
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclopalladated organosilane–tethered thiosemicarbazones: novel strategies for improving antiplasmodial activity
Muneebah
Adams
a,
Linley
Barnard
a,
Carmen
de Kock
b,
Peter J.
Smith
b,
Lubbe
Wiesner
b,
Kelly
Chibale
acd and
Gregory S.
Smith
*a
aDepartment of Chemistry, University of Cape Town, Private Bag, Rondebosch 7701, South Africa. E-mail: gregory.smith@uct.ac.za; Fax: +27-21-6505195; Tel: +27-21-6505279
bDivision of Pharmacology, Department of Medicine, University of Cape Town, K45, OMB, Groote Schuur Hospital, Observatory, 7925, South Africa
cInstitute of Infectious Disease and Molecular Medicine, University of Cape Town, Rondebosch 7701, South Africa
dSouth African Medical Research Council Drug Discovery and Development Research Unit, University of Cape Town, Rondebosch 7701, South Africa
First published on 25th February 2016
Abstract
Two series of ferrocenyl- and aryl-derived cyclopalladated organosilane thiosemicarbazone complexes were synthesised via C–H bond activation. Selected compounds were evaluated for in vitro antiplasmodial activity against the chloroquine-sensitive (NF54) and chloroquine-resistant (Dd2) strains of the human malaria parasite Plasmodium falciparum. Cyclopalladation of the thiosemicarbazones resulted in antiplasmodial activities in the low micromolar range.
Introduction
Malaria continues to be one of the most widespread parasitic infectious diseases in the world today. An estimated 198 million cases of malaria were reported in 2013, with a large percentage of people who die from the disease made up of children under the age of five.1 Most of the malaria infection cases are known to occur in sub-Saharan Africa.Chloroquine (CQ), a quinoline-based compound, was the most successful clinical drug of choice for the treatment of malaria, but the emergence of resistant strains of the causative agent Plasmodium falciparum has rendered CQ useless in many parts of the world. The current last line of defence against resistant parasites is the artemisinin combination therapy (ACT) regimen. Unfortunately, the recent emergence of resistance to this regimen in parts of Asia, warrants the search for novel drug regimens.2,3
Thiosemicarbazones (TSCs) are thioureas known to display a large spectrum of pharmacological properties, particularly as antiparasitic agents, and are therefore exploitable in the development of new chemotherapeutics.4–9 The precise antiplasmodial mode of action of TSCs is currently unknown. Thiosemicarbazones are proposed to inhibit cysteine proteases, which are integral in several parasite functions.10
The use of transition metals in drug discovery has become a popular strategy, particularly where metals have been combined with compounds of known therapeutic value in an attempt to combat drug resistance. This has been exemplified by the bioorganometallic compound ferroquine, with ferrocene incorporated into the lateral side-chain of chloroquine, allowing for greater transmembrane interactions owing to its more lipophilic nature.11–13 The most notable pioneering work is the rhodium(I)–chloroquine derivatives synthesised by Sánchez-Delgado and co-workers.14 These derivatives showed a reduction in parasitaemia in vivo to a greater extent than that of chloroquine, endorsing the use of metal-based compounds in malaria chemotherapy. For further reports on metalloantimalarials in the literature, several excellent recent reviews abound.12,13,15–17
The application of platinum group metals (PGMs) as chemotherapeutic agents has therefore emerged as a viable area of research. There are not many examples of transition metal complexes of TSCs reported with antiplasmodial activity. Within our research group, we have evaluated transition metals from the platinum group series as antiplasmodials.4,18–21 We have explored the chemistry associated with N,S- and O,N,S-chelated TSC systems, and to a limited extent the preparation of C,N,S-chelated TSC cyclometallated complexes. Cyclometallated complexes can be prepared via the oxidative addition of aryl halides; however, within our group cyclometallated complexes prepared via C–H activation is of interest.22,23 In particular, the biological evaluation of these types of cyclopalladated complexes especially, is relatively rare.
A recent strategy that we have pursued in the search for novel antimalarial metal-based drug leads is the incorporation of organosilane moieties into our structures.24–26 This strategy has been used with great success to enhance biological activity and reduce toxicity in an effort to increase the therapeutic value of drugs. Organosilane moieties also increase the lipophilicity of a drug molecule which in turn provides several physiological benefits, including improved tissue and cell penetration, which may result in an increase in permeability and ultimately bioavailability.27–32
As a follow-up to our recent studies,24–26 we were prompted to explore the effect of introducing silicon into cyclopalladated thiosemicarbazone complexes, in an attempt to identify potential new antimalarial compounds. To the best of our knowledge, there are no reports on the use of silicon-containing thiosemicarbazone cyclopalladated complexes in the field of bioorganometallic antiplasmodial research.
Herein, we report the synthesis and characterisation of a series of new cyclopalladated silicon-containing compounds based on a thiosemicarbazone scaffold along with their in vitro antiplasmodial activities.
