
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA very peculiar family of N-heterocyclic phosphines: unusual structures and the unique reactivity of 1,3,2-diazaphospholenes
D.
Gudat
Institut für Anorganische Chemie, University of Stuttgart, Pfaffenwaldring 55, 70550 Stuttgart, Germany. E-mail: gudat@iac.uni-stuttgart.de
First published on 28th January 2016
Abstract
This Perspective gives an account of the peculiar electronic and molecular structures of N-heterocyclic phosphines featuring either a single 1,3,2-diazaphospholene (DAP) ring with an exocyclic P-substituent X or two DAP rings linked by a P–P bond (bis-diazaphospholenyls), respectively, and their impact on the chemical properties of these molecules. The bonding situation in simple DAPs is epitomized by strong hyperconjugation between endocyclic π-type electrons and the exocyclic P–X bond. This interaction may induce a perceptible ionic polarization of the P–X bond which can persist even in the limit of a vanishing electronegativity gradient between P and X, and becomes visible in unusual geometric distortions of molecular structures and a unique chemical behaviour. Structural distortions are particularly evident in bond lengthening effects in P-halogen and P-phosphino derivatives R2P–DAP (with R2P ≠ DAP) which span the whole range from covalent molecules to contact ion pairs with a close relation to frustrated Lewis-pairs. The most significant impact on the chemical properties is found for P-phosphino- and P-hydrogen derivatives where reactions at substantially accelerated rates or totally new reaction modes can be observed, and new stoichiometric and first catalytic processes exploiting these features are currently emerging. The recently discovered bis-diazaphospholenyls differ from the simple derivatives as their central bond remains unpolarised as a consequence of the symmetric molecular structure. The occurrence of low-energy P–P bond homolysis that was nonetheless observed in one case is according to the results of thermochemical studies of P–P bond fission reactions attributable to the effects of steric congestion and induces chemical reactivity that can be considered complementary to that of the simple R2P–DAPs. Some concluding remarks will pay attention to a facet of DAP reactivity that has so far been widely neglected but is currently receiving increasing attention, namely well-defined ring-opening processes.
 D. Gudat | Dietrich Gudat received his Diploma and Doctoral degrees from the University of Bielefeld under the supervision of Prof. Edgar Niecke. After conducting postdoctoral research, including a period at Iowa State University with Prof. John G. Verkade, he went to the University of Bonn where he finished his Habilitation in 1995. Since 2002, he is a professor of Inorganic Chemistry at the University of Stuttgart, Germany. His research interests include the chemistry and application of compounds of main group elements and their complexes (with a strong focus on phosphorus compounds), and the application of multinuclear NMR spectroscopy in molecular inorganic chemistry. |
Introduction
Aminophosphines (or phosphinous amides) P(NR2)nR′3−n (n = 1–3) featuring directly bonded tervalent phosphorus and nitrogen atoms were discovered by Michaelis more than a century ago1 and have to date been developed into a class of compounds that joins structural diversity with profound chemical applications.2 Specific attention has been devoted to heterocyclic derivatives which combine the good chemical stability and synthetic accessibility of P–N bonded frameworks with well-defined conformations and molecular shapes imposed by the constraints of the cyclic structure. Prominent examples include phosphazanes3 with rings of alternating phosphorus and nitrogen atoms (e.g.I, IIScheme 1), and species whose rings are formed by fusing phosphazane type N–P–N units with Cn-moieties (e.g.III–V), and which we will further denote as N-heterocyclic phosphines (NHPs) in a narrower sense. Note that phosphazane rings are obviously even-membered while NHPs may have even- or odd-membered rings with saturated or unsaturated Cn-chains. Generally, 4- to 6-membered rings are most frequent and most stable, but larger ring systems are also known.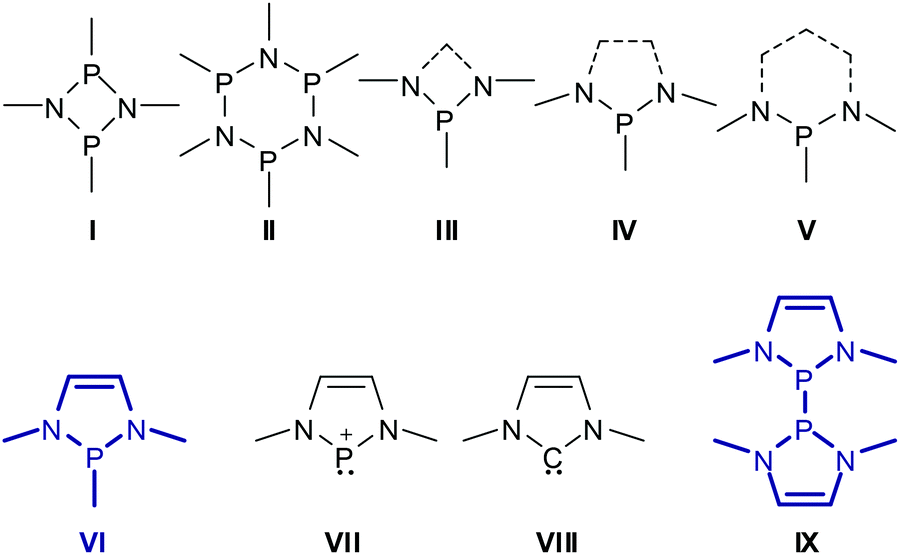 | ||
| Scheme 1 Molecular structures of generic phosphazanes (I, II) and organic N-heterocyclic phosphines (III–V) with four- to six-membered rings, N-heterocyclic phosphines covered in this review (VI, IX), and phosphenium ions derived from these by abstraction of a P-substituent (VII). Dashed lines may be single or double bonds. | ||
In this review, we will focus on NHPs featuring five-membered rings and C2-units with a double bond, the 1,3,2-diazaphospholenes (DAPs) VI. The first isolable DAPs were reported in the 1980s4 but initially received only moderate attention. This began to change when it was unveiled5 that P-halogen-substituted DAPs are useful precursors to N-heterocyclic phosphenium cations VII which are isoelectronic analogues of imidazoyl carbenes VIII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and they were intensely studied in the wake of the booming interest in carbene chemistry set off by the discovery of the first stable imidazoyl-ylidene.7 During these explorations, it became clear that neutral DAPs VI make an appealing research target on account of their own properties. Although the molecular structure of VI closely resembles at first glance (neglecting the extra double bond) that of NHPs IV with saturated C2-backbones, DAPs exhibit unusual structural features and chemical reactivity which differ in many cases greatly from those of the saturated analogues and give way to new reaction modes that are unprecedented for phosphines. The key to understanding this behaviour is based on the insight into the electronic interaction between the σ-bonding electron pair of the exocyclic P–X bond and delocalized π-type orbitals in the heterocycle, which may in turn induce ionic polarization of the former. The analysis of this effect by computational studies and its manifestation in observable structural trends – in particular for P-halogen- and P-phosphino-substituted DAPs – constitute the keynote of this review. Following an account of DAP synthesis, we will then reflect on the appealing chemical reactivity of P-phosphino- and P-hydrogen-substituted DAPs which gives rise to the development of new applications in synthetic chemistry and catalysis. Finally, we shall consider the special case of symmetrical bis-diazaphospholenyls (IX) and analyse the factors responsible for the easy homolytic cleavage of the central P–P bond which in this case cannot be attributed (for symmetry reasons) to ionic bond polarization. The advances in the chemistry of heterocyclic phosphenium ions VII were reviewed elsewhere8 and will not be repeated here.
6 and they were intensely studied in the wake of the booming interest in carbene chemistry set off by the discovery of the first stable imidazoyl-ylidene.7 During these explorations, it became clear that neutral DAPs VI make an appealing research target on account of their own properties. Although the molecular structure of VI closely resembles at first glance (neglecting the extra double bond) that of NHPs IV with saturated C2-backbones, DAPs exhibit unusual structural features and chemical reactivity which differ in many cases greatly from those of the saturated analogues and give way to new reaction modes that are unprecedented for phosphines. The key to understanding this behaviour is based on the insight into the electronic interaction between the σ-bonding electron pair of the exocyclic P–X bond and delocalized π-type orbitals in the heterocycle, which may in turn induce ionic polarization of the former. The analysis of this effect by computational studies and its manifestation in observable structural trends – in particular for P-halogen- and P-phosphino-substituted DAPs – constitute the keynote of this review. Following an account of DAP synthesis, we will then reflect on the appealing chemical reactivity of P-phosphino- and P-hydrogen-substituted DAPs which gives rise to the development of new applications in synthetic chemistry and catalysis. Finally, we shall consider the special case of symmetrical bis-diazaphospholenyls (IX) and analyse the factors responsible for the easy homolytic cleavage of the central P–P bond which in this case cannot be attributed (for symmetry reasons) to ionic bond polarization. The advances in the chemistry of heterocyclic phosphenium ions VII were reviewed elsewhere8 and will not be repeated here.
Structural features and bonding situation–experimental observations and computational insight
Analyses of electron densities or wave functions obtained from DFT or MP2 calculations with the NBO and NRT formalism suggest that the electronic structure of simple DAPs can be described in the framework of a VB picture by resonance between a covalent canonical structure A with a series of zwitterionic canonical structures, the leading ones of which were identified as B–D (Scheme 2; D does not apply to X = H).9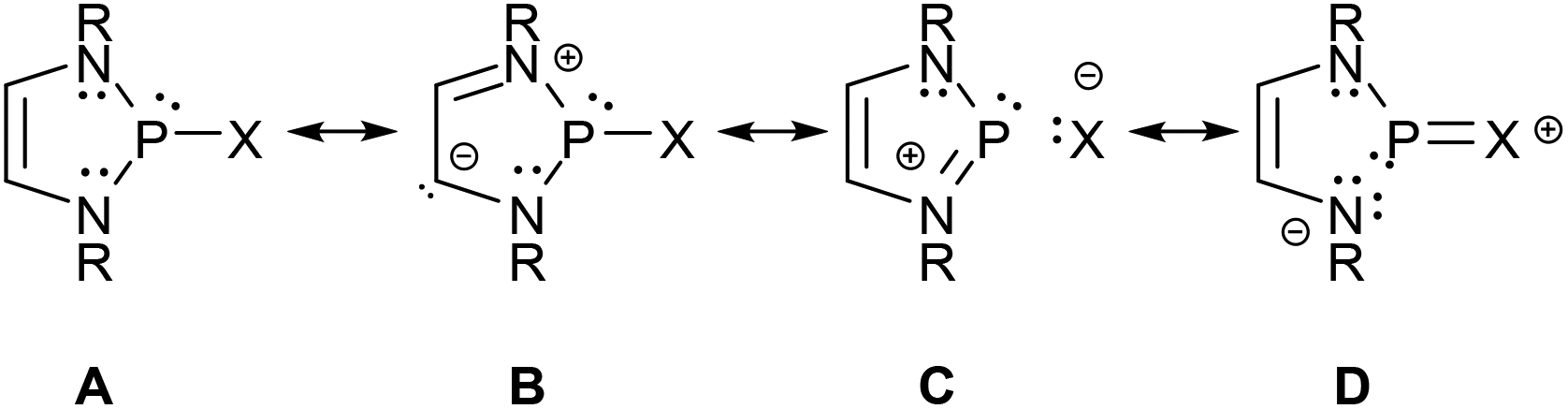 | ||
| Scheme 2 Leading canonical formulae for a VB-description of the bonding in DAPs. Mirror images of structures B–D were omitted for simplicity (see ref. 9 for further details). | ||
While the polar structure B reflects the effect of π-conjugation in the ene-diamine unit, the “no-bond” structures C and D embody n(N)–σ*(P–X) and n(X)–σ*(P–N) hyperconjugation interactions that tend to reduce covalent bond orders and increase the charge polarization in the P–X (in C) and P–N σ-bonds (in D), respectively. Similar no-bond resonance structures can also be devised for other aminophosphines and this description is thus quite generally applicable, but DAPs are special as the extent of n(N)–σ*(P–X) interaction is unusually large.† This trend has been related to the unique stability of the weakly aromatic6 1,3,2-diazaphospholenium ion which constitutes the cation fragment in structure C. Putting it simply, the possibility of cyclic 6π-delocalization in this fragment yields extra energetic stabilization that is unavailable for other types of aminophosphines but requires the incorporation of the σ*(P–X) fragment orbital into the 6π-system. The associated implicit transfer of electron density into this orbital lowers the covalent P–X bond order and transfers an extra negative partial charge to X so that the additional stabilization can only be reached at the expense of an increase in P–X bond iconicity. The degree of this electronic polarization varies strongly with the nature of the exocyclic substituent X (and, to a lesser extent, the substituents on other ring atoms), but the effect itself was considered as crucial in the explanation of the unique reactivity of all DAPs, regardless of the nature of X.9
Specific structural distortions related to the bonding situation in DAPs include averaging of endocyclic distances with respect to standard single and double bond lengths and a distinctive lengthening of P–X distances, and the recognition of these distortions was considered to be of a high diagnostic value in the verification of the proposed bonding model.9 Changes in the heterocycle geometry may be tiny and undetectable in X-ray diffraction data, in particular for DAPs with exocyclic bonds of a low intrinsic polarity (e.g. P–H, P–C), but distinct P–X bond lengthening has been reported even then (cf. Table 1).
