
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Permanently porous hydrogen-bonded frameworks of rod-like thiophenes, selenophenes, and tellurophenes capped with MIDA boronates†
Peng-Fei
Li
,
Chenxi
Qian
,
Alan J.
Lough
,
Geoffrey A.
Ozin
and
Dwight S.
Seferos
*
Department of Chemistry, University of Toronto 80 St. George, Toronto, ON M5S 3H6, Canada. E-mail: dseferos@chem.utoronto.ca
First published on 5th January 2016
Abstract
Permanently porous hydrogen-bonded organic frameworks comprising rod-like molecules with two MIDA boronate termini have been prepared. We show that MIDA boronates self-assemble through multiple hydrogen-bonding interactions. Thiophene-containing frameworks are fluorescent and have a 6.6% absolute quantum yield. The approach appears to be general and introduces new design rules for constructing hydrogen-bonded organic frameworks.
Porous materials such as metal organic frameworks,1–3 covalent organic frameworks (COFs),4–6 and porous aromatic frameworks7,8 have emerged as promising candidates for a range of uses. Porous organic materials include intrinsic and extrinsic porous structures. Organic cage compounds are a typical class of intrinsic porous crystalline solids.9–14 Extrinsic porous materials are formed by the assembly of nonporous small molecules including COFs and the recent revival of hydrogen-bonded organic frameworks (HOFs). HOFs are connected by non-covalent interactions such as hydrogen bonds and various π–π interactions. HOFs that incorporate solvent guest molecules typically lose their porosity after the solvent is removed. As a result relatively few HOFs exhibit permanent porosity.15–22 Permanently porous materials are important for a range of uses,23–26 yet designing building blocks for permanently porous HOFs is challenging.15 The most frequently used building blocks contain the 2,4-diaminotriazinyl group (DAT) and related nitrogen-containing heterocycles (Scheme 1).16 There are a few alternative motifs such as benzimidazolones18 and pyrazoles,19 however, the design rules for HOFs are still in their infancy and very important for the continued development of porous materials.
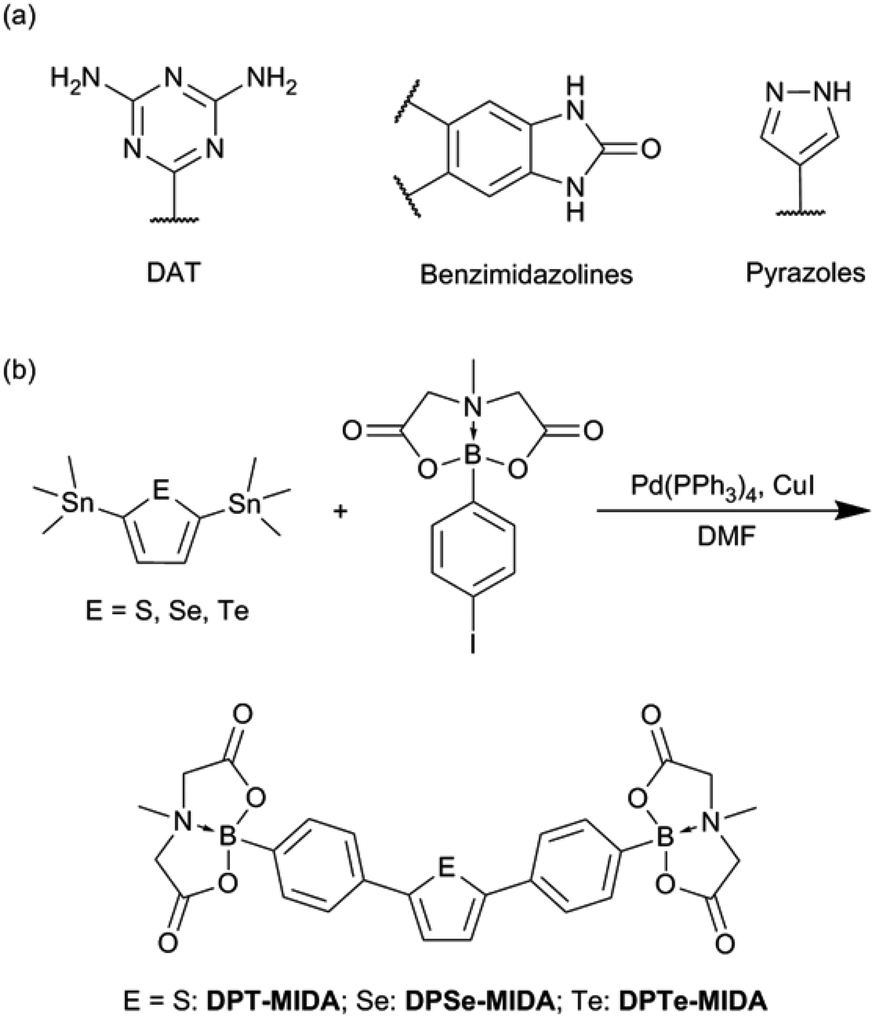 | ||
| Scheme 1 (a) Previously reported functional groups for constructing HOFs. (b) The synthesis of diphenylheterocycle MIDA boronates in this report. | ||