Results and discussion
Synthesis and characterisation
The ferrocenyl- and aryl-derived TSCs (1–3) containing either a methylene (1a–3a) or propyl (1b–3b) spacer were prepared by refluxing the corresponding Schiff-base dithiocarbamate and the appropriate amine (Scheme 1). This occurred via a nucleophilic substitution of the methanethiol group of the dithiocarbamates with the amine. The dithiocarbamates and the TSCs (1a–3a) are known compounds which were prepared following published methods.26,33,34 The dithiocarbamates were prepared via the Schiff-base condensation of methyl hydrazinecarbodithioate35 with either acetylferrocene or 3,4-dichloroacetophenone.33,34 The cyclopalladated complexes (4–6) were prepared via C–H activation of the cyclopentadienyl or aryl rings of the ferrocenyl- and aryl-derived TSCs, respectively, when reacted with the palladium precursor cis-[PdCl2(PTA)2] in the presence of triethylamine as the base (Scheme 1). These palladacycles are formed through an electrophilic substitution reaction. The palladium(II) metal centre inserts into the C–H bond to form a Pd–C bond with the ortho carbon atom of the ferrocenyl or aryl ring. The triethylamine facilitates the deprotonation of the ring. The incorporation of 1,3,5-triaza-7-phosphaadamantane (PTA) into complexes of biological interest has increased due to the improved solubility of PTA-containing complexes. Within the context of malaria, PTA-containing complexes have exhibited promising in vitro activities against P. falciparum.4,20,36–38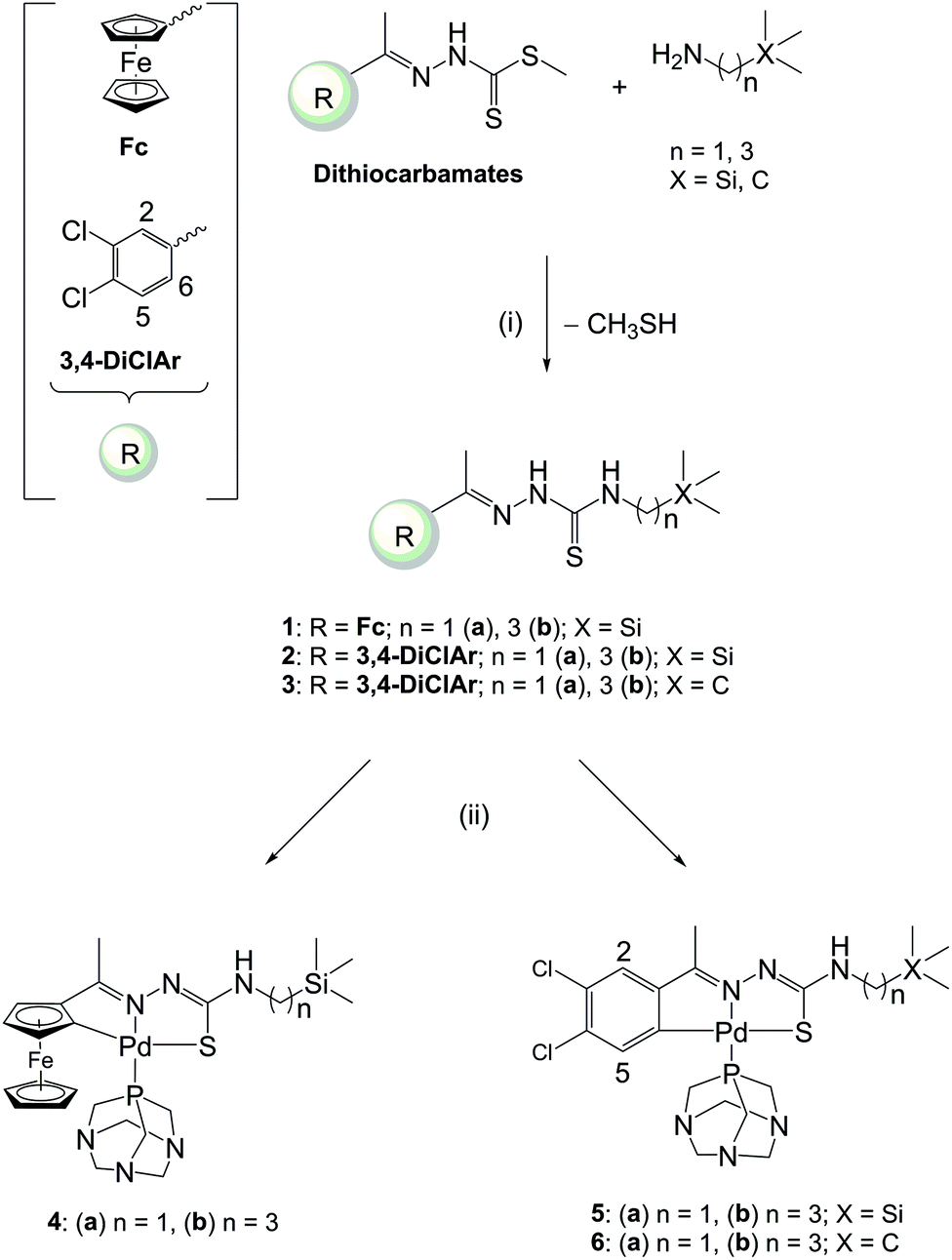 | ||
| Scheme 1 Reagents and conditions: (i) EtOH, reflux [7 h (1b); 24 h (2b, 3b)]; (ii) cis-[PdCl2(PTA)2], Et3N, EtOH, reflux, 24 h. | ||
Synthesis of the ferrocenyl- and aryl-derived TSCs was confirmed by the absence of the methanethiol (SCH3) protons and the appearance of signals for the newly incorporated amine in the 1H NMR spectra. The absence of a signal for a proton of the substituted cyclopentadienyl ring (4) or the C-6 position on the aryl ring (5; 6) confirmed the synthesis of the cyclopalladated complexes. Formation of the palladacycle was further confirmed by the absence of the hydrazinic proton, which suggests that the TSC chelates in the thiolate form. Furthermore, in the case of the ferrocenyl-derived cyclopalladated complexes (4a–b), the 1,2-disubstitution of the cyclopentadienyl ring results in the formation of planar chiral complexes. An AB spin system splitting pattern is observed for the PTA ligand.39–41 The NCH2N protons resonate as two doublets corresponding to the different environments experienced by the axial and equatorial protons, while the PCH2N protons resonate as a singlet (ca. 4.26 ppm), as expected for these types of cyclopalladated complexes.
