). 31P NMR (161.9 MHz, CD2Cl2): δ 11.70 (s, with satellites JPtP = 2502 Hz). 13C NMR (150.9 MHz, CD2Cl2): δ 178.5 (t, 2JPC = 7.8 Hz, with satellites JPtC = 492 Hz, C1), 143.5 (s, with satellites 2JPtC = 22.4 Hz, C2, C6), 137.1 (s, C5, C9), 133.8 (s, with satellites 3JPtC = 25.2 Hz, C3, C7), 116.6 (s, C4, C8), 14.28 (t, JPC = 33.0 Hz, with satellites 2JPtC = 17.0 Hz, P–CH2–), 8.2 (t, 2JPC = 10.0 Hz, P–CH2–CH3). 195Pt NMR (86.0 MHz, CD2Cl2): δ −4134 (t, JPtP = 2502 Hz). C, H, N analysis calculated for C21H36ClBF2N2P2Pt·0.3 C5H12: C, 39.77; H, 5.87; N, 4.12. Found: C, 39.44; H, 6.09; N, 4.06.trans-Iodo-(4,4-difluoro-4-bora-3a,4a-diaza-s-indacen-8-yl)-bis(triethylphosphine)-platinum(II) (Pt–I)
trans-Pt(BODIPY)(OTf)(PEt3)2 prepared as described above was dissolved in 5 ml of dry THF under N2 atmosphere. To this solution 0.08 ml of a freshly prepared 1 M MeMgI solution (1.4 eq.) in diethyl ether were added. The solution was stirred for 20 min, the precipitate was filtered off and the solution was evaporated to dryness.The crude product was purified by column chromatography (silica deprotonated with 5% NEt3, CH2Cl2:PE = 5:1, Rf = 0.46). After washing twice with 0.3 ml of n-pentane the yellow product was obtained in 58% yield. Single crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of a C6D6 solution. 1H NMR (400 MHz, C6D6): δ 7.81 (br s, 2H, H5, H9), 7.43 (d, 2H, 3JHH = 3.51 Hz, H3, H7), 6.19 (dd, 2H, 3JHH = 3.51 Hz, 3JHH = 1.89 Hz, H4, H8), 1.68 (m, 12H, P–CH2–), 0.72 (dt, 18H, 3JHH = 7.68 Hz, 3JPH = 15.89 Hz, P–CH2–CH3). 31P NMR (161.9 MHz, C6D6): δ 4.02 (s with satellites, JPtP = 2450 Hz). 13C NMR (100.6 MHz, C6D6): δ 181.1 (s, C1), 143.3 (s, C2, C6), 138.5 (s, C5, C9), 133.3 (s with satellites, 3JPtC = 25.0 Hz, C3, C7), 116.7 (s, C4, C8), 16.2 (t, JPC = 34.2 Hz, with satellites 2JPtC = 17.6 Hz, P–CH2–), 8.36 (t, 2JPC = 11.5 Hz, P–CH2–CH3). 195Pt NMR (86.0 MHz, C6D6): δ −4503.4 (t, JPtP = 2450 Hz). C, H, N analysis calculated for C21H36BF2IN3P2Pt: C, 33.66; H, 4.84; N, 3.74. Found: C, 33.85; H, 5.06; N, 3.86.
trans-κN-(4,4-difluoro-4-bora-3a,4a-diaza-s-indacen-8-yl)-(thiocyanato)-bis(triethylphosphine)-platinum(II) (Pt–NCS)
A solution of 30 mg (57 μmol, 1 eq.) of cis-Pt(BODIPY)Br(PEt3)2 and 19.5 mg (76 μmol, 1.4 eq.) of AgOTf in 0.8 ml of CH2Cl2 was refluxed for 5 min. The solution was filtered and 2.6 ml of a methanolic solution of NaSCN (9.2 mg, 114 μmol, 2 eq.) was added. After stirring the reaction mixture for 1.5 h all volatiles were removed and the crude product was extracted with CH2Cl2. The product was further purified by column chromatography (silica deprotonated with 5% NEt3, PE:EE = 3:1, Rf = 0.32). The product was washed with small amounts of n-pentane. Yield: 36%. Single crystals for X-ray diffraction analysis were obtained by slow evaporation of a CH2Cl2 solution. 1H NMR (400 MHz, C6D6): δ 7.80 (br s, 2H, H5, H9), 7.28 (d, 2H, 3JHH = 3.67 Hz, H3, H7), 6.18 (dd, 2H, 3JHH = 3.67 Hz, 3JHH = 2.00 Hz, H4, H8), 1.27 (m, 12H, P–CH2–), 0.68 (m, 18H, P–CH2–CH3). 31P NMR (161.9 MHz, C6D6): δ 12.50 ppm (s, with satellites JPtP = 2450 Hz). 13C NMR (150.9 MHz, C6D6): δ 174.3 (s, C1), 143.4 (s, C2, C6), 138.3 (s, C5, C9), 132.5 (s, with satellites 3JPtC = 22.5 Hz, C3, C7), 116.7 (s, C4, C8), 14.6 (t, JPC = 17.0 Hz, P–CH2–), 7.6 (br s, P–CH2–CH3), signal for C10 not detected. 195Pt NMR (129.0 MHz, C6D6): δ −4181.5 (t, JPtP = 2450 Hz). C, H, N analysis calculated for C22H36BF2N3P2PtS: C, 38.83; H, 5.33; N, 6.18. Found: C, 38.81; H, 5.59; N, 6.20.
