
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of oxime-based CO-releasing molecules, CORMs and their immobilization on maghemite nanoparticles for magnetic-field induced CO release†
Hajo
Meyer
a,
Markus
Brenner
a,
Simon-P.
Höfert
a,
Tim-O.
Knedel
a,
Peter C.
Kunz
a,
Annette M.
Schmidt
b,
Alexandra
Hamacher
c,
Matthias U.
Kassack
c and
Christoph
Janiak
*a
aInstitut für Anorganische Chemie und Strukturchemie, Universität Düsseldorf, 40204 Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de
bInstitut für Physikalische Chemie, Universität zu Köln, Luxemburger Str. 116, D-50939 Köln, Germany. E-mail: annette.schmidt@uni-koeln.de
cInstitut für Pharmazeutische und Medizinische Chemie, Universität Düsseldorf, 40204 Düsseldorf, Germany. E-mail: matthias.kassack@hhu.de
First published on 21st March 2016
Abstract
Oxime-based CO-releasing molecules (oximeCORMs) were immobilized with a catechol-modified backbone on maghemite iron oxide nanoparticles (IONPs) to give oximeCORM@IONP. The CO release from the free and immobilized oximeCORMs was measured using the standard myoglobin assay. The oximeCORM-nanoparticles were coated with dextran for improved water solubility and confined into an alginate shell for protection and separation from the surrounding myoglobin assay to allow for CO release studies by UV/Vis absorption without interference from highly-absorptive oximeCORM@IONP. Half-lifes of the oxime-based polymer-confined alginate@dextran@oximeCORM@IONPs were estimated at 20 °C to 814 ± 23 min, at 37 °C to 346 ± 83 min and at 50 °C to 73 ± 1 min. The alginate@dextran@oximeCORM@IONP composite showed a further decrease of the half-life of CO release to 153 ± 27 min at 37 °C through local magnetic heating of the susceptible iron oxide nanoparticles with application of an external alternating magnetic field (31.7 kA m−1, 247 kHz, 39.9 mTesla). The activation energy for the CO release from molecular dicarbonylchlorido(imidazole-2-carbaldehydeoxime)(alkoxycarbonyl)ruthenium(II) complexes is determined to be ∼100 kJ mol−1 for five different imidazole-oxime derivatives.
Introduction
The toxic physiological effects of carbon monoxide (CO) were first described by John Haldane in 1927.1 CO is generally known as a highly toxic gas which hinders the oxygen transport in the blood system, by strong coordination to the oxygen binding site in hemoglobin. Even breathing of small quantities (about 100 ppm in air) of this color- and odorless gas can be potentially harmful to the human organism.2,3 On the other hand, carbon monoxide is classified as a biochemical messenger molecule like hydrogen sulfide and nitrogen oxide.4–6 Human organisms express heme oxygenase enzymes,7 which are capable of producing carbon monoxide continuously in order to regulate the circadian rhythms, memory and hemodynamics.8–10 Due to its regulatory and cytoprotective effects CO has already been extensively investigated for medical applications.9 Yet, when CO is administered to a human organism as a gas, it is difficult to accumulate at a specific site (e.g. tumor or burned skin). Gaseous CO as a therapeutic agent features a half-life of 3 to 7 hours in the body and was well tolerated at 3 mg per kg for one hour. CO binds reversibly to cellular targets and the only way for CO to exit mammal bodies in a therapeutic approach was by lung exhalation.11,12 In fact the gas also reaches healthy tissue and the dose needs to be relatively high in order to achieve an effect on the desired tissue. In addition, the quantity needs to be carefully controlled and special administering equipment is necessary.CO releasing molecules (CORMs), primarily metal carbonyl derivatives were invented for specific targeting and controlled release of carbon monoxide. Metal carbonyl derivatives are investigated also as antibacterial compounds and for their anticancer therapeutic potential.13 However, the first CORMs were hardly water-soluble and released CO a way too fast for precise administering (half the gas load in less than one minute under physiological conditions).14 Motterlini and coworkers3,15 introduced in 2003 the water-soluble complex [Ru(CO)3(glycinate)] (CORM-3) which released CO under physiological conditions16 and showed vasodilatative, anti-inflammatory, renoprotective and anti-apoptosis effects in preclinical studies.17 Since the last decade, Ru-based CORMs have become a very important field of research with several studies of CORM-2 and the water-soluble CORM-3 (Scheme 1).9,14,16
 | ||
| Scheme 1 Scheme of the synthesized oxime-based CORMs from Oresmaa et al. with their half-lifes. The half-lifes were determined at ambient (room) temperature in a phosphate buffered saline solution (pH = 6.8).38 | ||
The half-life of CO release from CORM-3 via a pseudo-first-order reaction is 1 min at 37 °C in PBS buffer,18 2.3 min at 30 °C in PBS buffer19 and 3.6 min in human plasma,20 apparently due to CO-abstraction by heme proteins.21 Recent studies presented CORMs which liberate CO upon external stimuli like change in the pH,22,23 increasing temperature, UV irradiation24,25 and oxidative or enzymatic degradation.26,27 The immobilization of CORMs in28 and on29,30 other materials opens new methodologies in medicinal chemistry.31,32 However, different possible medical applications of CORMs will require CORMs with different release kinetics.
Drugs which are conjugated to polymers are restricted to the vascular system and can be transported directly to the target area. A passive accumulation of macromolecular systems in tumor tissues due to the so-called enhanced permeability and retention (EPR) effect was first described and investigated by Maeda et al.33,34
The growth of tumors (>2 mm) is ensured by fast growing blood vessels to supply nutrients and dioxygen. The blood vessels are destabilized due to vascular endothelial growth factors (VEGFs) into the extracellular space and have higher permeability than normal grown blood vessels.35 The resulting blood vessels lack tight junctions that are normally present between adjacent vascular endothelial cells. This deficiency leaves holes on an order of magnitude greater than normal vascular pores (healthy tissue has pores of 5 to 8 nm, tumor tissue can have pores of about 50 nm and inter-endothelial junctions of about 500 nm). The EPR effect refers to the properties of these large pores to allow polymers to enter the tumor interstitium and to remain and accumulate there because the polymers are not removed by the lymphatic system.36
The accumulation of functionalized polymers in tumor tissue with the ability to release CO is an additional concept of developing polymer–CORM conjugates. In such conjugates the use of a slow CO releaser from which the CO release can be triggered or activated by magnetic heating through alternating magnetic field should be of interest.
Different CORMs with ruthenium carbonyls are known and the number is still increasing.37 Oresmaa et al. described a simple synthetic route to oxime-based CORMs for which they reported a longer half-life for CO release than for CORM-3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 (Scheme 1).
38 (Scheme 1).
In our previous work, we presented a new concept of controlled CO release by covalently attaching the CO-releasing molecule (a CORM-3 analogue) to the surface of iron oxide nanoparticles which are susceptible to local heating through an external alternating current magnetic field.39 The increased temperature of the nanoparticles, due to the external alternating magnetic field, decreased the half-life from 13 ± 2 min to 7 ± 2 min in a buffered myoglobin-assay solution kept at 25 °C.
Here we target the covalent immobilization of relatively stable (long half-life) oxime-based CORMs on the surface of iron oxide nanoparticles. In addition a dextran coating is applied for better water solubility and as a barrier against heme proteins.
At the same time the strong absorptivity of the black IONP dispersion hampers CO release studies with the standard myoglobin assay in the same dispersion. The CO release by a myoglobin assay is followed by UV/Vis spectrometry. Therefore, we used an alginate confinement of “water-soluble” dextran@oximeCORM@IONP to achieve a spatial separation from the surrounding myoglobin assay.
Results and discussion
Synthesis
The covalent immobilization of the oxime-based CORMs on an iron oxide nanoparticle (IONP) surface is depicted in Scheme 2.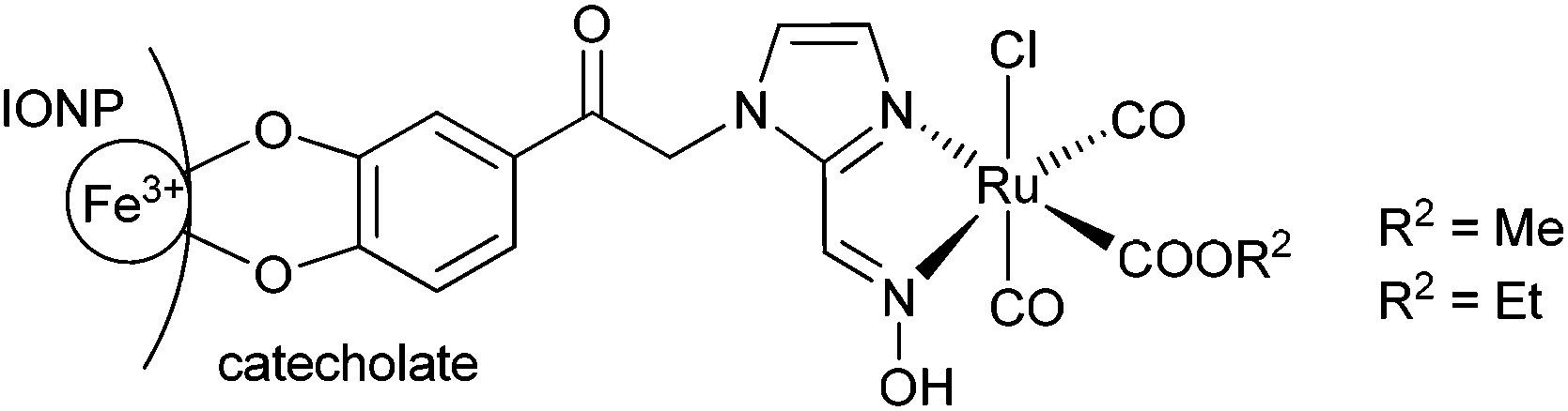 | ||
| Scheme 2 Covalent immobilization of oxime-based CORM on an iron oxide nanoparticle (IONP) surface through a catecholate group. | ||
Starting with the commercially available 2-chloro-3′,4′-dihydroxyacetophenone 3 (Scheme 3), we first tried unsuccessfully a simple condensation reaction with 1H-imidazole-2-carbaldehyde 4 to try to obtain the condensation product 1-(2-(3,4-dihydroxyphenyl)-2-oxoethyl)-1H-imidazole-2-carbaldehyde 5 in a single step. The single-step route failed so that the diol was first protected by an acetal group to form product 6 in good yields. The condensation of 6 with 4 to give 1-(2-(3,4-dihydroxyphenyl)-2-oxoethyl)-1H-imidazole-2-carbaldehyde 7 in acetonitrile with an equimolar ratio of a base proved successful. Acid cleavage of the acetal protecting group afforded the desired product 1-(2-(3,4-dihydroxyphenyl)-2-oxoethyl)-1H-imidazole-2-carbaldehyde 5 in high yields. Introducing the oxime function into compounds 7 and 5 was done similarly to ref. 38 (Scheme 3). The reaction times were extended up to 24 h to obtain good yields for (E)-1-(2-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)-2-oxoethyl)-1H-imidazol-2-carbaldehyde-oxime 8 (75%) and 1-(2-(3,4-dihydroxyphenyl)-2-oxoethyl)-1H-imidazole-2-carbaldehyde-oxime 9 (47%).
 | ||
| Scheme 3 Synthetic route (with yields) for the coupling of the catechol group with 1H-imidazol-2-carbaldehyde (4) to 1-(2-(3,4-dihydroxyphenyl)-2-oxoethyl)-1H-imidazole-2-carbaldehyde (5) and the acetate protected (8) and unprotected oxime (9). (a) Acetonitrile, sodium carbonate, 60 °C, 5 d; (b) dry toluene, dry acetone, phosphorus pentoxide, 75 °C, 4 h; (c) acetonitrile, sodium carbonate, 60 °C, 7 d; (d) acetic acid, hydrochloric acid, 100 °C, 2 h; (e) sodium carbonate, hydroxylammonium chloride, 70 °C, 24 h. | ||
The synthesis of the diol-protected ruthenium carbonyl oximes 10 and 11 was performed as described in the literature38 with [Ru(CO)3Cl2]2 (12) and an extended reaction time of 5 d (Scheme 4). To obtain the methoxy-compound 10 the reaction was carried out in methanol and for the ethoxy-compound 11 in ethanol. CORMs 10 and 11 precipitated from the solution as the modified backbone leads to lower solubility in polar solvents than for the parent compounds a and b (Scheme 1).
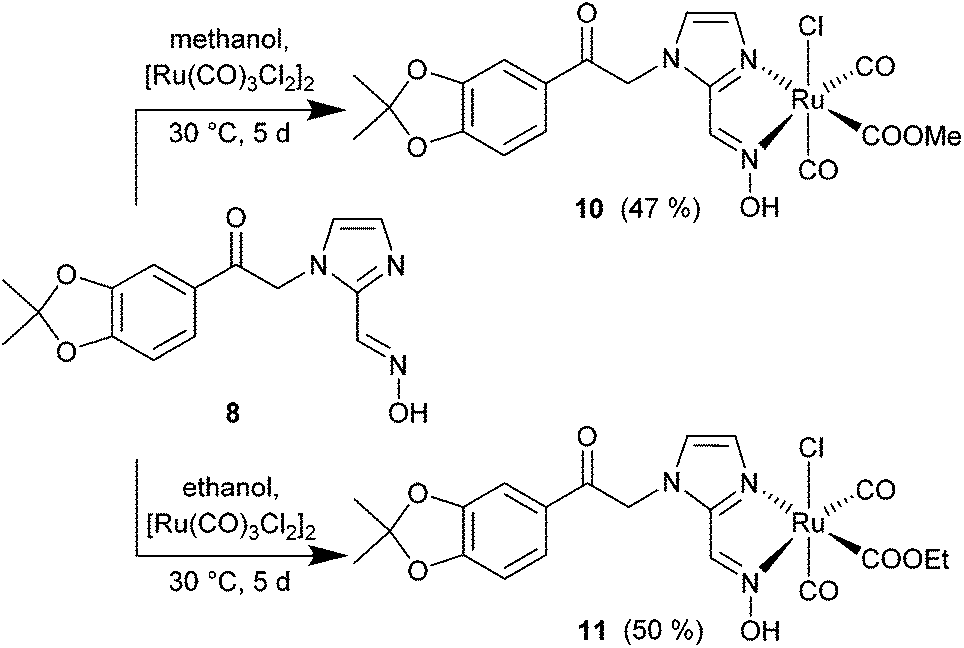 | ||
| Scheme 4 Synthesis of CORMs with the diol-protected catechol oxime 8 as methoxy- (10) and ethoxy- (11) ruthenium complexes (yields in parentheses). | ||
For immobilization on an iron oxide nanoparticle surface the dihydroxyphenyl-oxime 9 was reacted with [Ru(CO)3Cl2]2 (12) to give the ruthenium carbonyl oximes 13 and 14 as shown in Scheme 5.
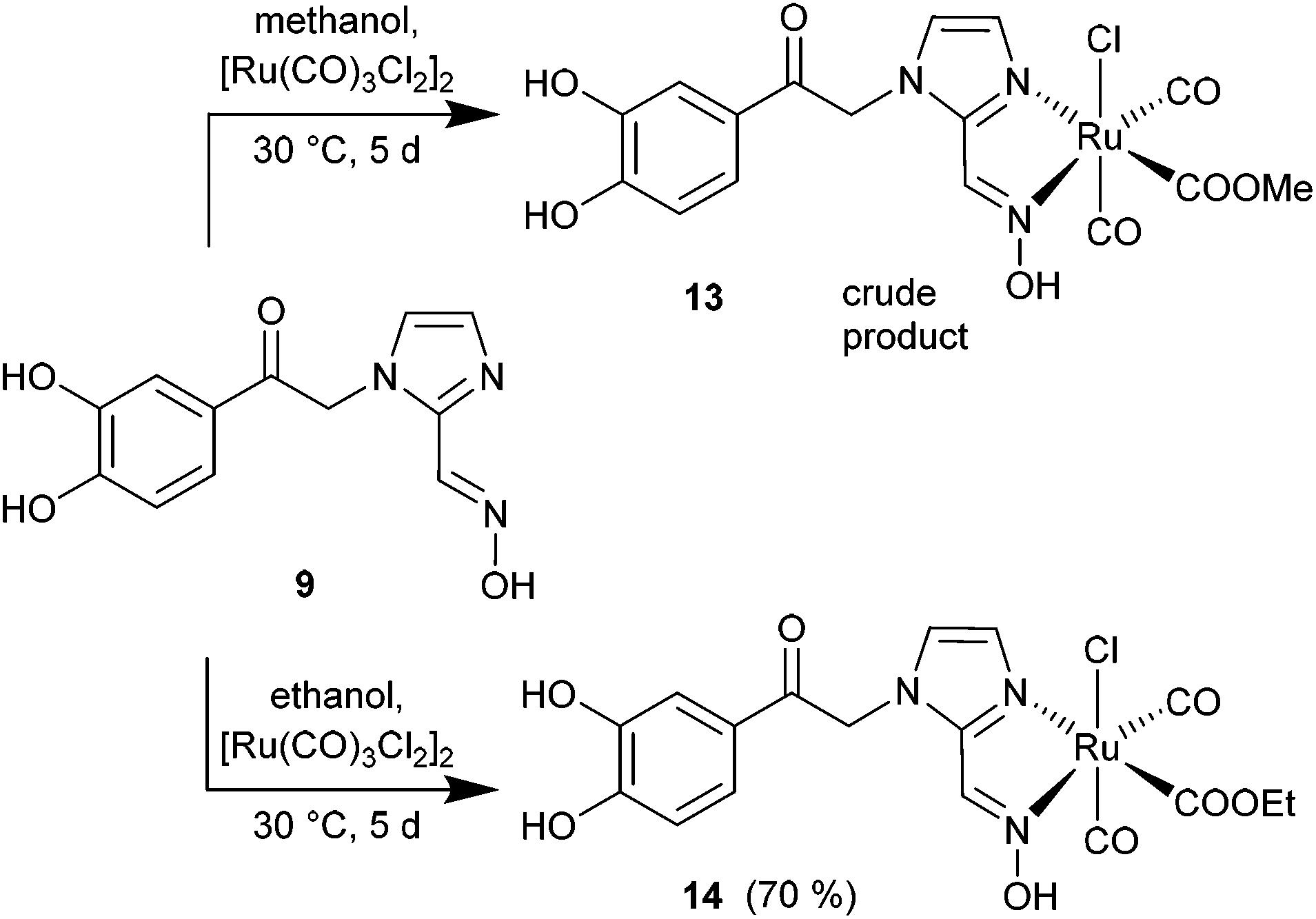 | ||
| Scheme 5 Synthesis of CORMs with the (unprotected) catechol oxime 9 as methoxy- (13) and ethoxy- (14) ruthenium complexes (yields in parentheses). | ||
Electron spray ionization mass spectrometric analysis of 10–14 in acetonitrile solution showed ligand lability (see Fig. S3, ESI†). For compounds 10 and 11 or 13 and 14, respectively, identical mass spectra were obtained and the isotopic pattern showed a ruthenium complex from which the methoxy (or ethoxy) group (OR−), a CO unit and/or a chloride ligand had been lost. In addition, a proton was abstracted, presumably from the N–OH group, which together with CO and OR− amounts to the liberation of formic acid ester or with OR− only to the liberation of the alcohol ROH. Instead, acetonitrile was coordinated under ESI-MS conditions. Hence, the dominant fragments (for 102Ru) of m/z = 499.1 (10, 11) or 459.2 (13, 14) correspond to [M − OR− − CO − Cl− − H+ + CH3CN]+ and m/z = 527.0 (10, 11) or 487.0 (13, 14) to [M − OR− − Cl− − H+ + CH3CN]+ (R = Me, Et). The lability of carbonyl complexes in ESI mass spectrometric analysis is known.40
IR spectroscopic measurements of 10–14 show the expected strong carbonyl absorptions (Fig. S1 in the ESI†) which agree well with the ones reported by Oresmaa et al.38 for a and b in the regions 2056–2059 cm−1 and 1980–1985 cm−1 (Table 1).
| Compound | νCO [cm−1] | νCO [cm−1] | Ref.d |
|---|---|---|---|
| a KBr disks. b FT-IR in DMSO. c Formula in Scheme 1. d This work with reference to the figure showing the IR spectrum, unless indicated otherwise. e We trace the third CO band to partial CO release during CORM deposition on IONP and inclusion in dextran in water (cf.Scheme 6). | |||
| 1a , | 2064 | 1990 | Fig. S2-1a |
| 2a , | 2067 | 1994 | Fig. S2-1a |
| 1a , , 2ab,c | 2056–2059 | 1980–1985 | 38 |
| 10 | 2075 | 1989 | Fig. S2-1b |
| 11 | 2065 | 1988 | Fig. S2-1b |
| 14 | 2077 | 2012 | Fig. S2-2 |
| 17 | 2064 | 1995, 1961e | Fig. S2-3 |
| 18 | 2061 | 1991 | Fig. S24 |
| 18 after CO release | 2071, 2015 | 1992 | Fig. S24 |
Maghemite nanoparticles (15) were synthesized by the known method from Ewijk et al.41 A catechol based anchor group is common for immobilizing molecules on an IONP surface.42 The unprotected catechol–oxime ruthenium complex 14 was immobilized on the surface of maghemite nanoparticles. Compound 14 was dissolved in water under basic conditions and stirred with maghemite nanoparticles. To prevent agglomeration of the nanoparticles the combined solutions of both compounds were kept below pH 8. After precipitation with acetone and washing with ethanol, the oximeCORM@IONP composite nanoparticles 16 were not able to build a stable dispersion in MOPS-buffer or water but sedimented within a few seconds (see the ESI† for DLS and TEM analysis of 16). To increase the solubility of the oximeCORM@IONP nanoparticles in water, they were coated by using dextran polymers to give dextran@oximeCORM@IONP (17). Water-soluble dextran (M = 500000 g mol−1) consists of glucose monomers with α-1,4- and α-1,6-connectivity and was added during the reaction of 14 with the iron oxide nanoparticles.
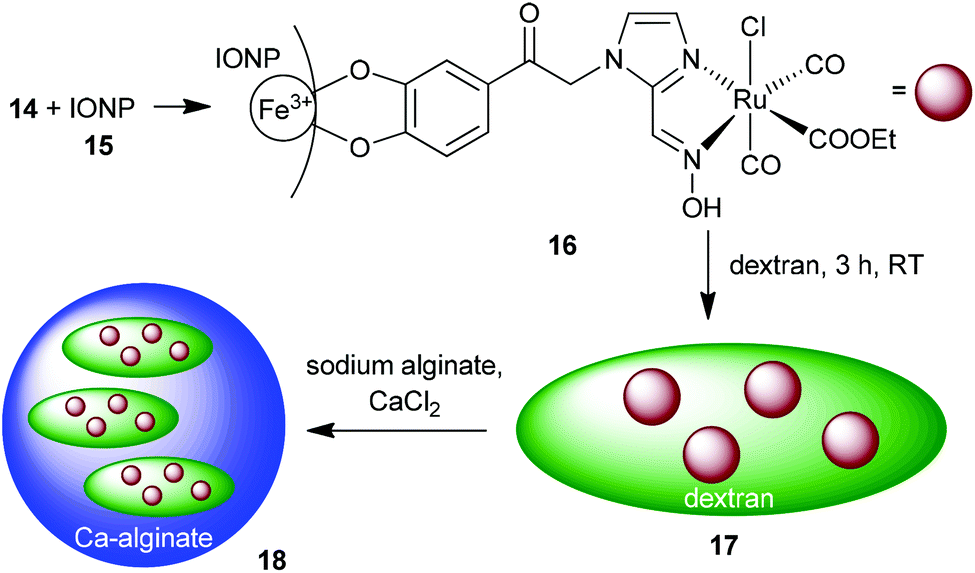 | ||
| Scheme 6 Synthesis of the composite material alginate@dextran@oximeCORM@IONP (18) by immobilization of the oximeCORM 14 on the maghemite nanoparticle surface 15. | ||
The IR spectrum of the composite material 17 (Fig. S4 in the ESI†) shows the OH-vibrations of the dextran at 3600–3000 cm−1 and the carbonyl region exhibits three strong absorptions at 2064, 1995 and 1961 cm−1.
The amount of ruthenium in the dextran@oximeCORM@IONP (17) composite was determined by atomic absorption spectroscopy (AAS) to an average of 0.60 wt% ruthenium and a molar Ru:Fe ratio of 1:25 (see Experimental section or the ESI† for details). This is the same molar Ru:Fe ratio as in our previous report on a CORM-3 analog on IONP (Ru:Fe 1:2539).
Further, composite 17 was incorporated into calcium alginate spheres. A mixture of 17 with sodium alginate was added dropwise to a calcium chloride solution. Calcium cross-linked alginate spheres were rapidly formed which almost quantitatively incorporate the dextran@oximeCORM@IONP substrate as is evident from the clear, non-colored surrounding solution (Fig. S26 in the ESI†). The IR spectrum of the composite material 18 (Fig. S28 in the ESI†) shows two strong absorptions at 2061 and 1991 cm−1, which are indicators for the immobilized carbonyls on the nanoparticle surface. Yet, the still necessary use of water to anchor 14 on IONPs to give 16 and in the dextran and alginate coating of 16 to 17 and further to 18 represents a drawback and already induces partial decomposition with a loss of CO (see below and Table S1 in the ESI†). Future work has to address replacing water by other solvents.
CO release
The half-lifes t1/2 of the CO release from compounds 1, 2, 10, 11 and 18 were measured using the myoglobin assay to elucidate differences in the CO release kinetics at 20, 37 and 50 °C (Fig. 1a). A 3-(N-morpholino)propanesulfonic acid buffer (pH = 7.4, 0.1 M) was used by us instead of the phosphate buffer which Oresmaa et al. described to prevent further agglomeration of the IONP-containing samples and to make the results comparable to our previously presented system.39 All measurements were done three times to ensure reproducibility. | ||
| Fig. 1 Time-dependent visible absorption spectra (left) and the plot of the intensity changes of selected wavelengths (right) of alginate@dextran@oximeCORM@IONP (18) with the myoglobin assay at 37 °C (a) without the influence of an external alternating magnetic (AC) field to t1/2 = 346 ± 83 min, and (b) with alternating field t1/2 = 155 ± 27 min. Arrows in the absorption spectra mark the selected wavelengths with the direction of their time-dependent change. | ||
The half-lifes of the CO release are listed in Table 2 (for kinetic data see Fig. S5–S25 in the ESI†). Oresmaa et al. described a slightly faster CO release from the methoxy-1a (t1/2 = 60 min) than from the ethoxy-2a oximeCORM (cf.Scheme 1) (t1/2 = 70 min) at ambient temperature, albeit not further specified temperature.38 From our thermostated measurement at 20 °C, 37 °C and 50 °C, the methoxy- (1a) and ethoxy- (2a) derivatives have the same half-life within the experimental error. We note that the oximeCORM compounds 1a and 2a show a slower CO release than CORM-3 at 37 °C (t1/2 ≤ 1 min) in the presence of the myoglobin assay.
| Compound | t 1/2 [min] | |||
|---|---|---|---|---|
| 20 °C | ∼25 °C | 37 °C | 50 °C | |
| a The work of Oresmaa et al.38 does not specify a temperature. Thus, ambient or room temperature can be assumed which can be – depending on season, climate and weather – somewhere between 15 °C to 30 °C. The half-life values from Oresmaa et al. must be traced to a temperature higher than 20 °C or to buffer influences. b With applied alternating magnetic field (31.7 kA m−1, 247 kHz, 39.9 mTesla). | ||||
| 1a | 220 ± 23 | 21 ± 2 | 5 ± 1 | |
| 1a | 60 | |||
| 2a | 226 ± 9 | 18 ± 1 | 6 ± 1 | |
| 2a | 70 | |||
| 10 | 172 ± 23 | 18 ± 3 | 4 ± 1 | |
| 11 | 155 ± 13 | 18 ± 1 | 4 ± 1 | |
| 14 | 207 ± 6 | 16 ± 1 | 3 ± 1 | |
| 18 | 814 ± 23 | 346 ± 83 | 73 ± 1 | |
| 18 in AC fieldb | 153 ± 27 | |||
The diol-protected catechol oximeCORMs 10, 11 and the unprotected catechol oximeCORM 14 also show similar half-lifes within the experimental error with a slightly faster release than from 1a and 2a at 20 °C, which might correlate to the modified backbone.
The dark “soluble” dextran-coated iron oxide nanoparticles 17 do not lend themselves to follow the CO release with the myoglobin assay due to their strong UV/Vis absorption.42 Dextran coating leads to a stable dispersion with no sedimentation anymore. Thus, the high background absorption from the dark color from the iron oxide nanoparticles masks the absorption changes of the myoglobin assay. The confinement of dextran@oximeCORM@IONP in calcium alginate spheres allowed for minimization of the spectral interference by separation within the myoglobin assay.42 We briefly note that there are other methods available for CO release studies from CORMs. Electrochemical sensing,43 fluorescent probes,44,45 gas-phase IR spectroscopy,43 or gas chromatography46,47 have been reported in the literature. Yet, the inexpensive and easy alginate-separation method allows for working with the widely accepted myoglobin assay.
The calcium alginate shell around the “soluble” dextran-coated iron oxide nanoparticles presents a membrane, which allows for diffusion of gaseous CO but prevents the interaction with the outside protein and sodium dithionite of the myoglobin assay.42
Previous CO-release studies from CORM-2 and CORM-3 showed that the rate depended on myoglobin and was accelerated by an increasing concentration of the reducing agent, sodium dithionite.21 The loss of carbon monoxide was shown when CORM-3 was preincubated with sodium dithionite in the absence of myoglobin. After reduction of the myoglobin and removal of the sodium dithionite in the assay, no liberation of CO from CORM-3 was seen.19 The interactions of CORM-3 with proteins result in the loss of a chloride ion, glycinate, and one CO ligand because of rapid formation of stable protein–Ru(CO2) adducts.21
For the oxime-based CORMs (1, 2) Oresmaa suggested “that the alkoxycarbonyl group plays an important role in CO release. Whether the role is merely to destabilize carbonyl ligands or act as a source of CO remains to be studied”.38
Furthermore, the alginate spheres containing the iron oxide nanoparticles were magnetic and could be retained at the bottom of the cuvette with a magnet to prevent them from moving into the light path during UV/Vis measurement (Fig. S22 in the ESI†). Spheres of the composite material 18 were transferred within 30 min after their synthesis into the cuvette under nitrogen to avoid contamination with dioxygen. MOPS-buffer and myoglobin solution (containing Na2S2O3) were added and the CO release measurements were started immediately by collecting time-dependent UV/Vis spectra at 20, 37 and 50 °C. The half-lifes were determined from the kinetic curves of the absorption changes with time (see the ESI†) and are listed in Table 2.
The CO release of the composite material 18 was also measured at 37 °C (surrounding solution temperature) under the influence of an external alternating magnetic (AC) field (31.7 kA m−1, 247 kHz, 39.9 mTesla) to determine the accelerated CO release due to magnetic heating of the iron oxide particles (Fig. 2). The estimated half-life at 37 °C with an AC field is 153 ± 27 min (ESI, Fig. S27†). The magnetic heating of the maghemite nanoparticles leads to an accelerated CO release by a factor 2.3 through the increase in the surface temperature of the IONP, which affects the attached CORM (Fig. 3a).
 | ||
| Fig. 2 Half-lifes for temperature-dependent CO release from alginate@dextran@oximeCORM@IONP (18) without and with applied alternating (AC) magnetic field (at 37 °C only). | ||
 | ||
| Fig. 3 (a) Correlation of ln(t1/2) versus the temperature for 18 for AC on and off to estimate the IONP surface temperature upon magnetic heating, thereby assuming a direct correlation between their half-life and temperature. The initial temperature position of the red dot (AC on) is the set solution temperature. The AC field then increases the temperature on the IONP surface as traced by the blue arrows. (b–d) Arrhenius plots for 18 (b), 11 (c) and 14 (d) of lnk = ln(ln(2)/t1/2) versus 1/TKelvin to determine the activation energy from the slope (−EA/R, R = 8.314 J mol−1 K−1) of the graph. | ||
We determined the AC effect only at the physiologically relevant temperature of 37 °C. We have shown recently with a CORM-3@IONP analog that the CO-release is accelerated from a half-life of 890 ± 70 min at 20 °C, 172 ± 27 min at 37 °C and 45 ± 7 min at 50 °C with an AC field to 155 ± 18 min at 20 °C, 65 ± 5 min at 37 °C and 30 ± 3 min at 50 °C.42 Thus, the acceleration of CO release or lowering of the CORM half-life by the AC field will be even larger at 20 °C.
IR spectroscopic measurements showed shifted carbonyl absorptions after the CO release of the composite material alginate@dextran@oximeCORM@IONP (18) from 2061 and 1991 cm−1 to 2071, 2015 and 1992 cm−1 (ESI, Fig. S28†). The complete disappearance of the carbonyl absorption bands could not be observed after the CO release studies with the myoglobin assay. Therefore, the release of both CO-ligands from oximeCORMs does not take place. For CORM-3 only one mol of CO can be released from [Ru(CO)3Cl(O2CCH2NH2)] in PBS buffered solution with myoglobin. After the release of one CO from CORM-3 an inactive form, termed iCORM is formed. The inactive iCORM-form of CORM-3 is not able to release further CO after the loss of one CO.16
The absolute amount of released CO was determined with the Lambert–Beer law and gave the formation of 0.2(1) mol carbon monoxide per mol of Ru from alginate@dextran@oximeCORM@IONP (18), 0.9(1) mol for 10, 0.8(1) mol for 11 and 0.9(1) mol for 14 in the myoglobin assay (see eqn (1) in the ESI†). The low CO amount per Ru in 18 results apparently from the CO loss during the synthesis. It is known from recent work that CORM-2 ([Ru(CO)3Cl2]2) will lose 1.8 CO equivalents as CO2 in water within 24 h.37 In order to anchor 14 on maghemite nanoparticles (15) and coat with dextran to yield dextran@oximeCORM@IONP (17), the mixture had to be stirred in water for 3 h. Inclusion of 17 in the alginate spheres, giving 18 added another ∼35 min of water contact before the CO release measurements. Attempts to replace water by methanol in the formation of 17 led to agglomeration of the IONPs.
In our previous work on a CORM-3@IONP analog we realized that ln(t1/2) shows a linear correlation with temperature and that the ln(t1/2) values at 37 °C (AC off) and 20 °C (solution temperature, AC on) were very close, with ln(172) = 5.15 and ln(155), respectively = 5.04. From this, we deduced that the AC field at 20 °C generates an IONP surface temperature of about 37 °C.42 The surface temperature of the IONP through the AC field was determined through a plot of ln(t1/2) versus the temperature (Fig. 3a). Under the assumption that the half-life for the same material is only a function of temperature, then the ln(t1/2) value reached under the AC magnetic field would correlate with an IONP surface temperature of 43 °C in the surrounding solution temperature of 37 °C (Fig. 3a). A determination of the activation energy EA for the CO release from the alginate composite material 18 with an Arrhenius plot of ln(ln(2)/t1/2) versus 1/T gives a value of 62(17) kJ mol−1 (AC off) (Fig. 3b). This activation energy (with its error margins) compares well to the value of 78 kJ mol−1 for the CO release from a CORM-3 analog embedded in a similar alginate@dextran@CORM@IONP composite.42 Furthermore, we determined the activation energy for the CO release from 1a to 100(3) kJ mol−1, from 2a to 96(12) kJ mol−1, from 10 to 99(1) kJ mol−1, from 11 to 96(1) kJ mol−1 and from 14 to 111(2) kJ mol−1 (see ESI, Fig. S39†). The low correlation for 18 is due to its heterogeneous nature, whereas the CO release from compounds 1a, 2a, 10, 11 and 14 occurred in homogeneous solutions.
Toxicity tests of dextran@oximeCORM@IONP (17) against the three cell lines A2780, Cal27 and HEK293 showed no growth inhibition up to a concentration of 17 of 100 μg mL−1 (see ESI, Fig. S35–S38†). The three reference compounds CORM-2, CORM-3, and 14 also showed no or low cytotoxic effects with 0–9.23% growth inhibition, respectively, at 10 μmol L−1. A higher concentration of 100 μmol L−1 showed moderate growth inhibition of 14 up to 33.9%. For comparison, 100 μmol L−1 cisplatin inhibited cell growth by >93% (Table 3).
Conclusions
We have presented the formation of an oxime-based CORM covalently anchored on iron oxide nanoparticles and “solubilized” by a dextran coating. For this, we implemented a synthesis for the immobilization and backbone modification of oxime-based CORMs on iron oxide nanoparticles with a catechol anchor group. A dextran polymer coating with a molar mass of 500000 g mol−1 was applied to achieve good water “solubility” or rather a stable suspension of the modified iron oxide nanoparticles. For the measurement of the CO release with the myoglobin assay, the dark-colored CORM-functionalized iron oxide nanoparticles had to be separated to allow for reliable UV/Vis absorption measurements. This in situ separation was achieved by incorporating the “soluble” dextran@oximeCORM@IONP into a calcium alginate sphere. The composite material alginate@dextran@oximeCORM@IONP shows an extended half-life for CO release at 37 °C of 346 ± 83 min compared to the molecular oximeCORMs with the diol-protected catechol backbone 10 of 18 ± ∼3 min and the unprotected catechol compound 14 of 16 ± ∼1 min in the myoglobin assay. An alternating current magnetic field (31.7 kA m−1, 247 kHz, 39.9 mTesla) leads to a decrease of the half-life at 37 °C from 346 ± 83 (without AC field) to 153 ± 27 (with AC field).
Experimental section
All chemicals were purchased from Acros®, Aldrich®, VWR® and Fluka® and used as received. [Ru(CO)3Cl2]2 (12) was synthesized according to literature procedures.48 Doubly-deionized (d.d.) water was obtained by Millipore Synergy with a conductivity of 0.05 μS cm−1.1H-NMR spectra were recorded with a 300 or 600 MHz Bruker FT-NMR Spectrometer Avance III. For measurements with CDCl3, tetramethylsilane (TMS) was used as an internal reference. Measurements in other solvents were referenced to the solvent signal (DMSO-d6 2.50 ppm, acetone-d6 2.04 ppm). All chemical shifts δ are given in ppm. Spectra were recorded at 300 K. 1H and 13C NMR spectra are displayed in Fig. S1a–i in the ESI† and illustrate the purity of the compounds.
Mass spectra were obtained with a HR-MS UHR-QTOF maXis 4G (Bruker Daltonics) or with an ESI Ion-Trap-API-Massenspektrometer Finnigan LCQ Deca (Thermo Quest). Ruthenium-containing fragments show the Ru-96, -98, -99, -100, -101, -102, and -104 isotope distribution patterns. The most intense peak of the isotope distribution is noted in the mass spectrometric assignments (based on 102Ru, mass 101.904 with 31.6% abundancy).
IR spectra were recorded on a Bruker Tensor 37 IR spectrometer as KBr discs or using an attenuated total reflectance (ATR) technique. Care was taken to keep the amount of the nanoparticle composites as low as possible by using only 1–2 mg composite material/30–50 mg KBr because of the strong absorption of the dark nanoparticles. In order to see the relevant carbonyl bands the amount of substance needed to be higher than for pure compounds because of the low ruthenium carbonyl percentage in dextran@oximeCORM@IONP (17) and alginate@dextran@oximeCORM@IONP (18). Attempted IR spectroscopic measurements of materials containing the black-colored nanoparticles in ATR mode gave only very small signal intensities. Only compounds without nanoparticles were measured by ATR. The integration time was set to 50 scans per wavenumber to obtain good quality spectra. The IR spectra are given in the ESI.†
Dynamic light scattering measurements were carried out with a Malvern Zetasizer Nanoseries. For determination of the nanoparticles size distribution, a dispersion (“solution”) with a minimum concentration of 1 mg mL−1 in doubly deionized water was prepared. Measurements at different concentrations of the dispersion yielded the same hydrodynamic diameter.
Alternating current (AC) field generator measurements were performed with a Hüttinger HF generator AXIO T5, equipped with a water-cooled copper induction coil. The system was operated at 247 kHz, with a magnetic field amplitude of 31.7 kA m−1 and 39.9 mTesla.
UV/Vis-absorption spectra were collected on a Specord S 600 UV/Vis-spectrometer of Analytik Jena Co. This work was carried out under nitrogen or argon and with thermostated cuvettes.
Atomic absorption spectroscopy (AAS) measurements were performed with a Perkin Elmer AAS AAnalyst200 by MicroLab Kolbe, Höhenweg 17, D-45470 Mülheim.
Transmission electron microscopy (TEM) images were obtained with a Zeiss 902 TEM at the Center for Advanced Imaging (CAi) in the University of Düsseldorf. For the sample preparation about 1 mg of the particles was dispersed by sonication and fast up-and-down pipetting using a 1 mL syringe in deionized water (2 mL). This dispersion was diluted 1:10 with deionized water to yield a very low concentration of the nanoparticles. One drop of this dispersion was placed on a holey thin film of amorphous carbon on a copper grid. After a contact time of 15 min, the grid was floated on a water surface and held vertically to rinse of the solvent.
Column chromatography was performed using a Reveleris chromatograph from Grace.
Toxicity tests
Cisplatin was purchased from Sigma-Aldrich, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) from Serva (Germany). All other reagents were supplied by PAN Biotech (Germany).Synthesis
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] = 2068, 1990. 1H NMR (300 MHz, CD3OD): δ [ppm] = 8.43 (s, 1H, C–CH
[cm−1] = 2068, 1990. 1H NMR (300 MHz, CD3OD): δ [ppm] = 8.43 (s, 1H, C–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 7.42 (s, 1H, imi-CHCH), 7.37 (s, 1H, imi-CHCH), 3.36 (s, 3H, CH3) (matching NMR in ref. 38).
[cm−1] = 2067, 1994. 1H NMR (300 MHz, CD3OD): δ [ppm] = 8.43 (s, 1H, C–CHN), 7.43 (s, 1H, imi-CHCH), 7.37 (s, 1H, imi-CHCH), 3.62 (q, 3J = 7.1 Hz, 2H, CH2), 1.20 (t, 3J = 7.1 Hz, 3H, CH3) (matching NMR in ref. 38).
O), 7.56 (t, 3/5J = 0.8, 1H, imi-CHCH), 7.35 (dd, 3J = 8.4 Hz, 4J = 2.2 Hz, 1H, Caryl–H), 7.30 (d, 3J = 0.8 Hz, 1H, imi-CHCH), 7.25 (d, 4J = 2.1 Hz, 1H, Caryl–H), 6.65 (d, 3J = 8.3 Hz, 1H, Caryl–H), 5.88 (s, 2H, C(O)–CH2–NN). 13C-NMR (see Fig. S1a†) (75 MHz, DMSO-d6): δ [ppm] = 189.95 (CO), 181.85 (CHO), 153.52 (aryl-COH), 146.15 (aryl-COH), 143.51 (N–CN), 130.82 (aryl-C–CO), 128.72 (Caryl–H), 124.99 (imi-CHCH), 121.52 (imi-CHCH), 115.35 (Caryl–H), 114.32 (Caryl–H), 53.39 (CH2).
O), 7.71 (dd, 3J = 8.2 Hz, 4J = 1.8 Hz, 1H, Caryl–H), 7.48 (t, 3/5J = 0.9 Hz, 1H, imi-CHCH), 7.40 (d, 4J = 1.8 Hz, 1H, Caryl–H), 7.30 (d, 3J = 0.9 Hz, 1H, imi-CHCH), 6.94 (d, 3J = 8.1 Hz, 1H, Caryl–H), 5.99 (s, 2H, C(O)–CH2–N), 1.72 (s, 6H, CH3). 13C NMR (Fig. S1d†) (75 MHz, acetone-d6): δ [ppm] = 190.56 (CO), 182.61 (CHO), 153.11 (aryl-COH), 148.99 (aryl-COH), 144.84 (N–CN), 131.82 (aryl-C–CO), 129.79 (Caryl–H), 129.02 (imi-CHCH), 124.87 (imi-CHCH), 120.67 (C–(CH3)2), 108.89 (Caryl–H), 108.04 (Caryl–H), 54.33 (CH2), 25.89 (CH3).
N), 7.65 (dd, 3J = 8.2 Hz, 4J = 2.4 Hz, 1H, Caryl–H), 7.44 (d, 4J = 1.7 Hz, 1H, Caryl–H), 7.24 (d, 3J = 0.9 Hz, 1H, imi-CHCH), 7.04 (d, 3J = 1 Hz, 1H, imi-CHCH), 7.02 (d, 3J = 8.35 Hz, 1H, Caryl–H), 5.86 (s, 2H, C(O)–CH2–N), 1.70 (s, 6H, CH3).
[cm−1] = 1692, 1472. High-resolution, HR-ESI MS: m/z = 262.0822, calc. for [M + H]+m/z = 262.0823, matching with the exact mass for the indicated fragment; the absence of other significant peaks (besides the isotope pattern) confirms the purity of compound 9 (see Fig. S4a†). 1H-NMR (Fig. S1f†) (600 MHz, DMSO-d6): δ [ppm] = 11.21 (s, 1H, N–OH), 9.99 (s, 1H, C–OH), 9.45 (s, 1H, C–OH), 7.96 (s, 1H, C–CHN), 7.44 (dd, 3J = 8.27 Hz, 4J = 2.1 Hz, 1H, Caryl–H), 7.37 (d, 4J = 2.1 Hz, 1H, Caryl–H), 7.23 (s br, 1H, imi-CHCH), 7.03 (s br, 1H, imi-CHCH), 6.87 (d, 3J = 8.26, Caryl–H), 5.80 (s, 2H, C(O)–CH2–N). 13C-NMR (Fig. S1g†) (125 MHz, DMSO-d6): δ [ppm] = 190.62 (CO), 151.22 (aryl-COH), 145.37 (aryl-COH), 141.44 (CHNOH), 140.41 (s, 1C, N–CN), 128.34 (aryl-C–CO), 126.30 (Caryl–H), 124.77 (imi-CHCH), 121.14 (imi-CHCH), 115.20 (Caryl–H), 114.69 (Caryl–H), 53.33 (CH2).
[cm−1] = 2074; 1985, IR (KBr): [cm−1] = 2075; 1989. ESI-MS: m/z = 527.0 [M − OMe− − Cl− − H+ + CH3CN]+, 499.1 [M − CO/OMe− − Cl− − H+ + CH3CN]+. 1H NMR (Fig. S1h†) (300 MHz, acetone-d6): δ [ppm] = 8.75 (“d” = 2s for different cis- and trans ligands, 1H, C–CHN), 7.72 (dd, 3J = 8.2, 1.8 Hz, 1H, Caryl–H), 7.51–7.46 (m, 2H, imi-CHCH), 7.42 (d, 4J = 1.8 Hz, 1H, Caryl–H), 6.97 (d, 3J = 8.2 Hz, 1H, Caryl–H), 6.05 (s br, 2H, C(O)–CH2–N), 3.75 (s, 3H, COOCH3), 1.72 (s, 6H, (CH3)2).
[cm−1] = 2059; 1986, IR (KBr): [cm−1] = 2065, 1988. ESI-MS (Fig. S3a†): m/z = 527.0 [M − OEt− − Cl− − H+ + CH3CN]+, 499.1 [M − CO/OEt− − Cl− − H+ + CH3CN]+. 1H NMR (Fig. S1i†) (300 MHz, acetone-d6): δ [ppm] = 8.73 (“d” = 2s for different cis- and trans ligands, 1H, C–CHN), 7.72 (dd, 3J = 8.2 Hz, 4J = 1.8 Hz, 1H, Caryl–H), 7.51–7.47 (m, 2H, imi-CHCH), 7.42 (d, 4J = 1.8 Hz, 1H, Caryl–H), 6.97 (d, 3J = 8.2 Hz, 1H, Caryl–H), 6.06 (s br, 2H, C(O)–CH2–N), 4.26 (t, 3J = 8 Hz, 2H, –OCH2–), 1.72 (s, 6H, C(CH3)2), 1.28 (t, 3J = 7 Hz, 3H, –CH3).
[cm−1] = 2064, 1995, 1678, 1478. ESI-MS (Fig. S3b†): m/z = 487.0 [M − OEt− − Cl− − H+ + CH3CN]+, 459.1 [M − CO/OEt− − Cl− − H+ + CH3CN]+. High-resolution, HR-ESI-MS: m/z = 486.9828 [M − OEt− − Cl− − H+ + CH3CN]+, 458.9877 [M − CO/OEt− − Cl− − H+ + CH3CN]+, 419.9768 [M − CO/OEt− − Cl˙ + H]+, all matching with the calculated patterns for the indicated fragment masses (see Fig. S4b–d†). 1H NMR (500 MHz, acetone-d6): δ [ppm] = 8.70 (s, 1H, C-CHN), 7.60–7.38 (m, 4H, imi-CHCH, 2Caryl–H), 6.99 (d, 3J = 8.2, 1H, Caryl–H), 6.11–5.88 (m, 2H, C(O)–CH2–N), 3.57 (q, 3J = 6.5, 2H, –OCH2–), 1.12 (t, 3J = 6.7, 3H, –CH3). Calcd for C17H16ClN3O8Ru (526.85 g mol−1): C 38.76, H 3.06, N 7.98, found: C 38.72, H 3.09, N 7.47%.
[cm−1] = 2038, 1916, 1633, 1487.
000 g mol−1) was dissolved in 5 mL of d.d. water, combined with the above solution and stirred for 3 h. After neutralizing with hydrochloric acid, 100 mL of acetone were added and the formed solid removed by centrifugation. The solid was washed with ethanol 3 × 5 mL and the brown solid, now called composite material dextran@oximeCORM@IONP (17) was dried under vacuum and stored for several weeks. Yield: 69 mg, IR (KBr): [cm−1] = 2064, 1995, 1961.
N), 7.42 (s, 1H, imi-CHCH), 7.37 (s, 1H, imi-CHCH), 3.36 (s, 3H, CH3) (matching NMR in ref. 38).
[cm−1] = 2067, 1994. 1H NMR (300 MHz, CD3OD): δ [ppm] = 8.43 (s, 1H, C–CHN), 7.43 (s, 1H, imi-CHCH), 7.37 (s, 1H, imi-CHCH), 3.62 (q, 3J = 7.1 Hz, 2H, CH2), 1.20 (t, 3J = 7.1 Hz, 3H, CH3) (matching NMR in ref. 38).
O), 7.56 (t, 3/5J = 0.8, 1H, imi-CHCH), 7.35 (dd, 3J = 8.4 Hz, 4J = 2.2 Hz, 1H, Caryl–H), 7.30 (d, 3J = 0.8 Hz, 1H, imi-CHCH), 7.25 (d, 4J = 2.1 Hz, 1H, Caryl–H), 6.65 (d, 3J = 8.3 Hz, 1H, Caryl–H), 5.88 (s, 2H, C(O)–CH2–NN). 13C-NMR (see Fig. S1a†) (75 MHz, DMSO-d6): δ [ppm] = 189.95 (CO), 181.85 (CHO), 153.52 (aryl-COH), 146.15 (aryl-COH), 143.51 (N–CN), 130.82 (aryl-C–CO), 128.72 (Caryl–H), 124.99 (imi-CHCH), 121.52 (imi-CHCH), 115.35 (Caryl–H), 114.32 (Caryl–H), 53.39 (CH2).
O), 7.71 (dd, 3J = 8.2 Hz, 4J = 1.8 Hz, 1H, Caryl–H), 7.48 (t, 3/5J = 0.9 Hz, 1H, imi-CHCH), 7.40 (d, 4J = 1.8 Hz, 1H, Caryl–H), 7.30 (d, 3J = 0.9 Hz, 1H, imi-CHCH), 6.94 (d, 3J = 8.1 Hz, 1H, Caryl–H), 5.99 (s, 2H, C(O)–CH2–N), 1.72 (s, 6H, CH3). 13C NMR (Fig. S1d†) (75 MHz, acetone-d6): δ [ppm] = 190.56 (CO), 182.61 (CHO), 153.11 (aryl-COH), 148.99 (aryl-COH), 144.84 (N–CN), 131.82 (aryl-C–CO), 129.79 (Caryl–H), 129.02 (imi-CHCH), 124.87 (imi-CHCH), 120.67 (C–(CH3)2), 108.89 (Caryl–H), 108.04 (Caryl–H), 54.33 (CH2), 25.89 (CH3).
N), 7.65 (dd, 3J = 8.2 Hz, 4J = 2.4 Hz, 1H, Caryl–H), 7.44 (d, 4J = 1.7 Hz, 1H, Caryl–H), 7.24 (d, 3J = 0.9 Hz, 1H, imi-CHCH), 7.04 (d, 3J = 1 Hz, 1H, imi-CHCH), 7.02 (d, 3J = 8.35 Hz, 1H, Caryl–H), 5.86 (s, 2H, C(O)–CH2–N), 1.70 (s, 6H, CH3).
[cm−1] = 1692, 1472. High-resolution, HR-ESI MS: m/z = 262.0822, calc. for [M + H]+m/z = 262.0823, matching with the exact mass for the indicated fragment; the absence of other significant peaks (besides the isotope pattern) confirms the purity of compound 9 (see Fig. S4a†). 1H-NMR (Fig. S1f†) (600 MHz, DMSO-d6): δ [ppm] = 11.21 (s, 1H, N–OH), 9.99 (s, 1H, C–OH), 9.45 (s, 1H, C–OH), 7.96 (s, 1H, C–CHN), 7.44 (dd, 3J = 8.27 Hz, 4J = 2.1 Hz, 1H, Caryl–H), 7.37 (d, 4J = 2.1 Hz, 1H, Caryl–H), 7.23 (s br, 1H, imi-CHCH), 7.03 (s br, 1H, imi-CHCH), 6.87 (d, 3J = 8.26, Caryl–H), 5.80 (s, 2H, C(O)–CH2–N). 13C-NMR (Fig. S1g†) (125 MHz, DMSO-d6): δ [ppm] = 190.62 (CO), 151.22 (aryl-COH), 145.37 (aryl-COH), 141.44 (CHNOH), 140.41 (s, 1C, N–CN), 128.34 (aryl-C–CO), 126.30 (Caryl–H), 124.77 (imi-CHCH), 121.14 (imi-CHCH), 115.20 (Caryl–H), 114.69 (Caryl–H), 53.33 (CH2).
[cm−1] = 2074; 1985, IR (KBr): [cm−1] = 2075; 1989. ESI-MS: m/z = 527.0 [M − OMe− − Cl− − H+ + CH3CN]+, 499.1 [M − CO/OMe− − Cl− − H+ + CH3CN]+. 1H NMR (Fig. S1h†) (300 MHz, acetone-d6): δ [ppm] = 8.75 (“d” = 2s for different cis- and trans ligands, 1H, C–CHN), 7.72 (dd, 3J = 8.2, 1.8 Hz, 1H, Caryl–H), 7.51–7.46 (m, 2H, imi-CHCH), 7.42 (d, 4J = 1.8 Hz, 1H, Caryl–H), 6.97 (d, 3J = 8.2 Hz, 1H, Caryl–H), 6.05 (s br, 2H, C(O)–CH2–N), 3.75 (s, 3H, COOCH3), 1.72 (s, 6H, (CH3)2).
[cm−1] = 2059; 1986, IR (KBr): [cm−1] = 2065, 1988. ESI-MS (Fig. S3a†): m/z = 527.0 [M − OEt− − Cl− − H+ + CH3CN]+, 499.1 [M − CO/OEt− − Cl− − H+ + CH3CN]+. 1H NMR (Fig. S1i†) (300 MHz, acetone-d6): δ [ppm] = 8.73 (“d” = 2s for different cis- and trans ligands, 1H, C–CHN), 7.72 (dd, 3J = 8.2 Hz, 4J = 1.8 Hz, 1H, Caryl–H), 7.51–7.47 (m, 2H, imi-CHCH), 7.42 (d, 4J = 1.8 Hz, 1H, Caryl–H), 6.97 (d, 3J = 8.2 Hz, 1H, Caryl–H), 6.06 (s br, 2H, C(O)–CH2–N), 4.26 (t, 3J = 8 Hz, 2H, –OCH2–), 1.72 (s, 6H, C(CH3)2), 1.28 (t, 3J = 7 Hz, 3H, –CH3).
[cm−1] = 2064, 1995, 1678, 1478. ESI-MS (Fig. S3b†): m/z = 487.0 [M − OEt− − Cl− − H+ + CH3CN]+, 459.1 [M − CO/OEt− − Cl− − H+ + CH3CN]+. High-resolution, HR-ESI-MS: m/z = 486.9828 [M − OEt− − Cl− − H+ + CH3CN]+, 458.9877 [M − CO/OEt− − Cl− − H+ + CH3CN]+, 419.9768 [M − CO/OEt− − Cl˙ + H]+, all matching with the calculated patterns for the indicated fragment masses (see Fig. S4b–d†). 1H NMR (500 MHz, acetone-d6): δ [ppm] = 8.70 (s, 1H, C-CHN), 7.60–7.38 (m, 4H, imi-CHCH, 2Caryl–H), 6.99 (d, 3J = 8.2, 1H, Caryl–H), 6.11–5.88 (m, 2H, C(O)–CH2–N), 3.57 (q, 3J = 6.5, 2H, –OCH2–), 1.12 (t, 3J = 6.7, 3H, –CH3). Calcd for C17H16ClN3O8Ru (526.85 g mol−1): C 38.76, H 3.06, N 7.98, found: C 38.72, H 3.09, N 7.47%.
[cm−1] = 2038, 1916, 1633, 1487.
000 g mol−1) was dissolved in 5 mL of d.d. water, combined with the above solution and stirred for 3 h. After neutralizing with hydrochloric acid, 100 mL of acetone were added and the formed solid removed by centrifugation. The solid was washed with ethanol 3 × 5 mL and the brown solid, now called composite material dextran@oximeCORM@IONP (17) was dried under vacuum and stored for several weeks. Yield: 69 mg, IR (KBr): [cm−1] = 2064, 1995, 1961.
CO detection
Acknowledgements
We thank Dr Juri Barthel and the Ernst Ruska-Centre (ER-C) for Microscopy and Spectroscopy with Electrons, the Jülich Research Centre and the RWTH Aachen University, 52425 Jülich (Germany) for help and access to the HR-TEM facilities. Dr Peter Tommes is thanked for collecting the HR-ESI mass spectra.Notes and references
- B. J. Haldane, Biochem. J., 1927, 21, 1068–1075 CrossRef PubMed.
- L. D. Prockop and R. I. Chichkova, J. Neurol. Sci., 2007, 262, 122–130 CrossRef CAS PubMed.
- T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. J. Green and R. Motterlini, Angew. Chem., Int. Ed., 2003, 42, 3722–3729 CrossRef CAS PubMed.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- D. S. Bredt and S. H. Snyder, Annu. Rev. Biochem., 1994, 63, 175–195 CrossRef CAS PubMed.
- B. E. Mann and R. Motterlini, Chem. Commun., 2007, 4197–4208 RSC.
- R. Tenhunen, H. S. Marver and R. Schmid, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 748–755 CrossRef CAS.
- D. Boehning and S. H. Snyder, Science, 2002, 298, 2339–2340 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nature, 2010, 9, 728–743 CAS.
- B. E. Mann, Organometallics, 2012, 31, 5728–5735 CrossRef CAS.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- R. Foresti and R. Motterlini, Curr. Drug Targets, 2010, 11, 1595–1604 CrossRef CAS PubMed.
- M. Patra, M. Wenzel, P. Prochnow, V. Pierroz, G. Gasser, J. E. Bandow and N. Metzler-Nolte, Chem. Sci., 2015, 6, 214–224 RSC.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, 17–24 CrossRef.
- R. Motterlini, B. E. Mann, T. R. Johnson, J. E. Clark, R. Foresti and C. J. Green, Curr. Pharm. Des., 2003, 9, 2525–2539 CrossRef CAS PubMed.
- J. E. Clark, P. Naughton, S. Shurey, C. J. Green, T. R. Johnson, B. E. Mann, R. Foresti and R. Motterlini, Circ. Res., 2003, 93, 2–8 CrossRef PubMed.
- R. Alberto and R. Motterlini, Dalton Trans., 2007, 1651–1660 RSC.
- R. Motterlini, B. E. Mann and R. Foresti, Expert Opin. Invest. Drugs, 2005, 14, 1305–1318 CrossRef CAS PubMed.
- S. McLean, B. E. Mann and R. K. Poole, Anal. Biochem., 2012, 427, 36–40 CrossRef CAS PubMed.
- T. R. Johnson, B. E. Mann, I. P. Teasdale, H. Adams, R. Foresti and C. J. Green, Dalton Trans., 2007, 1500–1508 RSC.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernandes, C. C. Romao and M. J. Romao, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- T. Pitchumony, B. Spingler, R. Motterlini and R. Alberto, Chimia, 2009, 62, 277–279 CrossRef.
- R. Motterlini, P. Sawle, J. Hammad, S. Bains, R. Alberto, R. Foresti and C. J. Green, FASEB J., 2005, 19, 284–286 CAS.
- N. E. Brückmann, M. Wahl, G. J. Reiß, M. Kohns, W. Wätjen and P. C. Kunz, Eur. J. Inorg. Chem., 2011, 4571–4577 CrossRef.
- W. Huber, R. Linder, J. Niesel, U. Schatzschneider, B. Spingler and P. C. Kunz, Eur. J. Inorg. Chem., 2012, 3140–3146 CrossRef CAS.
- S. Romanski, B. Kraus, U. Schatzschneider, J. M. Neudörfl, S. Amslinger and H. G. Schmalz, Angew. Chem., Int. Ed., 2011, 50, 2392–22396 CrossRef CAS PubMed.
- N. S. Sitnikov, Y. Li, D. Zhang, B. Yard and H.-G. Schmalz, Angew. Chem., Int. Ed., 2015, 54, 12314–12318 CrossRef CAS PubMed.
- M. Ma, H. Noei, B. Mienert, J. Niesel, E. Bill and M. Muhler, Chem. – Eur. J., 2013, 19, 6785–6790 CrossRef CAS PubMed.
- G. Dördelmann, T. Meinhardt, T. Sowik, A. Krüger and U. Schatzschneider, Chem. Commun., 2012, 48, 11528–11530 RSC.
- A. Ruggi and F. Zobi, Dalton Trans., 2015, 44, 10928–10931 RSC.
- C. Bohlender, S. Glaeser, M. Klein, J. Weisser, S. Thein, U. Neugebauer, J. Popp, R. Wyrwa and A. J. Schiller, J. Mater. Chem. B, 2014, 2, 1454–1463 RSC.
- U. Schatzschneider, Br. J. Pharmacol., 2015, 172, 1638–1650 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- L. D. D'Andrea, A. Romanelli, R. Di Stasi and C. Pedone, Dalton Trans., 2010, 39, 7625–7636 RSC.
- E. Neuse, Met.-Based Drugs, 2008, 1–19 CrossRef PubMed.
- J. D. Seixas, F. A. Marino, A. Mukhopadhyay, A. C. Coelho, P. M. Reis, L. F. Veiros, A. R. Marques, N. Penacho, A. M. L. Goncalves, M. J. Romao, G. J. L. Bernardes, T. Santos-Silva and C. C. Romao, Dalton Trans., 2015, 44, 5058–5075 RSC.
- L. Oresmaa, H. Tarvainen, K. Machal and M. Haukka, Dalton Trans., 2012, 41, 11170–11175 RSC.
- P. C. Kunz, H. Meyer, J. Barthel, S. Sollazzo, A. M. Schmidt and C. Janiak, Chem. Commun., 2013, 49, 4896–4898 RSC.
- B. F. G. Johnson and J. S. McIndoe, Coord. Chem. Rev., 2000, 200–202, 901–932 CrossRef CAS.
- G. A. van Ewijk, G. J. Vroege and A. P. J. Philip, J. Magn. Magn. Mater., 1999, 201, 31–33 CrossRef CAS.
- H. Meyer, F. Winkler, P. Kunz, A. M. Schmidt, A. Hamacher, M. U. Kassack and C. Janiak, Inorg. Chem., 2015, 54(23), 11236–11246 CrossRef CAS PubMed.
- M. Klein, U. Neugebauer, A. Gheisari, A. Malassa, T. M. Jazzazi, F. Froehlich, M. Westerhausen, M. Schmitt and J. Popp, J. Phys. Chem. A, 2014, 118, 5381–5390 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, L. Tan, K. B. Zheng and W. M. Huang, Angew. Chem., Int. Ed., 2013, 52, 1628–1630 CrossRef CAS PubMed.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- H. J. Vreman and D. K. Stevenson, Anal. Biochem., 1988, 168, 31–81 CrossRef CAS PubMed.
- M. Balazy and H. Jiang, Acta Haematol., 2000, 103, 78–83 CrossRef.
- A. Mantovani and S. Cenini, Inorg. Synth., 1976, 16, 51–53 CrossRef CAS.
- L. Marek, A. Hamacher, F. K. Hansen, K. Kuna, H. Gohlke, M. U. Kassack and T. Kurz, J. Med. Chem., 2013, 56, 427–436 CrossRef CAS PubMed.
- L. Oresmaa, H. Kotikoski, M. Haukka, J. Salminen, O. Oksala, E. Pohjala, E. Moilanen, H. Vapaatalo, P. Vainiotalo and P. Aulaskari, J. Med. Chem., 2005, 48, 4231–4236 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis for oxime-based CORMs, NMR, IR and ESI-mass spectra, myoglobin assay, leaching experiments, DLS experiments, TEM measurements, toxicity tests, and activation energy. See DOI: 10.1039/c5dt04888e |
| This journal is © The Royal Society of Chemistry 2016 |