DOI:
10.1039/C5DT03940A
(Paper)
Dalton Trans., 2016,
45, 667-680
Chirality at metal and helical ligand folding in optical isomers of chiral bis(naphthaldiminato)nickel(II) complexes†
Received
8th October 2015
, Accepted 5th November 2015
First published on 6th November 2015
Abstract
Enantiopure bis[{(R or S)-N-1-(Ar)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}]nickel(II) complexes {Ar = C6H5 (1R or 1S), p-OMeC6H4 (2R or 2S), and p-BrC6H4 (3R or 3S)} are synthesized from the reactions between (R or S)-N-1-(Ar)ethyl-2-oxo-1-naphthaldimine and nickel(II) acetate. Circular-dichroism spectra and their density-functional theoretical simulation reveal the expected mirror image relationship between the enantiomeric pairs 1R/1S and 3R/3S in solution. CD spectra are dominated by the metal-centered Λ- or Δ-chirality of non-planar four-coordinated nickel, this latter being in turn dictated by the ligand chirality. Single crystal structure determination for 1R and 1S shows that there are two symmetry-independent molecules (A and B) in each asymmetric unit that give a Z′ = 2 structure. Two asymmetric and chiral bidentate N^O-chelate Schiff base ligands coordinate to the nickel atom in a distorted square planar N2O2-coordination sphere. The conformational difference between the symmetry-independent molecules arises from the “up-or-down” folding of the naphthaldiminato ligand with respect to the coordination plane, which creates right- (P) or left-handed (M) helical conformations. Overall, the combination of ligand chirality, chirality at the metal and ligand folding gives rise to discrete metal helicates of preferred helicity in a selective way. Cyclic voltammograms (CV) show an oxidation wave at ca. 1.30 V for the [Ni(L)2]/[Ni(L)2]+ couple, and a reduction wave at ca. −0.35 V for the [Ni(L)2]/[Ni(L)2]− couple in acetonitrile.
Introduction
Chiral metal coordination complexes are the subject of continuous study due to their fascinating structures, and enormous potential applications such as in molecular recognition, non-linear optical materials, asymmetric catalysis, enantiomeric separation, and so on.1,2 One of the possible ways of generating enantiopure chiral-at-metal complexes is the employment of chiral chelating ligands, which may then transfer their chirality to the metal centre(s) generating right- (Δ) or left-handed (Λ) helical structures in a stereocontrolled way.3 The use of achiral chelating ligands usually results in a racemic mixture of left- and right-handed Δ/Λ-optical isomers.4 Of special interest in the context of supramolecular chemistry are metal helicates, that is, metal complexes where one or more ligand “strands” wrap around one or more metal centres in a helical fashion.5 In tetrahedral or distorted square-planar complexes the metal-centered chirality (Λ vs. Δ) can be induced with a C2-symmetric structure by the use of an asymmetric ligand N^O to give M(N^O)2 complexes (cf. Scheme 1).6,7 An enantiomerically pure chelate ligand (R or S-N^O) often leads to the preferential formation of one diastereomer with absolute configuration of Λ(R,R) or Δ(S,S), while a racemic ligand gives both diastereomers {e.g. Λ(R,R) and Δ(S,S)}.6–9
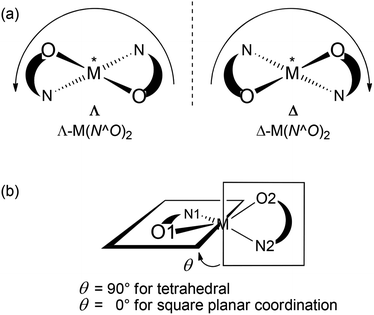 |
| | Scheme 1 Definition of (a) Λ/Δ chirality at the metal centre and (b) dihedral angle θ quantifying the deviation from tetrahedral or square planar geometry. | |
We have recently paid attention to the phenomenon of helicity (P vs. M) and induced metal-centered chirality (Λ vs. Δ) in the complexes formed by the reaction of enantiopure amino acids and Schiff base ligands with Rh(I)10,11 and Cu/Ni/Zn(II), respectively.12–15 The most noteworthy finding in our studies is that the achiral N-phenylglycinate ligand coordinates to [Rh(η4-cod)(acetate)]2 and gives a racemic chiral [Rh(η4-cod)(N-phenylglycinate)] complex with the nitrogen atom becoming a stereogenic center upon metal coordination.10,11 The crystal structures reveal a case of two-fold spontaneous resolution of the racemic mixture into two homochiral helical enantiomers (helicates), namely the right-(P) and left-handed (M) helical chain structures with S- and R-N-phenylglycinate, respectively. Later, we have reported some examples of induced chirality at-metal-center with the preferential formation of the Λ or Δ-M configuration in distorted square planar M(N,O-chelate)2 (N,O-chelate = enantiopure deprotonated Schiff base ligands; M = Cu, Ni, and Zn), which can be efficiently controlled by R- or S-chirality of the ligand.12–14 Similarly, induced chirality at-metal-center in distorted square planar Cu/Zn(II) complexes with enantiopure or racemic amino alcohol-based Schiff base ligands was reported.15 In particular, we have reported for the first time an example of induced chirality at-nickel-centre (Δ vs. Λ) in distorted octahedral, dinuclear μ-aqua-tetrakis{(R or S)-N-1-(Ar)ethyl-salicylaldiminato}-di-Λ- or Δ-nickel(II).13
The present paper reports the results of syntheses, spectroscopy, excited-state calculations and structural analyses of Z′ = 2 helical structures of bis{(R or S)-N-1-(Ar)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}nickel(II) {Ar = C6H5, p-OMeC6H4, and p-BrC6H4}. The present complexes are structurally simpler than the related octahedral dinuclear analogs,13 however, the specific folding of naphthaldiminato ligands around the metal centre gives rise to structurally intriguing discrete metal helicates.
Results and discussion
The enantiopure Schiff base ligands, (R or S)-N-1-(Ar)ethyl-2-oxo-1-naphthaldimine (R or S-HL), react with nickel(II)acetate to provide the bis[{(R or S)-N-1-(Ar)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}]nickel(II) complexes {Ar = C6H5 (Ni-R-L1; 1R or Ni-S-L1; 1S), p-OMeC6H4 (Ni-R-L2; 2R or Ni-S-L2; 2S), and p-BrC6H4 (Ni-R-L3; 3R or Ni-S-L3; 3S)} (Scheme 2). Vibrational spectra show the main characteristic band at 1617–1604 cm−1 for the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching.12–16,17 ESI-MS shows the parent ion peak at m/z 607 (1R or 1S) and 667 (2R or 2S) for [M + H]+ species, while EI-MS shows this peak at m/z 764 (3R or 3S) for [M]+. The spectra further show several ion peaks for [M − HL]+, [HL or HL + H]+ and different fragmented ligand species (Table S1†). 1H NMR spectra (Fig. S1†) in CDCl3 show a series of peaks expected for the C2-symmetric four coordinated Ni(II)-complexes with a square planar geometry (see the Experimental section and the ESI†).11,13,18–20 Notably, the imine (CHN) protons display large coordination shifts and appear as broad resonances above 11 ppm. The signal broadening, also seen to a smaller extent for other resonances, is probably due to solution paramagnetism. This is a well-known phenomenon for Ni(II) salicylideneaminato-complexes with bulky N-substituents and has been explained invoking a fast equilibrium between a dominant singlet species and a minor triplet species triggered by geometrical distortion (toward a tetrahedral geometry) and/or molecular association.21
N stretching.12–16,17 ESI-MS shows the parent ion peak at m/z 607 (1R or 1S) and 667 (2R or 2S) for [M + H]+ species, while EI-MS shows this peak at m/z 764 (3R or 3S) for [M]+. The spectra further show several ion peaks for [M − HL]+, [HL or HL + H]+ and different fragmented ligand species (Table S1†). 1H NMR spectra (Fig. S1†) in CDCl3 show a series of peaks expected for the C2-symmetric four coordinated Ni(II)-complexes with a square planar geometry (see the Experimental section and the ESI†).11,13,18–20 Notably, the imine (CHN) protons display large coordination shifts and appear as broad resonances above 11 ppm. The signal broadening, also seen to a smaller extent for other resonances, is probably due to solution paramagnetism. This is a well-known phenomenon for Ni(II) salicylideneaminato-complexes with bulky N-substituents and has been explained invoking a fast equilibrium between a dominant singlet species and a minor triplet species triggered by geometrical distortion (toward a tetrahedral geometry) and/or molecular association.21
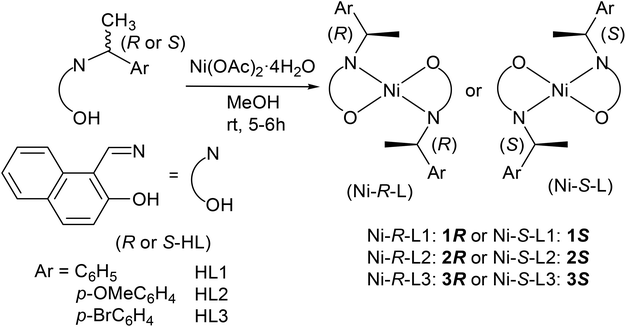 |
| | Scheme 2 Syntheses of the bis[{(R or S)-N-1-(Ar)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}]nickel(II). | |
Electronic spectra
UV-vis absorption and CD spectra of the complexes 1 and 3 measured in cyclohexane are shown in Fig. 1 and the spectral data are summarized in Table 1. All absorption spectra show consistent similarities over the whole measured range of 200–900 nm. In the visible region, there is a weak broad band around 600 nm followed by three bands of increasing intensity between 380–480, 330–380 and around 310 nm, respectively. They are followed by two more intense bands at 250–300 and below 250 nm. The band around 600 nm is due to the superposition of several metal-centred transitions typical of the Ni2+ core. The next bands in the UV region (<480 nm) involve a complex combination of several transitions, centred on both the metal and the ligands, as will be discussed below.