trans-(4,4-Difluoro-4-bora-3a,4a-diaza-s-indacen-8-yl)-(nitrito)-bis(triethylphosphine)-platinum(II) (Pt–NO2)
To trans-Pt(BODIPY)(OTf)(PEt3)2 prepared as described above from 40 mg (57 μmol, 1 eq.) of cis-Pt(BODIPY)Br(PEt3)2 in CH2Cl2, a solution of 7.9 mg (114 μmol, 2 eq.) of NaNO2 in 2.6 ml of MeOH was added. The orange solution was stirred for 1.5 h at r.t., the solvents were removed and the crude solid was extracted with CH2Cl2. The product was purified by column chromatography (silica deprotonated with 5% NEt3, PE:EE = 1:1, Rf = 0.41). After removing the solvent at low temperature the yellow product was washed two times with n-pentane and dried in vacuo. Yield: 26%. Single crystals for X-ray diffraction analysis were obtained by slow evaporation of a CH2Cl2 solution. 1H NMR (400 MHz, C6D6): δ 7.81 (br s, 2H, H5, H9), 7.58 (d, 2H, 3JHH = 3.65 Hz, H3, H7), 6.22 (dd, 2H, 3JHH = 3.65 Hz, 3JHH = 1.89 Hz, H4, H8), 1.29 (m, 12H, P–CH2–), 0.73 (m, 18H, P–CH2–CH3). 31P NMR (161.9 MHz, C6D6): δ 9.69 (s, with satellites JPtP = 2600 Hz). 13C NMR (150.9 MHz, C6D6): δ 174.9 (t, 2JPC = 8.8 Hz, with satellites JPtC = 409 Hz, C1), 143.5 (s, with satellites 3JPtC = 15.8 Hz, C2, C6), 138.6 (s, C5, C9), 133.2 (s, with satellites 3JPtC = 22.0 Hz, C3, C7), 116.7 (s, C4, C8), 14.7 (t, JPC = 32.5 Hz, with satellites 2JPtC = 17.0 Hz, P–CH2–), 7.6 (br s, P–CH2–CH3). 195Pt NMR (129.0 MHz, C6D6): δ −4081.6 (t, JPtP = 2600 Hz). C, H, N analysis calculated for C21H36BF2N3O2P2Pt: C, 37.74; H, 5.43; N, 6.29. Found: C, 37.78; H, 5.73; N, 6.40.
trans-(4,4-Difluoro-4-bora-3a,4a-diaza-s-indacen-8-yl)-(methyl)-bis(triethylphosphine)-platinum(II) (Pt–CH3)
A CH2Cl2 solution containing 40 mg (57 μmol, 1 eq.) of cis-Pt(BODIPY)Br(PEt3)2 and 19.5 mg (76 μmol, 1.4 eq.) of AgOTf was refluxed for 5 min, then filtered and evaporated to dryness. The crude solid was placed under nitrogen atmosphere, 10 ml of diethyl ether were added and the solution was cooled to −78° C. To this bright orange solution 0.5 ml of a 0.18 M solution of methyl lithium in diethyl ether was added dropwise. The reaction mixture turned green while it was allowed to warm to room temperature over 4 h. The mixture was filtered, the solvent was removed and the crude solid was purified by column chromatography (silica deprotonated with 5% NEt3, petroleum ether:ethyl acetate = 8:3, Rf = 0.73). The product was washed two times with n-pentane and dried in vacuo. Yield: 49%. Single crystals were obtained by slow diffusion of n-pentane in a saturated C6D6 solution. 1H NMR (600 MHz, C6D6): δ 7.92 (br s, 2H, H5, H9), 7.47 (d, 2H, 3JHH = 3.48 Hz, C3, C7), 6.31 (dd, 2H, 3JHH = 3.48 Hz, 3JHH = 2.05 Hz, H4, H8), 1.35 (m, 12H, P–CH2–), 0.70 (dt, 18H, 3JHH = 7.67 Hz, 3JPH = 15.68 Hz, P–CH2–CH3), −0.24 (t with satellites, 3H, 3JPH = 6.82 Hz, 2JPtH = 24.9 Hz, CH3). 31P NMR (161.9 MHz, C6D6): δ 9.18 (s, with satellites JPtP = 2692 Hz). 13C NMR (150.9 MHz, C6D6): δ 210.6 (t, 2JPC = 9.41 Hz, C1), 146.4 (s with satellites, 2JPtC = 22.0 Hz, C2, C6), 136.4 (s, C5, C9), 132.2 (s with satellites, 3JPtC = 37.0 Hz, C3, C7), 115.6 (s, C4, C8), 14.3 (t with satellites, JPC = 17.0 Hz, 2JPtC = 67.1 Hz, P–CH2–), 8.0 (s with satellites, 3JPtC = 25.5 Hz, P–CH2–CH3), −14.0 (t, 2JPC = 8.80 Hz, with satellites JPtC = 412.7 Hz, C10). 195Pt NMR (86.0 MHz, C6D6): δ −4396 (t, JPtP = 2692 Hz). C, H, N analysis calculated for C22H39BF2N2P2Pt: C, 41.46; H, 6.17; N, 4.39. Found: C, 41.34; H, 6.47; N, 4.51.