Herein we report that hydrogen-bonded porous frameworks can be constructed from rod-like molecules that are capped with N-methyliminodiacetic acid (MIDA) boronate termini. The rod-like molecules contain heterocycles that are typical of optoelectronically active materials, including thiophene, selenophene, and tellurophene.27–31 The MIDA boronate group has been applied to a range of cross-coupling synthetic methodologies.32–34 MIDA esters are easily handled, stable under air, and contain both hydrogen bond donors and acceptors. Here we demonstrate that this synthon is useful for the self-assembly of permanently porous materials.
MIDA-containing thiophene, selenophene, and tellurophene rod-like molecules (hereafter denoted DPT-MIDA, DPSe-MIDA, and DPTe-MIDA, respectively) were synthesized by a Pd(PPh3)4 and CuI catalyzed Stille-coupling reaction between 4-iodophenylboronic acid MIDA ester (3.5 eq.) and the corresponding 2,5-bis(trimethyltin)heterocycle35,36 (1.0 eq.) (Scheme 1). This reaction afforded yellow solids of DPT-MIDA, DPSe-MIDA, and DPTe-MIDA, respectively in high yields (84%–90%). Pure and crystalline products were obtained by recrystallization from acetonitrile. The MIDA boronates were characterized by FT-IR, NMR, and HR-MS (see the ESI†).
The single crystal structures of DPT-MIDA and DPSe-MIDA reveal that two molecules of DPT-MIDA or DPSe-MIDA are contained within each unit cell and the MIDA end groups are in the trans-configuration. DPT-MIDA crystallize through multiple C–H⋯O hydrogen bonding and C–H⋯π interactions (Fig. 1a). The C–H⋯O hydrogen bonding interactions between the oxygen atom in the carbonyl groups and hydrogen in the methylene groups of adjacent MIDA functionalities vary between 2.20 to 2.65 Å. There are two kinds of C–H⋯π interactions, one is a 2.85 Å interaction between the thiophene and the phenyl groups of adjacent molecules, the other is a 2.71 Å interaction between the phenyl group and the hydrogen in the methyl group of adjacent MIDA functional groups. DPSe-MIDA assembled in a similar fashion (see S4, ESI†). Since the only difference is the chalcogen atom, both of the microporous frameworks have a 7 Å × 10 Å pore size along the shortest distance across the channel. Solvent molecules are incorporated in the channel of the frameworks. Three molecules of acetonitrile are incorporated within the DPT-MIDA or DPSe-MIDA unit cells. In DPT-MIDA, one of the acetonitrile molecule is disordered (see S6, ESI†). In both frameworks, guest acetonitrile molecules are bonded with the wall of the channel through weak C–H⋯N hydrogen bonding and C–H⋯π interactions.
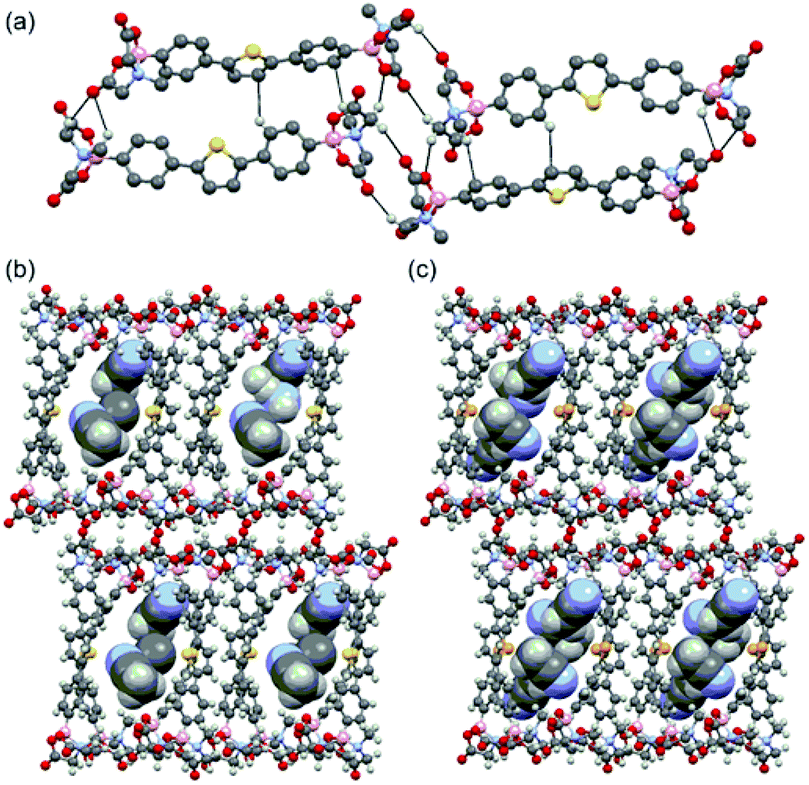 | ||
| Fig. 1 (a) Hydrogen-bonding interactions in DPT-MIDA, (b) and (c) microporous structure of DPT-MIDA and DPSe-MIDA with acetonitrile solvent incorporated. | ||
DPTe-MIDA was also recrystallized from acetonitrile, however only needle-like crystals could be obtained. In order to compare the structure of DPTe-MIDA with the other samples, powder X-ray diffraction patterns (PXRDs) were collected on all three compounds. For reference the simulated PXRD patterns of DPT-MIDA and DPSe-MIDA were generated from their single crystal data. These closely match the experimental PXRD data (see S8–S9, ESI†), indicating that the structure in the powders are similar to the single crystals. The PXRD scattering pattern of DPTe-MIDA is weak compared to the other two frameworks, which indicates low crystallinity. None-the-less, the main diffraction peaks are close to the other two frameworks (see S10, ESI†), which implies all three frameworks are assembled into a similar structure.
Thermogravimetric analysis (TGA) was performed on each sample under a N2 atmosphere. These experiments reveal that the incorporated acetonitrile molecules are removed at different temperatures. For S and Se, the guest-removal temperatures are around 150 °C; for Te, the temperature drops to 70 °C (see S11–S13, ESI†). The weight loss of DPT-MIDA, DPSe-MIDA, and DPTe-MIDA are 12%, 8.2%, and 6.1% respectively. This is close to the expected weight loss based on acetonitrile inclusion (10%, 9.4%, and 8.7% for DPT-MIDA, DPSe-MIDA, and DPTe-MIDA, respectively). This provides support that the degree of acetonitrile inclusion is similar in all frameworks. No additional weight loss was observed after the loss of acetonitrile until 350 °C, after which the frameworks began to decompose. The incorporation of acetonitrile in the frameworks was also observed in the 1H NMR spectra at δ = 2.07 ppm (see S14–S16, ESI†).
To investigate stability, variable-temperature PXRD patterns were collected on all three frameworks (Fig. 2 and see S20–S21, ESI†). Under air, structural changes were observed. DPT-MIDA is stable up to 100 °C. After that the structure begins to transform, the peaks at 8° and 8.8° become broad, and the peak at 12.2° shifts to 10.7° (Fig. 2). DPSe-MIDA is stable in air up to 125 °C. After that the structure begins to transform, the peaks at 5.4°, 8.2°, and 8.8° become broad, the intensity of peak 10.9° increased, while the peak at 12.3° attenuated (see S20, ESI†). Under vacuum, DPT-MIDA is stable up to 300 °C (see S22, ESI†). DPSe-MIDA is stable under vacuum as well, even when heated at 150 °C overnight, no obvious change was observed using optical microscopy (see S24, ESI†). Thermally induced transformation are difficult to observe for DPTe-MIDA due to the lower diffraction intensity of this less crystalline sample.
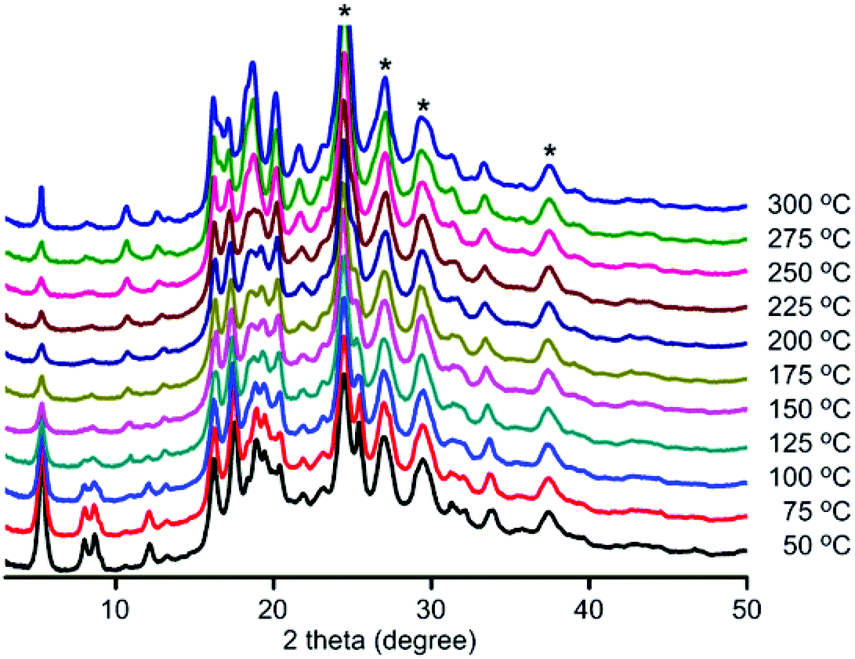 | ||
| Fig. 2 Variable-temperature PXRD patterns for DPT-MIDA. The asterisks (*) denotes the background signal from a polyether ether ketone layer that protects the detector. The temperature was recorded in 25 °C increments. | ||
Permanent porosity was evaluated by CO2 adsorption at 273 K. Activated frameworks have 267, 239 and 197 m2 g−1 BET surface areas for DPT-MIDA, DPSe-MIDA, and DPTe-MIDA, respectively. The surface area decreases with the increase in molecular weight, which is another sign of similar structure. A slight deviation from linearity is observed which could come from larger atomic radii and lower crystallinity of the Te frameworks (see S25, ESI†). The pore width distributions of DPT-MIDA and DPSe-MIDA have a peak at 5 Å, which is slightly smaller than the pore width expected from single crystal data, likely due to lattice shrinkage after solvent removal (see S26–S27, ESI†). For DPTe-MIDA, two peaks are observed, one at 5.5 Å, and one at 9.8 Å (see S28, ESI†). While the peak at 5.5 Å comes from the ideal crystal structure, the peak at 9.8 Å is almost twice as large and likely come from molecular vacancies in the lattice. DPT-MIDA and DPSe-MIDA take up 39 and 36 cm3 g−1 of CO2 (STP), which corresponds to 1 mole of CO2 adsorbed per mole of building block. DPTe-MIDA takes up 15 cm3 g−1 (STP) of CO2, corresponds to around 0.5 mole of CO2 adsorbed per mole of building block (Fig. 3).
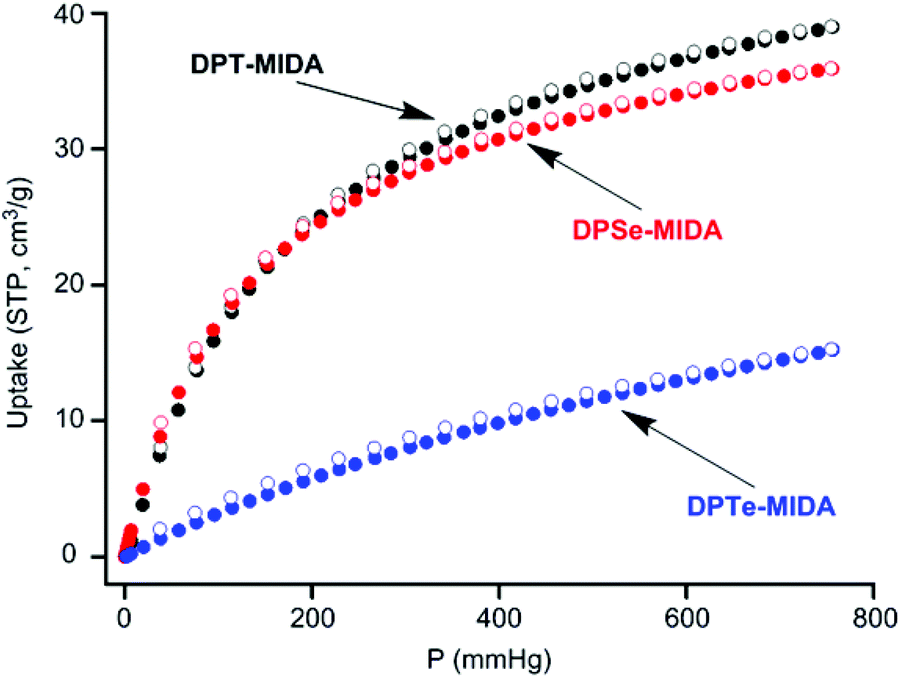 | ||
| Fig. 3 CO2 sorption isotherm of DPT-MIDA, DPSe-MIDA and DPTe-MIDA at 273 K. (filled circles: adsorption, open circles: desorption, STP = standard temperature pressure). | ||
Since thiophene, selenophene, and tellurophene are incorporated into the frameworks, it is interesting to examine their optical properties. The solution absorption maximum (λmax) of the solvated molecules shifts to longer wavelengths (lower energy) as one moves from S (334 nm) to Se (344 nm) to Te (356 nm) in acetonitrile. The emission spectra also red shift from S (412 nm) to Se (424 nm) to Te (439 nm) (see S29–S30, ESI†). The absolute quantum yield of the solid was measured using an integrating sphere.37 Here we chose to focus on DPT-MIDA because it exhibits the strongest fluorescence in solution. The absolute quantum yield of DPT-MIDA is 6.6%. This value is quite high for HOFs and greater than both HOF-111138 and HOF-5,22 which undergo aggregation induced emission. A powder of DPT-MIDA exhibits strong enough blue fluorescence that can be observed with the naked eye (see S31, ESI†).
In conclusion, we have discovered that rod-like molecules that possess two MIDA boronate termini self-assemble into permanently porous HOFs. The thiophene-containing framework exhibits strong photoluminescence in the solid state. Combining rod-like molecules and MIDA esters appears to be a straightforward way to develop porous solids and could be quite general for other types of building blocks with a variety of shapes and symmetry. Thus the design rules presented herein should be useful for new types of frameworks for a range of uses.
Acknowledgements
This work was supported by the National Science and Engineering Research Council of Canada and the Alfred P. Sloan Foundation. GAO is Government of Canada Research Chair in Materials Chemistry and Nanochemistry.Notes and references
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed.
- M. Mastalerz, Angew. Chem., Int. Ed., 2008, 47, 445–447 CrossRef CAS PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed.
- M. W. Schneider, I. M. Oppel, A. Griffin and M. Mastalerz, Angew. Chem., Int. Ed., 2013, 52, 3611–3615 CrossRef CAS PubMed.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 1516–1520 CrossRef CAS PubMed.
- J.-H. Fournier, T. Maris and J. D. Wuest, J. Org. Chem., 2004, 69, 1762–1775 CrossRef CAS PubMed.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7892–7894 CrossRef CAS PubMed.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. Š. Miljanić, Nat. Commun., 2014, 5 DOI:10.1038/ncomms6131.
- P. Li, Y. He, H. D. Arman, R. Krishna, H. Wang, L. Weng and B. Chen, Chem. Commun., 2014, 50, 13081–13084 RSC.
- W. Xiao, C. Hu and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 14200–14206 CrossRef CAS PubMed.
- H. Wang, B. Li, H. Wu, T.-L. Hu, Z. Yao, W. Zhou, S. Xiang and B. Chen, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS PubMed.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS PubMed.
- Y. Li, S. Zhang and D. Song, Angew. Chem., Int. Ed., 2013, 52, 710–713 CrossRef CAS PubMed.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- P.-F. Li, T. B. Schon and D. S. Seferos, Angew. Chem., Int. Ed., 2015, 54, 9361–9366 CrossRef CAS PubMed.
- J. Hollinger, A. A. Jahnke, N. Coombs and D. S. Seferos, J. Am. Chem. Soc., 2010, 132, 8546–8547 CrossRef CAS PubMed.
- A. A. Jahnke, G. W. Howe and D. S. Seferos, Angew. Chem., Int. Ed., 2010, 49, 10140–10144 CrossRef CAS PubMed.
- L. Li, J. Hollinger, A. A. Jahnke, S. Petrov and D. S. Seferos, Chem. Sci., 2011, 2, 2306–2310 RSC.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 539–547 CrossRef CAS PubMed.
- Y. S. Park, Q. Wu, C.-Y. Nam and R. B. Grubbs, Angew. Chem., Int. Ed., 2014, 53, 10691–10695 CrossRef CAS PubMed.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084–14085 CrossRef CAS PubMed.
- J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed.
- P. Trinchera, V. B. Corless and A. K. Yudin, Angew. Chem., Int. Ed., 2015, 54, 9038–9041 CrossRef CAS PubMed.
- D. P. Sweat and C. E. Stephens, Synthesis, 2009, 3214–3218 CAS.
- T. Earmme, Y.-J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960–14963 CrossRef CAS PubMed.
- D. O. Faulkner, J. J. McDowell, A. J. Price, D. D. Perovic, N. P. Kherani and G. A. Ozin, Laser Photon. Rev., 2012, 6, 802–806 CrossRef CAS.
- Z. Sun, Y. Li, L. Chen, X. Jing and Z. Xie, Cryst. Growth Des., 2015, 15, 542–545 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental information and characterization data. CCDC 1434463 (DPT-MIDA) and 1434461 (DPSe-MIDA). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04960a |
| This journal is © The Royal Society of Chemistry 2016 |