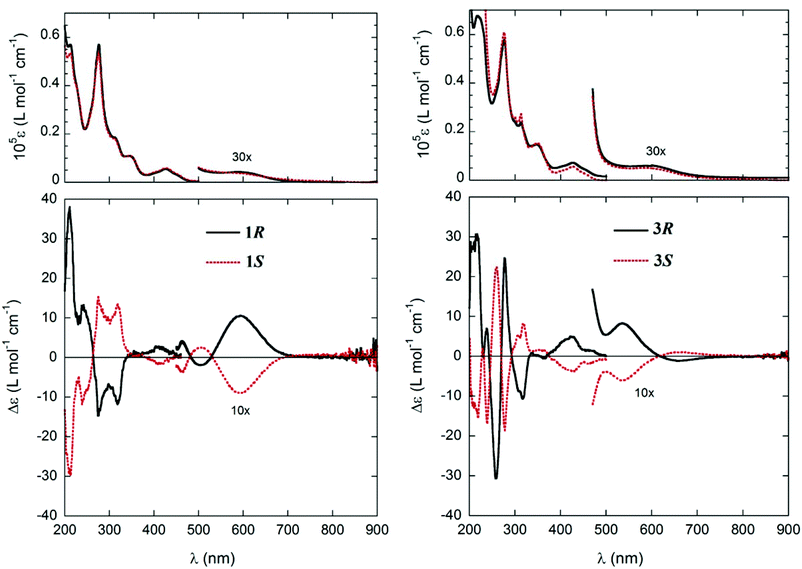 |
| | Fig. 1 UV-vis. and CD spectra of 1R (0.76 mM), 1S (0.79 mM) (left), and 3R (1.95 mM), 3S (1.96 mM) (right) in cyclohexane; cell path-length: 0.1 mm, 200–500 nm; 5 mm, 425–600 nm; 10 mm, 400–900 nm. | |
Table 1 UV-vis and CD spectral data of 1R/1S and 3R/3S in cyclohexane; cell path-length: 0.1 mm (200–500 nm); 5–10 mm (400–900 nm) at 20 °C
| Compounds |
Spectra |
Bandsa |
|
vw = very weak, w = weak, m = medium, s = strong, vs = very strong.
|
|
1R/1S |
UV-vis |
588 (vw), 427 (m), 345 (s), 312 (s), 276 (vs), and 212 (vs) nm |
|
3R/3S |
UV-vis |
595 (vw), 425 (m), 348 (s), 312 (s), 277 (vs), and 220 (vs) nm |
|
1R/1S |
CD |
590 (+/−, w), 505 (−/+, vw), 460 (+/−, vw), 410 (+/−, w), 317 (−/+, s), 276 (−/+, s), 240 (+/−, s), and 210 (+/−, vs) nm |
|
3R/3S |
CD |
660 (−/+, vw), 534 (+/−, w), 425 (+/−, s), 313 (−/+, s), 278 (+/−, vs), 256 (−/+, vs), 237 (+/−, m), and 213 (+/−, vs) nm |
Electronic CD spectra of the enantiomeric couples 1R/1S and 3R/3S show the expected mirror-image relationship (Fig. 1 and Table 1) in cyclohexane. CD spectra display a larger variation between the two compounds as compared to the absorption spectra and also in comparison with the analogue Cu(II)-complexes.12 In particular, in the visible range there is no distinctive feature immediately related to the ligand configuration for both 1 and 3. The only consistent signals are in the UV region, and they are (for R configuration, Fig. 1 and Table 1) the weak positive band between 400 and 450 nm, the stronger negative band at 300–330 nm, and the strong positive band below 220 nm.
We also measured solid-state CD spectra of crystalline samples of compound 1, but found them to be not fully reproducible. Moreover, in the case of the analogous Cu complexes,12 we demonstrated that solid-state CD spectra are dominated by inter-crystalline couplings and do not reflect in a simple way the molecular conformation.
Simulating the solution CD spectra of Ni complexes 1–3 by means of CD calculations is a laborious task because of the inherent complexity of the system22 and the uncertainty of the solution structures. Therefore, the work described in the following section aimed mainly at establishing the relationship between the observed CD spectra and the chirality at both carbon centres and the metal ion (if any), in analogy with our previous work on Cu analogues.12 Although the solid-state structures of 1R and 1S show only a faint chirality at the Ni atoms (see below), the situation in solution may in principle be different. The chirality at the metal centre is defined as shown in Scheme 1a, and the distortion from an ideal tetrahedral or square-planar geometry can be quantified by the dihedral angle θ between the two planes formed by the donor atoms with the metal atom, that is, N1–M–O1 and N2–M–O2 (Scheme 1b). Starting from the X-ray structure of 1R, we investigated the solution conformation of this complex by means of a conformational search using molecular mechanics and DFT geometry optimizations at the B3LYP/6-31G(d) level (see the Computational section). The resulting most stable structure showed a small but detectable angle θ = −10.8° corresponding to Λ-Ni-1R chirality, and resembled strongly molecule A found in the X-ray discussed below (inset in Fig. 2A); other conformations had much higher energies and were neglected. Starting from the same complex, the chirality at the metal centre was inverted and the same conformational search/geometry optimization procedure was applied. The set of structures with Δ-Ni-1R chirality thus obtained showed two low-energy conformations, one (more stable) with θ = 15.0° and the second (less stable by 0.16 kcal mol−1) with θ = 9.0° (see the ESI†). Interestingly, the lowest-energy Λ-Ni-1R structure was more stable by about 1.7 kcal mol−1 than the Δ-Ni-1R one. Thus, a small but non-negligible diastereomeric preference in favour of the Λ-Ni-1R isomer is predicted, similarly to what was observed for the Cu analogues.12 To confirm this finding, CD calculations were run with the TDDFT method23 at the B3LYP/TZVP level on the above described structures (Fig. 2). The most striking observation from Fig. 2 is that the CD spectra calculated for Λ-Ni-1R and Δ-Ni-1R are almost the mirror image of each other on a wide wavelength range. This demonstrates that the CD spectrum in solution is dominated by the metal chirality, this latter being in turn dictated by the ligand chirality. Second, the CD spectrum calculated for Λ-Ni-1R is in reasonable agreement with the experimental one for compound 1R. As mentioned above, a perfect agreement cannot be expected because of the system complexity and the very large number of transitions contributing to the spectrum (80 excited states were included in the TDDFT calculations, which are intrinsically poorly accurate for high-lying transitions).24 The comparison between experimental and calculated CD spectra allows us to confirm that the complex obtained from ligand R-L1 assumes a predominant Λ-Ni-1R configuration in solution, and the same is inferred for the remaining ligands.
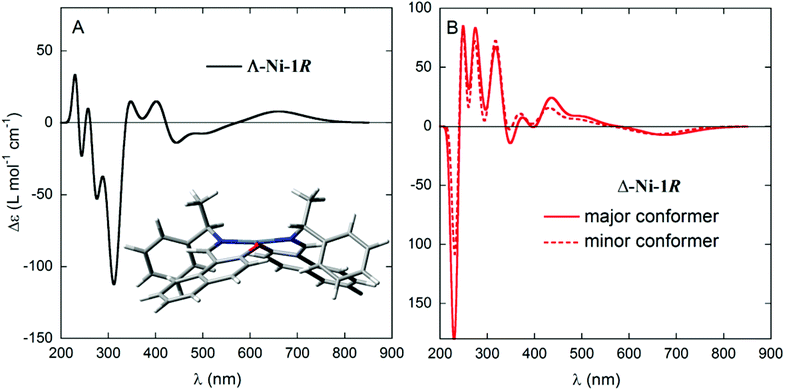 |
| | Fig. 2 CD spectra, calculated at the B3LYP/TZVP//B3LYP/6-31G(d) level, for the two diastereomers Λ-Ni-1R (left, conformation shown in the inset) and Δ-Ni-1R (right, two conformations shown in Fig. S3†). Gaussian band shape with exponential bandwidth σ = 0.2 eV. | |
We have already stressed12,13 that for d-metal complexes containing chromophoric ligands rich in transitions, such as compounds 1–3, a straightforward assignment of electronic spectra in terms of purely metal- or ligand-centred transitions is not possible. On the basis of orbital and population analyses of compound 1R at the B3LYP/TZVP level, the simplified assignment shown in Table 2 was reached, concerning only the transitions that contributed the most to the CD spectrum above 300 nm. It is important to notice, however, that every transition is derived from many single excitations, and there is no clear separation between metal-centred and ligand-centred transitions, because several metal-centred transitions occur deeply in the UV region of the spectrum.
Table 2 Main transitions calculated for the Λ-Ni-1R complex with B3LYP/TZVP//B3LYP/6-31G(d), contribution to the first observed CD bands, and simplified assignment
| Exc. state |
Calculated transition wavelength (nm) |
Observed CD band (maximum wavelength, nm, and sign) |
Assignmenta |
|
Legend: M–M, metal d–d; L–M, ligand-to-metal; M–L, metal-to-ligand; CT, charge transfer; in order of importance.
|
| 1 |
660 |
590 (+) |
M–M |
| 3 |
514 |
505 (−) |
M–M, L–M, CT |
| 4 |
498 |
CT, M–M |
| 5 |
428 |
M–M |
| 6 |
414 |
460 (+) |
M–M |
| 7 + 8 |
359 |
410 (+) |
M–M |
| 9 |
344 |
M–M, L–M |
| 10 |
336 |
M–M, L–M |
| 13 |
322 |
317 (−) |
M–M |
| 15 |
309 |
M–L |
Solid state structural studies
Single crystal X-ray structure determination for Ni-R-L1 (1R) and Ni-S-L1 (1S) shows that there are two symmetry-independent molecules in each asymmetric unit, that is, molecule A with Ni1 and molecule B with the Ni2 centre in each unit. In each structure, two bidentate N^O-chelate Schiff base ligands coordinate to the nickel atom with a square planar N2O2-coordination sphere around the metal atom (Fig. 3). The two nitrogen atoms (and subsequently the two oxygen atoms) are trans positioned. The Ni–O/N bond lengths and O–Ni–N bond angles are listed in Table 3 and are as expected for analogous Ni(II)-Schiff base complexes.8,13 Despite the presence of aromatic rings in the complexes Ni-R-L1 (1R) and Ni-S-L1 (1S), there are no detectable π–π interactions25 but intermolecular C–H⋯π contacts26 are evident in the packings; a detailed analysis is reported in the ESI.†
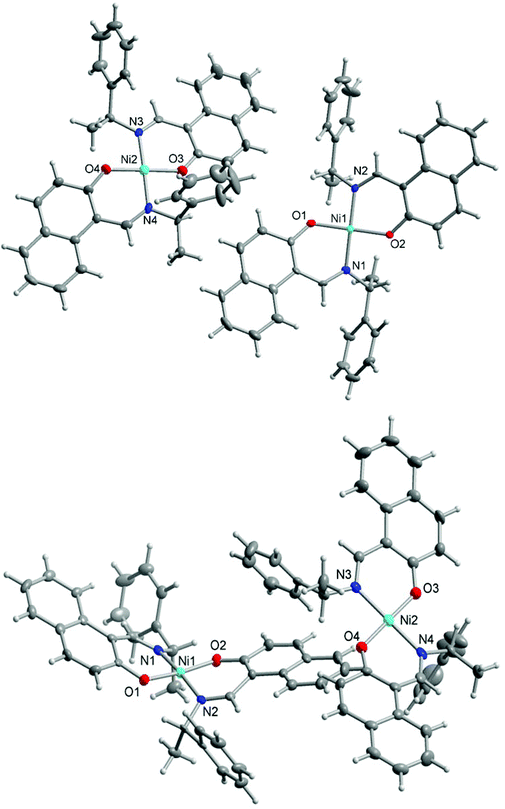 |
| | Fig. 3 Structures of the two symmetry-independent molecules of the enantiomeric couple Ni-R-L1 (1R) (above) and Ni-S-L1 (1S) (below). Thermal ellipsoids are at the 70% level. See Table 3 for bond lengths and angles. | |
Table 3 Selected bond lengths (Å) and angles (°) in the compoundsa
| Ni-R-L1 (1R) |
Ni-S-L1 (1S) |
|
1R (A) |
1S
(A)
|
|
A and B refer to the two symmetry independent molecules in each of the structure.
|
| Ni(1)–O(1) |
1.848(2) |
O(1)–Ni(1)–O(2) |
172.73(10) |
Ni(1)–O(1) |
1.840(3) |
O(1)–Ni(1)–O(2) |
172.94(8) |
| Ni(1)–O(2) |
1.850(2) |
O(1)–Ni(1)–N(1) |
92.16(11) |
Ni(1)–O(2) |
1.835(2) |
O(1)–Ni(1)–N(1) |
91.94(10) |
| Ni(1)–N(1) |
1.908(3) |
O(2)–Ni(1)–N(1) |
88.35(11) |
Ni(1)–N(1) |
1.909(3) |
O(2)–Ni(1)–N(1) |
88.44(10) |
| Ni(1)–N(2) |
1.914(3) |
O(1)–Ni(1)–N(2) |
88.39(11) |
Ni(1)–N(2) |
1.901(3) |
O(1)–Ni(1)–N(2) |
88.43(10) |
|
|
|
O(2)–Ni(1)–N(2) |
92.01(11) |
|
|
O(2)–Ni(1)–N(2) |
92.08(10) |
|
|
|
N(1)–Ni(1)–N(2) |
172.85(11) |
|
|
N(1)–Ni(1)–N(2) |
172.72(12) |
| |
|
1R
(B)
|
1S
(B)
|
| Ni(2)–O(3) |
1.818(2) |
O(3)–Ni(2)–O(4) |
179.39(12) |
Ni(2)–O(3) |
1.817(3) |
O(3)–Ni(2)–O(4) |
179.33(10) |
| Ni(2)–O(4) |
1.828(2) |
O(3)–Ni(2)–N(3) |
92.08(11) |
Ni(2)–O(4) |
1.823(3) |
O(3)–Ni(2)–N(3) |
92.01(10) |
| Ni(2)–N(3) |
1.946(3) |
O(4)–Ni(2)–N(3) |
88.52(11) |
Ni(2)–N(3) |
1.935(3) |
O(4)–Ni(2)–N(3) |
88.6(1) |
| Ni(2)–N(4) |
1.963(3) |
O(3)–Ni(2)–N(4) |
88.74(11) |
Ni(2)–N(4) |
1.953(3) |
O(3)–Ni(2)–N(4) |
88.8(1) |
|
|
|
O(4)–Ni(2)–N(4) |
90.66(11) |
|
|
O(4)–Ni(2)–N(4) |
90.6(1) |
|
|
|
N(3)–Ni(2)–N(4) |
178.02(12) |
|
|
N(3)–Ni(2)–N(4) |
178.11(11) |
Two symmetry-independent Ni-Schiff base molecules or, more correctly, two identical chemical formula units were found here in the structural asymmetric unit27 to give a Z′ = 2 structure. The definition of Z′ is that it refers to the number of formula units in the unit cell (here 4) divided by the number of independent general positions (here 2).28 Different reasons can lead to such Z′ > 1 structures:29 a structure stuck en-route to a more stable form,28 that is, a crystal “on the way”,27,30–32 or strong and special supramolecular (e.g. hydrogen bonding) interactions between the two (or more) symmetry-independent units.33–38 A high Z′ is also obtained when the molecule has different equi-energetic conformations, with these conformations co-existing in the crystal.39,40 The chance for Z′ > 1 is higher in non-centrosymmetric space groups with (enantiopure) chiral molecules which have difficulties in packing efficiently in the absence of centrosymmetry.41
Different from our previous studies on four-coordinated Cu(II) with asymmetric (N^O) and chiral Schiff base ligands12,42 the distortion from square-planar geometry, as assessed by the dihedral angle θ (cf. Scheme 1b) at the Ni atoms in 1R and 1S is very small. The dihedral angle θ is only 10° for molecules A and less than 2° for molecules B. The experimental angle of about 10° agrees well with the angle of 10.8° obtained from DFT geometry optimizations at the B3LYP/6-31G(d) level for Λ-Ni-R-L1 (1R) (see above). This difference in the dihedral angle θ could explain the formation of the two independent molecules A and B. The two molecules A and B, after relaxation of the hydrogen atoms at the B3LYP/6-31G(d) level, have different DFT energies, molecule A being more stable by 1.8 kcal mol−1. This is similar to the difference found between fully optimized Λ- and Δ-Ni-1R structures described above.
Yet, the conformational difference between the two symmetry independent molecules A and B is better explained with the help of Scheme 3. Each Ni-naphthaldiminato half does not assume a planar arrangement. Instead, each six-membered NiNOC3 chelate ring is folded “up-or-down” hinging on the N⋯O vector to assume an envelope conformation.43 In each molecule A and B, the folding of the two chelate rings occurs on the same side of the NiN2O2 plane. The folding angles ϕ, that is, the angles defined by the NiN2O2 plane and each of the naphthaldiminato planes (Scheme 3), are listed in Table 4. The folding in molecule A with Ni1 is more pronounced with very similar folding angles of ϕ ∼ 21° than in molecule B with Ni2 which has smaller and two different folding angles of 5° and 16°. An overlay of both molecules in each structure illustrates the different folding directions (Fig. 4). The folding angles measured for DFT-optimized structures are also listed in Table 4, and they agree well with the values measured for molecule A in the crystals.
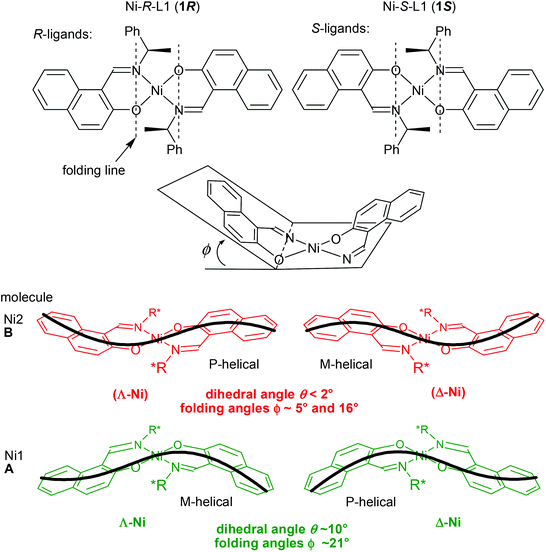 |
| | Scheme 3 Schematic illustration of the non-planar, skewed bis(naphthaldiminato) N2O2 ligand arrangement around the nearly square-planar coordinated Ni atom in 1R (left) and 1S (right) and definition of the folding angle ϕ. The thick black line depicts the P- or M-helical arrangement from the envelope conformation in the NiNOC3 chelate ring together with the curvature in the naphthaldiminato ligand. The round brackets indicate a faint chirality at Ni. | |
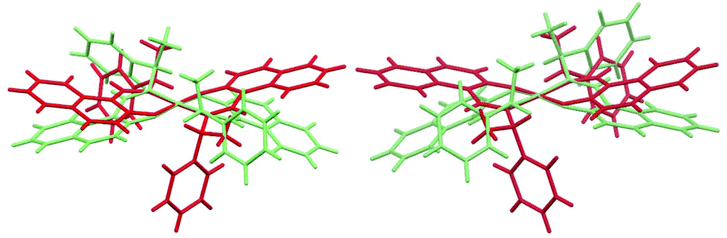 |
| | Fig. 4 Overlay of the two symmetry independent molecules in Ni-R-L1 (1R) (left) and Ni-S-L1 (1S) (right). The Ni1 molecule A is shown in green, the Ni2 molecule B in red. The five atoms NiN2O2 are pairwise specified to orient the overlay which is managed with the “Structure overlay” option in Mercury 3.5.1 (copyright CCDC 2001–2014, http://www.ccdc.cam.ac.uk/mercury/). | |
Table 4 Dihedral and folding angles for the two symmetry independent molecules (A, B) in Ni-R-L1 (1R) and Ni-S-L1 (1S), respectively
| Complexes (molecule) |
Chirality at Nia |
θ (°)b exp. |
θ (°)b calc.c |
ϕ (°)d exp. |
ϕ (°)d calc.c |
|
The round brackets indicate a faint chirality at Ni.
Dihedral angle θ between the two planes is formed by the donor atoms with the metal atom (cf. Scheme 1), that is, N1–Ni–O1 and N2–Ni–O2 (molecule A) and respective numbering in molecule B.
By DFT geometry optimizations at the B3LYP/6-31G(d) level; the calculated chirality at Ni is also given.
Folding angle is defined as the angle between O–Ni–N and O–C–C–C–N (cf. Scheme 3) with all these atoms forming the six-membered NiNOC3 chelate ring. Each chelate ring has a folding angle, hence, there are two independent folding angles in a molecule.
|
|
1R
(A, Ni1) |
Λ
|
10.0(1) |
10.8 Λ |
20.6(1) |
21.8(10) |
25.3 Λ |
|
1R
(B, Ni2) |
(Λ) |
1.80(9) |
5.1(1) |
16.1(1) |
|
1S
(A, Ni1) |
Δ
|
9.98(9) |
10.8 Δ |
20.8(1) |
21.8(1) |
25.3 Δ |
|
1S
(B, Ni2) |
(Δ) |
1.73(9) |
5.28(8) |
16.4(1) |
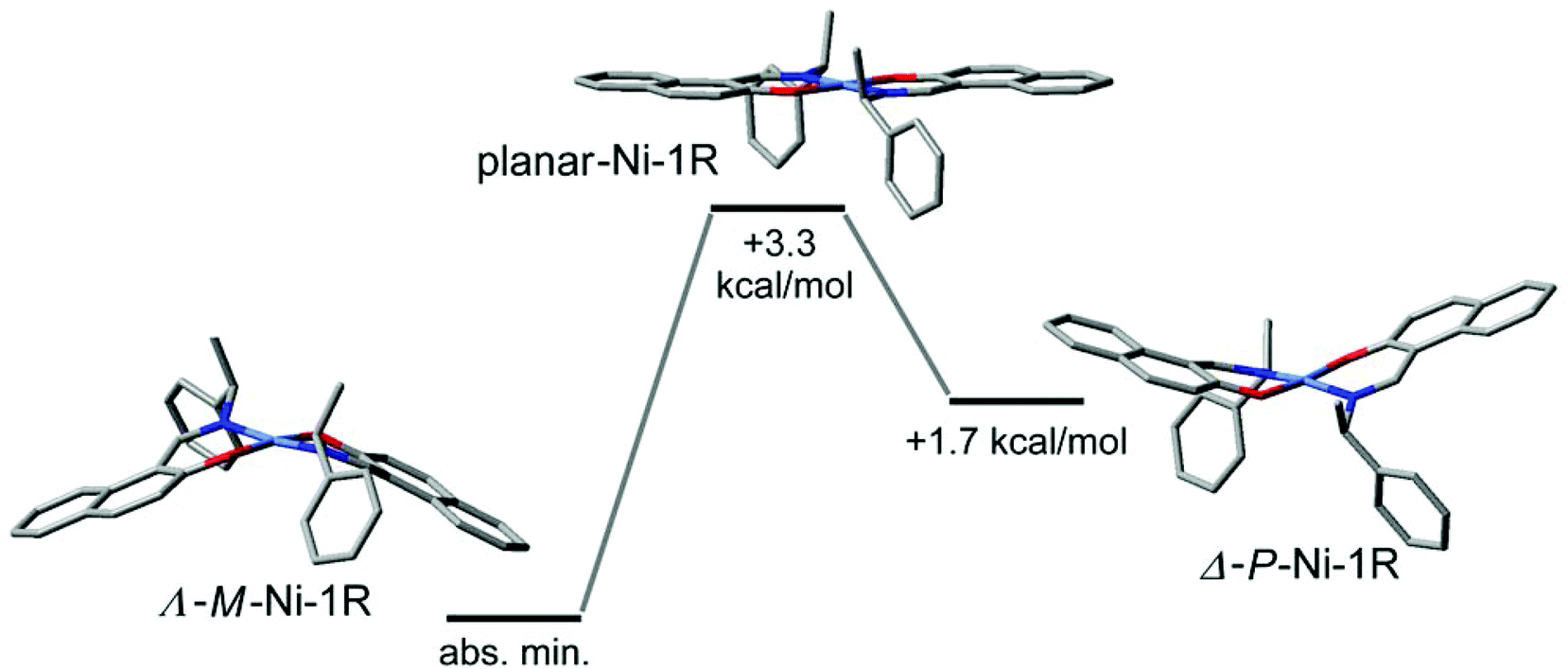
The combination of the folding in the NiNOC3 ring, the intrinsic curvature in the naphthaldiminato ligand, and the concurrence of these two phenomena for the two ligands, creates an overall complex conformation around the nickel atom which has a helical appearance. The helicity can be differentiated into right-handed (P) or left-handed (M) as shown in Scheme 3. With their different “up-or-down” folding the two symmetry-independent molecules A and B found in the crystal assume a different helicity. For example, in Ni-R-L1 (1R) the molecule with Ni1 is M-helical, the molecule with Ni2 is P-helical, and vice versa in Ni-S-L1 (1S) (Scheme 3). This P- or M-conformational helicity is in addition to the Λ- or Δ-metal-centred chirality. It is noteworthy that the left-handed M-conformation with a large folding angle ϕ of about 21° goes together with a more distinct dihedral angle θ of 10° for the clearly Λ-chiral molecule A in 1R (and conversely the right-handed P with the clearly Δ-chiral molecule A in 1S). However, the right-handed P-conformation with the smaller and uneven folding angles ϕ of 5° and 16° combines with the barely recognizable Λ-chiral molecule B in 1R which has only a very small dihedral angle θ < 2°, that is, essentially square planar (and it is the other way round for M which combines with the barely recognizable Δ-chiral molecule B in 1S) (see Scheme 3). The two angles θ and ϕ, describing together the deviation from an ideal square planar metal geometry for all-planar naphthaldiminato ligands, appear to be correlated with each other. The essentially square-planar Ni configuration in the B molecules in 1R and 1S may be seen as an effort of the P- (in 1R) and M-configurations (in 1S) to invert the Ni configuration. Evidently P prefers to have Δ at Ni and M prefers having Λ. This is confirmed by DFT results and by CD spectroscopy. Our DFT structures (see the Electronic Spectra section and Table 4) show a strong prevalence for the Δ(Ni)/P (or Λ(Ni)/M) combination. The DFT lowest energy structure has large dihedral θ and folding ϕ angles, with absolute values very similar to those measured for molecule A. In turn, solution CD spectra and CD calculations give clear evidence that the Λ- (in 1R) and Δ-configurations (in 1S) are mostly retained (although they could partly invert upon the conformational rearrangement).14,41 Further studies are necessary to detect and quantify any inversion phenomenon occurring in solution. Possibly, the P- and M-conformational helicities may invert dynamically in solution akin to a bird's flap with its wings, as sketched in Fig. 5. The structure with an all-planar Ni(bis-naphthaldiminato) moiety (in the middle of Fig. 5), though not necessarily coinciding with the real transition state, offers an estimate of the conversion barrier (around 3.5 kcal mol−1) between the P- and M-conformational isomers of compound 1R, allowing for a very fast process. Very likely, the unfavorable P (in 1R) and M-configurations (in 1S) in the crystals invert upon dissolution. It should be noted that the chelate-ring folding, when idealized, maintains the C2-symmetry of the molecule.
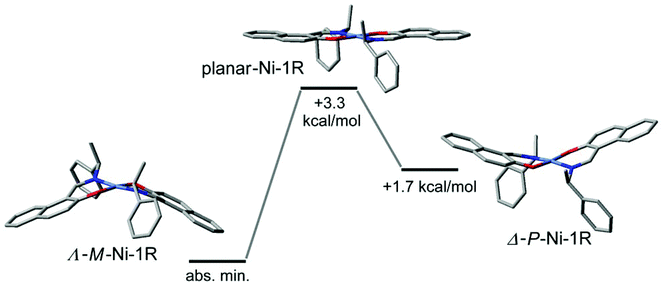 |
| | Fig. 5 Sketch of the interconversion process between Λ-Ni-1R (M-helical) and Δ-Ni-1R (P-helical) structures in solution, passing through a planar intermediate (devoid of metal chirality and helicity). Geometry optimizations and energies are calculated at the B3LYP/6-31G(d) level; the middle structure is optimized by constraining the Ni(bis-naphthaldiminato) moiety into a planar conformation. Hydrogen atoms are omitted for clarity. | |
Thermally induced structural phase transformation
Thermally induced structural phase transformation has been reported for transition metal–chiral N,O-chelate complexes, accompanying a change from a distorted square planar/tetrahedral geometry in the solid state to a regular square planar/tetrahedral geometry in the isotropic liquid phase.8a–d,12,13,15,21,44–46 Differential Scanning Calorimetry (DSC) has successfully been used to study the phenomenon. Thus DSC heating curves of the present compounds show an exothermic peak at ca. 190 °C for 1S, 3R, 3S and at 141 °C for 2R (Fig. 6 and Table 5), while cooling curves show no corresponding peak in the reverse direction. The results demonstrate a thermally induced irreversible phase transformation as reported for the analogous Ni(II)-N,O-chelate complexes.8c,d,13
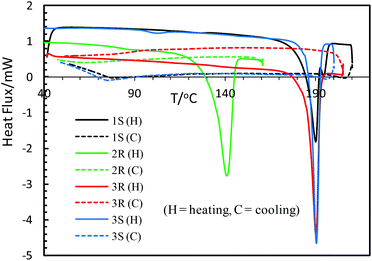 |
| | Fig. 6 DSC analysis curves of heating and cooling for the compounds. | |
Table 5 Thermally induced structural phase transformation data for the compounds
| Compounds (physical appearance) |
Peaks (T/°C) |
ΔH (kJ mol−1) |
|
The reported value is 183 °C for the analogous [(R)-N-1-(phenyl)ethyl-X-salicylaldiminato]Ni(II).8d
|
|
1S (dark brown block shaped crystals) |
190.1a |
−33.01 |
|
2R (greenish microcrystals) |
140.8 |
−28.03 |
|
3R (greenish microcrystals) |
190.2 |
−36.34 |
|
3S (greenish microcrystals) |
190.7 |
−42.62 |
Cyclic voltammetry
Cyclic voltammograms (CV) of the free Schiff base (S-HL1) and compounds 1S and 3R were recorded in the range of −0.50 to 1.50 V versus Ag/AgCl in acetonitrile using different switching potentials at varying scan rates (Fig. 7 and the ESI†). The free Schiff base solution shows three anodic waves at ca. Ea1 = 0.73, Ea2 = 1.00 and Ea3 = 1.20 V (Fig. 7a). Among them the former two waves are also seen for the electrolyte (TBAP) solution (Fig. 7a, dashed line), which shows the corresponding reduction waves at ca. Ec1 = 0.70 and Ec2 = 0.90 V. Hence, the latter wave (Ea3) is due to the oxidation of the free Schiff base ligand. Voltammograms of 1S, 2R and 3R (Fig. 7b and the ESI†) are identical and show an additional anodic wave at ca. 1.30 V (Ea4), which becomes more significant at higher scan rates, and is associated with the oxidation wave for the [Ni(L)2]/[Ni(L)2]+ couple.47,48 However, the corresponding cathodic wave is not detected even at higher scan rates, because of the instability of the cationic species ([Ni(L)2]+) which undergoes a rapid irreversible reaction to produce an electrode-inactive species.47a The voltammograms in the cathodic region (i.e., 0 to −0.50 V) show a reduction wave at ca. −0.35 V (Ec) for the [Ni(L)2]/[Ni(L)2]− couple (Fig. 7b and the ESI†), which overlaps with the decomposition peak of the electrolyte (TBAP) at higher scan rates.48a In fact, a corresponding poor oxidation wave is seen in the reverse scan, which undergoes rapid chemical transformation due to the instability of the [Ni(L)2]− species. Analysis of voltammograms at varying scan rates (0.02 to 0.40 V s−1) demonstrates a linear relationship between the anodic peak current (Ia) at ca. 1.30 V and the square root of the scan rate (ν1/2, Fig. 8), indicating a diffusion-controlled electrochemical process.
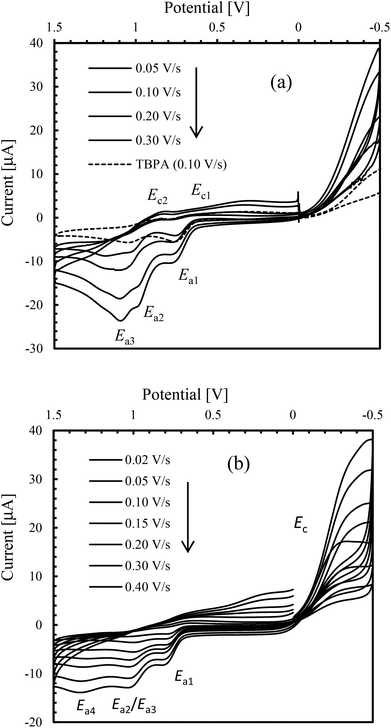 |
| | Fig. 7 Cyclic voltammograms of (a) S-HL1 (1.5 mmol L−1) and (b) 1S (1.0 mmol L−1); TBAP (0.1 mol L−1) at varying scan rates in acetonitrile at 25 °C. | |
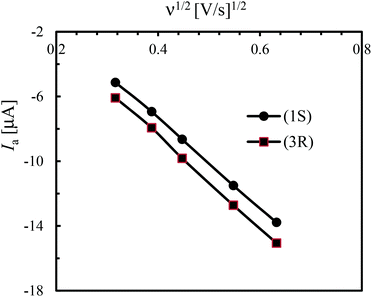 |
| | Fig. 8 Change of anodic current (Ia) at ca. 1.30 V vs. square root of the scan rate, (ν1/2). | |
Conclusions
We have reported the synthesis, characterization, X-ray structure, and computational study of a series of chiral bis(naphthaldiminato)Ni(II) complexes 1–3 with a distorted square planar geometry around the metal ion. These complexes show a remarkable stereochemistry because the ligand configuration is selectively and concurrently transferred to the metal chirality and to the conformational helicity of the ligands. Thus, a ligand with R configuration favors the formation of a complex with Λ chirality at the Ni and M ligand helicity. The stereochemical behavior of our series of complexes may be of interest in different fields based on the multiplication and amplification of chirality. First, the chiral folding of the naphthaldiminato moieties imparts the complexes with a discrete helicity which may be the starting point to obtain supramolecular helicates in a very selective and efficient way.5 Second, the complexes are possibly expected to induce high twisting powers when used as dopants for cholesteric liquid crystals.49
Experimental section
IR-spectra were recorded on a Nicolet iS10 spectrometer as KBr discs at ambient temperature. UV-Vis. spectra were obtained with a Shimadzu UV 1800 spectrophotometer in cyclohexane at 25 °C. Elemental analyses were performed on a Vario EL instrument from Elementar Analysensysteme. An Epsilon™ Instruments (BASi) electrochemical analyzer was used for cyclic voltammetry (CV) experiments containing tetra-N-butyl-ammonium-hexafluorophosphate (TBAP) as the supporting electrolyte in acetonitrile at 25 °C. The three-electrode measurement was carried out with a platinum disc working electrode, a platinum wire auxiliary electrode and an Ag/AgCl reference electrode. The solution containing the compounds and TBAP was deoxygenated for 10 minutes by passing nitrogen gas prior to running the experiments. CD spectra were obtained with a JASCO spectropolarimeter (J715) in cyclohexane on 2–2.5 mM samples. For each solution sample, three distinct spectra were recorded using cells with different path-lengths (0.1 mm, 5 mm and 10 mm) to cover the whole spectral range from 190–200 to 900 nm by keeping the absorbance below 1.5 AU. 1H-NMR spectra were run on a Bruker Avance DPX 400 spectrometer (operating at 400 MHz, 1H) in CDCl3 (δ 7.25 ppm) at 20 °C. EI-MS: Thermo-Finnigan TSQ 700. Isotopic distribution patterns for 58/60Ni (in 1R/S and 2R/S) or combined 63/65Ni + 79/81Br (in 3R/S) containing ions are clearly visible in the mass spectra.
General procedure to synthesise the complexes
Two equivalents of enantiopure (R or S)-N-1-(C6H5)ethyl-2-oxo-1-naphthaldimine (R or S-HL1) (341 mg, 1.24 mmol) dissolved in 5 mL methanol were added into a 5 mL hot methanolic solution of Ni(O2CCH3)2·4H2O (154 mg, 0.62 mmol) and the solution was stirred for 6–8 h at room temperature. The color changes from light green to greenish brown in clear solution. The volume of the solvent was reduced to ca. 50%, and this clear solution was left standing for crystallization via slow solvent evaporation of the solvent at room temperature. Dark brown block shaped crystals of Ni-R-L1 (1R) or Ni-S-L1 (1S), suitable for X-ray measurement, were obtained within 5–6 d. The crystals were separated, washed with methanol (2 × 2 mL), and dried in air for 3–4 d. The same procedure was followed for the syntheses of Ni-R-L2 (2R) or Ni-S-L2 (2S) using the Schiff bases of (R or S)-N-1-(p-OMeC6H4)ethyl-2-oxo-1-naphthaldimine (R or S-HL2), and greenish microcrystals were obtained after 5–6 d, that were not suitable for X-ray measurement. For the syntheses of Ni-R-L3 (3R) or Ni-S-L3 (3S) using (R or S)-N-1-(p-BrC6H4)ethyl-2-oxo-1-naphthaldimine (R or S-HL3), a green precipitate was formed within 30 min of stirring the solution. Stirring was continued for 6–8 h, and this solution was left standing for 8–10 h until complete precipitation. The solution was filtered off, and the precipitate was washed with methanol (2 × 2 mL). Green microcrystals of Ni-R-L3 (3R) or Ni-S-L3 (3S) were obtained after drying the sample in air for 3–4 d.
Bis{(R)-N-1-(C6H5)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}nickel(II) (Ni-R-L1; 1R).
Dark brown block shaped crystals. Yield 275 mg (73%). IR (KBr, cm−1): ν = 3057, 3027, 2971w (H–C), 1617, 1605vs (CN), and 1541s (CC). 1H NMR (400 MHz, CDCl3): δ = 1.97 (d, J = 6.5 Hz, 6H, CH3), 6.64 (d, J = 9.2 Hz, 2H), 6.78 (t, J = 7.4 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 4H), 7.42 (t, J = 7.4 Hz, 4H), 7.60 (m, 6H), 7.74 (d, J = 8.8 Hz, 2H), and 11.05 (s, 2H, CHN). ESI-MS: 607 (30) [M + H]+, 331 (58) [M − HL1]+, 228 (100) [C10H6(CHNH)(O)Ni] and 105 (33) [CH(C6H5)(CH3)]+. C38H32N2O2Ni (607.40): calcd C 75.14, H 5.31, N 4.61; found C 75.35, H 5.04, N 4.36.
Bis{(S)-N-1-(C6H5)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}nickel(II) (Ni-S-L1; 1S).
Dark brown block shaped crystals. Yield 260 mg (67%). IR (KBr, cm−1): ν = 3055, 3032, 2970w (H–C), 1616, 1605vs (CN), and 1540s (CC). 1H NMR (400 MHz, CDCl3): δ = 1.97 (d, J = 6.5 Hz, 6H, CH3), 6.62 (d, J = 9.2 Hz, 2H), 6.76 (t, J = 7.4 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 7.32 (d, J = 8.4 Hz, 4H), 7.41 (t, J = 7.4 Hz, 4H), 7.59 (m, 6H), 7.72 (d, J = 8.8 Hz, 2H), and 11.02 (br, 2H). ESI-MS: 607 (20) [M + H]+, 331 (45) [M − HL1]+, 228 (100) [C10H6(CHNH)(O)Ni] and 105 (25) [CH(C6H5)(CH3)]+. C38H32N2O2Ni·H2O (625.41): calcd C 72.98, H 5.48, N 4.48; found C 71.46, H 4.86, N 4.34.
Bis{(R)-N-1-(p-OMeC6H4)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}nickel(II) (Ni-R-L2; 2R).
Greenish microcrystals. Yield 300 mg (72%). IR (KBr, cm−1): ν = 3052, 3043w (H–Ar), 1624, 1605vs (CN), and 1541s (CC). 1H NMR (400 MHz, CDCl3): δ = 2.01 (d, J = 6.5 Hz, 6H, CH3), 3.83 (s, 6H, OCH3), 6.56 (d, J = 7.2 Hz, 2H), 6.96 (m, 6H), 7.09 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 7.4 Hz, 4H), 7.61 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.8 Hz, 2H), and 12.81 (br, 2H). ESI-MS: 667 (15) [M + H]+, 306 (10) [HL2 + H]+, 135 (100) [CH(CH3)(C6H4OCH3)]+ and 105 (15) [CH(C6H5)(CH3)]+. C40H36N2O4Ni (667.45): calcd C 71.98, H 5.44, N 4.20; found C 71.32, H 5.29, N 4.12.
Bis{(S)-N-1-(p-OMeC6H4)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}nickel(II) (Ni-S-L2; 2S).
Greenish microcrystals. Yield 280 mg (68%). IR (KBr, cm−1): ν = 3050, 3025w (H–Ar), 1642, 1611vs (CN), and 1541s (CC). ESI-MS: 667 (5) [M + H]+, 306 (10) [HL2 + H]+, 135 (100) [CH(CH3)(C6H4OCH3)]+ and 105 (20) [CH(C6H5)(CH3)]+. C40H36N2O4Ni (667.45): calcd C 71.98, H 5.44, N 4.20; found C 71.43, H 5.30, N 4.09.
Bis{(R)-N-1-(p-BrC6H4)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}nickel(II) (Ni-R-L3; 3R).
Greenish microcrystals. Yield 360 mg (72%). IR (KBr, cm−1): ν = 3051, 3027, 2973w (H–C), 1616, 1607vs (CN), and 1541s (CC). 1H NMR (400 MHz, CDCl3): δ = 1.94 (d, J = 6.0 Hz, 6H, CH3), 6.63 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 7.2 Hz, 2H), 7.12 (t, J = 6.8 Hz, 2H), 7.37 (d, J = 7.4 Hz, 4H), 7.55 (d, J = 8.4 Hz, 6H), 7.67 (d, J = 7.0 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), and 11.27 (br, 2H). C38H30N2O2Br2Ni·2H2O (801.24): calcd C 56.96, H 4.28, N 3.50; found C 56.24, H 3.86, N 3.40.
Bis{(S)-N-1-(p-BrC6H4)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}nickel(II) (Ni-S-L3; 3S).
Greenish microcrystals. Yield 350 mg (70%). IR (KBr, cm−1): ν = 3057, 3030, 2972w (H–C), 1616, 1605vs (CN), and 1541s (CC). EI-MS (70 eV): m/z (%) = 764 (10) [M]+, 411 (100) [M − HL3]+, 353 (30) [HL3]+, 229 (20) [C10H6(O)(CHNH)Ni + H]+, 183 (35) [C6H4(Br)(CHCH3)]+, 170 (60) [C10H6(O)(CHNH)]+, and 104 (60) [CH3CHC6H5–H]+ (isotopic distribution pattern resulting from the combination of 58/60Ni+79/81Br containing ions is clearly visible following the peaks at 764, 411, 229 while for 79/81Br containing ions at 353, and 183). C38H30N2O2Br2Ni·2H2O (801.24): calcd C 56.96, H 4.28, N 3.50; found C 56.04, H 3.66, N 3.42).
X-ray crystallography
Single-crystals of enantiomeric pairs Ni-R-L1 (1R) and Ni-S-L1 (1S) were carefully selected under a polarizing microscope and mounted on a loop. Data collection: Bruker APEX II CCD diffractometer with multi-layer mirror-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 203(2) K; ω-scans (see Table 6). Data collection and cell refinement with APEX2,50 data reduction with SAINT (Bruker).51Structure analysis and refinement: the structures were solved by direct methods (SHELXS-97),52 refinement was done by full-matrix least squares on F2 using the SHELXL-97 program suite,52 empirical (multi-scan) absorption correction with SADABS (Bruker).53 All non-hydrogen positions were refined with anisotropic temperature factors. Hydrogen atoms for aromatic CH, aliphatic or olefinic CH, CH2 and OH groups were positioned geometrically (C–H = 0.94 Å for aromatic CH, C–H = 0.94 Å for olefinic CH, 0.99 for aliphatic CH and 0.97 Å for CH3), and refined using a riding model (AFIX 43 for aromatic CH, AFIX 13 for aliphatic CH, AFIX 137 for CH3), with Uiso(H) = 1.2Ueq(CH, CH2) and Uiso(H) = 1.5Ueq(CH3). Details of the X-ray structure determination and refinements are provided in Table 6. Graphics were drawn with DIAMOND (Version 3.2).54 Computations on the supramolecular interactions were carried out with PLATON for Windows.55 The structural data for this paper have been deposited with the Cambridge Crystallographic Data Center (CCDC numbers 1405162 and 1405163).
Table 6 Crystal data and structure refinement for the compounds
| |
Ni-R-L1 (1R) |
Ni-S-L1 (1S) |
|
Largest difference peak and hole.
R
1 = [∑(‖Fo| − |Fc‖)/∑|Fo|]; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2.
Goodness-of-fit = [∑[w(Fo2 − Fc2)2]/(n − p)]1/2.
Absolute structure parameter.59
|
| Empirical formula |
C38H32N2O2Ni |
C38H32N2O2Ni |
|
M/g mol−1 |
607.37 |
607.37 |
| Crystal size/mm3 |
0.072 × 0.072 × 0.067 |
0.56 × 0.36 × 0.11 |
| Temperature/K |
95 |
100 |
|
θ range/°(completeness) |
1.97–27.50 (99.8%) |
1.66–25.23 (99.1%) |
|
h; k; l range |
−8,12; −15, 19; ±26 |
±11; ±18; ±24 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21 (no. 4) |
P21 (no. 4) |
|
a/Å |
9.5282(16) |
9.5328(9) |
|
b/Å |
15.355(3) |
15.2970(16) |
|
c/Å |
20.655(4) |
20.560(2) |
|
β/° |
90.146(7) |
90.192(6) |
|
V/Å3 |
3021.93(10) |
2998.1(5) |
|
Z/Dcalc(g cm−3) |
4/1.335 |
4/1.346 |
|
μ (Mo Kα)/mm−1 |
0.679 |
0.685 |
|
F(000) |
1272 |
1272 |
| Max./min. transmission |
0.956/0.953 |
0.700/0.928 |
| Reflections collected |
28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 227 227 |
25081 |
| Independent reflect. (Rint) |
12217 (0.0617) |
10664 (0.0573) |
| Data/restraints/parameters |
12217/13/779 |
10664/1/779 |
| Max./min. Δρa/e Å−3 |
0.688/−0.948 |
0.394/−0.661 |
|
R
1/wR2 [I > 2σ(I)]b |
0.0494/0.0944 |
0.0384/0.0922 |
|
R
1/wR2 (all data)b |
0.0678/0.1016 |
0.0438/0.0945 |
| Goodness-of-fit on F2c |
0.991 |
1.006 |
| Flack parameterd |
0.008(10) |
0.040(8) |
Computational section
Conformational searches and geometry optimizations were run with Spartan'14 (Wave function, Inc. Irvine, CA). Excited-state CD calculations were run with Gaussian09.56 Initial structures were generated starting from the available X-ray structure of the Λ-Ni-1R complex. An initial structure with an opposite configuration at the metal, Δ-Ni-1R, was obtained by mirror inversion of the whole complex, followed by a second inversion of the carbon chirality centers only. Conformational searches were run with molecular mechanics, using the Molecular Merck Force Field (MMFF). The geometry around the metal was kept fixed by constraining the O–Ni and N–Ni bond lengths and the O–Ni–O and N–Ni–N bond angles at their respective X-ray values. All the remaining rotatable bonds were included in the conformational search (i.e., varied systematically) and optimized with MMFF. All structures thus obtained were fully re-optimized with DFT using the B3LYP functional with the 6-31G(d) basis set on all atoms.57 Excited state calculations were run with the TDDFT method. A preliminary set of calculations was run to test the performance of various DFT functionals and basis sets, including a limited number of excited states (roots). The following functionals were tested: B3LYP, CAM-B3LYP, M06, PBE1PBE, wB97X-D and the two basis sets SVP and TZVP.56 Final calculations including 80 roots were run with the B3LYP/TZVP combination. The spectra were generated using the program SpecDis58 by applying a Gaussian band shape with a 0.2 eV exponential half-width. Rotational strengths calculated with a dipole-length gauge were employed, the differences between dipole-velocity values being negligible for most transitions.
Acknowledgements
We acknowledge the financial support from the Ministry of Science and Technology (MOST) under the project 2012/13, Dhaka, Bangladesh. We acknowledge the “Wazed Miah Science Research Centre” at Jahangirnagar University, Dhaka, Bangladesh for obtaining CV and elemental data. G.P. thanks Prof. Fabio Marchetti and Dr Francesco Zinna for useful discussions.
Notes and references
-
(a) J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CrossRef CAS PubMed;
(b) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CrossRef CAS PubMed;
(c) L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205 CrossRef CAS PubMed;
(d) N. Chen, M.-X. Li, P. Yang, X. He, M. Shao and S.-R. Zhu, Cryst. Growth Des., 2013, 13, 2650 CrossRef CAS;
(e) Z. Chen, S. Qin, D. Liu, Y. Shen and F. Liang, Cryst. Growth Des., 2013, 13, 3389 CrossRef CAS;
(f) W.-W. He, J. Yang, Y. Yang, Y.-Y. Liu and J.-F. Ma, Dalton Trans., 2012, 41, 9737 RSC;
(g) K. Kano and H. Hasegawa, J. Am. Chem. Soc., 2001, 123, 10616 CrossRef CAS PubMed;
(h) W.-G. Lu, J.-Z. Gu, L. Jiang, M.-Y. Tan and T.-B. Lu, Cryst. Growth Des., 2008, 8, 192 CrossRef CAS;
(i) L.-L. Wen, D.-B. Dang, C.-Y. Duan, Y.-Z. Li, Z.-F. Tian and Q.-J. Meng, Inorg. Chem., 2005, 44, 7161 CrossRef CAS PubMed;
(j) D.-R. Xiao, E.-B. Wang, H.-Y. An, Y.-G. Li, Z.-M. Su and C.-Y. Sun, Chem. – Eur. J., 2006, 12, 6528 CrossRef CAS PubMed;
(k) Y. Cui, O. R. Evans, H. L. Ngo, P. S. White and W. Lin, Angew. Chem., Int. Ed., 2002, 41, 1159 CrossRef CAS.
-
(a) H. Hou, X. Meng, Y. Song, Y. Fan, Y. Zhu, H. Lu, C. Du and W. Shao, Inorg. Chem., 2002, 41, 4068 CrossRef CAS PubMed;
(b) A. C. Kathalikkattil, K. K. Bisht, N. Aliaga-Alcalde and E. Suresh, Cryst. Growth Des., 2011, 11, 1631 CrossRef CAS;
(c) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed;
(d) S. J. Lee, A. Hu and W. Lin, J. Am. Chem. Soc., 2002, 124, 12948 CrossRef CAS PubMed;
(e) W. Lin, Z. Wang and L. Ma, J. Am. Chem. Soc., 1999, 121, 11249 CrossRef CAS;
(f) X. Meng, Y. Song, H. Hou, Y. Fan, G. Li and Y. Zhu, Inorg. Chem., 2003, 42, 1306 CrossRef CAS PubMed;
(g) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS PubMed;
(h) P. J. Deuss, R. den Heeten, W. Laan and P. C. J. Kamer, Chem. – Eur. J., 2011, 17, 4680 CrossRef CAS PubMed;
(i) J. Crassous, Chem. Soc. Rev., 2009, 38, 830 RSC.
-
(a) E. V. Anokhina and A. J. Jacobson, J. Am. Chem. Soc., 2004, 126, 3044 CrossRef CAS PubMed;
(b) S. P. Anthony and T. P. Radhakrishnan, Chem. Commun., 2004, 1058 RSC;
(c) J. Heo, Y.-M. Jeon and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 7712 CrossRef CAS PubMed;
(d) L. Jiang, X.-L. Feng, C.-Y. Su, X.-M. Chen and T.-B. Lu, Inorg. Chem., 2007, 46, 2637 CrossRef CAS PubMed; L. Jiang, T.-B. Lu and X.-L. Feng, Inorg. Chem., 2005, 44, 7056 Search PubMed;
(e) L. Ohrstrom, K. Larsson, S. Borg and S. T. Norberg, Chem. – Eur. J., 2001, 7, 4805 CrossRef CAS PubMed;
(f) A. Roth, D. Koth, M. Gottschaldt and W. Plass, Cryst. Growth Des., 2006, 6, 2655 CrossRef CAS.
-
(a) C. S. Isfort, T. Kreickmann, T. Pape, R. Froehlich and F. E. Hahn, Chem. – Eur. J., 2007, 13, 2344 CrossRef CAS PubMed;
(b) H.-J. Kim, E. Lee, H.-S. Park and M. Lee, J. Am. Chem. Soc., 2007, 129, 10994 CrossRef CAS PubMed;
(c) R. Wang, Y. Zhou, Y. Sun, D. Yuan, L. Han, B. Lou, B. Wu and M. Hong, Cryst. Growth Des., 2005, 5, 25 CrossRef;
(d) S.-T. Wu, Y.-R. Wu, Q.-Q. Kang, H. Zhang, L.-S. Long, Z. Zheng, R.-B. Huang and L.-S. Zheng, Angew. Chem., Int. Ed., 2007, 46, 8475 CrossRef CAS PubMed;
(e) Y. Zhang, J. Li, J. Chen, Q. Su, W. Deng, M. Nishiura, T. Imamoto, X. Wu and Q. Wang, Inorg. Chem., 2000, 39, 2330 CrossRef CAS PubMed.
-
(a) J.-M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565 CrossRef CAS PubMed;
(b) M. Albrecht, Chem. Soc. Rev., 2001, 101, 3457 CrossRef CAS;
(c) C. Biswas, M. G. B. Drew, M. Estrader and A. Ghosh, Dalton Trans., 2009, 5015 RSC;
(d) F. Lin, H.-Y. Peng, J.-X. Chen, D. T. W. Chik, Z. Cai, K. M. C. Wong, V. W. W. Yam and H. N. C. Wong, J. Am. Chem. Soc., 2010, 132, 16383 CrossRef CAS PubMed.
-
(a) U. Knof and A. von Zelewsky, Angew. Chem., Int. Ed., 1999, 38, 302–322 CrossRef;
(b) A. von Zelewsky and O. Mamula, J. Chem. Soc., Dalton Trans., 2000, 219–231 RSC;
(c) P. J. Steel, J. Org. Chem., 1991, 408, 395–402 CrossRef CAS;
(d) P. Hayoz and A. von Zelewsky, Tetrahedron Lett., 1992, 33:36, 5165–5168 CrossRef;
(e) R. Vadavi, E. D. Conrad, D. I. Arbuckle, T. S. Cameron, E. Essoun and M. A. S. Aquino, Inorg. Chem., 2011, 50, 11862–11864 CrossRef CAS PubMed;
(f) B. R. Groves, D. I. Arbuckle, E. Essoun, T. L. Lundrigan, R. Wang and M. A. S. Aquino, Inorg. Chem., 2013, 52, 11563–11572 CrossRef CAS PubMed;
(g) L. J. Boudreau, T. L. Clarke, A. H. Murray, K. N. Robertson, T. S. Cameron and M. A. S. Aquino, Inorg. Chim. Acta, 2013, 394, 152–158 CrossRef CAS;
(h) E. C. Constable, G. Zhang, C. E. Housecroft, M. Neuburger and J. A. Zampese, Chem. Commun., 2010, 3077–3079 RSC;
(i) E. C. Constable, C. E. Housecroft, T. Kulke, C. Lazzarini, E. R. Schofield and Y. Zimmermann, J. Chem. Soc., Dalton Trans., 2001, 2864–2871 RSC.
-
(a) K. Warnmark, J. A. Thomas, O. Heyke and J.-M. Lehn, Chem. Commun., 1996, 701–702 RSC;
(b) F. M. MacDonnell and S. Bodige, Inorg. Chem., 1996, 35, 5758–5759 CrossRef CAS;
(c) S. Bodige, A. S. Torres, D. J. Maloney, D. Tale, G. R. Kinsel, A. K. Walker and F. M. MacDonnell, J. Am. Chem. Soc., 1997, 119, 10364–10369 CrossRef CAS;
(d) K. Wärnmark, O. Heyke, J. A. Thomas and J.-M. Lehn, Chem. Commun., 1996, 2603–2604 RSC;
(e) T. J. Rutherford, M. G. Quagliotto and F. R. Keene, Inorg. Chem., 1995, 34, 3857–3858 CrossRef CAS;
(f) D. Tzalis and Y. Tor, J. Am. Chem. Soc., 1997, 119, 852–853 CrossRef CAS.
-
(a) T. Akitsu and Y. Einaga, Polyhedron, 2005, 24, 2933–2943 CrossRef CAS;
(b) G.-P. Li, Q.-C. Yang, Y.-Q. Tang, Y.-D. Guan and Z.-H. Shang, Acta Chim. Sin., 1987, 45, 421–425 CrossRef CAS;
(c) T. Akitsu, Polyhedron, 2007, 26, 2527–2535 CrossRef CAS;
(d) T. Akitsu and Y. Einaga, Polyhedron, 2005, 24, 1869–1877 CrossRef CAS;
(e) T. Akitsu and Y. Einaga, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m640–m642 Search PubMed;
(f) H. Sakiyama, H. Okawa, N. Matsumoto and S. Kida, J. Chem. Soc., Dalton Trans., 1990, 2935–2939 RSC;
(g) C. Evans and D. Luneau, J. Chem. Soc., Dalton Trans., 2002, 83–86 RSC;
(h) H. Sakiyama, H. Okawa, N. Matsumoto and S. Kida, Bull. Chem. Soc. Jpn., 1991, 64, 2644–2647 CrossRef CAS.
-
(a) F. Wang, H. Zhang, L. Li, H.-Q. Hao, X.-Y. Wang and J.-G. Chen, Tetrahedron: Asymmetry, 2006, 17, 2059–2063 CrossRef CAS;
(b) H. S. Chow, E. C. Constable, R. Frantz, C. E. Housecroft, J. Lacour, M. Neuburger, D. Rappoport and S. Schaffner, New J. Chem., 2009, 33, 376–385 RSC;
(c) G. Brewer, C. Brewer, R. J. Butcher, G. T. Robichaux and C. Viragh, Inorg. Chim. Acta, 2014, 410, 171–177 CrossRef CAS;
(d) E. C. Constable, C. E. Housecroft, V. Jullien, M. Neuburger, I. Poleschak, S. Reymann, S. Saxer and S. Schaffner, Polyhedron, 2007, 26, 5519–5526 CrossRef CAS;
(e) E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637–1651 RSC.
- M. Enamullah, A. Sharmin, M. Hasegawa, T. Hoshi, A.-C. Chamayou and C. Janiak, Eur. J. Inorg. Chem., 2006, 2146–2154 CrossRef CAS.
- C. Janiak, A.-C. Chamayou, A. K. M. Royhan Uddin, M. Uddin, K. S. Hagen and M. Enamullah, Dalton Trans., 2009, 3698–3709 RSC.
- M. Enamullah, A. K. M. Royhan Uddin, G. Pescitelli, R. Berardozzi, G. Makhloufi, V. Vasylyeva, A.-C. Chamayou and C. Janiak, Dalton Trans., 2014, 43, 3313–3329 RSC.
- M. Enamullah, M. A. Quddus, M. R. Hasan, G. Pescitelli, R. Berardozzi, G. J. Reiß and C. Janiak, Eur. J. Inorg. Chem., 2015, 2758–2768 CrossRef CAS.
- A.-C. Chamayou, S. Lüdeke, V. Brecht, T. B. Freedman, L. A. Nafie and C. Janiak, Inorg. Chem., 2011, 50, 11363–11374 CrossRef CAS PubMed.
-
(a) M. Enamullah, V. Vasylyeva and C. Janiak, Inorg. Chim. Acta, 2013, 408, 109–119 CrossRef CAS;
(b) M. Enamullah and M. K. Islam, J. Coord. Chem., 2013, 66, 4107–4118 CrossRef CAS.
-
(a) M. Enamullah, W. Linert, V. Gutmann and R. F. Jameson, Monatsh. Chem., 1994, 125, 1301–1309 CrossRef CAS;
(b) M. Enamullah, W. Linert and V. Gutmann, Vib. Spectrosc., 1995, 9, 265–271 CrossRef CAS;
(c) M. Enamullah, F. Renz, U. El-Ayaan, G. Wiesinger and W. Linert, Vib. Spectrosc., 1997, 14, 95–104 CrossRef CAS;
(d) M. Enamullah and W. Linert, J. Coord. Chem., 1996, 40, 193–201 CrossRef CAS.
-
(a) M. Enamullah, J. Coord. Chem., 2011, 64, 1608–1616 CrossRef;
(b) M. Enamullah, J. Coord. Chem., 2012, 65, 911–922 CrossRef CAS;
(c) M. Enamullah, A. K. M. Royhan Uddin and G. Hogart, J. Coord. Chem., 2012, 65, 4263–4276 CrossRef CAS.
- M. Enamullah, A.-C. Chamayou and C. Janiak, Z. Naturforsch., 2007, 62b, 807–817 Search PubMed.
- M. Enamullah, A. K. M. Royhan Uddin, G. Hogarth and C. Janiak, Inorg. Chim. Acta, 2012, 387, 173–180 CrossRef CAS.
-
M. Enamullah, R. Ahmed, G. Makhloufi and C. Janiak unpublished results.
- S. Yamada, Coord. Chem. Rev., 1999, 190–192, 537–555 CrossRef CAS.
- A. Ipatov, F. Cordova, L. J. Doriol and M. E. Casida, J. Mol. Struct., 2009, 914, 60–73 CrossRef CAS.
-
(a)
J. Autschbach, in Comprehensive Chiroptical Spectroscopy, ed. N. Berova, R. W. Woody, P. Polavarapu and K. Nakanishi, Wiley, New York, 2012, vol. 1, pp. 593–642 Search PubMed;
(b) H. Cox and A. J. Stace, Int. Rev. Phys. Chem., 2010, 29, 555–588 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
-
(a) M. Nishio, CrystEngComm, 2004, 6, 130 RSC;
(b)
M. Nishio, M. Hirota and Y. Umezawa, The CH/π interaction (Evidence, Nature and consequences), Wiley-VCH, 1998 Search PubMed;
(c) Y. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, Bull. Chem. Soc. Jpn., 1998, 71, 1207 CrossRef CAS;
(d) C. Janiak, S. Temizdemir, S. Dechert, W. Deck, F. Girgsdies, J. Heinze, M. J. Kolm, T. G. Scharmann and O. M. Zipffel, Eur. J. Inorg. Chem., 2000, 1229 CAS.
- G. S. Nichol and W. Clegg, CrystEngComm, 2007, 9, 959–960 RSC.
- J. W. Steed, CrystEngComm, 2003, 5, 169–179 RSC.
- A. Gavezotti, CrystEngComm, 2008, 10, 389–398 Search PubMed.
- G. R. Desiraju, CrystEngComm, 2007, 9, 91–92 RSC.
- J. Ruiz, V. Rodríguez, N. Cutillas, A. Hoffmann, A.-C. Chamayou, K. Kazmierczak and C. Janiak, CrystEngComm, 2008, 10, 1928–1938 RSC.
- V. Vasylyeva, T. Kedziorski, N. Metzler-Nolte, C. Schauerte and K. Merz, Cryst. Growth Des., 2010, 10, 4224–4226 CAS.
- G. Althoff, J. Ruiz, V. Rodríguez, G. López, J. Pérez and C. Janiak, CrystEngComm, 2006, 8, 662–665 RSC.
- X. Hao, S. Parkin and C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 689–699 Search PubMed.
- N. J. Babu and A. Nangia, CrystEngComm, 2007, 9, 980–983 RSC.
- A.-C. Chamayou, C. Biswas, A. Ghosh and C. Janiak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m311–m313 CAS.
- G. Makhloufi, K. Schütte and C. Janiak, Z. Kristallogr. – New Cryst. Struct., 2014, 229, 429–430 CAS.
- M. T. Kirchner, D. Bläser, R. Boese and G. R. Desiraju, CrystEngComm, 2009, 11, 229–231 RSC.
- S. Roy, R. Banerjee, A. Nangia and G. J. Kruger, Chem. – Eur. J., 2006, 12, 3777–3788 CrossRef CAS PubMed.
-
(a) J. O. Hernandez, J. Portilla, J. Cobo and C. Glidewell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 363–368 Search PubMed;
(b) B. K. Sarojini, H. S. Yathirajan, E. C. Hosten, R. Betz and C. Glidewell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 59–64 CAS.
- K. M. Anderson, K. Afarinkia, H.-W. Yu, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2006, 6, 2109–2113 CAS.
- A.-C. Chamayou, G. Makhloufi, L. A. Nafie, C. Janiak and S. Lüdeke, Inorg. Chem., 2015, 54, 2193–2203 CrossRef CAS PubMed.
- For examples of metal-chelate ring folding see: B. M. Barry, B. W. Stein, C. A. Larsen, M. N. Wirtz, W. E. Geiber, R. Wateman and R. E. Kemp, Inorg. Chem., 2013, 52, 9875–9884 CrossRef CAS PubMed.
- M. Enamullah, M. A. Quddus, M. A. Halim, M. K. Islam, V. Vasylyeva and C. Janiak, Inorg. Chim. Acta, 2015, 427, 103–111 CrossRef CAS.
-
(a) N. Arai, M. Sorai and S. Seki, Bull. Chem. Soc. Jpn., 1972, 45, 2398 CrossRef CAS;
(b) T. Ashida, S. Iwata, T. Yamane, M. Kakudo, A. Takeuchi and S. Yamada, Bull. Chem. Soc. Jpn., 1976, 49, 3502 CrossRef CAS.
- M. Enamullah, M. K. Islam, M. A. Halim and C. Janiak, J. Mol. Struct., 2015, 1099, 154–162 CrossRef CAS.
-
(a) S. Dilli, A. M. Maitra and E. Patsalides, Inorg. Chem., 1982, 21, 2832–2838 CrossRef CAS;
(b) M. S. El-Shahawi and W. E. Smith, Analyst, 1994, 119, 327–331 RSC.
-
(a) M. Tumer, D. Ekinci, F. Tumer and A. Bulut, Spectrochim. Acta, Part A, 2007, 67, 916–929 CrossRef PubMed;
(b) N. Mondala, S. Mitra, V. Gramilich, S. O. Ghodsi and K. M. Abdul Malik, Polyhedron, 2001, 20, 135–141 CrossRef.
-
(a) H. Sato, A. Yamagishi, J. Yoshida, H. Nakano and N. Hoshino, Jpn. J. Appl. Phys., 2005, 44, 4067–4072 CrossRef CAS;
(b) S. Zahn, G. Proni, G. P. Spada and J. W. Canary, Chem. – Eur. J., 2001, 7, 88–93 CrossRef CAS PubMed.
- APEX2, data collection program for the CCD area-detector system, Version 2.1-0, Bruker Analytical X-ray Systems, Madison (WI), 2006.
- SAINT, data reduction and frame integration program for the CCD area-detector system, Bruker Analytical X-ray Systems, Madison (WI), 2006.
- G. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Program SADABS, University of Göttingen, Göttingen, Germany, 1996.
- K. Brandenburg, Diamond (Version 3.2), Crystal and Molecular Structure Visualization, Crystal Impact, K. Brandenburg & H. Putz Gbr, Bonn, Germany, 2009.
-
(a) A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed;
(b) A. L. Spek, PLATON – A multipurpose crystallographic tool, Utrecht University, Utrecht, The Netherlands, 2005.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Wallingford CT, 2009 Search PubMed.
- All references about DFT functionals and basis sets can be found in the on-line documentation for Gaussian'09 at http://www.gaussian.com/g_tech/g_ur/g09help.htm.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
-
(a) H. D. Flack, M. Sadki, A. L. Thompson and D. J. Watkin, Acta Crystallogr., Sect. A, 2011, 67, 21–34 CAS;
(b) H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed;
(c) H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A, 1999, 55, 908–915 CrossRef PubMed;
(d) H. D. Flack, Acta Crystallogr., Sect. A, 1983, 39, 876–881 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS data, 1H-NMR spectra and data, cyclic voltammograms, analysis of supramolecular interactions, and CIF files reported in this paper. CCDC 1405162 and 1405163. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03940a |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence