
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insertion of tBuNC into thorium–phosphorus and thorium–arsenic bonds: phosphaazaallene and arsaazaallene moieties in f element chemistry†
Andrew C.
Behrle
and
Justin R.
Walensky
*
Department of Chemistry, University of Missouri, Columbia, MO 65211, USA. E-mail: walenskyj@missouri.edu
First published on 28th April 2016
Abstract
The reactivity of thorium–phosphido and thorium–arsenido bonds was probed using tert-butyl isocyanide, tBuNC. Reaction of (C5Me5)2Th[E(H)R]2, E = P, As; R = 2,4,6-iPr3C6H2, 2,4,6-Me3C6H2 with tBuNC affords the first phosphaazaallene and arsaazaallene moieties with an f-element.
Introduction
Hydroelementation reactions such as hydrophosphination1,2 are atom efficient processes which are important in developing building blocks containing phosphorus. For example, tertiary phosphines are of interest as ligands3–5 and for various applications.6–8 However, the development of these reactions in organoactinide chemistry9 has been attenuated by a lack of starting materials.Despite the intensity with which complexes with actinide–nitrogen bonds have been studied,10–13 there exists a tremendous knowledge gap with respect to the heavier pnictogen elements. To date, twenty actinide–phosphido or phosphinidene bonds14–26 and five actinide–arsenido bonds27–29 have been reported. Of these, only nine thorium–phosphorus and one thorium–arsenic bond are known.
Due to the dearth of actinide–phosphorus and actinide–arsenic bonds, the reactivity of these bonds is unknown. Migratory insertion, the initial step in many catalytic cycles, has been used historically to probe reactivity. In transition metals, primary phosphido complexes are also sparse,30–37 but Waterman's group has investigated the reactivity of zirconium–phosphorus bonds with tBuNC.38 Interestingly, a proton migration from the phosphorus to the carbon of the isocyanide occurs. Here we describe the synthesis of new thorium phosphido and arsenido complexes and their reactivity with tBuNC, which differs from their transition metal analogs.
Results and discussion
The synthesis of (C5Me5)2Th[P(H)Tipp]2, Tipp = 2,4,6-iPr3C6H2, the first primary phosphido complex of thorium, was recently described.26 In the same vein, we have begun investigating the reactivity of primary phosphido and arsenido complexes. In an effort to expand the scope of thorium–pnictogen complexes, we synthesized (C5Me5)2Th[P(H)Mes]2, 1, Mes = 2,4,6-Me3C6H2, from the stoichiometric salt metathesis reaction between (C5Me5)2ThCl2 and KP(H)Mes, eqn (1). Complex 1 was isolated as a vibrant orange crystalline solid in 54% yield. The diagnostic spectroscopic features include the doublet P–H resonance and the νPH stretch centered at 3.80 ppm with 1JP–H = 224 Hz and 2304 cm−1, respectively. The large 1JP–H coupling constant reflects the large amount of s-character in the P–H bond.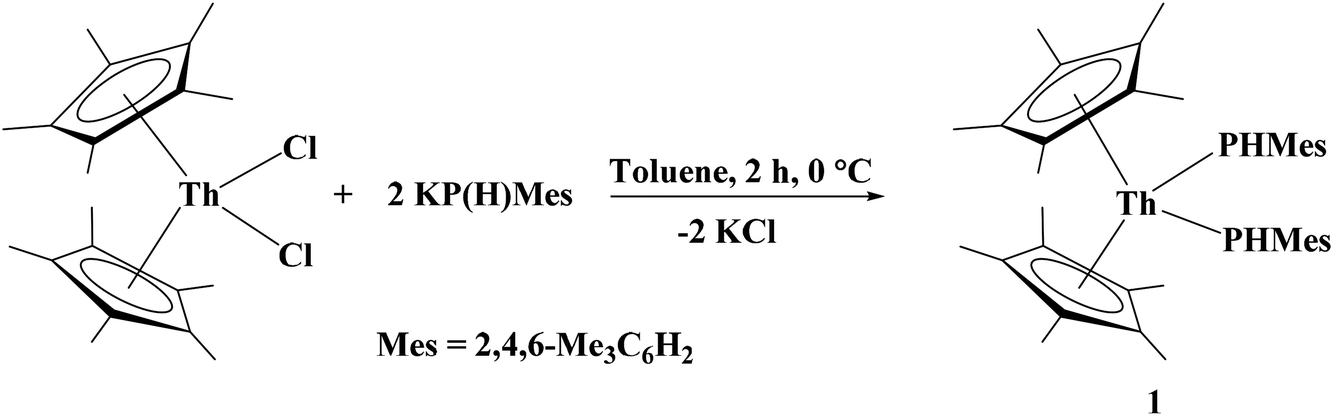 | (1) |
The 31P{1H} resonance is located at 15.37 ppm and compares well to the 31P{1H} resonance of a structurally similar thorium–phosphido compound, (C5Me5)2Th[P(H)Tipp]2, located at 1.66 ppm. The molecular structure of 1 is shown in Fig. 1 and mimics the bond distances and angles of (C5Me5)2Th[P(H)Tipp]2.
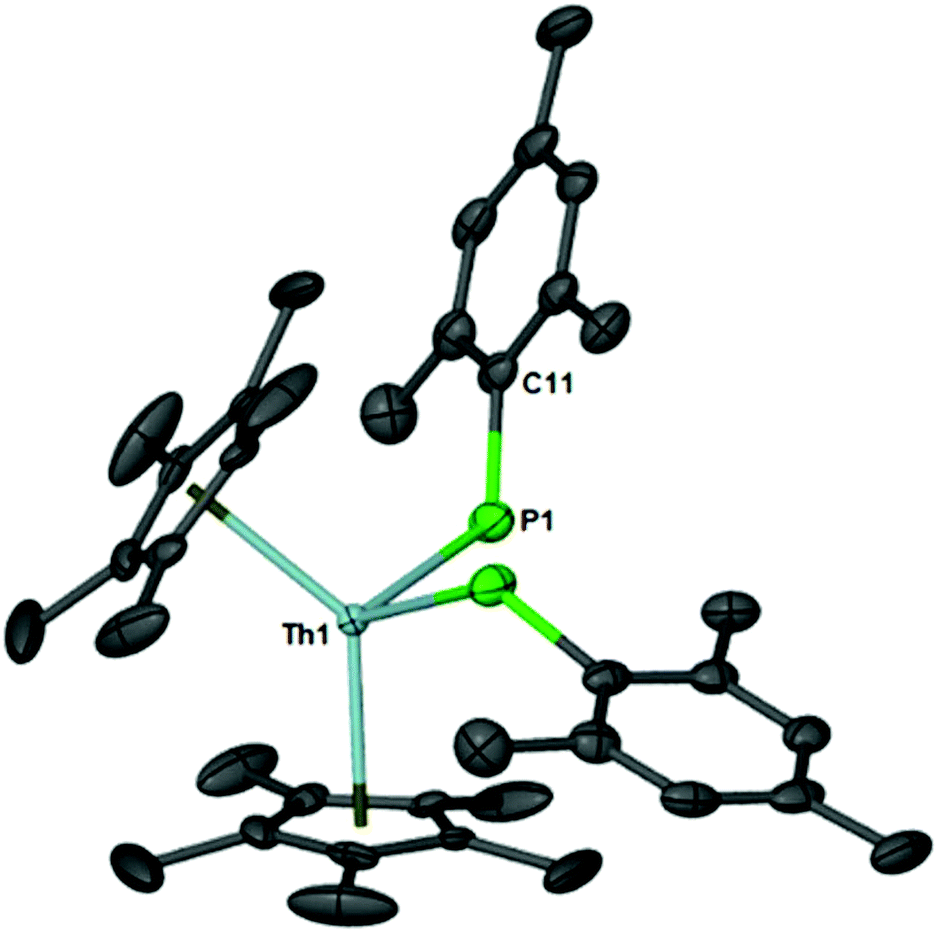 | ||
| Fig. 1 Thermal ellipsoid plot of 1 at the 50% probability level. Hydrogens have been omitted for clarity. Selected bond distances (Å) and angles (°): Th1–P1, 2.872(5); P1–C11, 1.829(3); P1–Th–P1*, 102.68(2); Th1–P1–C11, 128.45(9). | ||
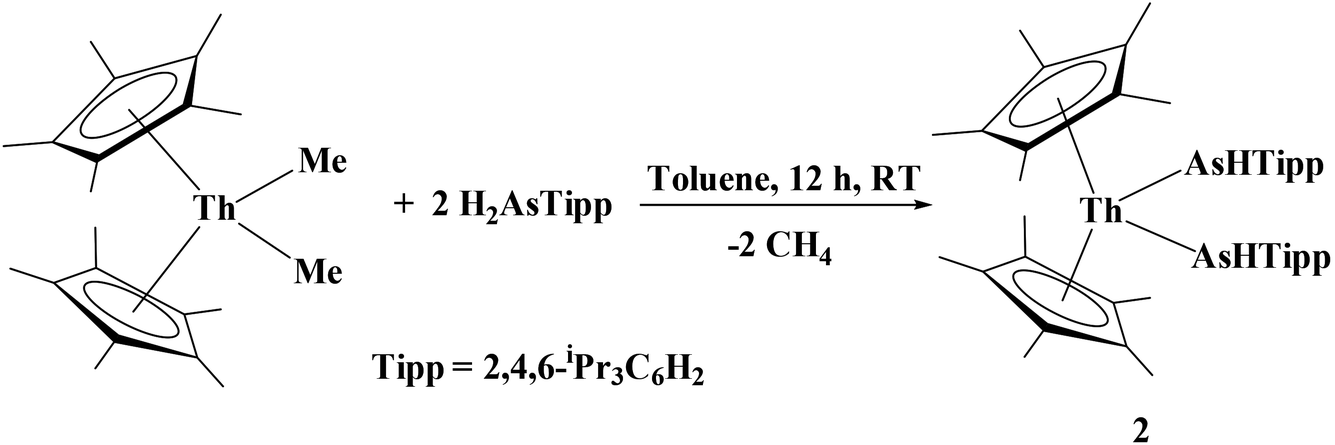
While actinide–phosphido complexes are few in number, the number of structurally characterized actinide–arsenido compounds is five: a bimetallic thorium poly-arsenide cluster, [Cp′2Th(μ–η2:1:2:1-As6)ThCp′2] (Cp′ = C5H3tBu2),29 and a series of uranium(IV) complexes: [{U(TrenTIPS)}2(μ–η2:η2-As2H2)],27 [U(TrenTIPS)(AsH2)], [U(TrenTIPS)(AsH)][K(B15C5)2], and [{U(TrenTIPS)(AsK2)}4] TrenTIPS = N(CH2CH2NSiPri3).28 We hypothesized that the 2,4,6-iPr3C6H2 framework would be sterically large enough to stabilize an actinide metal center such as thorium. Using room temperature σ-bond metathesis between (C5Me5)2ThMe2 and two equivalents of H2AsTipp, we successfully isolated the first organothorium primary arsenido complex, (C5Me5)2Th[As(H)Tipp]2, 2, eqn (2):
 | (2) |
Compound 2 was isolated as a ruby red crystalline solid in 70% yield. The diagnostic spectroscopic handles include the As–H resonance at δ 2.61 in the 1H NMR spectrum and the νAsH stretch at 2089 cm−1 in the IR spectrum. The IR stretching frequencies compare well to the νAsH stretches at 2061 cm−1 and 2052 cm−1 for zirconium(IV) and uranium(IV) primary arsenido complexes, [(N3N)ZrAsHR] (R = Mes and Ph; N3N = N(CH2CH2NSiMe3)33−),39 [U(TrenTIPS)(AsH2)]28 reported by Waterman and Liddle's groups, respectively. The molecular structure of 2 is shown in Fig. 2. The Th1–As1 bond length is 3.0028(6) Å and is slightly longer than the sum of the single bond covalent radii for thorium and arsenic (2.96 Å).40 The Th1–As1–C11 bond angle is 116.53(15)°.
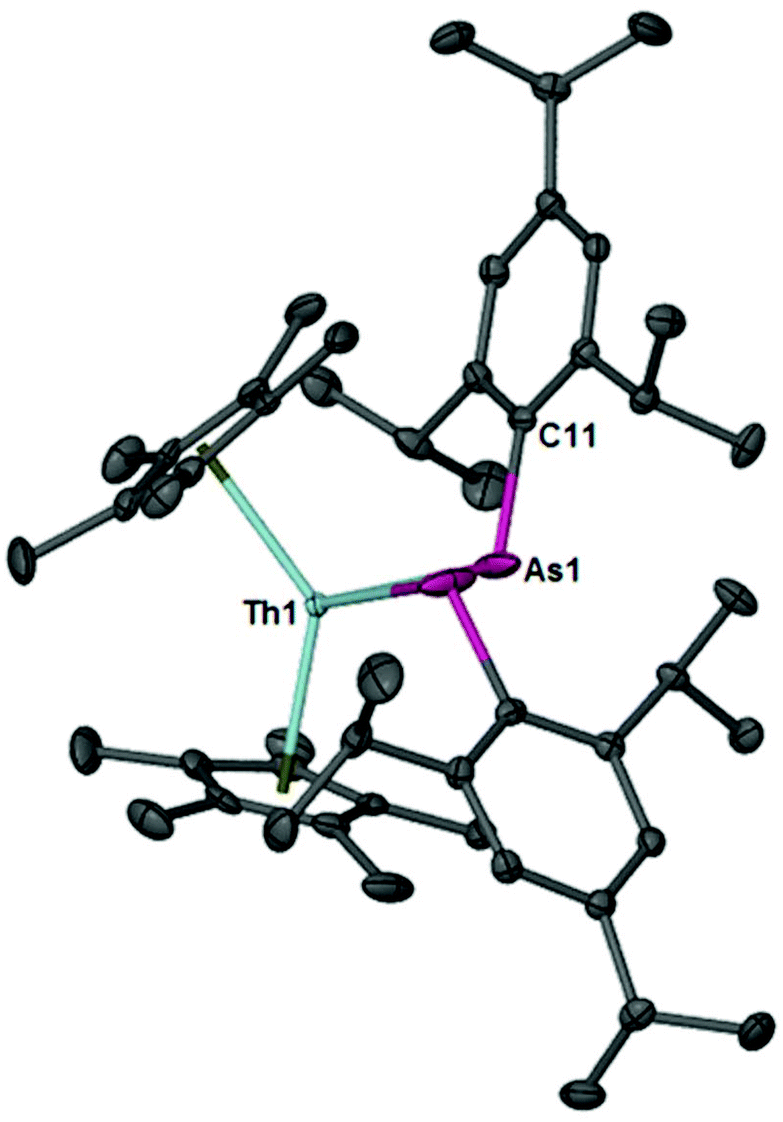 | ||
| Fig. 2 Thermal ellipsoid plot of 2 at the 50% probability level. Hydrogens have been omitted for clarity. Selected bond distances (Å) and angles (°): Th1–As1, 3.0028(6); As1–C11, 1.959(5); As1–Th1–As1*, 88.02(2); Th1–As1–C11, 116.53(15). | ||
We sought to investigate the reactivity of 1 and 2 through insertion reaction with CO surrogates. Waterman's group has reported on the generation of phosphaalkenes and arsaalkenes from the reaction between a primary phosphido/arsenido organometallic complex and an alkyl isocyanide.38,39 We anticipated a similar reactivity with our thorium complexes. When tert-butyl isocyanide was added to (C5Me5)2Th[P(H)Tipp]2 or 1 the solution underwent a color change to yellow, eqn (3). Initial spectroscopic experiments showed that one equivalent of the primary phosphine had been formed in the reaction. After recrystallization from a concentrated methylcyclohexane solution, the η2-(N,C)-phosphaazaallene thorium complexes [(C5Me5)2Th(CNtBu)(η2-N,C)-(tBuNC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PTipp)], 3, and [(C5Me5)2Th(CNtBu)(η2-N,C)-(tBuNCPMes)], 4, were isolated as yellow solids.
PTipp)], 3, and [(C5Me5)2Th(CNtBu)(η2-N,C)-(tBuNCPMes)], 4, were isolated as yellow solids.
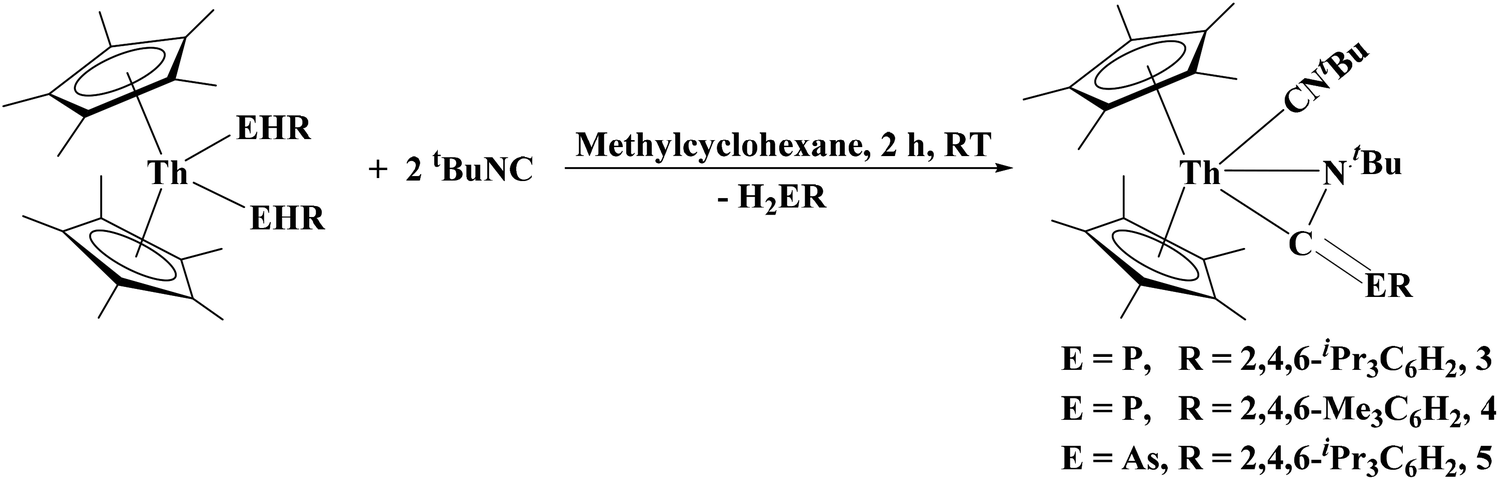 | (3) |
The diagnostic spectroscopic features associated with 3 and 4 include the stretches at 2181 and 2186 cm−1, and 1600 and 1602 cm−1, which can be assigned to the νCN and νCP stretches, respectively. The 31P NMR resonances shifted slightly upfield from the starting material to −21.28 and −10.70 ppm for 3 and 4, respectively. Additionally, the 13C NMR resonance of the central carbon of the phosphaazaallene was found at 150.85 and 151.05 ppm with 1JP−C = 152.3 Hz and 103.0 Hz, for 3 and 4, respectively. Compound 2 exhibited a similar reactivity to yield a η2-(N,C)-arsaazaallene thorium complex [(C5Me5)2Th(CNtBu)(η2-N,C)-(tBuNCAsTipp)], 5, as an orange solid. The spectroscopic features of 5 can be found in Table 1.
| 31P{1H} (δ) | 13C{1H} central allene carbon (δ) | ν CN (cm−1) | ν CE (E = P, As) (cm−1) | |
|---|---|---|---|---|
| 3 | −21.28 | 150.85, 1JP–C = 152.3 Hz | 2181 | 1600 |
| 4 | −10.70 | 151.05, 1JP–C = 103.0 Hz | 2186 | 1602 |
| 5 | 154.25 | 2182 | 1513 |
The solid-state structures of 4 and 5 were determined using X-ray diffraction studies, Fig. 3. Table 2 lists selected bond distances (Å) and angles (°). Compounds 4 and 5 are isostructural with one another and represent the first examples of actinide phospha- and arsaazaallene complexes. As with transition metals, such complexes are very rare as only two phosphaazaallene compounds have been isolated: (η1-Nacnac)Ti(CNtBu)(η2-N,C)-tBuNCPMes*)41 (Nacnac = [2,6-iPr2C6H3]NC(CH3)CHC(CH3)N[2,6-iPr2C6H3], Mes* = 2,4,6-tBu3C6H2) and Cp′2Nb(Cl)(η2-N,C)-PhNCPMes*).42
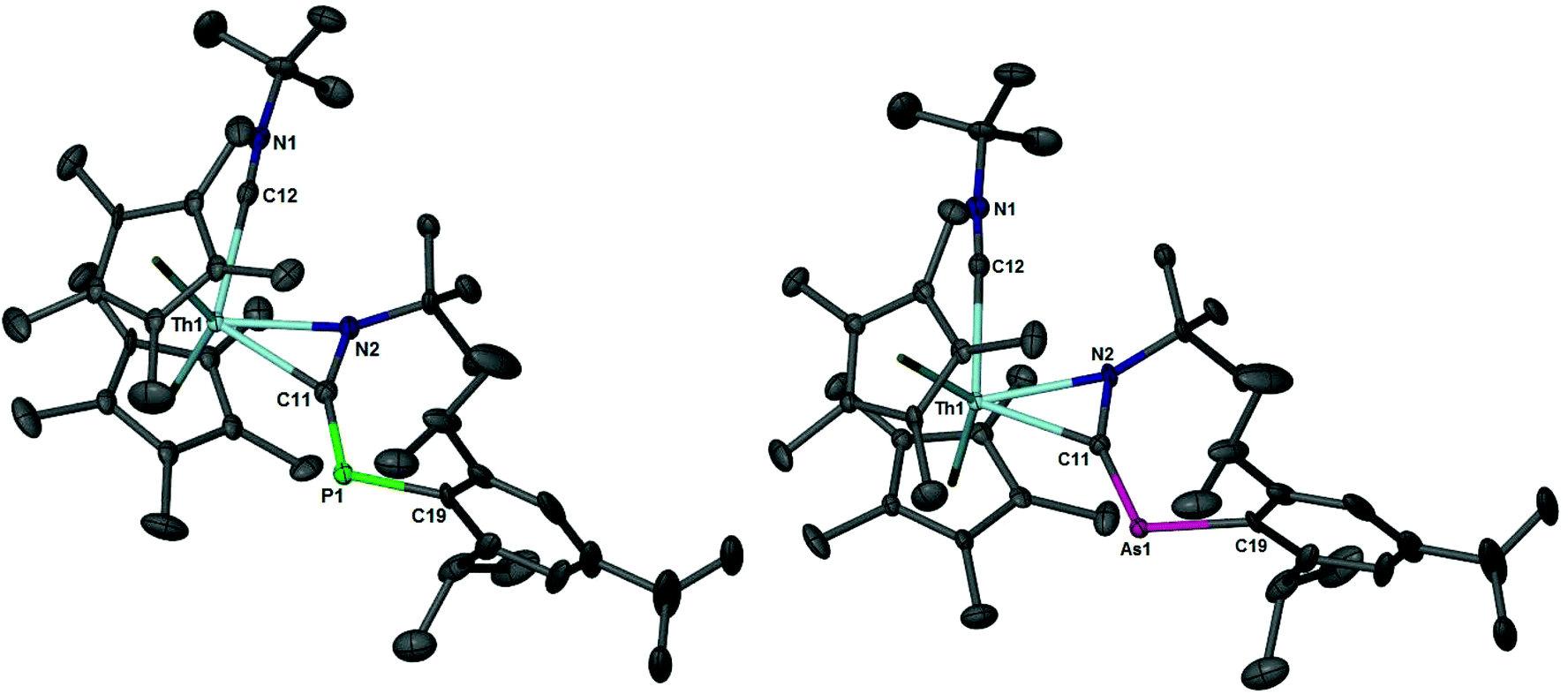 | ||
| Fig. 3 Thermal ellipsoid plots of 4 and 5 at the 50% probability level. Hydrogens have been omitted for clarity. | ||
| Th1–C11 | Th1–N2 | Th1–C12 | N1–C12 | N2–C11 | C11–E1 | N2–C11–E1 | C11–E1–C19 | |
|---|---|---|---|---|---|---|---|---|
| 4 | 2.430(6) | 2.346(5) | 2.643(6) | 1.131(8) | 1.348(8) | 1.691(6) | 152.1(5) | 115.8(3) |
| 5 | 2.419(5) | 2.364(4) | 2.638(6) | 1.128(7) | 1.347(7) | 1.822(5) | 150.3(4) | 114.5(2) |
There is a substantial elongation of the N–C bond (N2–C11) to 1.348(8) and 1.347(7) Å in 4 and 5, respectively, compared to a metal free heterocumulene such as PhNCPMes*, with a N–C bond distance of 1.210 Å.43 The N–C bonds in 4 and 5 are also longer than those found in products of isocyanide insertion into actinide–alkyl bonds. For example, 1.276(7) Å was observed in (C5Me5)(C8H8)U[η2-(N,C)-C(Ph)NtBu].44 Additionally the N–C–E bond angle is decreased to 152.1(5) and 150.3(4)°, respectively, relative to the N–C–E bond angle of 171.0° of PhNCPMes*. The CP bond distance in 4 of 1.691(6) Å is only slightly longer than the CP of 1.651(1) Å in PhNCPMes* and matches the CP bond length of 1.688(19) Å in (C5H4SiMe3)2Nb(Cl)[η2-(N,C)-PhNCP(2,4,6-tBu3C6H2)]. The Th1–N2 bond distances of 2.346(5) and 2.364(4) Å in 4 and 5, respectively, compare well to other thorium–amido bond lengths of 2.389(2) Å, [η5-1,2,4-(Me3C)3C5H2]2Th(Cl)[N(p-tolyl)SiH2Ph];45 2.322(5) Å, [η5-1,2,4-(Me3C)3C5H2]2Th[N(p-tolyl(Se–Se))];45 and 2.256(8) Å, (C5Me5)2Th[NC(Ph)(CH2Ph)]2.46
The formation of 4 and 5 is expected to occur through a 1,1 insertion of the alkyl isocyanide in the Th–P bond. Unlike the [(N3N)ZrEHR] (E = P, As; R = Cy, Ph) complexes which can undergo 1,2 rearrangement to phospha/arsa-azaalkenes, 4 and 5 do not undergo rearrangement, rather a double reduction of the alkyl isocyanide with the concomitant release of H2ER (E = P, As; R = Tipp, Mes). There are two conceivable reaction pathways for the generation of 4 and 5, Fig. 4. The first involves a transient terminal thorium–phosphinidene intermediate. There is a literature precedent for this route as Mindiola's group have reported the reaction of a terminal titanium phosphinidene, (η1-Nacnac)Ti(CH2tBu)(PMes*), with a tert-butyl isocyanide to yield the titanium phosphaazaallene complex, (η1-Nacnac)Ti(CNtBu)(η2-N,C)-tBuNCPMes*).41 The other route is 1,1 insertion of the isocyanide to form an η2-iminoacyl, followed by an intramolecular deprotonation. To investigate the possible reaction pathway, we attempted the addition of tert-butyl isocyanide to (C5Me5)2Th[P(H)Tipp]2 at −200 °C and slowly warmed the reaction while monitoring the reaction progress using 31P NMR. At −80 °C we observed the formation of 4, H2PTipp, (C5Me5)2Th[P(H)Tipp]2, and a singlet at −26.5 ppm. Upon heating to −70 °C the reaction was complete with disappearance of the resonance at −26.5 ppm. This resonance at −26.5 ppm has not been identified but it is possible that it is 4 without a coordinated isocyanide. We saw no evidence of a transient terminal phosphinidene as no resonance >100 ppm was observed (see ESI†).
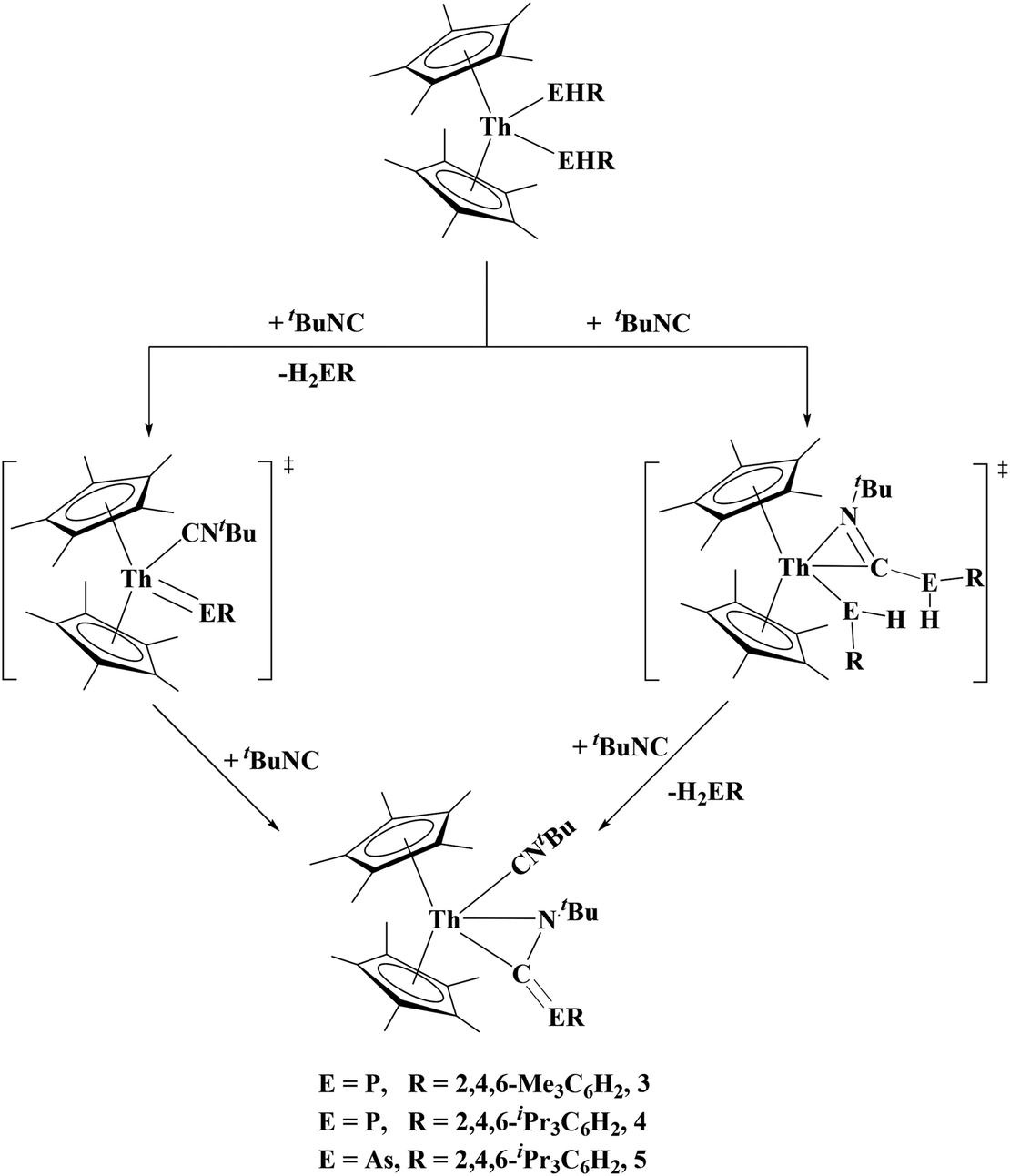 | ||
| Fig. 4 Possible reaction pathways for the generation of 3, 4 and 5. | ||
Conclusion
In summary we have broadened the scope of actinide–pnictogenide complexes by the isolation and characterization of new thorium primary phosphido and arsenido compounds. Both compounds exhibited spectroscopic diagnostic features in the infrared and heteronuclear NMR experiments. Insertion reactions of an alkyl isocyanide into the thorium–primary pnictogenide bond resulted in the formation of phospha/arsaazaallene complexes that do not exhibit any type of rearrangement. Further investigation is required to elucidate whether this reactivity is unique to the actinides or Lewis acids coordinated to two primary phosphido or arsenido ligands. Therefore group IV and alternative actinide metals are under investigation.Experimental
General considerations
The syntheses and manipulations described below were conducted using standard Schlenk and glovebox techniques. All the reactions were conducted in a Vacuum Atmospheres inert atmosphere (N2) glovebox or a double-manifold Schlenk line. Toluene, 1,2-dimethoxyethane, diethyl ether and hexane were purchased anhydrous, stored over activated 4 Å molecular sieves, and sparged with nitrogen prior to use. Methylcyclohexane was dried over activated 4 Å molecular sieves and sparged with nitrogen for thirty minutes prior to use. tert-Butyl isocyanide was dried over 4 Å molecular sieves, freeze–evacuate–thawed three times, distilled, and stored under nitrogen. All available reactants were purchased from suppliers and used without further purification. ThCl4(DME)2,47 (C5Me5)2ThCl2,48 (C5Me5)2ThMe2,48 TippPCl2,49 H2PTipp,50 MesPCl2,51 MesPH2,52 and (C5Me5)2Th[P(H)Tipp]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 (Tipp = 2,4,6-iPr3C6H2, Mes = 2,4,6-Me3C6H2) were synthesized as previously described. KPH(Mes) was prepared from H2PMes and KN(SiMe3)2 in THF. Benzene-d6 and toluene-d8 (Cambridge Isotope Laboratories) were dried over molecular sieves and degassed with three freeze–evacuate–thaw cycles. All 1H and 13C NMR spectra were obtained on a 500 or 600 MHz DRX Bruker spectrometer. All 31P NMR spectra were obtained on a 300 MHz ARX spectrometer at 121 MHz. 1H NMR shifts given were referenced internally to the residual solvent peak at δ 7.16 ppm (C6D5H). 13C NMR shifts given were referenced internally to the residual peak at δ 128.0 ppm (C6D6). 31P NMR spectra were externally referenced to 0.00 ppm with 5% H3PO4 in D2O. Infrared spectra were recorded as KBr pellets on a Perkin-Elmer Spectrum One FT-IR spectrometer. Elemental analyses were performed at the University of California, Berkeley Microanalytical Facility using a Perkin-Elmer Series II 2400 CHNS analyzer.
26 (Tipp = 2,4,6-iPr3C6H2, Mes = 2,4,6-Me3C6H2) were synthesized as previously described. KPH(Mes) was prepared from H2PMes and KN(SiMe3)2 in THF. Benzene-d6 and toluene-d8 (Cambridge Isotope Laboratories) were dried over molecular sieves and degassed with three freeze–evacuate–thaw cycles. All 1H and 13C NMR spectra were obtained on a 500 or 600 MHz DRX Bruker spectrometer. All 31P NMR spectra were obtained on a 300 MHz ARX spectrometer at 121 MHz. 1H NMR shifts given were referenced internally to the residual solvent peak at δ 7.16 ppm (C6D5H). 13C NMR shifts given were referenced internally to the residual peak at δ 128.0 ppm (C6D6). 31P NMR spectra were externally referenced to 0.00 ppm with 5% H3PO4 in D2O. Infrared spectra were recorded as KBr pellets on a Perkin-Elmer Spectrum One FT-IR spectrometer. Elemental analyses were performed at the University of California, Berkeley Microanalytical Facility using a Perkin-Elmer Series II 2400 CHNS analyzer.
Crystallographic data collection and structure determination
The selected single crystal was mounted on nylon cryoloops using viscous hydrocarbon oil. X-ray data collection was performed at 100(2) K. X-ray data were collected on a Bruker CCD diffractometer with monochromated Mo-Kα radiation (λ = 0.71073 Å). The data collection and processing were performed using a Bruker Apex2 suite of programs.53 The structures were solved by direct methods and refined by full-matrix least-squares methods on F2 using the Bruker SHELX-2014/7 program.54 All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed at calculated positions and included in the refinement using a riding model. Thermal ellipsoid plots were prepared using X-seed55 with 50% of probability displacements for non-hydrogen atoms. Crystal data and details of data collection for complexes 1, 2, 3, and 5 are provided in Table 3.| 1 | 2 | 3 | 5 | TippAsCl2 | |
|---|---|---|---|---|---|
| CCDC deposit number | 1455163 | 1455164 | 1455165 | 1455166 | 1455345 |
| Empirical formula | C38H54P2Th | C50H78As2Th | C45H71N2PTh | C45H71N2AsTh | C15H23AsBr0.55Cl1.45 |
| Formula weight (g mol−1) | 804.79 | 1061.00 | 903.04 | 946.99 | 349.15 |
| Crystal habit, color | Prism, orange | Prism, red | Needle, yellow | Needle, orange | Prism, colorless |
| Temperature (K) | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Space group | Pbcn | C2/c | Pnma | Pnma |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic | Orthorhombic | Triclinic |
| Volume (Å3) | 3598.3(5) | 4809.0(7) | 5659.3(6) | 5712.6(6) | 813.6(2) |
| a (Å) | 11.0230(9) | 23.195(2) | 28.9553(18) | 29.0966(19) | 8.3739(12) |
| b (Å) | 15.3941(13) | 12.1067(10) | 14.5673(9) | 14.5564(9) | 9.1197(13) |
| c (Å) | 21.2051(18) | 17.8732(15) | 13.4169(9) | 13.4878(9) | 11.6208(16) |
| α (°) | 90.00 | 90.00 | 90.00 | 90.00 | 75.718(2) |
| β (°) | 90.00 | 106.6350(10) | 90.00 | 90.00 | 71.5280(10) |
| γ (°) | 90.00 | 90.00 | 90.00 | 90.00 | 81.712(2) |
| Z | 4 | 4 | 4 | 4 | 2 |
| Calculated density (Mg m−3) | 1.486 | 1.465 | 1.060 | 1.101 | 1.425 |
| Absorption coefficient (mm−1) | 4.257 | 4.497 | 2.687 | 3.208 | 2.400 |
| Final R indices [I > 2σ(I)] | R = 0.0211 | R = 0.0195 | R = 0.0317 | R = 0.0330 | R = 0.0627 |
| R W = 0.0431 | R W = 0.0437 | R W = 0.0823 | R W = 0.0741 | R W = 0.1803 |
Acknowledgements
We gratefully acknowledge the U.S. Department of Energy, Office of Science, Early Career Research Program under award DE-SC-0014174 for support of this work.References
- V. Koshti, S. Gaikwad and S. H. Chikkali, Coord. Chem. Rev., 2014, 265, 52 CrossRef CAS.
- L. Rosenberg, ACS Catal., 2013, 3, 2845 CrossRef CAS.
- K. B. Dillon, F. Mathey and J. F. Nixon, in Phosphorus: The Carbon Copy, Wiley, Chichester, 1998 Search PubMed.
- L. Ackermann, Synthesis, 2006, 1557 CrossRef CAS.
- P. Garcia, P. R. Payne, E. Chong, R. L. Webster, B. J. Barron, A. C. Behrle, J. A. R. Schmidt and L. L. Schafer, Tetrahedron, 2013, 69, 5737 CrossRef CAS.
- N. R. Price and J. Chambers, in The Chemistry of Organophosphorus Compounds, ed. F. R. Hartley, Chichester, 1990, vol. 1 Search PubMed.
- D. E. C. Corbridge, in Phosphorus: Chemistry, Biochemistry, and Technology, CRC Press, Boca Raton, FL, 2013, 6th edn, p. 1473 Search PubMed.
- E. Rafter, T. Gutmann, F. Low, G. Buntkowsky, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2013, 3, 595 CAS.
- I. S. R. Karmel, M. Tamm and M. S. Eisen, Angew. Chem., Int. Ed., 2015, 54, 12422 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed.
- N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919 CrossRef CAS PubMed.
- N. H. Anderson, H. Yin, J. J. Kiernicki, P. E. Fanwick, E. J. Schelter and S. C. Bart, Angew. Chem., Int. Ed., 2015, 54, 9386 CrossRef CAS PubMed.
- B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 4484 CrossRef CAS PubMed.
- D. S. J. Arney, R. C. Schnabel, B. C. Scott and C. J. Burns, J. Am. Chem. Soc., 1996, 118, 6780 CrossRef CAS.
- M. R. Duttera, V. W. Day and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 2907 CrossRef CAS.
- D. Patel, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 13334 CrossRef CAS PubMed.
- N. Tsoureas, A. F. R. Kilpatrick, O. T. Summerscales, J. F. Nixon, F. G. N. Cloke and P. B. Hitchcock, Eur. J. Inorg. Chem., 2013, 2013, 4085 CrossRef CAS.
- A. S. P. Frey, F. G. N. Cloke, P. B. Hitchcock and J. C. Green, New J. Chem., 2011, 35, 2022 RSC.
- O. J. Scherer, B. Werner, G. Heckmann and G. Wolmershäuser, Angew. Chem., Int. Ed., 1991, 30, 553 CrossRef.
- S. W. Hall, J. C. Huffman, M. M. Miller, L. R. Avens, C. J. Burns, A. P. Sattelberger, D. S. J. Arney and A. F. England, Organometallics, 1993, 12, 752 CrossRef CAS.
- P. J. Hay, R. R. Ryan, K. V. Salazar, D. A. Wrobleski and A. P. Sattelberger, J. Am. Chem. Soc., 1986, 108, 313 CrossRef CAS.
- D. A. Wrobleski, R. R. Ryan, H. J. Wasserman, K. V. Salazar, R. T. Paine and D. C. Moody, Organometallics, 1986, 5, 90 CrossRef CAS.
- J. M. Ritchey, A. J. Zozulin, D. A. Wrobleski, R. R. Ryan, H. J. Wasserman, D. C. Moody and R. T. Paine, J. Am. Chem. Soc., 1985, 107, 501 CrossRef CAS.
- A. Formanuik, F. Ortu, R. Beekmeyer, A. Kerridge, R. W. Adams and D. P. Mills, Dalton Trans., 2016, 45, 2390 RSC.
- A. C. Behrle, L. Castro, L. Maron and J. R. Walensky, J. Am. Chem. Soc., 2015, 137, 14846 CrossRef CAS PubMed.
- B. M. Gardner, G. Balázs, M. Scheer, A. J. Wooles, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 15250 CrossRef CAS PubMed.
- B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, J. L. McInnesEric, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2015, 7, 582 CrossRef CAS PubMed.
- O. J. Scherer, J. Schulze and G. Wolmershäuser, J. Organomet. Chem., 1994, 484, c5 CrossRef CAS.
- B. A. Vaughan, E. M. Arsenault, S. M. Chan and R. Waterman, J. Organomet. Chem., 2012, 696, 4327 CrossRef CAS.
- A. J. Roering, S. N. MacMillan, J. M. Tanski and R. Waterman, Inorg. Chem., 2007, 46, 6855 CrossRef CAS PubMed.
- M. R. Douglass, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 2001, 123, 10221 CrossRef CAS PubMed.
- A. V. Firth and D. W. Stephan, Inorg. Chem., 1998, 37, 4732 CrossRef CAS PubMed.
- N. C. Zanetti, R. R. Schrock and W. M. Davis, Angew. Chem., Int. Ed., 1995, 34, 2044 CrossRef CAS.
- J. Ho, Z. Hou, R. J. Drake and D. W. Stephan, Organometallics, 1993, 12, 3145 CrossRef CAS.
- J. Ho and D. W. Stephan, Organometallics, 1992, 11, 1014 CrossRef CAS.
- G. A. Vaughan, G. L. Hillhouse and A. L. Rheingold, Organometallics, 1989, 8, 1760 CrossRef CAS.
- A. J. Roering, L. T. Elrod, J. K. Pagano, S. L. Guillot, S. M. Chan, J. M. Tanski and R. Waterman, Dalton Trans., 2013, 42, 1159 RSC.
- A. F. Maddox, J. J. Davidson, T. Shalumova, J. M. Tanski and R. Waterman, Inorg. Chem., 2013, 52, 7811 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef PubMed.
- F. Basuli, L. A. Watson, J. C. Huffman and D. J. Mindiola, Dalton Trans., 2003, 4228 RSC.
- J. B. Alexander, D. S. Glueck, G. P. A. Yap and A. L. Rheingold, Organometallics, 1995, 14, 3603 CrossRef CAS.
- M. Yoshifuji, T. Niitsu, K. Toyota, N. Inamoto, K. Hirotsu, Y. Odagaki, T. Higuchi and S. Nagase, Polyhedron, 1988, 7, 2213 CrossRef CAS.
- W. J. Evans, M. K. Takase, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 5802 CrossRef CAS.
- E. Zhou, W. Ren, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Organometallics, 2015, 34, 3637 CrossRef CAS.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Organometallics, 2004, 23, 4682 CrossRef CAS.
- T. Cantat, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2010, 46, 919 RSC.
- P. J. Fagan, J. M. Manriquez, E. A. Maatta, A. M. Seyam and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6650 CrossRef CAS.
- V. Chandrasekhar, P. Sasikumar, R. Boomishankar and G. Anantharaman, Inorg. Chem., 2006, 45, 3344 CrossRef CAS PubMed.
- Y. van den Winkel, H. M. M. Bastiaans and F. Bickelhaupt, J. Organomet. Chem., 1991, 405, 183 CrossRef CAS.
- S. T. Liddle and K. Izod, Organometallics, 2004, 23, 5550 CrossRef CAS.
- J. D. Masuda, K. C. Jantunen, O. V. Ozerov, K. J. T. Noonan, D. P. Gates, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 2408 CrossRef CAS PubMed.
- APEX2 Suite, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1455163–1455166, 1455345. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00776g |
| This journal is © The Royal Society of Chemistry 2016 |