Formation of the complexes is further confirmed by the 13C{1H} NMR spectra. The imine carbon atom, which resonates at ca. 147 ppm for the free TSCs, shifts significantly downfield (ca. 162 ppm) for the complex. Furthermore, the imine carbon atom resonates as a doublet (J = 7.4 Hz), which is ascribed to coupling with the PTA phosphorus. A similar trend is observed for the carbon atom to which the palladium atom is bonded. The carbon atom resonates as a doublet (7.4–8.8 Hz) at 99.5 and ca. 136 ppm for the ferrocenyl- and aryl-derivatives, respectively. This further confirms C–H activation, and thus formation of the palladacycle.
31P{1H} NMR spectroscopic analysis reveals one singlet for the phosphorus nuclei, resonating upfield at approximately −41.6 ppm for the ferrocenyl complexes (4) and approximately −49.7 ppm for complexes 5 and 6. This confirms the presence of one phosphorus species. This is consistent with the literature values for analogous cyclopalladated compounds containing a PTA ligand.20,21 Infrared spectral analysis of the of the palladium-free compounds revealed an absorption band at ca. 1610 cm−1 which corresponds to the imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching frequency.
N) stretching frequency.
Upon formation of the cyclopalladated complex, two absorption bands at around 1578 and 1553 cm−1 are observed which correspond to two CN stretching frequencies. The absorption band observed at the lower wavenumber is assigned to the palladium-coordinated imine, while the absorption band at the higher wavenumber corresponds to the newly formed CN bond in the cyclopalladated complexes. The formation of these complexes is further confirmed by mass spectrometry. The mass spectra for complexes 4–6 displayed peaks corresponding to either the molecular ion [M]+ or the protonated form [M + H]+.
Metal-based compounds which are stable in solution are important when ensuring that the tested compound is the compound responsible for the observed biological activity. Therefore, as a model system, the stability of complex 5a was investigated and monitored by 1H NMR spectroscopy over a 72 h period at 37 °C. The 1H NMR spectra (Fig. 1) of complex 5a was recorded in DMSO-d6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O (9:1, v/v). The spectra remain unchanged over the time period for the assay, suggesting that compound 5a may be the compound responsible for any observed activity.
:D2O (9:1, v/v). The spectra remain unchanged over the time period for the assay, suggesting that compound 5a may be the compound responsible for any observed activity.
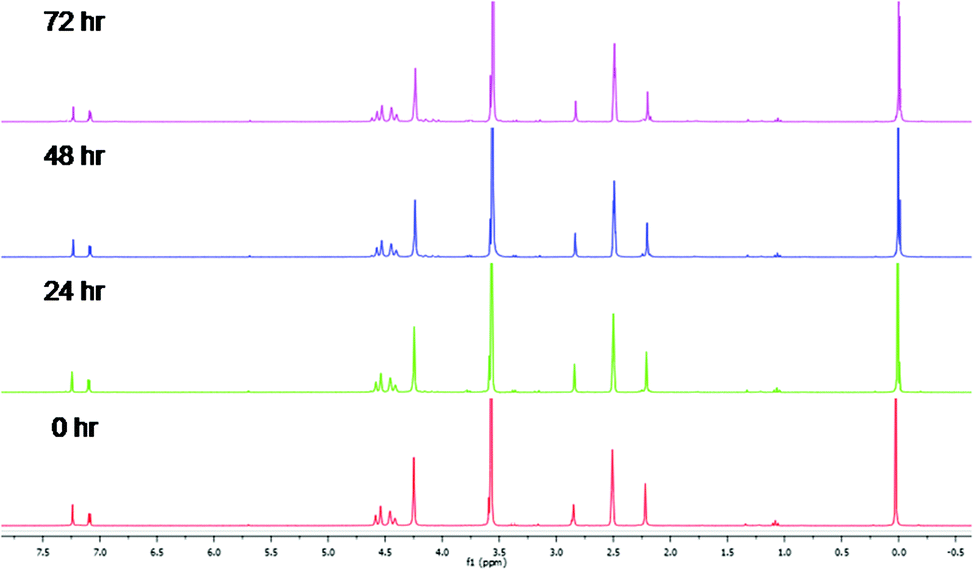 | ||
| Fig. 1
1H NMR stability study of complex 5a in DMSO-d6:D2O (9:1, v/v) at 37 °C over 72 h. | ||
In vitro antiplasmodial and cytotoxicity evaluation
The dithiocarbamates, thiosemicarbazones (1–3) and the cyclopalladated complexes (4–6) were evaluated for their in vitro antiplasmodial activity against the CQ-sensitive (NF54) and CQ-resistant (Dd2) strains of Plasmodium falciparum. The cytotoxicity of selected compounds was evaluated against the Chinese Hamster Ovarian (CHO) cell-line. Chloroquine (CQDP) and artesunate were used as the control drugs for the parasite strains and emetine for the CHO cell-line. The IC50 values are listed in Table 1.| Compound | IC50 (μM) | RIa | SI1b |
SI2c |
||
|---|---|---|---|---|---|---|
| NF54 | Dd2 | CHO | ||||
| a RI = IC50(Dd2)/IC50(NF54). b SI1 = IC50(CHO)/IC50(NF54). c SI2 = IC50(CHO)/IC50(Dd2); ND = not determined. | ||||||
| Fc Dithiocarbamate | 3.76 ± 1.35 | 13.70 ± 1.32 | 0.49 ± 0.21 | 3.64 | 0.13 | 0.036 |
| 3,4-DiClAr dithiocarbamate | 2.59 ± 0.89 | 10.95 ± 1.06 | 1.20 ± 0.41 | 4.23 | 0.46 | 0.11 |
| 1a | 7.92 ± 1.99 | ND | ND | — | — | — |
| 1b | 1.88 ± 0.58 | 2.43 ± 0.26 | ND | 1.29 | — | — |
| 2a | 2.24 ± 0.26 | 2.40 ± 0.57 | 29.28 ± 0.89 | 1.07 | 13.07 | 12.20 |
| 2b | 14.61 ± 2.07 | ND | ND | — | — | — |
| 3a | 175.74 ± 43.03 | ND | ND | — | — | — |
| 3b | 8.72 ± 3.41 | ND | ND | — | — | — |
| 4a | 1.43 ± 0.29 | 1.26 ± 0.03 | 3.93 ± 0.26 | 0.88 | 2.75 | 3.12 |
| 4b | 1.52 ± 0.09 | 1.07 ± 0.01 | ND | 0.70 | — | — |
| 5a | 0.55 ± 0.10 | 0.29 ± 0.05 | 3.54 ± 0.21 | 0.53 | 6.44 | 12.21 |
| 5b | 0.58 ± 0.14 | 0.64 ± 0.13 | ND | 1.10 | — | — |
| 6a | 0.83 ± 0.12 | 0.34 ± 0.08 | 2.73 ± 0.34 | 0.41 | 3.29 | 8.03 |
| 6b | 1.60 ± 0.48 | 1.49 ± 0.26 | ND | 0.93 | — | — |
| Ferroquine | 0.0249 ± 0.0028 | 0.017 ± 0.0006 | ND | 0.70 | — | — |
| CQDP | 0.0097 ± 0.0039 | 0.19 ± 0.06 | ND | 19.59 | — | — |
| Artesunate | 0.0104 ± 0.0026 | 0.05 ± 0.02 | ND | 4.81 | — | — |