Notes
The authors declare no competing financial interest.Acknowledgements
We thank the state of Baden-Württemberg and the Deutsche Forschungsgemeinschaft for providing us with access to the computational facilities of the bwFor computer cluster JUSTUS.References
- A. Treibs and F.-H. Kreuzer, Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577–6595 CrossRef CAS.
- F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001–10017 CrossRef CAS.
- J. Chen, A. Burghart, A. Derecskei-Kovacs and K. Burgess, J. Org. Chem., 2000, 65, 2900–2906 CrossRef CAS PubMed.
- F. E. Alemdaroglu, S. C. Alexander, D. Ji, D. K. Prusty, M. Börsch and A. Herrmann, Macromolecules, 2009, 42, 6529–6536 CrossRef CAS.
- M. T. Whited, P. I. Djurovich, S. T. Roberts, A. C. Durrell, C. W. Schlenker, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2011, 133, 88–96 CrossRef CAS PubMed.
- R. Ziessel, G. Ulrich, A. Haefele and A. Harriman, J. Am. Chem. Soc., 2013, 135, 11330–11344 CrossRef CAS PubMed.
- T. Lazarides, T. M. McCornick, K. C. Wilson, S. Lee, D. W. McCamant and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 350–364 CrossRef CAS PubMed.
- M. Cordaro, P. Mineo, F. Nastasi and G. Magazzu, RSC Adv., 2014, 4, 43931–43933 RSC.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed.
- X.-F. Zhang and X. Yang, J. Phys. Chem. B, 2013, 117, 5533–5539 CrossRef CAS PubMed.
- W. Wu, J. Zhao, J. Sun, L. Huang and X. Yi, J. Mater. Chem. C, 2013, 1, 705–716 RSC.
- W. Wu, J. Zhao, H. Guo, J. Sun, S. Ji and Z. Wang, Chem. – Eur. J., 2012, 18, 1961–1968 CrossRef CAS PubMed.
- J. Sun, F. Zhong, X. Yi and J. Zhao, Inorg. Chem., 2013, 52, 6299–6310 CrossRef CAS PubMed.
- F. Geist, A. Jackel and R. F. Winter, Inorg. Chem., 2015, 54, 10946–10957 CrossRef CAS PubMed.
- P. Batat, M. Cantuel, G. Jonusauskas, L. Scarpantonio, A. Palma, D. F. O'Shea and N. D. McClenaghan, J. Phys. Chem. A, 2011, 115, 14034–14039 CrossRef CAS PubMed.
- X.-D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- W. Wu, J. Sun, X. Cui and J. Zhao, J. Mater. Chem. C, 2013, 1, 4577–4589 RSC.
- S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, CA – Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- W. H. Lam, E. S.-H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2013, 135, 15135–15143 CrossRef CAS PubMed.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC.
- P.-T. Chou, Y. Chi, M.-W. Chung and C.-C. Lin, Coord. Chem. Rev., 2011, 255, 2653–2665 CrossRef CAS.
- F. Geist, A. Jackel and R. F. Winter, Dalton Trans., 2015, 44, 3974–3987 RSC.
- T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335–422 CrossRef CAS.
- T. J. McCarthy, R. G. Nuzzo and G. M. Whitesides, J. Am. Chem. Soc., 1981, 103, 3396–3403 CrossRef CAS.
- S. Lentijo, J. A. Miguel and P. Espinet, Inorg. Chem., 2010, 49, 9169–9177 CrossRef CAS PubMed.
- P. Nilsson, F. Plamper and O. F. Wendt, Organometallics, 2003, 22, 5235–5242 CrossRef CAS.
- R. Bardi and A. M. Piazzesi, Inorg. Chim. Acta, 1981, 47, 249–254 CrossRef CAS.
- C. Albrecht, C. Wagner, K. Merzweiler, T. Lis and D. Steinborn, Appl. Organomet. Chem., 2005, 19, 1155–1163 CrossRef CAS.
- S. J. Anderson and R. J. Goodfellow, J. Chem. Soc., Dalton Trans., 1977, 1683–1686 RSC.
- J. L. Burmeister and F. Basolo, Inorg. Chem., 1964, 3, 1587–1593 CrossRef CAS.
- E. Hartmann and R. M. Gschwind, Angew. Chem., Int. Ed., 2013, 52, 2350–2354 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- C.-W. Hsu, C.-C. Lin, M.-W. Chung, Y. Chi, G.-H. Lee, P.-T. Chou, C.-H. Chang and P.-Y. Chen, J. Am. Chem. Soc., 2011, 133, 12085–12099 CrossRef CAS PubMed.
- C.-C. Hsu, C.-C. Lin, P.-T. Chou, C.-H. Lai, C.-W. Hsu, C.-H. Lin and Y. Chi, J. Am. Chem. Soc., 2012, 134, 7715–7724 CrossRef CAS PubMed.
- Y.-C. Chang, K.-C. Tang, H.-A. Pan, S.-H. Liu, I. O. Koshevoy, A. J. Karttunen, W.-Y. Hung, M.-H. Cheng and P.-T. Chou, J. Phys. Chem. C, 2013, 117, 9623–9632 CAS.
- E. Yu-Tzu Li, T.-Y. Jiang, Y. Chi and P.-T. Chou, Phys. Chem. Chem. Phys., 2014, 16, 26184–26192 RSC.
- W. Wu, C. Cheng, W. Wu, H. Guo, S. Ji, P. Song, K. Han, J. Zhao, X. Zhang, Y. Wu and G. Du, Eur. J. Inorg. Chem., 2010, 2010, 4683–4696 CrossRef.
- W. Wu, W. Wu, S. Ji, H. Guo, P. Song, K. Han, L. Chi, J. Shao and J. Zhao, J. Mater. Chem., 2010, 20, 9775–9786 RSC.
- Y. Liu, H. Guo and J. Zhao, Chem. Commun., 2011, 47, 11471–11473 RSC.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Dalton Trans., 2011, 40, 5953–5963 RSC.
- H. Sun, H. Guo, W. Wu, X. Liu and J. Zhao, Dalton Trans., 2011, 40, 7834–7841 RSC.
- H. Xiang, L. Zhou, Y. Feng, J. Cheng, D. Wu and X. Zhou, Inorg. Chem., 2012, 51, 5208–5212 CrossRef CAS PubMed.
- H. Wasserman and R. W. Murray, Singlet Oxygen, Academic Press, New York, 1979 Search PubMed.
- H. H. Wasserman and J. L. Ives, Tetrahedron, 1981, 37, 1825–1852 CrossRef CAS.
- S.-Y. Takizawa, R. Aboshi and S. Murata, Photochem. Photobiol. Sci., 2011, 10, 895–903 CAS.
- Y. Usui, Chem. Lett., 1973, 2, 743–744 CrossRef.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS.
- J. N. Chaon, G. R. Jamieson and R. S. Sinclair, Chem. Phys. Lipids, 1987, 43, 81–99 CrossRef.
- W. Herrendorf and W. Bärnighausen, X-Area, Version 1.06, Stoe, Darmstadt, Karslruhe, Gießen, 1999 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Solution and Refinement, Universität Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- P. R. Edgington, P. McCabe, C. F. Macrae, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- M. J. Frisch, G. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Blonio, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. M. Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- O. Gunnarsson and B. I. Lundqvist, Phys. Rev. B: Solid State, 1976, 13, 4274 CrossRef CAS.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7532 CrossRef.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- P. H. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Enzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E. Cancés, B. Menucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- E. Runge and K. U. G. E, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- T. Keith and J. Millam, GaussView, Version 3, Shawnee Mission, KS, USA, 2009 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchinson, J. Cheminf., 2012, 4, 17 CAS.
- N. Adarsh, M. Shanmugasundaram, R. R. Avirah and D. Ramaiah, Chem. – Eur. J., 2012, 18, 12655–12662 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Multinuclear NMR spectra of the complexes, packing diagrams with short interatomic contacts, absorption, emission and excitation spectra recorded at different excitation wavelengths, figures displaying oxygen quenching of the phosphorescence emission, changes of the absorption spectra of DHN in the presence of catalytic amounts of MB or Pt-I and plots showing the absence of the reaction in the dark; table with the cell parameters and structure refinement data for the complexes; atomic positions for the geometry-optimized structures in the S0 and the T1 states and comparison with the experimental structure parameters. CCDC 1474955–1474959. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01623e |
| This journal is © The Royal Society of Chemistry 2016 |