| Substituent X | P–X [Å] | |
|---|---|---|
| In DAPsa | Reference data | |
| a Data from ref. 9 if not stated otherwise. b Average and standard deviation for P–Cl distances in diaminochlorophosphanes (R2N)2ClP, data from ref. 12. c Data for 2-chloro-1,3,2-diazaphospholidines from ref. 13. d Average and standard deviation for P–H distances in three-coordinate phosphorus compounds HnY3−nP, data from ref. 14. e P–H distance in a diazaphospholidine, data from ref. 15. f Data for alkynyl phosphines from ref. 16. g Includes Ph2P, phospholyl, and 1,2,4-triphospholyl substituents. h Average and standard deviation for P–P distances in diphosphines X2PPX2 (X = any substituents; data from ref. 17. i Distances in violet phosphorus and fibrous phosphorus, data from ref. 18. | ||
| Cl | 2.24–2.76 | 2.13 ± 0.06b |
| 2.20–2.31c | ||
| H | 1.48–1.51 | 1.288 ± 0.09d |
| 1.34e | ||
C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR CR |
1.80–1.8316 | 1.72–1.80f |
| PR2g |
2.31–2.79 | 2.21 ± 0.02h |
| 2.18–2.31i | ||
In the following section, we will deal more specifically with the structural features of P-chloro- and P-phosphino-DAPs which shed light on the effects of hyperconjugation in the limiting cases of high or negligible electronegativity gradients in the P–X bond, respectively.
The P–Cl and P–N distances in P-chloro-substituted DAPs 112 and analogous 1,3,2-diazaphospholidines 213 obtained from XRD studies display not only a notable variability but also an inverse correlation (Fig. 1) which supports the view that the changes reflect the tuning of n(N)–σ*(P–Cl) hyperconjugation by different substituents.‡ The magnitudes and variance of P–Cl distances in DAPs (Table 1) suggest that the impact of hyperconjugation is obviously much larger than in the CC-saturated reference compounds.12 A computational study supported this argument, but also revealed that a quantitative assessment of the observed P–Cl distances is only feasible if besides intramolecular effects intermolecular interactions like solvation or dipolar polarization by neighbouring molecules in the crystal are also taken into account.19 These considerations were backed by the experimental finding that the P–Cl distance in 1a (Fig. 1, R′ = H, R = tBu) changes by 6 pm upon incorporation of a solvate molecule (toluene) into the crystal lattice20 and led to the conclusion that the P–Cl bonds in DAPs exhibit a lower degree of covalency than in other diaminochlorophosphines and are thus “softer” (i.e. more easily polarizable).12 Based on this assumption, P-chloro-DAPs were described as Lewis pairs consisting of a phosphenium cation and a chloride anion connected by a “dative” (coordinative) P–Cl bond.20
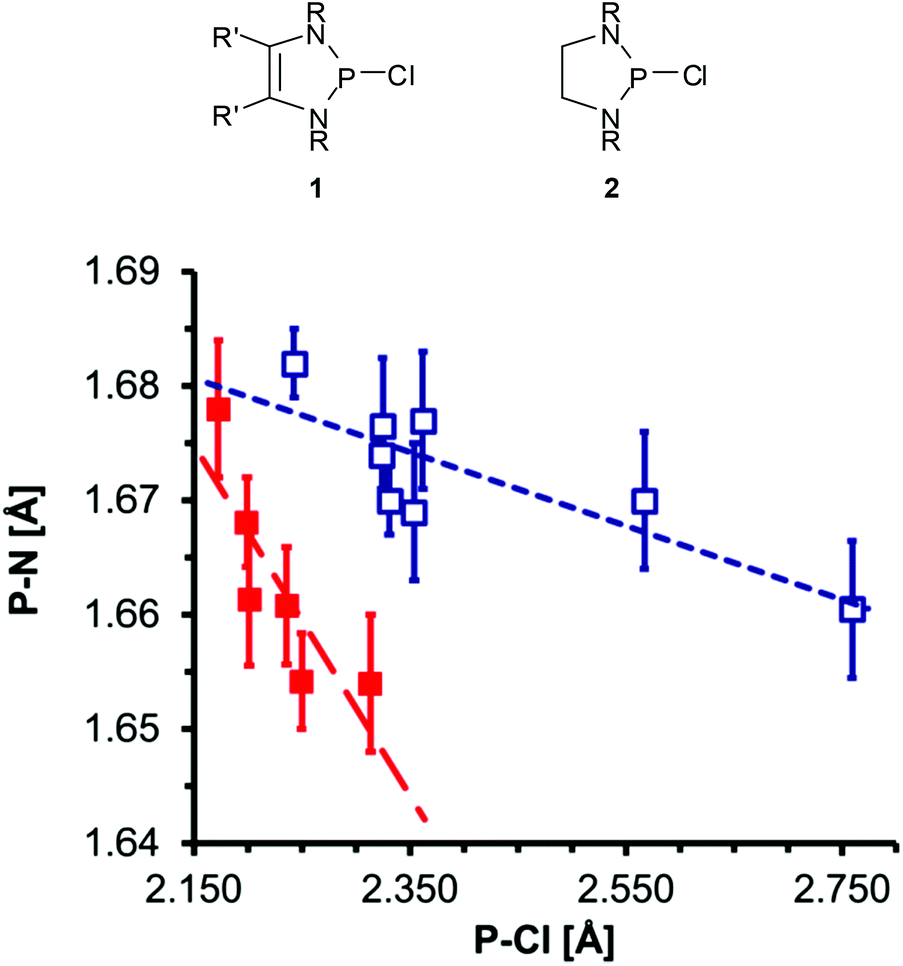 | ||
| Fig. 1 Plot of PCl distances vs. average PN distances in selected P-chloro-1,3,2-diazaphospholenes 1 (open squares; R = alkyl, aryl, R′ = H, Me) and 1,3,2-diazaphospholidines 2 (filled squares; R = alkyl, aryl). Error bars denote ranges of ±3σ; dashed lines represent the results of linear regression analyses. | ||
While P-fluoro-derivatives were considered to be closer to the orthodox picture of covalent molecules,20 a similar description was also adopted for P-bromo- and P-iodo-DAPs.21 A unique example is the P-iodo derivative 3 (Fig. 2) whose solid state structure was portrayed as containing no isolated molecules but one-dimensional arrays of cations and anions which engage in Lewis acid–base interactions to form a coordination polymer. Computational studies confirm that the P⋯I bonding is mainly ionic and that aromatisation of the cation fragment contributes significantly to the bond polarization but can neither fully compensate for the electrostatic destabilising effect of the charge separation nor account alone for the observed bond lengthening in the solid state.
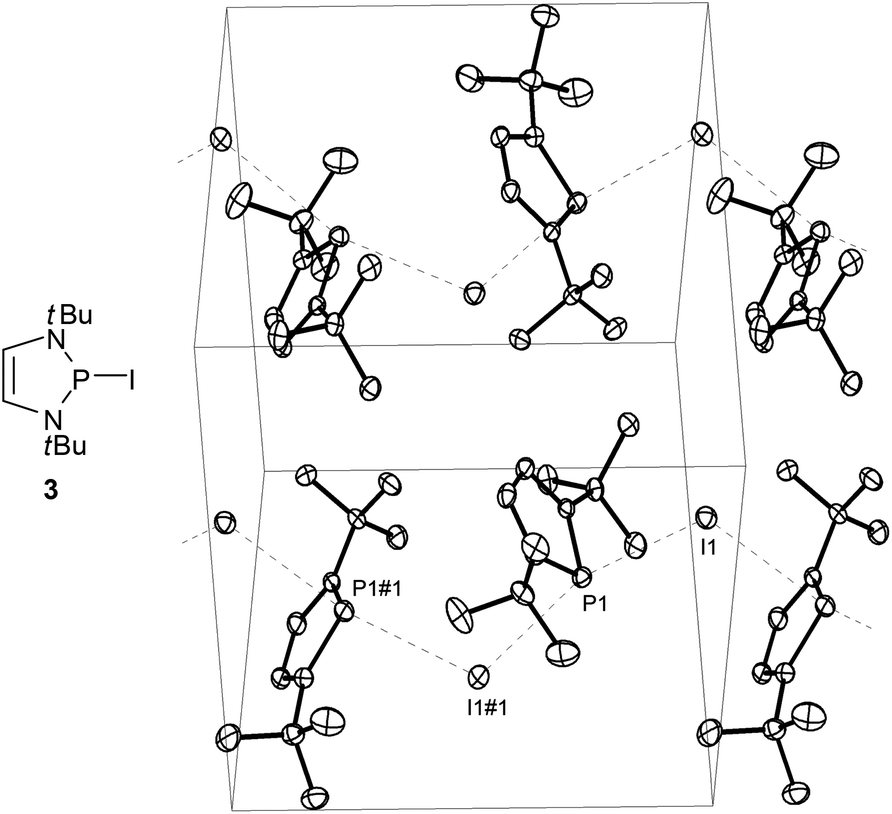 | ||
| Fig. 2 Molecular structure and representation of the unit cell of crystalline 2-iodo-1,3-di-t-butyl-1,3,2-diazaphospholene (3). Interactions between DAP fragments and iodine atoms parallel to the crystallographic b-axis are represented as dashed lines. Selected distances (Å) I(1)–P(1) 3.426(1), I(1)–P(1)#1 3.558(1). Reproduced with permission from ref. 21. Copyright 2009 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The P–P bonds in phosphino-DAPs show a similar overall variability as the P–Cl bonds in the chloro-derivatives (Table 1) but can be grouped into several subsets with different types of substituents (PR2 = PPh2, phospholyl, triphospholyl). The P–P distances in Ph2P-DAPs cover a narrow range (2.33–2.35 Å(ref. 22 and 23)) and are only moderately longer than the distances in crystalline red phosphorus modifications (cf. Table 1) or the sterically encumbered diphosphine [{(Me3Si)2CH}2P]2 (2.310 Å (ref. 24)). These features were taken as an indication that conjugative stabilization of the imidazolium fragment can enhance the weight of ionic resonance structures (C in Scheme 2, X = PR2) and introduce a certain degree of n(N)–σ*(P–X) hyperconjugation and P(δ+)–X(δ−) charge separation in a mainly covalent P–P bond even in the absence of intrinsic P–X bond polarity.22
Contrasting the structural rigidity of the PPh2-derivatives, the P–P distances in P-phospholyl-DAPs (2.31–2.70 Å (ref. 25)) are more flexible and can even exceed those in compounds with “one-electron bonds” (2.43–2.63 Å (ref. 26)) that formally join two phosphorus atoms by a single bonding electron. In connection with further geometrical distortions, the variability was interpreted as reflecting a greatly enhanced contribution of ionic resonance structures to the bonding which is driven by the gain of extra aromatic stabilization energy from 6π-delocalization in the phospholide unit.25 Collapsing all conceivable P–P non-bonded canonical formulae into a single delocalized “pseudo” resonance structure, this situation can be portrayed in the iconic form as shown in Scheme 3. Since the observable bond length distribution in the phosphole rings of phospholyl-DAPs remains closer to a conjugated diene than to an aromatic heterocycle, the covalent canonical structure E must still be regarded as the dominant one. The P–P bond may then be viewed as an intermediate case between covalent and dative bonding which combines a notable degree of covalency typical for a genuinely molecular species with the polarizability of a donor–acceptor bond in a Lewis pair.25 As in the case of P-halogeno-DAPs, this conjecture is backed by experimental findings, e.g. an unmatched variation in P–P distances in response to subtle crystal packing effects, and the occurrence of significant deviations of 1JPP couplings between solid-state and solution NMR spectra that are in part attributable to solvent-induced bond length relaxation in solution.25
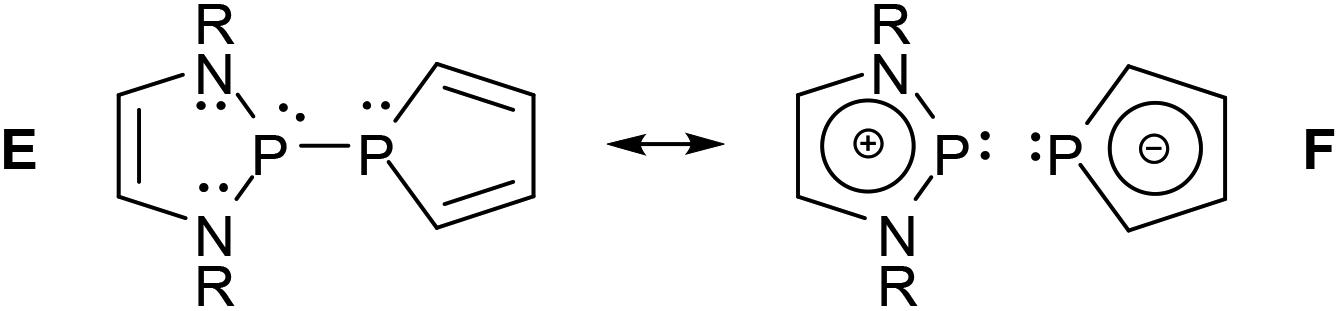 | ||
| Scheme 3 Covalent (E) and zwitterionic resonance structures (F) describing the bonding situation in P-phospholyl DAPs. The “pseudo” resonance structure F is meant to represent a superposition of several localized canonical formulae with differing electron distribution in order to highlight the effect of electron delocalization in the cation and anion fragments. | ||
A further shift to a predominantly ionic bonding situation was established for DAPs with sterically encumbered triphospholyl substituents (4a,b, Fig. 3).27 The extensive breakdown of covalent P–P bonding in this case was related to two factors, namely the higher aromatic stabilization energy of a triphospholide compared to a simple phospholide anion, and steric constraints which impede the formation of a strong covalent P–P bond by preventing a sufficiently close approach of the molecular fragments. Experimental evidence to support this bonding description includes a further rise in P–P distances (≥2.79 Å), the observation of small or even vanishing 1JPP coupling constants which exclude a strong P–P interaction in the solid state and in solution, and the occurrence of a dynamic intermolecular exchange of phosphenium and triphospholide fragments in solution. Adopting the use of arrows which has recently come into vogue to describe the bonding in main-group element compounds,284a can be portrayed as a phosphenium-triphospholide Lewis pair held together by a homonuclear donor–acceptor (or dative) bond, whereas 4b is best viewed as a tight contact ion pair that lacks any specific localized bonding interaction (according to computational predictions, the P–P distances exceed 3.2 Å (ref. 27)).
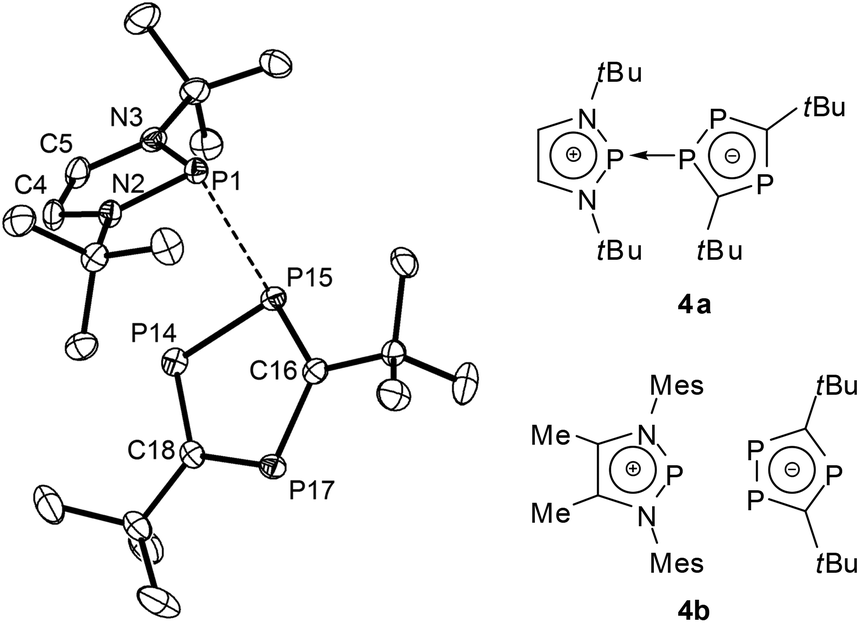 | ||
| Fig. 3 Representation of the structure of a crystalline diazaphospholenium triphospholide 4a (left, hydrogen atoms have been omitted for clarity; relevant bond distance: P1–P15 2.793(1) Å (ref. 27)) and molecular formulae of 4a,b (computed PP distances for 4b at the B3LYP/6-31+G* level range between 3.208 and 3.648 Å (ref. 27)). Structure reproduced with permission from ref. 27. Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
To sum it up, we note that the structural diversity of P-chloro- and P-phosphino-substituted DAPs evidenced by the unique flexibility of P–Cl and P–P distances depicts different stages of the way leading from covalently bonded molecules to molecular ion pairs. The unusually strong n(N)–σ*(P–X) hyperconjugation driven by the high stability of the diazaphospholenium cation fragment adds decisively to this effect but is not the only driving force – the full extent of the structural distortions can only be explained when other factors like the intrinsic bond polarity in chloro-DAPs, aromaticity of anion fragments and steric constraints in phosphino-DAPs, and intermolecular dipole interactions, are considered as well. An important corollary of this situation is that large structural distortions in specific classes of DAPs may help to detect and analyse the impact of hyperconjugation but are not inevitable, and the effect of hyperconjugation on chemical reactivity may still be visible in derivatives showing only limited structural distortion.
Synthetic aspects
Synthetically, DAPs are usually accessed from 1,4-diazadienes (α-diimines). The most widely used protocol is a two-step process consisting of reduction of the diazadiene to its dianion and subsequent metathetic ring closure (either directly as salt elimination29 or after quenching of the dianion with an acid as base-induced condensation,14 reaction (a) in Scheme 4) with a suitable electrophile. The reductant in the first step is typically lithium or sodium metal, but electrochemical reduction30 or an organometallic reagent (Cp2Co)31 have also been employed. Electrophilic ring closure can in principle be achieved with any dichlorophosphine RPCl2,30,32 but most frequently phosphorus trihalides were used to produce 2-halogeno-DAPs.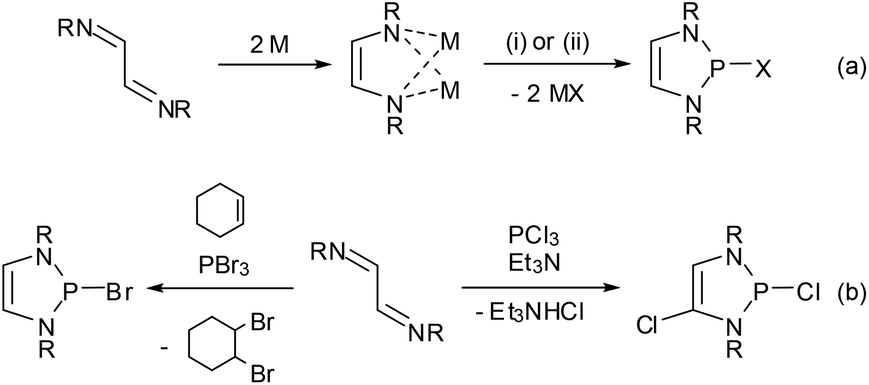 | ||
| Scheme 4 Generic syntheses of 2-halo-DAPs (a) in a two-step reaction via an intermediate diazadiene-dianion, (b) in one step from diazadienes and PX3 in the presence of auxiliary reagents (X = Cl, Br; R = alkyl, aryl; M = Li, Na). Reaction conditions: (i) PCl3; (ii) 2 Et3NHCl, then PCl3/2 Et3NHCl. | ||
Following an alternative approach, these products can also be generated in one step from diazadienes and phosphorus trihalides in the absence of a reductant. Using PCl320,33 or PBr334–36 together with auxiliary reagents like tertiary amines or cyclohexene gives di- or monohalogenated products, respectively (Scheme 4, reaction (b)).§ Reactions with PBr3 may also proceed without an auxiliary reagent.35,36 A recent computational study36 suggests that both reactions commence with an oxidative [4 + 1] addition to give cyclic phosphoranes (GCl,Br) which spontaneously ionize to yield di-halogeno-phosphonium halides (HCl,Br) as key intermediates (Fig. 4).
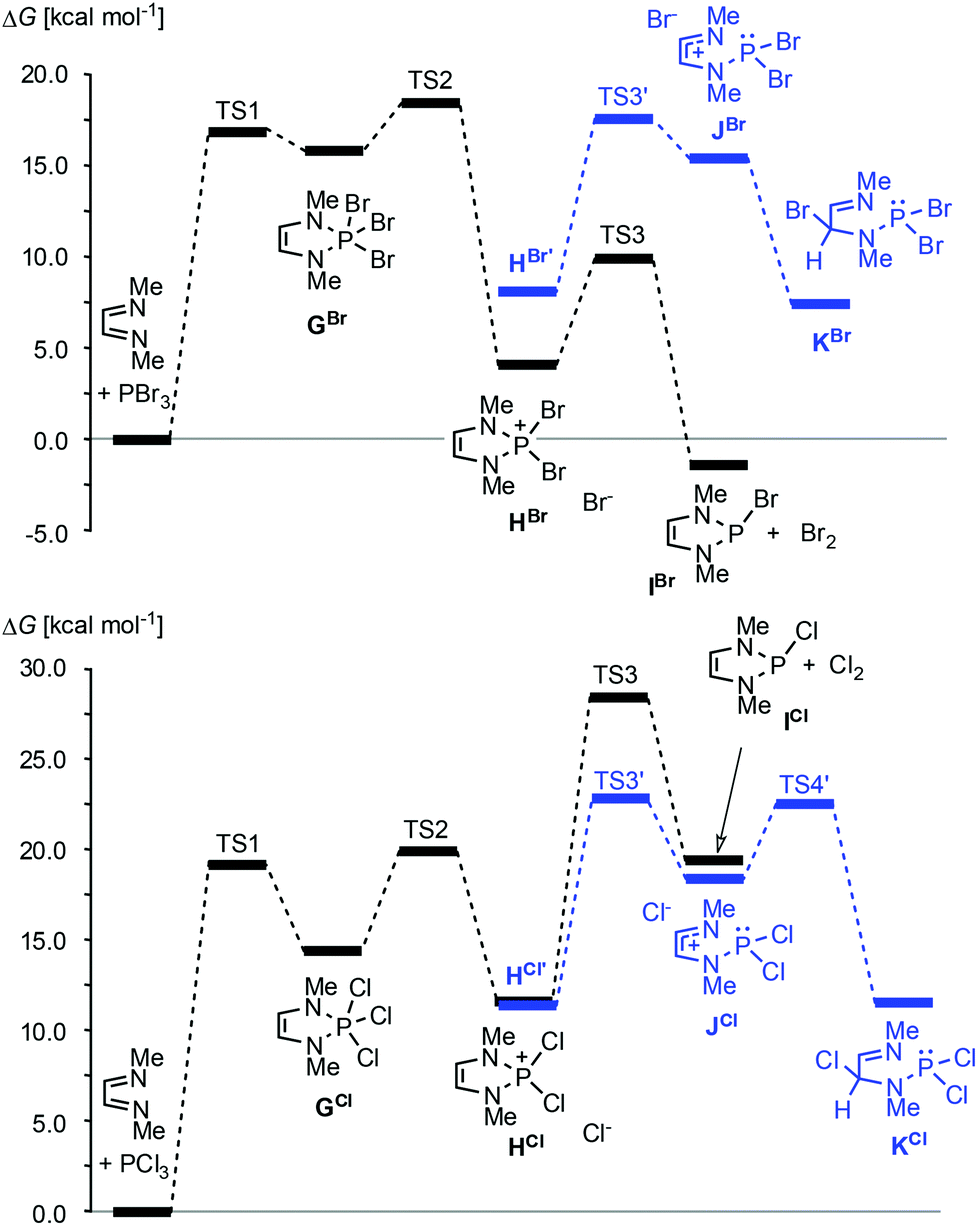 | ||
Fig. 4 Calculated minimum free energy pathways for the reactions of MeN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CHNMe with PBr3 (top) and PCl3 (bottom) at the B3LYP/def2-tzvp level. Solvent effects were modelled using a PCM approach. HX and HX’ (X = Br, Cl) denote contact ion pairs with different ion arrangements. Data from ref. 36. CH–CHNMe with PBr3 (top) and PCl3 (bottom) at the B3LYP/def2-tzvp level. Solvent effects were modelled using a PCM approach. HX and HX’ (X = Br, Cl) denote contact ion pairs with different ion arrangements. Data from ref. 36. | ||
Up to this stage, the reaction is an aza-variant of the well-known McCormack reaction. 2-Halogeno-DAPs (ICl,Br) can then be formed via a nucleophilic attack of the halide anion on a covalently bound halogen atom in the cation and the formation of X2. Alternatively, halogen migration from the phosphorus to the carbon atom may yield ring-opened products (KCl,Br) which may then cyclize to 2,4-dihalogeno-DAPs. The differing reactivity of PCl3 and PBr3 is due to the fact that the former channel is energetically unfavourable for reactions with PCl3.
The 2-halogeno-DAPs may further be subjected to electrophile induced halide abstraction yielding carbene-analogue phosphenium ions VII (Scheme 1),8a,9b or react with suitable nucleophiles under halide displacement to give products with a variety of functional P-substituents (e.g. H,14,38 PR2,22,23,25,27 NR2,10,11 OR,40 R,16,41 halogen20,21). Intriguing examples of nucleophilic substitution reactions of DAPs are the syntheses of phosphaketene 5 which serves as an entry for unique ring transformation reactions (vide infra)39 and boranyl derivatives 642 (Scheme 5). The latter may be viewed as formal III/V-analogues of tetraazafulvalenes or, alternatively, donor–acceptor adducts between NHC-analogue, boryl anions and phosphenium cations, but structural and computational studies indicate that they are best considered as boranyl phosphines with a covalent B–P single bond that has neither any dative nor a multiple bond character.
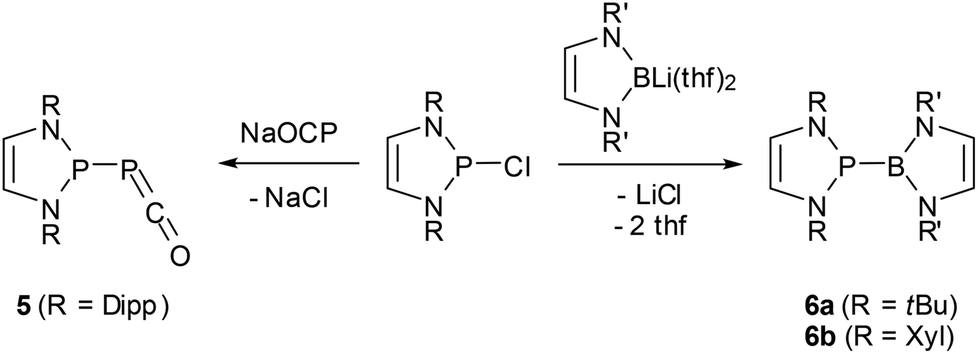 | ||
| Scheme 5 Syntheses of phosphaketene 539 and boranyl-DAPs 6a,b42 by nucleophilic halide displacement at P-chloro-DAPs. (Dipp = 2,6-di-isopropyl-phenyl; Xyl = 2,6-dimethylphenyl). | ||
The nucleophilic displacement of C-halogeno-substituents40 is also known but has apparently not found wide applications. In some cases, P-substituents other than halogens were replaced. Reactions of alkynyl-16 and cyclopentadienyl-DAPs41 under PC-bond cleavage and selective transfer of the organic residues to other substrates are examples of such processes which are of limited synthetic use but nonetheless conceptually interesting.
Reactivity of phosphino-DAPs – σ-bond metatheses, addition reactions, and more
The most prominent chemical asset of P-phosphino DAPs is their ability to react with various nucleophilic, electrophilic, or ambiphilic reactants under P–P bond fission. An example is the hydrolytic P–P bond cleavage (Scheme 6(i)) which occurs often spontaneously at or below ambient temperature.22,39 It is worthwhile to note that this reaction mode, like the other examples discussed in this section, is in principle well-known for diphosphines, and that the P–P bond polarization merely enhances this reactivity to enable reactions at appreciably increased rates and under unprecedentedly mild conditions. Even if high sensitivity toward hydrolysis can sometimes be a disadvantage (causing interference in reactions with other substrates), the H2O-induced P–P bond cleavage of 5 was fruitfully employed in the synthesis of a secondary phosphine oxide39 whose molecular structure matches that of derivatives with CC-saturated rings which are valuable pre-ligands for catalysis.43 Similar reactions occur with alcohols,22 and Grignard reagents39 or chloro-alkanes22 initiate P–P bond heterolysis under alkylation of the electrophilic or nucleophilic fragment, respectively. “Self-activation” was observed in the reaction of a phosphino-DAP with BH3-thf44 where the thermally stable substrates combine first to a transient phosphine-borane complex which then decays below room temperature via P–P bond fission and intramolecular hydride transfer (Scheme 6). The lability of the transient complex was attributed to the fact that coordination of the electrophile improves the stabilization of the negative charge on the PPh2-group and thus further facilitates ionic polarization and subsequent cleavage of the P–P bond.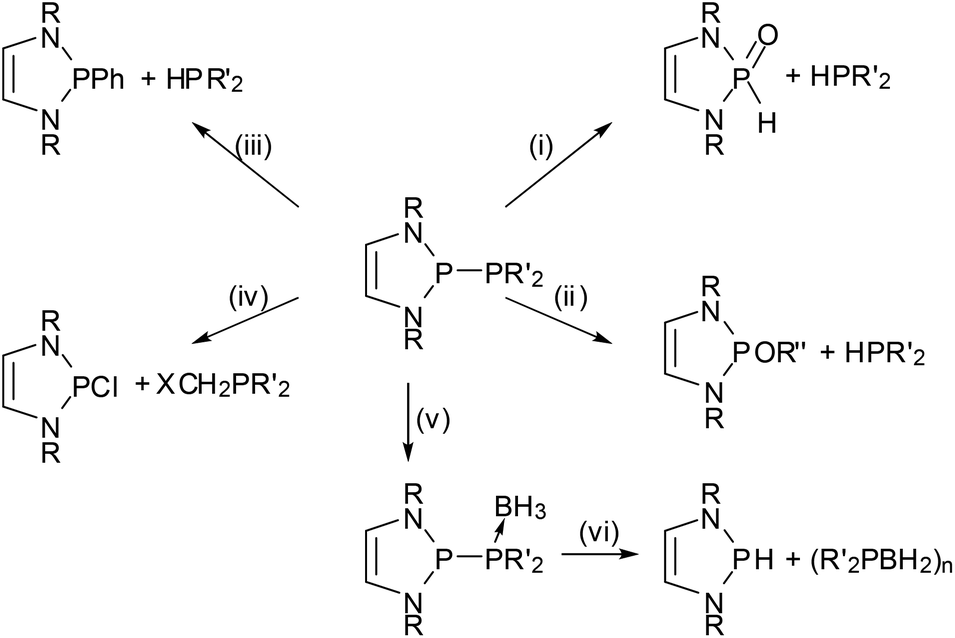 | ||
| Scheme 6 Selected metathesis reactions of phosphino-DAPs. Reagents and conditions: (i) H2O, r.t (R = Dipp, PR′2 = PCO); (ii) R′′OH, r.t. (R = tBu, Mes, R′ = Ph, R′′ = alkyl); (iii) PhMgBr, r.t. (R = Dipp, PR′2 = PCO); (iv) ClCH2X, r.t. (R = tBu, Mes; R′ = Ph, X = Cl); (v) BH3-thf, −10 °C (R = tBu, R′ = Ph), (vi) r.t. (R = tBu, R′ = Ph). | ||
The metathesis of phosphino-DAPs with chloroalkanes can be combined with the re-generation of the phosphino-DAP by condensation of the formed chloro-DAP with a trimethylsilyl-phosphine to yield a reaction cycle enabling a high yielding organocatalytic PC cross coupling reaction (Scheme 7).45
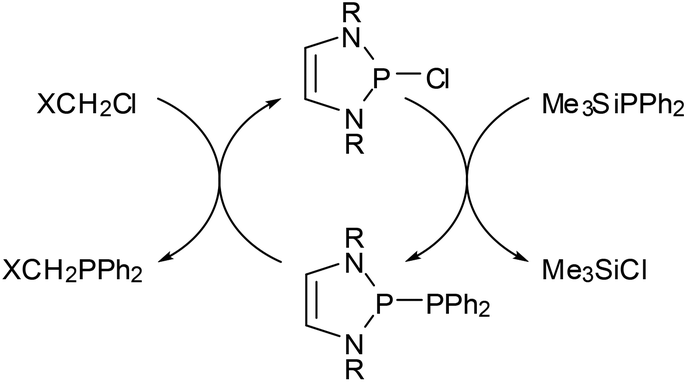 | ||
| Scheme 7 DAP-catalyzed phosphorus carbon cross coupling reaction (according to ref. 45; X = Cl, CN, Ph, C3H7, yields 80–99%). | ||
As the feasibility of substituent exchange is closely connected with the high stability of the diazaphospholenium fragment, this reaction was considered akin to the ligand exchange on phosphenium centres first reported by the Burford group.46 It is further remarkable as it represents the first catalytic transformation depending on the electrophilic rather than the nucleophilic character of an organophosphorus compound.52
Reactions of phosphino-DAPs with electron poor alkenes and alkynes occur via addition to the multiple bonds (“phosphino-phosphination”) and offer one-step access to unsymmetrical bis-phosphines from simple precursors (Scheme 8).22,23,47,48 As addition to di-activated alkynes is also known for other types of diphosphines,49 the special aspect here is that the unique reactivity of phosphino-DAPs widens the scope to include terminal alkynes as well as alkenes as new substrate classes. Additions to unsymmetrical alkenes and alkynes proceed regiospecifically in accordance with the P–P bond pre-polarization, and those to internal alkynes are Z-stereospecific. Stereospecific addition to activated internal alkenes is feasible if the double bond configuration is fixed by embedding into a ring. Acyclic alkenes undergo cis-trans isomerization, and the reaction gives mixtures of diastereomeric addition products some of which epimerized upon reaction with a Pd(II) salt to yield chelate complexes with a uniform backbone configuration.48 The isolation of a complex with an anionic bidentate ligand formed by deprotonation at the carbon atom carrying the activating (electron withdrawing) substituent provided a hint about the epimerization mechanism and indicated that the formed bis-phosphines are not inert.23
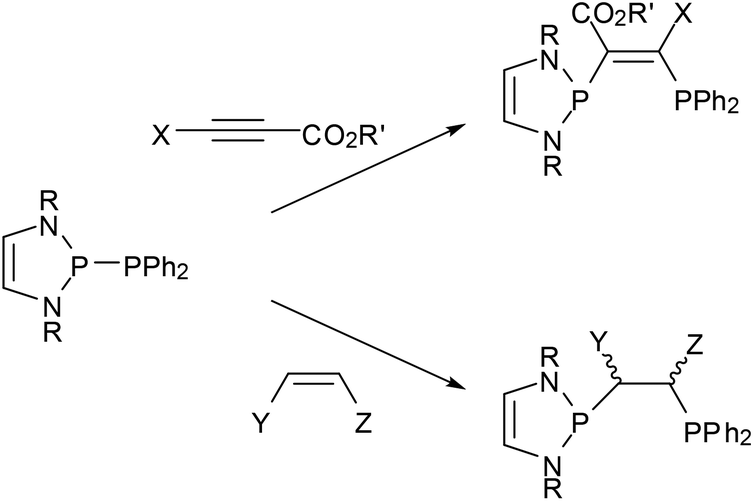 | ||
| Scheme 8 Addition of P-phosphino-DAPs (R = tBu, Mes, CH(Cy)Me) to activated alkynes (R′ = Me, Et; X = H, CO2R′) and alkenes (Y = CO2Me, Z = CO2Me, H; Y = CN, Z = H). | ||
Reactions of P-phosphino-DAPs with main-group or transition metal chlorides are also characterized by fragmentation to give chloro-DAPs and metal phosphides or phospholides,22,39,44,50 while chloride-free transition metal complexes with labile ligands react preferably via insertion of a metal fragment into the P–P bond.47,50 As a joint coordination of a nucleophilic and an electrophilic (phosphenium) P-based ligand leaves the formal metal oxidation state unchanged, these reactions were viewed to represent a process that had previously been termed51 as “non-oxidative addition”. An appealing case is the reaction of 7, acetonitrile, and [W(CO)4(1.5-cyclooctadiene)] where formation of the insertion product 9 is preceded by P–P addition to the nitrile triple bond and creation of a chelate complex 8 with a PCNP-backbone (Scheme 9).47 Even if the diazaphospholidine analogue of 7 with a CC-saturated ring showed quite similar reactivity,50 isolation of the addition product 8 was only feasible under mild reaction conditions granted by double activation of both the nitrile (by metal coordination) and the diphosphine (by P–P bond polarization). The phosphino-DAP offers in this respect no qualitatively new reactivity, but the simple acceleration of known reaction modes still suffices to produce interesting new results.
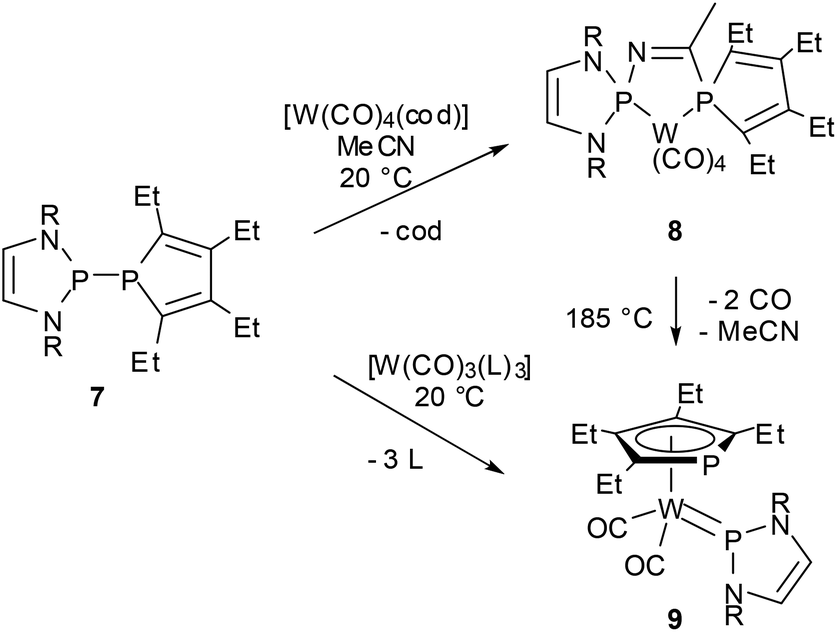 | ||
| Scheme 9 Metal insertion into the P–P bond of a phosphino-DAP (R = Mes, cod = 1,5-cyclooctadiene, L = MeCN). | ||
Reactivity of secondary DAPs – molecular hydrides
One of the earliest manifestations of the chemical consequences of the bonding situation in DAPs was the realization that the (N)-σ*(P–X) hyperconjugation in P-hydrogen-substituted derivatives (H-DAPs) suffices to suppress the weak Brønsted-acid character that is typical for secondary phosphines and bring the character of a hydride to the fore.38 Although such a substituent-induced change in effective bond polarity has precedence in the chemistry of other elements – e.g., reaction of R3GeH with carbonyl compounds can yield a germylcarbinol (for R = Cl) or a germyl ether (for R = Et), reflecting either the “proton” or the “hydride” character of the Ge–H bond53 – it had previously been unobserved for phosphines. In this respect, the discovery of stoichiometric and catalytic processes proves that “Umpolung”38 of the P–H bond reactivity in DAPs is not simply a perfection of already well-known chemistry, but a qualitatively new reaction mode. Accordingly, reactions with Brønsted acids or a triphenylcarbenium ion do not proceed as usual via quaternization at phosphorus but yield a phosphenium salt and H2 or triphenylmethane, respectively, and aldehydes and aryl ketones are reduced to phosphorodiamidites (alkoxy-DAPs) instead of forming α-phosphino-carbinols (Scheme 10).14,38 Lately, it has been shown that H-DAPs also enable the reductive hydrophosphination of CO2 (Scheme 10).54 Their potential as hydride donors is further illustrated by the occurrence of a reversible hydride exchange with phosphenium salts and their capability to react with other element halides via hydride/halide metathesis (cf. e.g. the selective reduction of SiCl4 to HSiCl3 shown in Scheme 10).14 Halides of main-group or transition metals may also be reduced to the element (e.g., SnCl2 or the SnCl3− salt shown in Scheme 10 can be cleanly reduced to elemental tin14) or to ligand-stabilized metal(0) complexes like 1055 (Scheme 10). Finally, the addition of the P–H bond across the NN double bond of azobenzenes (see Scheme 11) reveals that H-DAPs also have some potential to react with unpolar substrates.
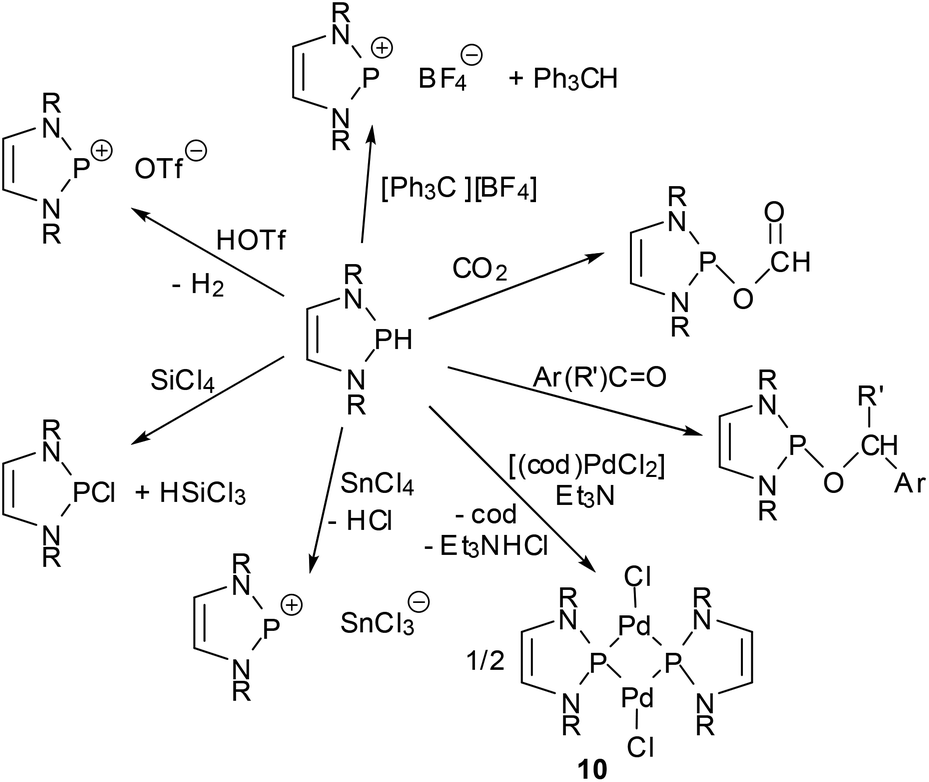 | ||
| Scheme 10 Selected stoichiometric reactions of P-hydrogen DAPs (R = alkyl, aryl; R′ = aryl, H; cod = 1,5-cyclooctadiene). | ||
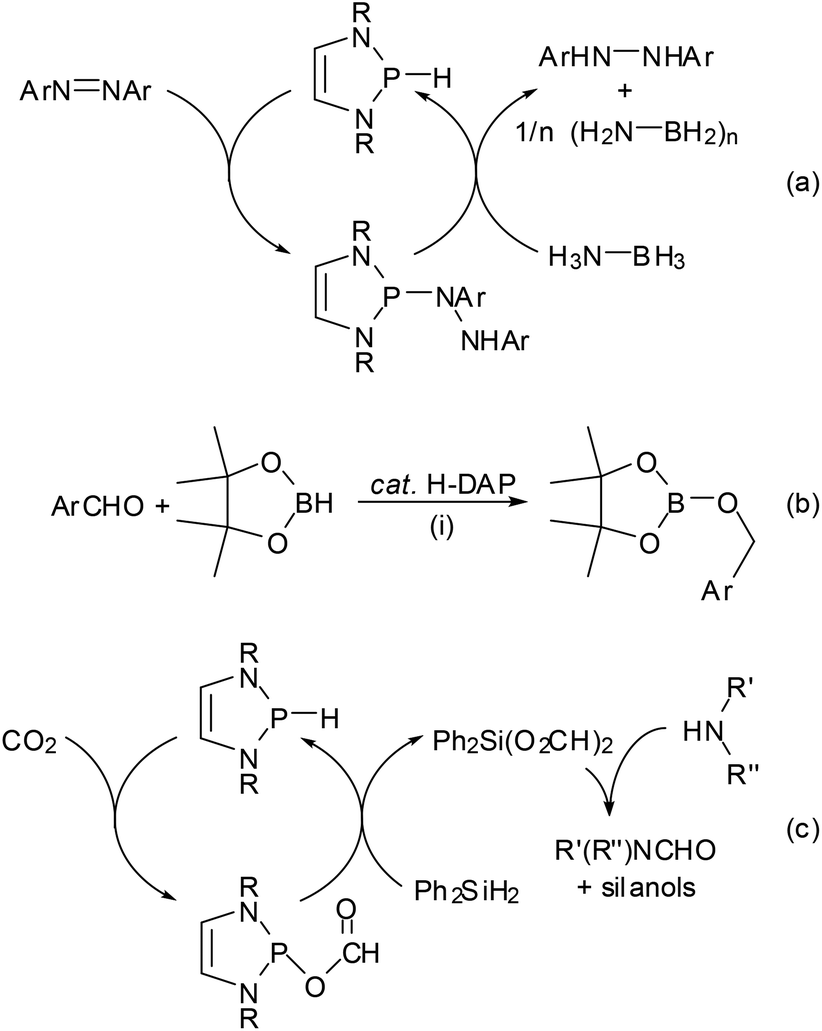 | ||
| Scheme 11 Organocatalytic reduction processes employing H-DAPs as the catalyst (R = tBu, Ar = aryl, R′, R′′ = alkyl, aryl; reaction conditions: (i) 0.5 mol% cat., r.t. for aldehydes and 10 mol% cat., 90 °C for ketones). | ||
As in the case of the previously mentioned PC cross coupling process (Scheme 7), reaction variants using H-DAPs as catalytic rather than stoichiometric reagents come in principle into reach if the actual reduction step can be coupled to a second process allowing the in situ re-generation of the reductant. Several elegant implementations of this concept were lately reported by the group of Kinjo. The reaction cycle displayed in Scheme 11(a) combines the hydrophosphination of azobenzenes with the reductive cleavage of the formed phosphine hydrazide by ammonia-borane under regeneration of the P–H bond and thus enables an organocatalytic transfer-hydrogenation of the azo-function with ammonia-borane as the hydrogen source.56 DFT-calculations suggest that the H-DAP may be formed via a concerted double hydrogen transfer which bears some similarity to that encountered in bifunctional metal–ligand catalysis57 with transition-metal based catalysts. The catalytic hydroboration of aldehydes and ketones (Scheme 11(b)) is based on the initial reduction of the substrate by the H-DAP (cf. Scheme 10) and metathesis of the formed alkoxy-DAP with pinacolborane to re-generate the reductant and release the final product;58 note that the high chemoselectivity of the reduction (H-DAPs reduce aldehydes and diarylketones much faster than aryl-alkylketones14) enables an unprecedented selective hydroboration of the CHO–group in 4-acetyl-benzaldehyde. Lately, it has also been shown that the product of the reductive hydrophosphination of CO2 reacts with diphenylsilane via formyl transfer to yield a mixture of the original H-DAP and a silyl formate Ph2Si(O2CH)2, and that both reactions can be combined to allow the catalytic hydrosilylation of CO2 (Scheme 11(c)). Quenching the silyl formate with amines permits further catalytic N-formylation of amines under metal-free and ambient conditions.54
Symmetrical bis-diazaphospholenyls
Bis-diazaphospholenyls (IX, Scheme 1) with two directly linked DAP rings differ in important aspects from simple phosphino-DAPs as their symmetric molecular structure must be assumed to inhibit substantial charge polarization of the central bond and limit the extent of the n(N)–σ*(P–P) hyperconjugation. It was, however, argued, that diphosphines IX offer an alternative mechanism to induce a P–P bond activating effect, namely by stabilizing the radical fragments arising from homolytic bond fission by delocalizing the unpaired electron in the π-electron system of the ring.59 In the following section, we will give an account of the exploration of the chemistry of the diphosphines IX which focused initially on the syntheses and studies of the bond breaking process and the electronic structure of the radicals, but shifted lately to pay more attention to the appealing chemical properties of these systems.Symmetrical bis-diazaphospholenyls were first obtained via the reductive coupling of Cl-DAP59 or a NHP cation precursor60 with magnesium or sodium, respectively. More recently, it has been found that these species are alternatively accessible through the photochemical dehydrocoupling of H-DAPs.61 Both reactions (Scheme 12) are not specific for DAPs but are more widely applicable, above all for syntheses of bis-diazaphospholidinyls without endocyclic double bonds.13,61 Computational studies61 suggest that the dehydrocoupling starts from a transient dimeric, weakly P⋯P bonded aggregate which is then photochemically excited and decays – possibly via an excimer – under cleavage of H2 and formation of the diphosphane.¶
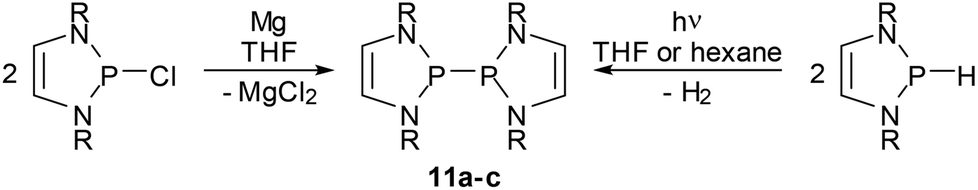 | ||
| Scheme 12 Syntheses of bis-diazaphospholenyls. Reactions are carried out at ambient temperature (R = tBu (11a), Mes (11b), Dipp (11c). | ||
Crystallographic studies reveal that bis-diazaphospholenyls can adopt trans- or gauche-conformations and that P–P distances (2.24–2.32 Å (ref. 59 and 62)) are similar as in bis-diazaphospholidinyls (2.24–2.32 Å (ref. 15)) and slightly shorter than in unsymmetrical Ph2P-DAPs (2.33–2.35 Å (ref. 22 and 23)). Although specific computational analyses of hyperconjugation effects are as yet lacking, these data suggest that their impact is presumably not very large.
The finding that 11c already dissociates at room temperature visibly into spectroscopically detectable radicals 11′c was viewed as the first qualitative evidence for low-energy P–P bond fission (cf. Scheme 13) of bis-diazaphospholenyls.59 However, even if the EPR hyperfine coupling pattern and computations support the description of 11′c as a 7π-radical,59,63 the markedly higher thermal stability of 11a,b (EPR signals of 11′a were only detected above 80 °C) and the observation of an even more extensive dissociation for an acyclic diphosphine (14b,63Scheme 13) imply that π-delocalization in the radical is not the major energetic driving force for facile P–P bond fission.
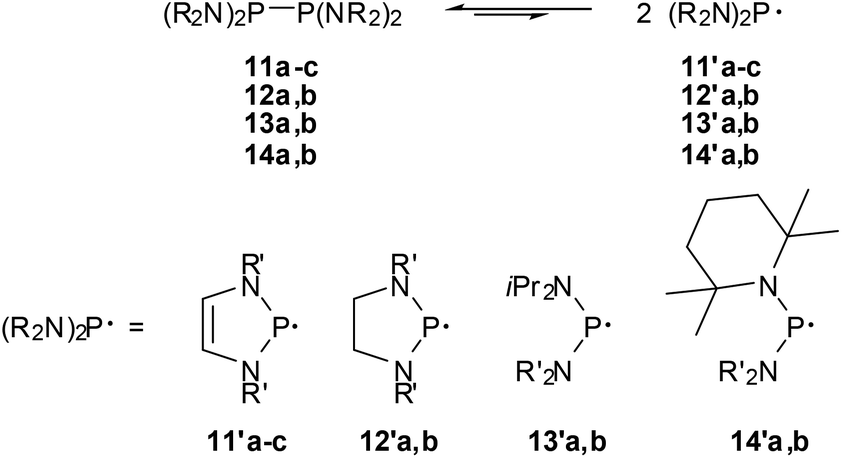 | ||
| Scheme 13 P–P bond dissociation equilibria for tetraamino-diphosphines (R′ = tBu (11a), 2,6-Me2C6H3 (11b), 2,6-iPr2C6H3 (11b, 12a), 2-tBu2C6H4 (12b), iPr (13a, 14b), SiMe3 (13b), Et (14a). | ||
This conjecture was confirmed by comparing the experimentally determined thermochemical parameters for the bond fission in 11c62 and a series of reference compounds63 (cf. Table 2). Analysis of trends in the data63 in connection with computational studies suggests that the evident shifts in the dissociation equilibria reflect both enthalpy and entropy effects, respectively, and that especially the extensive dissociation of 11c and 14b is driven by large entropy terms. A preference for radical formation arises chiefly from steric factors, namely a gain in the conformational flexibility of the radicals which increases the entropy term, and conformational changes (adaptation of bond angles and – in acyclic species – torsional angles in order to minimize steric congestion) which lower ΔHDiss, while electronic stabilization by π-conjugation is by far less important than in aminophosphenium cations. Consequently, the energetic impact of 7π-delocalization in the radical, which is assumed to account for most of the lowering in ΔHDiss in 11c (by 23–25 kJ mol−1 relative to the bis-diazaphospholidinyls 12), stands back behind the energetic (some 45 kJ mol−1 for 11–14) and entropic effects (changes in TΔS are 19–36 kJ mol−1 at 295 K) of steric congestion. Even if a further delineation of explicit energetic aspects of the low-energy dissociation and P–P bond strength of the studied diphosphines is tempting, it must be borne in mind that the greatest impact on the balance of the dissociation equilibria comes according to recent DFT-studies from entropy effects and the offsetting of any radical stabilizing influences by strong attractive dispersion forces,63,64 and the significance of specific electronic and steric substituent effects must not be overestimated.
| ΔG295Diss [kJ mol−1] | pK295Dissa |
ΔHDiss [kJ mol−1] | ΔSDiss [J (mol K)−1] | |
|---|---|---|---|---|
| a pK295Diss = −log (KDiss(295 K)/[mol l−1]). | ||||
| 11c | 26.8(8) | 4.8(8) | 78.8(8) | 176(15) |
| 12a | 69.4(21) | 12.3(22) | 102.3(11) | 112(4) |
| 12b | 88.6(17) | 15.7(28) | 104.9(10) | 55(3) |
| 13a | 70.2(21) | 12.4(4) | 108.9(12) | 131(4) |
| 13b | 69.6(28) | 12.3(5) | 95.4(14) | 87(5) |
| 14a | 66.9(23) | 11.9(4) | 99.5(12) | 110(4) |
| 14b | 13(7) | 2.3(12) | 64.2(30) | 174(13) |
Regardless of its origin, the facile dissociation of 11c into radicals provides for some interesting chemistry. Thus, its reactivity towards alkynes complements that of polarized phosphino-DAPs. Whereas the latter undergo cis-addition to electron poor alkynes but are unreactive towards unactivated substrates, 11c shows specific trans-addition to the triple bond of tolane. The observed stereochemistry suggests that this reaction follows not a dipolar but a radical mechanism (the reactivity of 11c towards electron poor alkynes differs likewise from that of phosphino-DAPs, but will be dealt with in the next section).62 The immediate availability of radicals was also established as crucial in explaining the reaction of 11c with Et3NHCl under formal disproportionation into hydrogen- and chloro-substituted DAPs, which enabled the development of a new access route to H-DAPs via reduction of chloro-substituted precursors with Et3NHCl/Mg (Scheme 14(a)).15 As radical formation is mainly promoted by steric rather than electronic factors, this method is also applicable to the synthesis of sterically congested secondary diazaphospholidines.
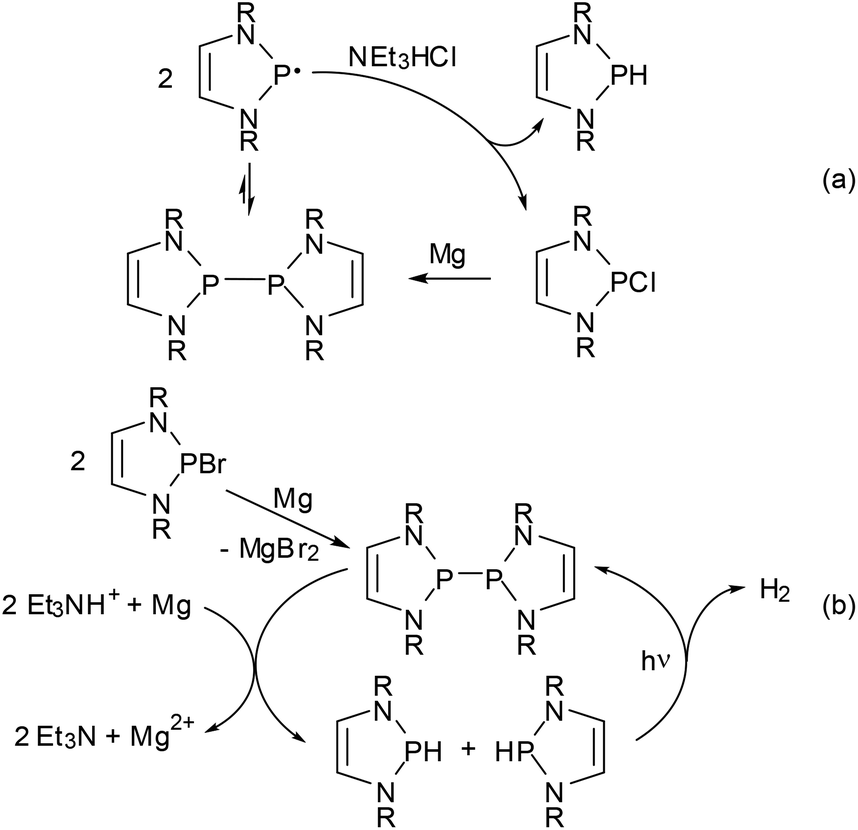 | ||
| Scheme 14 (a) Reduction of Cl-DAPs to H-DAPs using Mg/Et3NHCl via disproportionation of diazaphospholonyl radicals as the key step (R = Dipp, 2-tBuC6H4);15 (b) photocatalytic reductive generation of H2 (R = Dipp).61 Both reactions can also be performed with CC-saturated NHPs. Generation of the bis-diazaphospholenyl in (b) can be performed in situ from a Br-NHP precursor. | ||
Combination of the reductive diphosphane splitting involved in this transformation with the photochemical dehydrocoupling of secondary NHPs (Scheme 12) led to the construction of another reaction cycle which enables the reductive generation of dihydrogen using a tertiary ammonium ion as the proton source and magnesium metal as the stoichiometric reductant (Scheme 14(b)).61 This process carries in essence the well-known reduction of H-acidic substrates by base metals to a basic and aprotic milieu by employing the H-DAP/bis-diazaphospholenyl couple as a smart photocatalyst that is needed to overcome the strong kinetic inhibition observed under these conditions and can be switched on and off by controlling the radiation source. CC-saturated NHPs can also be used as catalysts but are less active, the difference reflecting presumably the superior radical stabilizing efficiency of the DAP-based system.
One aspect that has so far gone unnoticed in the discussion of the influence of 7π-delocalization on diazaphospholenyl radical stability is that the unpaired electron occupies an antibonding orbital, and its removal should result in an extra stabilizing effect due to a net increase in aromatic stabilization energy. In other words, diazaphospholenyls or their dimers can also be considered as potent reducing agents. The validity of this hypothesis was first demonstrated in the reactions of complexes with divalent group-10 metals with bis-diazaphospholenyls (Scheme 15).55,65 The products formed are best represented as phosphenium-metal(0)-halides, and the reactions may thus be viewed as redox processes involving formal two-electron reduction of the metal and oxidation of two diazaphospholenyl radicals to the corresponding cation fragments.
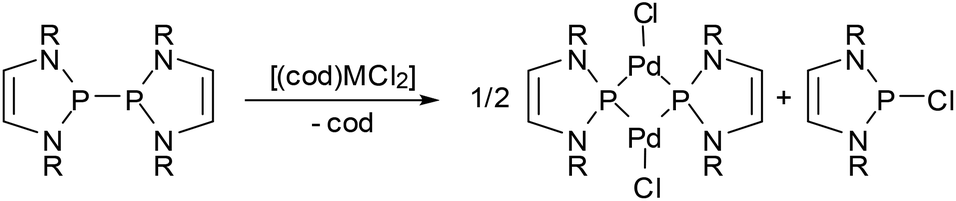 | ||
| Scheme 15 (a) Reduction of complex group-10 metal dihalides (M = Pd, Pt) with a bis-diazaphospholenyl to phosphenium-metal(0)-halides (R = Dipp). | ||
To new frontiers – ring opening reactions
Most reported reactions of DAPs occur under conservation of the heterocycle which has also been shown to withstand the presence of HCl16 or water,39 even if the hydrolytic cleavage of P–N bonds is of course feasible under more forcing conditions. However, a notable resistance to ring-opening is sometimes also found for other NHPs and is mostly attributable to kinetic stabilization by bulky N-substituents rather than the high intrinsic stability of the heterocycle. Since the DAP framework is on the other hand predestined for electrocyclic reactions,11,36,37,66 there should be ample room for ring transformation processes. Following theoretical predictions66 and mass spectrometric evidence11 of controlled cycloreversion reactions of DAPs, recently the first direct observations of such processes have been reported (Scheme 16). Thus, the bis-diazaphospholenyl 11c reacted with an activated alkyne via stepwise metathesis of both rings to give an unprecedented bis-1,3,2-azaphospholenyl instead of the expected P–P bond activation product.62 The phosphaketene 5 rearranged in solution to give cyclo-triphosphane 16via a postulated intermediate 15 that is formed by an attack of the electrophilic ketene moiety on one of the DAP carbon atoms and could be trapped with dimethylbutadiene.39 Finally, the reductive ring opening of a transient iodo-DAP 17 was involved in the magnesium reduction of a bicyclic diazaphospholenium ion to give a 4,6-diimino-1,3,5-diazaphosphinan-2-one anion. Interestingly, no reductive coupling of 17 to a bis-diazaphospholenyl was observed in this case, and the starting cation could be regenerated by oxidation of the anionic product with iodine.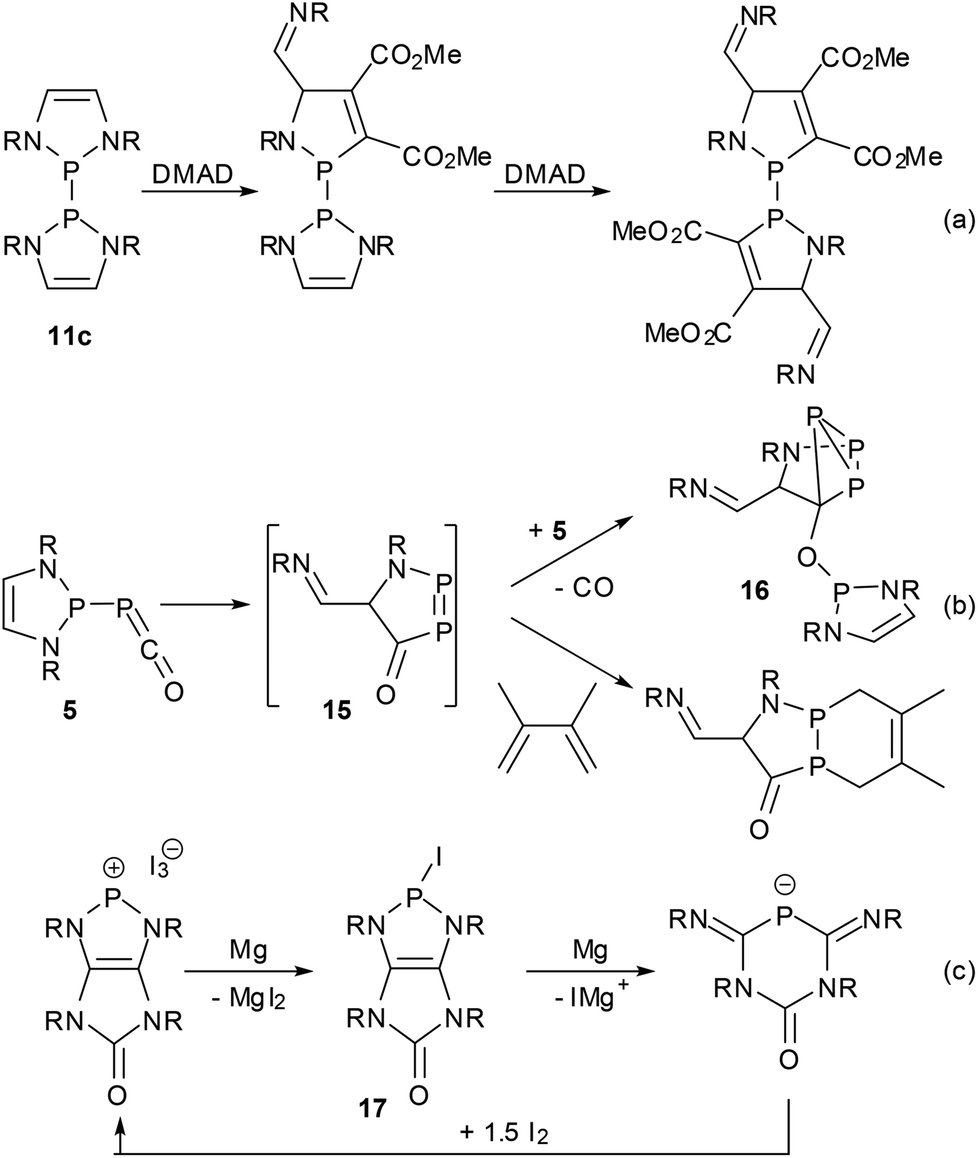 | ||
| Scheme 16 Ring opening reactions of DAPs (R = Dipp (a, b), Cy (c); DMAD = dimethyl acetylenedicarboxylate). | ||
Conclusions
Substituted diazaphospholenes with specific P-substituents – in particular hydrogen and phosphorus-based groups (PR2, phospholyl, PCO) and bis-diazaphospholenyls – continue to offer interesting chemical properties that are connected with the unique bonding situation in the heterocycle. Polarization of the exocyclic P–X bonds can bring about quantitative advances (i.e. increased reaction rates) which extend the scope of known reactions, but may also give rise to qualitatively new reaction modes. The potential of the latter is emphasized by first catalytic applications of phosphino- and – in particular – H-substituted DAPs, and it can be foreseen that the enduring exploration of this field and its further extension – e.g. by exploiting the potential as reductants in transition-metal chemistry – leaves much room for future development. Bis-diazaphospholenyls offer appealing perspectives for radical chemistry, even if their most interesting properties in this respect arise not in the first place from electronic peculiarities but from steric constraints, and one can envisage that diphosphines based on different molecular frameworks may show similar properties. The performance of the DAP-based system in photocatalytic H2 generation indicates, however, that the interplay between steric and – however tiny they may be – electronic effects may still make a decisive contribution which makes it worthwhile to continue paying special attention to DAPs. Last, but not least, first reports on specific ring-opening and ring-metathesis reactions reveal that DAPs also have some potential to undergo electrocyclic and cycloaddition reactions that have as yet been widely neglected and can be considered as good for further surprising discoveries in the future.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft, the Institut für Anorganische Chemie and the German Academic Exchange Service (DAAD) is gratefully acknowledged. The author is grateful to the many highly motivated graduate and PhD students as well as collaborators who contributed to this research. Their names appear in the references.Notes and references
- A. Michaelis, Annu. Chim., 1903, 326, 129 CrossRef CAS.
- Selected reviews: (a) M. Alajarín, C. López-Leonardo and P. Llamas-Lorente, Top. Curr. Chem., 2005, 250, 77 Search PubMed; (b) J. Gopalakrishnan, Appl. Organomet. Chem., 2009, 23, 291 CrossRef CAS.
- Selected reviews: (a) M. S. Balakrishna, V. S. Reddy, S. S. Krishnamurthy, J. F. Nixon and J. C. T. R. B. St. Laurent, Coord. Chem. Rev., 1994, 129, 1 CrossRef CAS; (b) L. Stahl, Coord. Chem. Rev., 2000, 210, 203 CrossRef CAS; (c) M. S. Balakrishna, J. Organomet. Chem., 2010, 695, 925 CrossRef CAS.
- (a) A. Schmidpeter and K. Karaghiosoff, Z. Naturforsch., B: J. Chem. Sci., 1981, 36, 1273 Search PubMed; (b) A. M. Kibardin, Yu. B. Mikhailov, T. V. Gryaznova and A. N. Pudovik, Izv. Akad. Nauk SSSR, Ser. Khim., 1986, 960 CAS.
- (a) I. A. Litvinov, V. A. Naumov, T. V. Gryaznova, A. N. Pudovik and A. M. Kibardin, Dokl. Akad. Nauk SSSR, 1990, 312, 623 CAS; (b) M. K. Denk, S. Gupta and A. J. Lough, Eur. J. Inorg. Chem., 1999, 41 CrossRef CAS.
- (a) D. Gudat, Eur. J. Inorg. Chem., 1998, 1087 CrossRef CAS; (b) H. M. Tuononen, R. Roesler, J. L. Dutton and P. J. Ragogna, Inorg. Chem., 2007, 46, 10693 CrossRef CAS PubMed.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- (a) D. Gudat, Coord. Chem. Rev., 1997, 163, 71 CrossRef CAS; (b) H. Nakazawa, Adv. Organomet. Chem., 2004, 50, 107 CrossRef; (c) L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606 CrossRef CAS.
- The earlier literature on this topic has been reviewed, see: (a) D. Gudat, Acc. Chem. Res., 2010, 43, 1307 CrossRef CAS PubMed; (b) D. Gudat, Top. Heterocycl. Chem., 2010, 21, 63 CAS , and the cited literature.
- S. Burck, D. Gudat, F. Lissner, K. Nättinen, M. Nieger and Th. Schleid, Z. Anorg. Allg. Chem., 2005, 631, 2738 CrossRef CAS.
- S. Burck, D. Gudat, M. Nieger, C. A. Schalley and T. Weilandt, Dalton Trans., 2008, 3478 RSC.
- S. Burck, D. Gudat, K. Nättinen, M. Nieger, M. Niemeyer and D. Schmid, Eur. J. Inorg. Chem., 2007, 5112 CrossRef CAS.
- O. Puntigam, I. Hajdók, M. Nieger, S. Strobel and D. Gudat, Z. Anorg. Allg. Chem., 2011, 637, 988 CrossRef CAS.
- S. Burck, D. Gudat, M. Nieger and W.-W. Du Mont, J. Am. Chem. Soc., 2006, 128, 3946 CrossRef CAS PubMed.
- O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2041 CrossRef CAS.
- M. Gediga, S. Burck, J. Bender, D. Förster, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2014, 1818 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- M. Ruck, D. Hoppe, B. Wahl, P. Simon, Y. Wang and G. Seifert, Angew. Chem., Int. Ed., 2005, 44, 7616 CrossRef CAS PubMed.
- S. Burck and D. Gudat, Inorg. Chem., 2008, 47, 315 CrossRef CAS PubMed.
- D. Gudat, A. Haghverdi, H. Hupfer and M. Nieger, Chem. – Eur. J., 2000, 6, 3414 CrossRef CAS.
- S. Burck, D. Gudat, M. Nieger, Z. Benkő, L. Nyulászi and D. Szieberth, Z. Anorg. Allg. Chem., 2009, 635, 245 CrossRef CAS.
- S. Burck, D. Gudat and M. Nieger, Angew. Chem., Int. Ed., 2004, 43, 4801 CrossRef CAS PubMed.
- D. Förster, M. Nieger and D. Gudat, Organometallics, 2011, 30, 2628 CrossRef.
- S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. Voigt, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, J. Am. Chem. Soc., 2001, 123, 9045 CrossRef CAS PubMed.
- S. Burck, K. Götz, M. Kaupp, M. Nieger, J. Weber, J. Schmedt auf der Günne and D. Gudat, J. Am. Chem. Soc., 2009, 131, 10763 CrossRef CAS PubMed.
- Y. Canac, D. Bourissou, A. Baceiredo, H. Gornitzka, W. W. Schoeller and G. Bertrand, Science, 1998, 279, 2080 CrossRef CAS PubMed.
- Z. Benkő, S. Burck, D. Gudat, M. Hofmann, F. Lissner, L. Nyulászi and U. Zenneck, Chem. – Eur. J., 2010, 16, 2857 CrossRef PubMed.
- G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040 CrossRef CAS PubMed ; But see also: D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370 ( Angew. Chem. , 2014 , 53 , 6047 ) CrossRef PubMed.
- C. J. Carmalt, V. Lomeli, B. G. McBurnett and A. H. Cowley, Chem. Commun., 1997, 2095 RSC.
- T. V. Gryaznova, Yu. G. Budnikova and O. G. Sinyashin, Russ. J. Electrochem., 2007, 43, 1151 CrossRef CAS.
- A. L. Brazeau, N. D. Jones and P. J. Ragogna, Dalton Trans., 2012, 41, 7890 RSC.
- A. M. Kibardin, A. N. Pudovik and T. V. Gryaznova, Izv. Akad. Nauk SSSR, Ser. Khim., 1987, 1676 CAS.
- (a) A. M. Kibardin, T. V. Gryaznova, K. M. Enikeev, Yu. B. Mikhailov and A. N. Pudovik, Izv. Akad. Nauk SSSR, Ser. Khim., 1987, 1684 CAS; (b) A. M. Kibardin, I. A. Litvinov, V. A. Naumov, J. T. Struchkov, T. V. Gryaznova, J. B. Mikhailov and A. N. Pudovik, Dokl. Akad. Nauk SSSR, 1988, 298, 369 CAS.
- J. W. Dube, G. J. Farrar, E. L. Norton, K. L. Szekely, B. F. T. Cooper and C. L. B. MacDonald, Organometallics, 2009, 28, 4377 CrossRef CAS.
- J. T. Price and P. J. Ragogna, Chem. – Eur. J., 2013, 19, 8473 CrossRef CAS PubMed.
- D. Herrmannsdörfer, M. Kaaz, O. Puntigam, J. Bender, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2015, 4819 CrossRef.
- G. Reeske and A. H. Cowley, Inorg. Chem., 2007, 46, 1426 CrossRef CAS PubMed.
- D. Gudat, A. Haghverdi and M. Nieger, Angew. Chem., Int. Ed., 2000, 39, 3084 CrossRef CAS.
- Z. Li, X. Chen, M. Bergeler, M. Reiher, C.-Y. Su and H. Grützmacher, Dalton Trans., 2015, 44, 6431 RSC.
- A. M. Kibardin, T. V. Gryaznova and A. N. Pudovik, Zh. Obshch. Khim., 1996, 66, 372 CAS.
- S. Burck, D. Gudat, M. Nieger and J. Tirrée, Dalton Trans., 2007, 1891 RSC.
- M. Kaaz, J. Bender, D. Förster, W. Frey, M. Nieger and D. Gudat, Dalton Trans., 2014, 43, 680 RSC.
- L. Ackermann, Synlett, 2007, 507 CrossRef CAS.
- S. Burck, D. Gudat, M. Nieger and D. Vinduš, Eur. J. Inorg. Chem., 2008, 704 CrossRef CAS.
- S. Burck, D. Förster and D. Gudat, Chem. Commun., 2006, 2810 RSC.
- N. Burford, T. S. Cameron, P. J. Ragogna, E. Ocando-Mavarez, M. Gee, R. McDonald and R. E. Wasylishen, J. Am. Chem. Soc., 2001, 123, 7947 CrossRef CAS PubMed.
- S. Burck, D. Gudat and M. Nieger, Angew. Chem., Int. Ed., 2007, 46, 2919 CrossRef CAS PubMed.
- I. Hajdók, F. Lissner, M. Nieger, S. Strobel and D. Gudat, Organometallics, 2009, 28, 1644 CrossRef.
- (a) D. L. Dodds, M. F. Haddow, A. G. Orpen, P. G. Pringle and G. Woodward, Organometallics, 2006, 25, 5937 CrossRef CAS; (b) D. Förster, I. Hartenbach, M. Nieger and D. Gudat, Z. Naturforsch., 2012, 67b, 765 Search PubMed.
- S. Burck, D. Gudat and M. Nieger, Organometallics, 2009, 28, 1447 CrossRef CAS.
- N. J. Hardman, M. B. Abrams, M. A. Pribisko, T. M. Gilbert, R. L. Martin, G. J. Kubas and R. T. Baker, Angew. Chem., Int. Ed., 2004, 43, 1955 CrossRef CAS PubMed.
- Use of phosphorus-based cations as electrophilic catalysts is currently gaining attention, see: (a) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS PubMed; (b) M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed.
- P. Rivière, M. Rivière-Baudet and J. Satgé, in Comprehensive Organometallic Chemistry, ed. G. Wilkinson, F. G. A. Stone and E. W. Abel, Pergamon, Oxford, 1982, vol. 2, p. 399 ff Search PubMed.
- C. C. Chong and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 12116 CrossRef CAS PubMed.
- D. Förster, J. Nickolaus, M. Nieger, Z. Benkő, A. W. Ehlers and D. Gudat, Inorg. Chem., 2013, 52, 7699 CrossRef PubMed.
- C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 3342 CrossRef CAS PubMed.
- K. Muñiz, Angew. Chem., Int. Ed., 2005, 44, 6622 CrossRef PubMed.
- C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190 CrossRef CAS PubMed.
- R. Edge, R. J. Less, E. J. L. McInnes, K. Müther, V. Naseri, J. M. Rawson and D. S. Wright, Chem. Commun., 2009, 1691 RSC.
- D. Heift, Z. Benkő and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 6757 CrossRef CAS PubMed.
- O. Puntigam, L. Könczöl, L. Nyulászi and D. Gudat, Angew. Chem., Int. Ed., 2015, 54, 11567 CrossRef CAS PubMed.
- D. Förster, H. Dilger, F. Ehret, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2012, 3989 CrossRef.
- M. Blum, O. Puntigam, S. Plebst, F. Ehret, J. Bender, M. Nieger and D. Gudat, Dalton Trans., 2016, 45, 1987 RSC.
- J.-D. Guo, S. Nagase and P. P. Power, Organometallics, 2015, 34, 2028 CrossRef CAS.
- J. Nickolaus, J. Bender, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2014, 3030 CrossRef CAS.
- Z. Benkö, D. Gudat and L. Nyulászi, Chem. – Eur. J., 2008, 14, 902 CrossRef PubMed.
Footnotes |
| † The bonding in P-amino-DAPs seems to be ruled by a balance between n(N)–σ*(P–X) and n(X)–σ*(P–N) hyperconjugation, but the n(N)–σ*(P–X) interaction and P–X charge separation grow with the increasing capability of X to stabilize a negative charge.10,11 |
| ‡ The inverse relationship between P–Cl and P–N bond lengths holds also for other diaminochlorophosphines (R2N)2PCl and indicates that this concept is indeed more general.12 |
| § The corresponding reaction with PI3 yields no 2-iodo-DAPs but rather diazaphospholenium triiodides37 and follows a somewhat different mechanism.36 |
| ¶ This contrasts with the known photochemical behaviour of PH3 where the first excited state of the monomer decays under PH bond fission leading to the formation of H and H2P radicals which may then react further to yield the final products H2 and P2H4.61 |
| This journal is © The Royal Society of Chemistry 2016 |