| Emetine | ND | ND | 0.13 ± 0.0062 | — | — | — |
In general, introduction of the cyclopalladated entity resulted in an overall enhancement of activity (IC50 values below 2 μM) against the NF54 strain of P. falciparum, for the complexes (4–6) in comparison with the dithiocarbamates and TSCs. Comparing the three pairs of complexes (4–6), reveals that the Si-containing 3,4-dichloroacetophenoneTSC palladium complexes (5) were the most potent.
An IC50 value of below 5 μM was selected as the cut-off value for identifying potential lead compounds for further testing. Therefore, only those compounds displaying activity below 5 μM against the NF54 strain were further tested for their activity against the Dd2 strain. A decrease in activity was observed for the dithiocarbamates against the resistant strain, whereas the palladium-free TSCs were generally equipotent against the NF54 and Dd2 strains. However, incorporation of the palladium-PTA fragment, generally results in a slight increase in potency against the resistant Dd2 strain. These cyclopalladated complexes were not as active as ferroquine (Table 1).
The resistance indices [RI = IC50 (Dd2)/IC50 (NF54)] were calculated for compounds displaying activity against the Dd2 strain. A value below or close to 1 suggests that the compounds are more likely to be active against resistant strains. The RI values for the cyclopalladated complexes were generally below 1, indicating that incorporation of the palladium-PTA fragment, delivers complexes likely to be active against resistant strains relative to chloroquine (19.59) and artesunate (4.81). Overall, the thiosemicarbazone compounds displayed lower RI values than the controls.
The compounds containing a methylene spacer (1a–6a) were generally more potent, and thus only those compounds were further tested for their cytotoxicity against the CHO cell-line. Selectivity indices [SI = IC50 (CHO)/IC50 (NF54 or Dd2)] calculated for the dithiocarbamates revealed low SI values (<1) suggesting a lack of selectivity. As seen in Table 1, the thiosemicarbazone compounds (2a, 4a, 5a, 6a) are more effective at killing parasitic cells as opposed to the mammalian CHO cells, with SI values higher than that observed for the dithiocarbamates. In terms of the cyclopalladated complexes, the Si-containing 3,4-dichloroacetophenoneTSC palladium complex (5a) displayed the best selectivity.
Conclusions
Two series of ferrocenyl- and aryl-derived organosilane TSCs were prepared, as well as their cyclopalladated complexes via C–H activation of the ring. The TSCs and cyclopalladated complexes were fully characterised using various spectroscopic and analytical techniques. Evaluation of the compounds against two strains of the malaria parasite Plasmodium falciparum revealed an enhancement of activity upon formation of the cyclopalladated complexes. The TSC compounds also displayed selective antiplasmodial activity. A preliminary β-haematin (synthetic haemozoin) inhibition study of the cyclopalladated complex 5b revealed that the formation of β-haematin, which is a target of antimalarial drugs, was not inhibited. Therefore, investigating other processes such as cysteine protease binding, or conducting ROS studies, could give insight into potential mechanistic routes by which these cyclopalladated complexes inhibit the parasite.Materials and methods
Chemicals
All reagents and solvents were obtained from commercial sources (Sigma-Aldrich, Merck, Kimix, UkrOrgSyntez and FCH group) and, unless otherwise stated, were used as received. The Schiff-base dithiocarbamates,33,42 TSCs (1a, 2a, 3a)26 and PdCl2(PTA)243 were prepared following literature methods. Nuclear magnetic resonance (NMR) spectra were recorded using a Varian Mercury 300 spectrometer (1H at 300.08 MHz), a Bruker 400 Biospin GmbH spectrometer (1H at 400.200 MHz, 13C{1H} at 100.600 MHz, 31P{1H} at 161.80 MHz) or a Bruker 600 FT spectrometer (1H at 600.100 MHz, 13C{1H} at 150.60 MHz) at 30.0 °C. Coupling constants are reported in Hertz. Infrared (IR) spectra were determined using a Perkin Elmer Spectrum 100 FT-IR spectrometer and was carried out using an Attenuated Total Reflectance (ATR) unit. High resolution (HR) ESI-mass spectrometry was carried out using a Waters API Quattro instrument in the positive mode. EI-mass spectrometry was carried out in the positive mode using a JEOL GCmateII apparatus. Elemental analyses (C, H and N) were recorded on a Thermo Flash 1112 Series CHNS-O Analyser. Melting points were recorded using a Buchi B-540 melting point apparatus and are uncorrected.
Synthesis of thiosemicarbazones
:70, v/v) as eluent. Compound 1b was isolated as an oily-brown solid (0.352 g, 62%). Anal. Calcd (%) for C19H29FeN3SiS·¼H2O: C 54.34; H 7.08; N 10.01; Found C 54.66; H 7.27; N 9.90. IR (ATR) νmax/cm−1: 1599w (CN). 1H NMR (400.22 MHz, DMSO-d6): δ (ppm) = 9.84 (1H, s, NNH); 8.22 (1H, t, 3JHH = 6.0 Hz, NH); 4.79 (2H, t, 3JHH = 3.6 Hz, C5H4); 4.37 (2H, t, 3JHH = 3.6 Hz, C5H4); 4.18 (5H, s, C5H5); 3.52 (2H, m, CH2); 2.19 (3H, s, CH3); 1.58 (2H, m, CH2); 0.48 (2H, m, CH2); 0.01 (9H, s, Si(CH3)3). 13C{1H} NMR (150.60 MHz, DMSO-d6): δ (ppm) = 177.6 (CS); 150.7 (CN); 83.7, 70.2, 69.5, 67.7 (Fc); 47.0, 23.9, 15.5 (CH2); 13.7 (CH3); −1.09 (Si(CH3)3). EI+-MS (m/z) 415.03 ([M]+, 100%).
General method for the synthesis of 2b and 3b
Anhydrous ethanol (10.0 mL) was added to the amine under N2, followed by the addition of 3,4-dichloroacetophenone dithiocarbamate. The solution was refluxed for 24 h. The addition of water (10.0 mL) resulted in the precipitation of a white solid which was collected by suction filtration.N). 1H NMR (399.95 MHz, DMSO-d6): δ (ppm) = 10.18 (1H, s, NNH); 8.66 (1H, t, 3JHH = 6.0 Hz, NH); 8.18 (1H, d, 4JHH = 2.2 Hz, H-2); 7.90 (1H, dd, 3,4JHH = 8.5, 2.2 Hz, H-5); 7.64 (1H, d, 3JHH = 8.5 Hz, H-6); 3.55 (2H, m, CH2); 2.28 (3H, s, CH3); 1.58 (2H, m, CH2); 0.49 (2H, t, 3JHH = 6.4 Hz, CH2); 0.01 (9H, s, Si(CH3)3). 13C{1H} NMR (100.64 MHz, DMSO-d6): δ (ppm) = 178.4 (CS); 145.7 (CN); 138.8, 132.1, 131.8, 130.7, 128.6, 127.2 (Ar–C); 47.2, 23.7, 14.4 (CH2); 13.8 (CH3); −1.09 (Si(CH3)3). EI+-MS (m/z) 377.06 ([M + H]+, 50%).
N). 1H NMR (399.95 MHz, DMSO-d6): δ (ppm) = 10.19 (1H, s, NNH); 8.64 (1H, t, 3JHH = 8.0 Hz, NH); 8.20 (1H, d, 4JHH = 2.2 Hz, H-2); 7.91 (1H, dd, 3,4JHH = 8.0, 2.2 Hz, H-5); 7.65 (1H, d, 3JHH = 8.0 Hz, H-6); 3.56 (2H, m, CH2); 2.30 (3H, s, CH3); 1.60 (2H, m, CH2); 1.19 (2H, t, 3JHH = 6.4 Hz, CH2); 0.89 (9H, s, C(CH3)3). 13C{1H} NMR (100.64 MHz, DMSO-d6): δ (ppm) = 178.4 (CS); 145.6 (CN); 138.8, 132.1, 131.8, 130.7, 128.6, 127.2 (Ar–C); 45.0, 41.1 (CH2); 30.5 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (CH3)3), 29.9 (C(H3)3); 24.6 (CH2); 14.4 (CH3). EI+-MS (m/z) 359.09 ([M]+, 100%).
(CH3)3), 29.9 (C(H3)3); 24.6 (CH2); 14.4 (CH3). EI+-MS (m/z) 359.09 ([M]+, 100%).
Synthesis of cyclopalladated complexes
N); 1530w (CN). 1H NMR (399.95 MHz, DMSO-d6): δ (ppm) = 6.56 (1H, t, 3JHH = 6.0 Hz, NH); 4.65 (3H, d, 4JHP = 12.4 Hz, NCH2(eq)N); 4.44 (4H, m, NCH2(ax)N & C5H3); 4.38 (1H, d, 3JHH = 2.4 Hz, C5H3); 4.32 (1H, t, 3JHH = 2.0 Hz, C5H3); 4.24 (6H, s, PCH2N); 4.11 (5H, s, C5H5); 2.79 (2H, d, 3JHH = 6.0 Hz, CH2); 2.14 (3H, s, CH3); 0.038 (9H, s, Si(CH3)3). 13C{1H} NMR (100.64 MHz, DMSO-d6): δ (ppm) = 163.0 (CN); 99.5 (d, 2JCP = 12.9 Hz, C–Pd); 95.2 (–CN); 75.4 (Fc); 72.5 (d, 3JCP = 6.7 Hz, NCH2N); 69.8, 68.5, 66.7 (Fc); 52.7 (d, 1JCP = 16.1 Hz, PCH2N); 37.3 (CH2); 13.4 (CH3); −1.35 (Si(CH3)3). 31P{1H} NMR (162.01 MHz, DMSO-d6): δ (ppm) = −41.6. EI+-MS (m/z) 647.97 ([M]+, 75%).
N); 1546s (CN). 1H NMR (400.22 MHz, DMSO-d6): δ (ppm) = 6.71 (1H, s, NH); 4.65 (3H, d, 4JHP = 12.8 Hz, NCH2(eq)N); 4.44 (4H, m, NCH2(ax)N & C5H3); 4.39 (1H, d, 3JHH = 2.4 Hz, C5H3); 4.33 (1H, t, 3JHH = 2.4 Hz, C5H3); 4.24 (6H, s, PCH2N); 4.12 (5H, s, C5H5); 3.16 (2H, m, CH2); 2.12 (3H, s, CH3); 1.51 (2H, m, CH2); 0.45 (2H, m, CH2); −0.014 (9H, s, Si(CH3)3). 13C{1H} NMR (100.64 MHz, DMSO-d6): δ (ppm) = 95.1 (C–CN); 75.5 (Fc); 72.4 (d, 4JCP = 7.4 Hz, PCH2N); 69.6, 68.6, 66.8 (Fc); 52.8 (d, 1JCP = 16.2 Hz, NCH2N); 48.0, 23.9, 13.9 (CH2); 13.4 (CH3); −1.07 (Si(CH3)3). 31P{1H} NMR (121.47 MHz, DMSO-d6): δ (ppm) = −41.7. ESI+-HRMS (m/z) 677.0923 ([M + H]+, 100%).
N); 1560s (CN). 1H NMR (300.07 MHz, DMSO-d6): δ (ppm) = 7.25 (2H, m, H-2 & NH); 7.11 (1H, d, 4JHP = 3.6 Hz, H-5); 4.58 (3H, d, 4JHP = 12.6 Hz, NCH2(eq)N); 4.44 (3H, d, 4JHP = 12.9 Hz, NCH2(ax)N); 4.27 (6H, s, PCH2N); 2.86 (2H, d, 3JHH = 6.0 Hz, CH2); 2.23 (3H, s, CH3); 0.039 (9H, s, Si(CH3)3). 13C{1H} NMR (100.64 MHz, DMSO-d6): δ (ppm) = 163.3 (d, 3JCP = 7.4 Hz, CN); 153.2 (Ar–C); 136.3 (d, 2JCP = 8.0 Hz, C–Pd); 130.3, 127.0, 126.9, 126.6 (Ar–C); 72.4 (d, 3JCP = 7.2 Hz, NCH2N); 51.5 (d, 1JCP = 15.4 Hz, PCH2N); 37.9 (CH2); 13.5 (CH3); −1.39 (Si(CH3)3). 31P{1H} NMR (121.47 MHz, DMSO-d6): δ (ppm) = −49.6. EI+-MS (m/z) 609.93 ([M + H]+, 2%).
N); 1562w (CN). 1H NMR (399.95 MHz, DMSO-d6): δ (ppm) = 7.23 (1H, s, H-2); 7.12 (1H, d, 4JPH = 3.0 Hz, H-5); 4.60 (3H, d, 4JPH = 13.2 Hz, NCH2(eq)N); 4.47 (3H, d, 4JPH = 13.2 Hz, NCH2(ax)N); 4.28 (6H, s, PCH2N); 3.27 (2H, m, CH2); 2.24 (3H, s, CH3); 1.52 (2H, m, CH2); 0.48 (2H, t, 3JHH = 8.4 Hz, CH2); −0.01 (9H, s, Si(CH3)3). 13C{1H} NMR (100.64 MHz, DMSO-d6): δ (ppm) = 175.2 (C–S); 163.8 (Ar–C); 162.4 (d, 2JCP = 7.4 Hz, CN); 153.2 (Ar–C); 136.4 (d, 2JCP = 8.2 Hz, C–Pd); 130.6, 127.2, 127.1 (Ar–C); 72.6 (d, 3JCP = 7.3 Hz, NCH2N); 51.8 (d, 1JCP = 15.3 Hz, PCH2N); 49.3, 23.9, 14.0 (CH2), 13.4 (CH3); −1.12 (Si(CH3)3). 31P{1H} NMR (121.47 MHz, DMSO-d6): δ (ppm) = −49.8 ppm. ESI+-HRMS (m/z) 637.0488 ([M + H]+, 80%).
N); 1558s (CN). 1H NMR (399.95 MHz, DMSO-d6): δ (ppm) = 7.26 (1H, s, H-2); 7.17 (1H, br s, NH); 7.11 (1H, d, 4JHP = 3.6 Hz, H-5); 4.58 (3H, d, 4JHP = 12.8 Hz, NCH2(eq)N); 4.45 (3H, d, 4JHP = 12.8 Hz, NCH2(ax)N); 4.28 (6H, s, PCH2N); 3.14 (2H, d, 3JHH = 6.4 Hz, CH2); 2.24 (3H, s, CH3); 0.88 (9H, s, C(CH3)3). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 165.5 (Ar–C); 161.6 (d, 3JCP = 7.4 Hz, CN); 152.3 (Ar–C); 135.7 (d, 2JCP = 8.8 Hz, C–Pd); 131.7, 128.1, 127.2 (Ar–C); 72.4 (d, 3JCP = 7.2 Hz, NCH2N); 58.0 (CH2); 51.5 (d, 1JCP = 15.4 Hz, PCH2N); 32.3 ((CH3)3); 27.3 (C(H3)3); 13.4 (CH3). 31P{1H} NMR (121.47 MHz, DMSO-d6): δ (ppm) = −49.6. EI+-MS (m/z) 592.09 ([M]+, 1.5%).
N); 1562s (CN). 1H NMR (399.95 MHz, CDCl3): δ (ppm) = 7.10 (1H, s, H-2); 7.32 (1H, d, 4JHP = 4.80 Hz, H-5); 4.98 (1H, br s, NH); 4.59 (6H, s, NCH2N); 4.32 (6H, s, PCH2N); 3.36 (2H, m, CH2); 2.28 (3H, s, CH3); 1.56 (2H, m, CH2); 1.22 (2H, m, CH2); 0.89 (9H, s, C(CH3)3). 13C{1H} NMR (100.64 MHz, CDCl3): δ (ppm) = 174.2 (C–S); 165.5 (Ar–C); 161.8 (d, 3JCP = 7.0 Hz, CN); 152.4 (Ar–C); 135.8 (d, 2JCP = 9.0 Hz, C–Pd); 131.7, 128.1, 127.2 (Ar–C); 73.4 (d, 3JCP = 7.0 Hz, NCH2N); 52.4 (d, 1JCP = 15.4 Hz, PCH2N); 47.4, 41.3 (CH2); 30.2 ((CH3)3), 29.3 (C(H3)3); 24.8 (CH2); 13.3 (CH3). 31P{1H} NMR (162.01 MHz, CDCl3): δ (ppm) = −50.8. EI+-MS (m/z) 620.13 ([M]+, 1.1%).
DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :D2O stability study
:D2O stability study
Cyclopalladated complexes 5a was dissolved in DMSO-d6:D2O (9:1, v/v) and the 1H NMR spectrum recorded at 0 h. The solution was warmed at 37 °C, and the stability monitored by 1H NMR spectroscopy at 24, 48 and 72 h time intervals to confirm stability of compound during the in vitro assay time period.
Cytotoxicity assay
The MTT-assay is used as a colorimetric assay for cellular growth and survival, and compares well with other available assays.46,47 The test samples were tested in triplicate on one occasion. The same stock solutions prepared for the antiplasmodial activity testing were used for the cytotoxicity tests. Dilutions were prepared on the day of the experiment in complete medium. Emetine was used as the reference drug. The initial concentration of emetine was 100 μg ml−1, which was serially diluted in complete medium with 10-fold dilutions to give 6 concentrations, the lowest being 0.001 μg ml−1. The same dilution technique was applied to the all test samples. The highest concentration of solvent to which the cells were exposed to had no measurable effect on the cell viability (data not shown). The IC50 values were obtained from full dose response curves, using a non-linear dose–response curve fitting analysis via GraphPad Prism v.4 software.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the University of Cape Town, the National Research Foundation (NRF) of South Africa and the Medical Research Council (MRC) of South Africa is gratefully acknowledged. Even though the work is supported by the MRC, the views and opinions expressed are not those of the MRC but of the authors of the material produced or publicised. The South African Research Chairs initiative of the Department of Science and Technology administered through the South African National Research Foundation is gratefully acknowledged for support (KC).Notes and references
- The World Health Organisation Malaria Report http://www.who.int/malaria/media/world_malaria_report_2014/en/.
- World Health Organisation, Global Plan for Artemisinin Resistance Containment (GPARC), 2011 Search PubMed.
- J. N. Burrows, E. Burlot, B. Campo, S. Cherbuin, S. Jeanneret, D. Leroy, T. Spangenberg, D. Waterson, T. N. Wells and P. Willis, Parasitology, 2014, 141, 128–139 CrossRef PubMed.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, C. M. Clavel, P. J. Dyson, C. de Kock, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2012, 31, 5791–5799 CrossRef CAS.
- R. B. de Oliveira, E. M. de Souza-Fagundes, R. P. P. Soares, A. A. Andrade, A. U. Krettli and C. L. Zani, Eur. J. Med. Chem., 2008, 43, 1983–1988 CrossRef CAS PubMed.
- A. Chipeleme, J. Gut, P. J. Rosenthal and K. Chibale, Bioorg. Med. Chem., 2007, 15, 273–282 CrossRef CAS PubMed.
- A. Moreno-Rodríguez, P. M. Salazar-Schettino, J. L. Bautista, F. Hernández-Luis, H. Torrens, Y. Guevara-Gómez, S. Pina-Canseco, M. B. Torres, M. Cabrera-Bravo, C. M. Martinez and E. Pérez-Campos, Eur. J. Med. Chem., 2014, 87, 23–29 CrossRef PubMed.
- L. P. de Carvalho, M. A. G. B. Gomes, B. S. Rocha, R. R. de Oliveira, E. J. Maria and E. J. T. de Melo, J. Dev. Drugs, 2013, 3, 126–132 Search PubMed.
- N. C. Fonseca, L. F. da Cruz, F. D. S. Villela, G. A. do Nascimento Pereira, J. L. de Siqueira-Neto, D. Kellar, B. M. Suzuki, D. Ray, T. B. de Souza, R. J. Alves, P. A. Sales Júnior, A. J. Romanha, S. M. F. Murta, J. H. McKerrow, C. R. Caffrey, R. B. de Oliveira and R. S. Ferreira, Antimicrob. Agents Chemother., 2015, 59, 2666–2677 CrossRef CAS PubMed.
- S. P. Fricker, Metallomics, 2010, 2, 366–377 RSC.
- C. Biot, G. Glorian, L. A. Maciejewski and J. S. Brocard, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- M. Navarro, W. Castro and C. Biot, Organometallics, 2012, 31, 5715–5727 CrossRef CAS.
- C. Biot, W. Castro, C. Botté and M. Navarro, Dalton Trans., 2012, 41, 6335–6349 RSC.
- R. A. Sánchez-Delgado, M. Navarro, H. Pérez and J. A. Urbina, J. Med. Chem., 1996, 39, 1095–1099 CrossRef PubMed.
- P. F. Salas, C. Herrmann and C. Orvig, Chem. Rev., 2013, 113, 3450–3492 CrossRef CAS PubMed.
- R. W. Brown and C. J. T. Hyland, MedChemComm, 2015, 6, 1230–1243 RSC.
- W. A. Wani, E. Jameel, U. Baig, S. Mumtazuddin and L. T. Hun, Eur. J. Med. Chem., 2015, 101, 534–551 CrossRef CAS PubMed.
- M. Adams, Y. Li, H. Khot, C. de Kock, P. J. Smith, K. Land, K. Chibale and G. S. Smith, Dalton Trans., 2013, 42, 4677–4685 RSC.
- P. Chellan, T. Stringer, A. Shokar, P. J. Dornbush, G. Vazquez-Anaya, K. M. Land, K. Chibale and G. S. Smith, J. Inorg. Biochem., 2011, 105, 1562–1568 CrossRef CAS PubMed.
- M. Adams, C. de Kock, P. J. Smith, K. Chibale and G. S. Smith, J. Organomet. Chem., 2013, 736, 19–26 CrossRef CAS.
- P. Chellan, S. Nasser, L. Vivas, K. Chibale and G. S. Smith, J. Organomet. Chem., 2010, 695, 2225–2232 CrossRef CAS.
- J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2571 CrossRef CAS PubMed.
- T. S. Lobana, P. Kumari, R. J. Butcher, T. Akitsu, Y. Aritake, J. Perles, F. J. Fernandez and M. C. Vega, J. Organomet. Chem., 2012, 701, 17–26 CrossRef CAS.
- Y. Li, C. de Kock, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2014, 33, 4345–4348 CrossRef CAS.
- Y. Li, C. de Kock, P. J. Smith, H. Guzgay, D. T. Hendricks, K. Naran, V. Mizrahi, D. F. Warner, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 141–150 CrossRef CAS.
- M. Adams, C. de Kock, P. J. Smith, K. M. Land, N. Liu, M. Hopper, A. Hsiao, A. R. Burgoyne, T. Stringer, M. Meyer, L. Wiesner, K. Chibale and G. S. Smith, Dalton Trans., 2015, 44, 2456–2468 RSC.
- J. S. Mills and G. A. Showell, Expert Opin. Invest. Drugs, 2004, 13, 1149–1157 CrossRef CAS PubMed.
- P. Englebienne, A. V. Hoonacker and C. V. Herst, Drug Des. Rev. – Online, 2005, 2, 467–483 CrossRef CAS.
- R. Tacke, T. Heinrich, R. Bertermann, C. Burschka, A. Hamacher and M. U. Kassack, Organometallics, 2004, 23, 4468–4477 CrossRef CAS.
- W. Bains and R. Tacke, Curr. Opin. Drug Discovery. Dev., 2003, 6, 526–543 CAS.
- G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551–556 CrossRef CAS PubMed.
- R. Tacke, T. Schmid, M. Penka, C. Burschka, W. Bains and J. Warneck, Organometallics, 2004, 23, 4915–4923 CrossRef CAS.
- B. K. Srivastava, S. K. Srivastava, O. P. Pandey and S. K. Sengupta, Indian J. Chem. A Inorg. Bioinorg. Phys. Theor. Anal. Chem., 1996, 35A, 57–59 CAS.
- C. Biot, B. Pradines, M.-H. Sergeant, J. Gut, P. J. Rosenthal and K. Chibale, Bioorg. Med. Chem. Lett., 2007, 17, 6434–6438 CrossRef CAS PubMed.
- D. L. Klayman, J. F. Bartosevich, T. S. Griffin, C. J. Mason and J. P. Scovill, J. Med. Chem., 1979, 22, 855–862 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- A. D. Phillips, L. Gonsalvi, A. Romerosa, F. Vizza and M. Peruzzini, Coord. Chem. Rev., 2004, 248, 955–993 CrossRef CAS.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- A. M. Kirillov, P. Smoleński, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2007, 2007, 2686–2692 CrossRef.
- P. Smoleński and A. J. L. Pombeiro, Dalton Trans., 2008, 87–91 RSC.
- P. Smoleński, M. Kirillova, M. Guedes Da Silva and A. Pombeiro, Dalton Trans., 2013, 42, 10867–10874 RSC.
- C. Biot, D. Taramelli, I. Forfar-Bares, L. A. Maciejewski, M. Boyce, G. Nowogrocki, J. S. Brocard, N. Basilico, P. Olliaro and T. J. Egan, Mol. Pharm., 2005, 2, 185–193 CrossRef CAS PubMed.
- A. M. M. Meij, S. Otto and A. Roodt, Inorg. Chim. Acta, 2005, 358, 1005–1011 CrossRef CAS.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS.
- M. T. Makler, J. M. Ries, J. A. Williams, J. E. Bancroft, R. C. Piper, B. L. Gibbins and D. J. Hinrichs, Am. Soc. Trop. Med. Hyg., 1993, 48, 739–741 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- L. V. Rubinstein, R. H. Shoemaker, K. D. Paull, R. M. Simon, S. Tosini, P. Skehan, D. A. Scudiero, A. Monks and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1113–1118 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |