
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-based drugs that break the rules
Claire S.
Allardyce
and
Paul J.
Dyson
*
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch
First published on 28th January 2016
Abstract
Cisplatin and other platinum compounds have had a huge impact in the treatment of cancers and are applied in the majority of anticancer chemotherapeutic regimens. The success of these compounds has biased the approaches used to discover new metal-based anticancer drugs. In this perspective we highlight compounds that are apparently incompatible with the more classical (platinum-derived) concepts employed in the development of metal-based anticancer drugs, with respect to both compound design and the approaches used to validate their utility. Possible design approaches for the future are also suggested.
 Professor Paul Dyson (right) being presented with his 2015 Royal Society of Chemistry Bioinorganic Chemistry Award by Dr Ben Murray at the University of Hull. | Paul Dyson is the director of the Institute of Chemical Sciences and Engineering at the Swiss Federal Institute of Technology in Lausanne (EPFL). Following positions in the UK (Imperial College of Science, Technology and Medicine and the University of York) he joined the EPFL in 2002. His research is focused on organometallic chemistry, in particular the application of organometallics in catalysis and biology. In 2015 he received the Bioinorganic Chemistry Award from the Royal Society of Chemistry. |
Introduction
In 1967, when Rosenberg set out to investigate the effect of an electrical field on bacterial growth, he observed elongation of the bacteria with the inhibition of cell division. Following a series of careful studies he found that the effect was not due to the field, but to the platinum electrodes producing cis-dichlorodiamineplatinum(II), i.e. cisplatin (Fig. 1).1 The complex was released into the medium where it entered the cells and formed adducts with DNA, primarily intra-strand cross-links,2 preventing replication. The discovery came at a time when anticancer compound screening focused on small organic molecules. It marked the entry of inorganic complexes (in the case of cisplatin, a carbon-free compound) into the arena. Cisplatin has gone on to revolutionize cancer treatment rendering formerly fatal disease, such as testicular cancer, largely curable.3 As a result, the intense research effort that preceded cisplatin's discovery shifted focus to include inorganic complexes with the objective of both widening the spectrum of chemotherapy of cisplatin and improving its clinical profile; after all, the (chance) discovery of cisplatin suggested that some rational design could lead to improved metal-based drugs. | ||
| Fig. 1 Structures of (from the left) cisplatin, carboplatin, oxaliplatin, and (bottom) triplatin tetranitrate, i.e. BBR3464 (or CT-3610). | ||
Today platinum drugs (cisplatin, carboplatin and oxaliplatin – Fig. 1) are used in an estimated 50–70% of cancer treatment regimens.4 The reason that cisplatin and its derivatives are not used in all cancer treatments is that some tumour types are resistant at the maximum tolerable dose. Dosage is limited by general toxicity, which manifests as severe side effects, including nausea, vomiting, loss of sensation in the extremities and nephrotoxicity. Some important types of cancer that are resistant to cisplatin include types of breast cancer, the most common cancer among women in Europe, and prostate cancer, which is the most common cancer among men in the United States and Europe.5 In addition cisplatin is largely inactive on colorectal cancers, which have increasing incidences in many countries, and many late stage cancers are also unresponsive, possibly due to epigenetic modifications.6 Other cancers can acquire cisplatin resistance during treatment.7
Drug design concepts related to the chemistry of the complexes
Relatively soon after the entry of cisplatin into the clinic a wide range of related compounds were prepared and evaluated. Neutral Pt(II) complexes with a square-planar geometry, including two cis-coordinated leaving groups, were defined as critical structural features for anticancer activity. As cisplatin is administered intravenously into the bloodstream, various transportation mechanisms have been proposed.8 Nevertheless, the coordination complex is believed to remain in a neutral, unchanged state during circulation and, after entering cells where intracellular chloride levels are lower than in blood, exchange of one or both chloride ligands by more labile ligands occurs thereby activating the complex. These findings led to a further rule being defined, i.e. slow ligand exchange rates relative to the rates of cell division processes. The rules was deemed necessary to render the complex inactive during transport and implies the use of mostly second and third row transition metal ions.9Various other rules were proposed as the mechanism of action of clinically successful platinum complexes became better understood and certain rules were abandoned and replaced by other rules and concepts. It was also found that compounds that were apparent ‘rule-breakers’ often had highly relevant and unique anticancer properties. For example, highly active di- and tri-nuclear cationic Pt(II) agents, in which the metal centres are linked by flexible diamine chains, do not contain cis-leaving groups.10 The complexes are active against a broad spectrum of tumours, including cisplatin-resistant cell lines, with apparently stronger DNA binding leading to these effects. The lead complex in this class of multinuclear platinum complexes is a trinuclear compound called triplatin tetranitrate or BBR3464 (also called CT-3610, Fig. 1), which has completed phase II clinical trials for melanoma, pancreatic, lung and ovarian cancers.11 Unfortunately, the results were mixed leading to reformulation. Nevertheless, other related complexes have subsequently been developed,12,13 including platinum bis-capronate (CT-47518) and platinum bis-butyrate (CT-47463), which have improved stability profiles in human plasma compared to the parent compound.14
A significant step forward in the field came in the form of compounds that do not primarily target DNA, with trans-[tetrachlorobis(1H-indazole)ruthenate(III)], KP1019, its sodium salt NKP-1339 and trans-[tetrachloro(dimethylsulfoxide)(imidazole)ruthenate(III)], NAMI-A (Fig. 2)15,16 being prominent examples of compounds that preferentially target other biomolecules. Although developed without a specific target in mind their conception did follow a prominent rule, i.e. slow ligand exchange kinetics, with that of Ru(III) complexes not being too dissimilar to Pt(II) compounds.17 Both these compounds are not particularly cytotoxic to cancer cells and in general are much less cytotoxic than cisplatin.18,19 Thus, the notion that the selection of drug candidates should be based on cytotoxicity studies was shown to be only partially valid and other cellular assays could be equally, if not more, valuable.20 Moreover, while both these Ru(III) complexes can bind to DNA, such binding is not believed to be relevant to their biological action, raising the question of the bias in the field towards DNA damaging agents. Compounds that target DNA, a ubiquitous target present in diseased and healthy cells, tend to be more systemically toxic than those with cancer-specific targets. When cisplatin was introduced into the clinic, acceptable therapeutic windows were narrower than those required today. Hence, today drugs that interact with cancer-specific targets should potentially provide more interesting therapies. This rationale does not negate approaches to selectively deliver cisplatin to the tumour site or the application of cisplatin prodrugs that only release cisplatin where needed21 and lipoplatin, a liposomal formulation of cisplatin, is in a number of advanced clinical trials.22
 | ||
| Fig. 2 Structures of (from the left) KP1019 and NAMI-A. | ||
The problem of following rules is exemplified by Ru(II) arene complexes, although it should be noted that to date none have entered clinical trials (a Ru(II) polypyridyl photosensitizer has apparently entered clinical trials).23 The low toxicity of Ru(III) complexes has been attributed, in part, to the ability of Ru(III) to mimic iron binding to serum proteins,24,25 which should increase the amount of compound that reaches cancer cells compared to healthy cells as the former tend to have higher levels of transferrin receptors on their membrane. However, this mechanism of drug delivery remains contentious, with recent studies suggesting albumin-mediated transport mechanisms due to the thermodynamically more stable complexes that tend to be formed with this protein.26 Cell uptake is a critical factor in activity, with the higher cytotoxicity of KP1019 compared to NAMI-A attributed to more complex entering the cells, which has been proposed to involve both passive diffusion27 and transferrin-dependent mechanisms.28 It has also been proposed that following cellular uptake Ru(III) complexes are activated by reduction to Ru(II) complexes,29 a process more pronounced in the hypoxic environment of tumour cells, thus offering selectivity and presumably lowering toxicity. There is recently published evidence to indicate that such a mechanism of activation may not take place.27 Nevertheless, the mechanisms mentioned above seemed, at the time, to justify the selection of Ru(III) anticancer compounds over Ru(II), which presumably would be unstable in a physiological environment due to ligand exchange kinetics and also would have higher general toxicities. However, these hypotheses were incorrect and there is now a vast body of data which show Ru(II) arene compounds have many ideal properties for the treatment of different cancers.30 Certainly, several Ru(II) arene complexes are very cytotoxic, equivalent or more cytotoxic than cisplatin, and lack the ability to distinguish between cancerous and healthy cells leading to high general toxicity in vivo and presumably limited therapeutic applications.31 However, other Ru(II) arene complexes not only appear to be non-toxic, but they display unique antitumour properties.32
Ru(II) arene complexes incorporating the amphiphilic 1,3,5-triaza-7-phosphaadamantane (pta) ligand, i.e. Ru(η6-toluene)(pta)Cl2, RAPTA-T, and Ru(η6-p-cymene)(pta)Cl2, RAPTA-C (Fig. 3), display very low toxicity in vivo and are not cytotoxic, but do have relevant antitumour properties. Instead of targeting DNA, RAPTA-C binds to the histone protein core in chromatin.33 Binding follows aquation of the chloride ligands and translates to mild growth inhibition on primary tumours in vivo.34 RAPTA-C also exerts a strong antiangiogenic effect in vivo35 and sections of primary tumours taken from treated mice have significantly fewer blood vessels.33 Interestingly, this activity is combined with antimetastatic activity,36 unlike other antiangiogenic compounds which are also prometastatic.37 The antiangiogenic and antimetastatic effects appear to be due to interactions with the cell membrane, with the majority of the compound localising on the membrane and relatively little entering the cell.38 The combined effects of RAPTA compounds appear to be unique and, importantly, when applied in combinations with other drugs, RAPTA-C leads to efficient inhibition of tumour growth at very low drug doses in the absence of toxic side effects.39
 | ||
| Fig. 3 The structures of (from the left) RAPTA-T (top left), RAPTA-C (top right), RDC11 (bottom left) and RAED-C (bottom right). RDC11 and RAED-C are formulated as PF6− salts. | ||
Other Ru(II) complexes shown to have clinically interesting properties include an organometallic ruthenium species termed RDC11 (Fig. 3).40 This complex has been tested against xenografted A2780 ovarian cancer cells or U87 glioblastoma cells implanted into nude mice in both cases demonstrating a 45% smaller tumour volume compared to control mice and similar tumour reduction compared to cisplatin, but with fewer side effects. The complex [Ru(η6-arene)(en)Cl]+ (en = ethylenediamine, RAED-C, Fig. 3), in which the en ligand was included in analogy to the two NH3 ligands in cisplatin, shows promising anticancer properties in different in vivo models.41 Many derivatives have since been studied, including ones based on osmium, and some have been evaluated in vivo.42
Concepts related to the biochemistry of cancer cells
Differences in the biochemistry of cancer cells can also be exploited to achieve selective activation or targeting. In principal, the hypoxic environment of a tumour can be exploited to selectively activate chemotherapeutics and based on this feature a series of cobalt complexes were developed with cytotoxicity depending on a hypoxia-released nitrogen mustard ligand (Fig. 4).43 The complex is administered as a prodrug with the cytotoxic group coordinated to the metal. This work shows the validity of metal drugs based on first row transition metal complexes and ingeniously exploits the very different exchange kinetics of cobalt in its different oxidation states through an appreciation of the unique biochemical characteristics of cancer cells. In a hypoxic environment, a one-electron reduction of Co(III) to Co(II) takes place which facilitates the release of the toxic ligand.40 Related cobalt44 and copper45 complexes that are activated by reduction have been developed, including very recently a Co(III) complex that releases a curcumin ligand upon reduction in a hypoxic environment (Fig. 4).46 It should be noted that a diverse range of curcumin-containing metal complexes have been reported that exhibit promising properties47 such as highly selective cytotoxicity to cancer cells over non-tumourigenic cells.48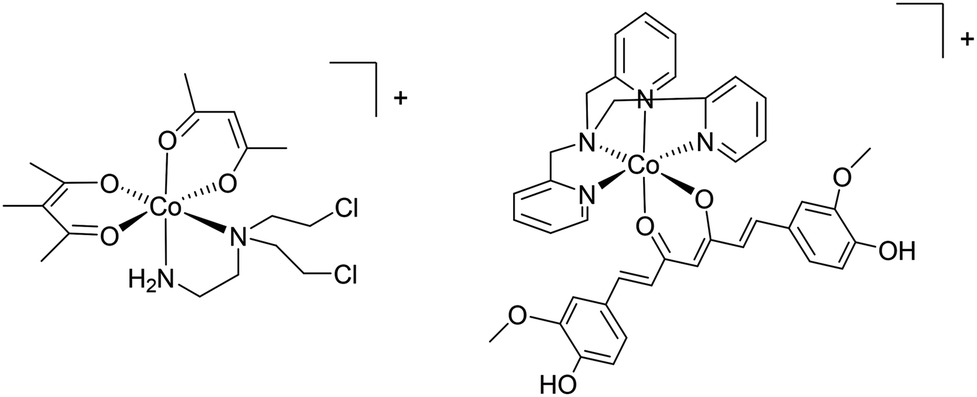 | ||
| Fig. 4 Examples of (from the left) a complex that releases a nitrogen mustand ligand and a curcumin ligand in hypoxic cancer tissue. Both complexes are formulated as ClO4− salts. | ||
Interestingly, in vivo studies in which tumours were oxygenated via normalization with axitinib showed that the Ru(II) arene compound, RAPTA-C, could inhibit tumour growth considerably more effectively than in a non-oxygen elevated tumour and was even markedly more active than doxorubicin.49 Since doxorubicin exhibits nanomolar levels of cytotoxicity on cancer cells, whereas RAPTA-C is essentially non-toxic in vitro, this switch in activity is quite remarkable.
Cancer cells are frequently characterized by elevated glutathione (GSH), glutathione transferase (GST) and gamma-glutamyl transferase (γGT) levels, which are generally considered to deactivate drugs. Indeed, the efficacy of cisplatin is modulated by interactions with glutathione with conjugation leading to efflux from cancer cells.50 However, GST and γGT overexpression actually provides the opportunity to target resistant tumours with GST and γGT-activated prodrugs, i.e. GST catalysed β-elimination may be harnessed to release a toxic drug molecule from a drug-glutathione conjugate.51 Such a concept is currently showing much potential with 4-(N-(S-glutathionylacetyl) amino) phenylarsenoxide (GSAO), a GST-activated arsenic-based agent, and with S-dimethylarsino-glutathione (dainaparsin), a γGT-activated arsenic-based prodrug (Fig. 5). These compounds are active against chemoresistant tumours such as pancreatic cancer and are undergoing clinical trials.51,52 Arsenic trioxide is a highly toxic compound that has been reintroduced into the clinic for the treatment of a number of cancers including acute promyelocytic leukemia when retinoid or anthracycline chemotherapy has not been effective, and multiple myeloma, chronic myelogenous leukemia, and acute myelogenous leukemia.53 A range of quite severe side-effects, albeit manageable ones, occur in patients treated with arsenic trioxide and more selective derivatives would be advantageous.
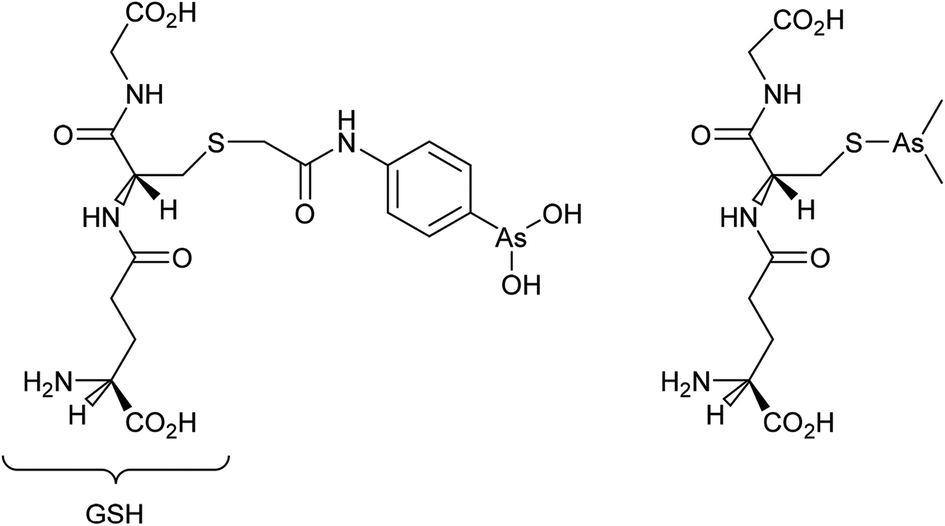 | ||
| Fig. 5 The structures of (from the left) GSAO and darinaparsin. | ||
As mentioned above, GSAO and darinaparsin are glutathione-conjugated arsenic complexes both assessed in clinical trials.54,55 A Phase I/II study showed that darinaparsin is well tolerated56 and a subsequent Phase II efficacy study showed promising disease-dependant results with lymphoma,55 leading to orphan drug designation for peripheral T-cell lymophoma.57 Darinaparsin is now being developed as both a monotherapy and in combination therapy (registered as NCT01139359) for T-cell lymphoma and as an oral formulation against solid tumours (registered as NCT01139346). Contrary to being eliminated from cancer cells, the modified GSH ligand facilitates cancer cell selective uptake in a multi-step process.51,58 First, the GSH conjugate of darinaparsin is processed by the external γGT and dipeptidase to afford dimethylarsinocysteine. Subsequently, this species is imported into the cell via cysteine transporters.59 Consequently, drug activity is dependent on the expression and efficacy of both the γGT and cysteine importing systems.59 In certain tumour types, these mechanisms are more active, such as pancreatic tumours including those of the pancreatic duct, affording a degree of selectivity.60
The majority of recently approved organic drugs target specific proteins that are over-expressed or even unique to cancer cells.61 Similarly, the mechanisms of many metal-based compounds are increasingly implicated with enzyme inhibition.62 Auranofin (Fig. 6), a gold-based drug used occasionally in the clinic in the treatment of rheumatoid arthritis, has been shown to induce apoptosis in cisplatin-resistant cell lines.63 The mechanism of auranofin has been studied and a number of enzymatic targets have been implicated in its mode of action61,64 However, it would appear that auranofin inhibits most isolated enzymes to some extent, with inhibition concentrations typically in the high nanomolar to low micromolar range. To some extent auranofin, and possibly most gold-based drugs, can be classified as likely frequent hitters for in vitro screens, i.e. inhibiting to some extent any target investigated. A similar phenomenon has been observed for certain organic compounds, which has been attributed to drug aggregates causing a clinically irrelevant cytotoxicity as oppose to the drug itself.65 Given the propensity of gold complexes to aggregate via the formation of gold–gold interactions66 the same rationale may apply.
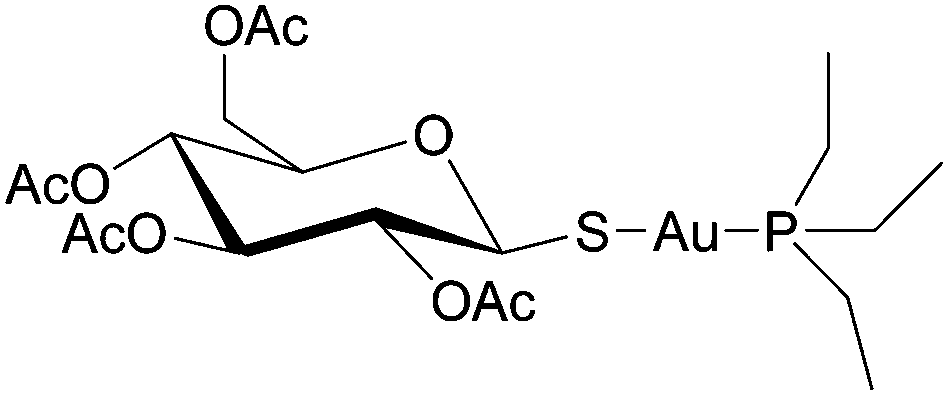 | ||
| Fig. 6 The structure of auranofin. | ||
Ascending the transition metal groups
Some examples of metal-based drugs comprising first row transition metals were mentioned in the previous section and many more classes of such compounds could be envisaged. The role of the metal in some drugs is not always to coordinate to a specific biomolecular target such as DNA or a protein. An example of such a compound undergoing clinical evaluation is a palladium–porphyrin complex that acts as a photosensitizer. The complex in question, TOOKAD-soluble (Fig. 7), has progressed to phase III clinical trials for the photodynamic treatment of prostate cancer (registered as NCT01875393). The role of the palladium ion in TOOKAD-soluble appears to be two-fold, first, helping to provide the ideal photophysical properties to the porphyrin and, second, being sufficiently inert so as not to be displaced during therapy.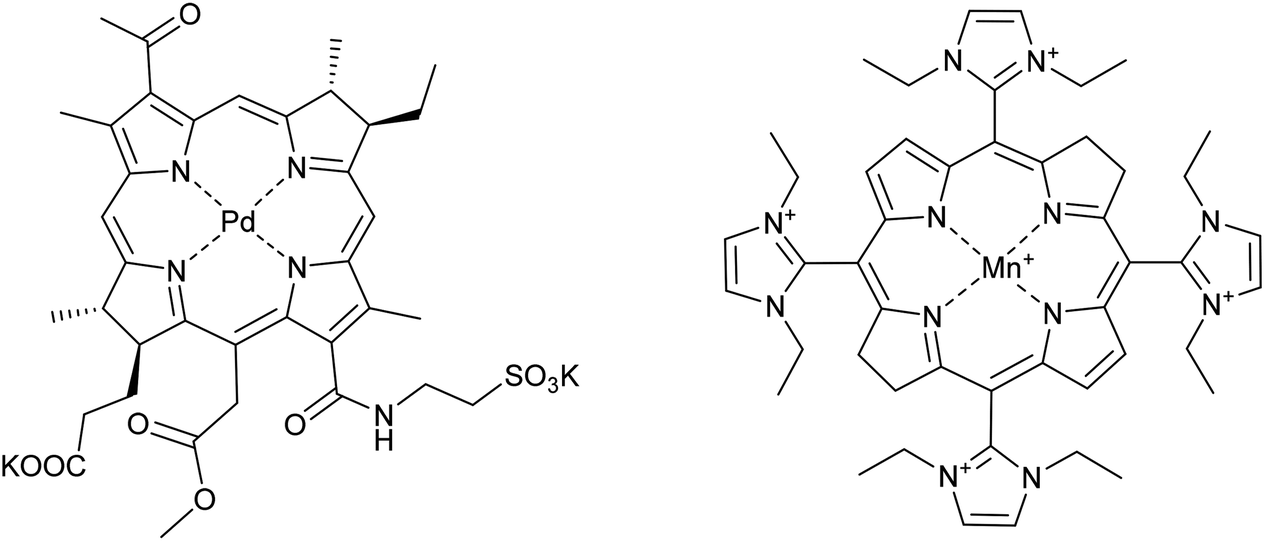 | ||
| Fig. 7 The structure of (from the left) TOOKAD-soluble and AEOL-10150 (formulated as the chloride salt). | ||
Photodynamic therapy focusses drug toxicity in the tumour environment by administering a photosensitizer that is activated by light – with the tumour environment being selectively illuminated. Under irradiation with the appropriate wavelength of light the photosensitizer reacts with oxygen to generate reactive oxygen species (ROS) that modulate the function of surrounding tissue, leading to various responses according to the level of ROS, but typically damaging blood vessels and causing them to close, thereby shutting off the supply of nutrients to a tumour.67 Singlet oxygen was initially thought critical for the PDT effect,68 but attributing the photodynamic effect exclusively to singlet oxygen may have been an oversimplification. Recent clinical studies demonstrate effective PDT using TOOKAD-soluble as the photosensitizer in the absence of singlet oxygen generation.69 Instead, the mechanism hinges on the production of hydroxyl radicals in the vascular system feeding the tumour tissue. Nevertheless, the net result is vascular shutdown which isolates the cancer and leads to cell death. Other possible mechanisms of Pd-containing porphyrins depend on the compartmentalisation of the photosensitiser, e.g. in the mitochondrial outer membrane70 or the endoplasmic reticulum (ER),71 with compartmentalisation depending on both the nature of the photosensitiser and the time between administration of this complex and treatment.
Photosenstisers have been coupled to known anticancer drug complexes such as cisplatin or oxaliplatin.72 The rationale for this approach is to combine the cytotoxicity of platinum-based drugs with PDT to reduce the amount of platinum drug administered for an effective dose, thereby reducing side effects. Co-administration may also overcome some types of drug resistance and further developments involve tethering PDT agents to other metal drugs such as those based on ruthenium.73
Combination therapies are used to provide stronger therapies for aggressive cancers and also to broaden the spectrum of activity of drugs and a well-established combination therapy comprises the application of cisplatin with radiotherapy. The rationale behind this particular combination is that heavy metal ions absorb X-rays more efficiently than biological tissues leading to the release of low energy electrons to give focussed treatments around the heavy metal centre.74 However, radiation damage to surrounding healthy tissue is frequently observed in patients that undergo this combination therapy. Drugs initially designed to treat radiation sickness, i.e. for use in the event of a nuclear catastrophe could be co-administered with cisplatin when used in combination with radiotherapy. Take, for example, the manganese porphyrin compound AEOL-10150 (Fig. 7). This compound is an antioxidant mimicking the catalytic site of superoxide dismutase. Although the complex is not in itself an anticancer drug, it could prove highly beneficial in anticancer combination therapies, and represents an interesting area for future research. AEOL-10150 is already in clinical trials for a number of diseases linked to oxidation damage, such as amyotropic lateral sclerosis and spinal cord injury, and if applied with cisplatin–radiotherapy combinations could reduce oxidative damage to health tissue.75 Indeed, many porphyrin complexes containing first row transition metal are under evaluation as catalytic antioxidants for the treatment of many different types of diseases.76
Outlook
Cisplatin is a broad acting anticancer drug used to treat a multitude of different cancers. One cannot overstate the success of this compound. And yet the success of cisplatin has certainly biased the screening of metal-based drugs towards compounds that are active against many cancer types, often with little thought as to which types or even sub-types of cancers may be relevant. Such an approach, when successful, undoubtedly affords a drug that helps a large number of patients. However, such an approach is highly risky and, not surprisingly, few new metal-based anticancer drugs have reached the clinic. The development of therapies that are effective on a small subset of cancers, and that target specific abnormalities in these cancers, potentially represents a more successful route and in the future should lead to new drugs that induce a greater tumour response for individual patients. Hence, designing drugs based on cancer biology of specific tumour types, while not ignoring the basic inorganic/organometallic chemistry involved, could improve the drug development process.New drugs that target specific abnormalities of certain tumours, whether a general phenomenon, such as hypoxia, or a single biomolecular target, such as a unique enzyme, could increasingly be based on less toxic metals or even metals that are essential to living systems. For catalytically active metal-based drugs, first row transition metals should be less generally toxic and also tend to display superior catalytic activities. Furthermore, if a metal complex is designed with a specific three-dimensional shape that interacts selectively with an enzyme and the metal itself is not reactive, it follows that non-toxic metal centres would be advantageous, assuming they afford inert compounds.77 Rational drug design in organic chemistry focuses on shape and often is inspired by nature. Similarly, medicinal inorganic chemists could do the same. As pointed out by others,78 the synthesis of transition metal complexes with complex three dimensional structures is often more facile than that of organic molecules and, if combined with suitable electronic and/or ligand exchange properties, may lead to new drugs that inhibit so called undruggable targets such as transcription factors and non-enzymatic proteins including scaffolds.79
In the clinic, most chemotherapeutic regimens used to treat cancer involve the application of drug combinations with each drug targeting a specific pathway or process. In the recent phase I/II clinical trial of NAMI-A the ruthenium compound was applied together with gemcitabine on patients with non-small cell lung cancer.80 Unfortunately, convincing efficacy results were not obtained and the likelihood of further clinical studies of NAMI-A are uncertain. NAMI-A was not specifically designed for application in this type of cancer which could have led to the poor outcome and the specific drug combination was chosen based on complementary mechanisms. Finding synergistic drug combinations is clearly critical and a highly challenging procedure, often relying on data from prior clinical studies. Alternative methods, such as a phenotypic-based feedback system control approach, i.e. high-throughput cytotoxicity screening combined with a mathematical model that reduces the number of experiments required, has been used on RAPTA-C with a series of antiangiogenic agents.39 All the resulting drug combinations that showed synergies contained RAPTA-C and the in vitro data was subsequently validated in vivo. This and related strategies may facilitate the process of finding synergistic drug combinations and should improve the chances of a positive outcome of metal-based drugs in phase II clinical studies, which are often based on drug combinations comprising the new compound with an established drug. Moreover, combinations that include broad acting compounds, such as platinum-based compounds, are increasingly likely to employ macroscopic drug delivery systems that help to target the compound to the tumour. Delivery systems include the liposomal formulation of cisplatin mentioned earlier, but increasingly more sophisticated systems that target tumours via cancer selective compounds or exploit natural delivery systems are being developed.81
In conclusion, it could be argued that the rules used to guide the design of metal-based drugs are entirely valid and should be followed, since the drugs that have made it into the clinic to date follow these rules. However, there are numerous examples of compounds which have been studied on advanced pre-clinical models or are currently undergoing clinical trials, e.g. the arsenic–glutathione prodrugs that target certain tumours as well as others described above, to suggest we should not be inhibited by these rules. There is clearly a long way to go before we can say that certain classes of metal complexes should be excluded from medicinal inorganic chemistry and a better understanding of biology and the use of assays other than cytotoxicity screening are essential to guide metal-based drug design in the future.
References
- B. Rosenberg, E. Renshaw, L. Vancamp, J. Hartwick and J. Drobnik, J. Bacteriol., 1967, 93, 716–721 CAS.
- F. Coste, J.-M. Malinge, L. Serre, M. Leng and C. Zelwer, Nucleic Acids Res., 1999, 27, 1837–1846 CrossRef CAS PubMed.
- L. C. Richardson, A. J. Neri, E. Tai and J. D. Glenn, Urol. Oncol., 2012, 30, 95–101 CrossRef CAS PubMed; R. S. Go and A. A. Adjei, J. Clin. Oncol., 1999, 17, 409–422 Search PubMed; B. Desoize and C. Madoulet, Crit. Rev. Oncol. Hematol., 2002, 42, 317–325 CrossRef PubMed.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- V. Brabec and J. Kasparkova, Drug Resist. Updat., 2005, 8, 131–146 CrossRef CAS PubMed; T. Torigoe, H. Izumi, H. Ishiguchi, Y. Yoshida, M. Tanabe, T. Yoshida, T. Igarashi, I. Niina, T. Wakasugi, T. Imaizumi, Y. Momii, M. Kuwano and K. Kohno, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 15–27 CrossRef PubMed.
- D. Matei, F. Fang, C. Shen, J. Schilder, A. Arnold, Y. Zeng, W. A. Berry, T. Huang and K. P. Nephew, Cancer Res., 2012, 72, 2197–2205 CrossRef CAS PubMed.
- D. Chen, V. Milacic, M. Frezza and Q. P. Dou, Curr. Pharm. Des., 2009, 15, 777–791 CrossRef CAS PubMed; L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef PubMed; A.-M. Florea and D. Büsselberg, Cancers, 2011, 3, 1351–1371 CrossRef PubMed.
- S. Harrach and G. Ciarimboli, Front. Pharmacol., 2015, 6, 85–92 Search PubMed.
- J. Reedjik, Platinum Met. Rev., 2008, 52, 1–9 Search PubMed.
- N. Farrell, Met. Ions Biol. Syst., 2004, 41, 252–296 Search PubMed.
- D. Jodrell, T. Evans, W. Steward, D. Cameron, J. Prendiville, C. Aschele, C. Noberasco, M. Lind, J. Carmichael and N. Dobbs, Eur. J. Cancer, 2004, 40, 1872–1877 CrossRef CAS PubMed.
- C. Billecke, S. Finniss, L. Tahash, C. Miller, T. Mikkelsen, N. P. Farrell and O. Bögler, Neurol Oncol., 2006, 8, 215–226 CrossRef CAS PubMed.
- T. Shingu, V. C. Chumbalkar, H. S. Gwak, K. Fujiwara, S. Kondo, N. P. Farrell and O. Bogler, Neurol Oncol., 2010, 12, 1269–1277 CAS.
- L. Gatti, P. Perego, R. Leone, P. Apostoli, N. Carenini, E. Corna, C. Allievi, U. Bastrup, S. De Munari, S. Di Giovine, P. Nicoli, M. Grugni, M. Natangelo, G. Pardi, G. Pezzoni, J. W. Singer and F. Zunino, Mol. Pharm., 2010, 7, 207–216 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525–1535 CrossRef CAS PubMed; E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Met. Ions Biol. Syst., 2004, 42, 323–351 Search PubMed.
- J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611–3616 CrossRef CAS PubMed.
- A. Herman, J. M. Tanski, M. F. Tibbetts and C.-M. Anderson, Inorg. Chem., 2008, 47, 274–280 CrossRef CAS PubMed.
- D. Pluim, R. C. A. M. van Waardenburg, J. H. Beijnen and J. H. M. Schellens, Cancer Chemother. Pharmacol., 2004, 54, 71–78 CrossRef CAS PubMed.
- A. Amin and M. A. Buratovich, Mini-Rev. Med. Chem., 2009, 9, 1489–1503 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed; S. van Zutphen and J. Reedijk, Coord. Chem. Rev., 2005, 249, 2845–2853 CrossRef.
- M. Fantini, L. Gianni, C. Santelmo, F. Drudi, C. Castellani, A. Affatato, M. Nicolini and A. Ravaioli, Chemother. Res. Pract., 2011, 125192 Search PubMed.
- http://investorintel.com/biotech-intel/theralase-progresses-to-high-purity-anti-cancer-drug-poised-for-human-trials accessed 11.23, 6 October 2015.
- F. Kratz and L. Messori, J. Inorg. Biochem., 1993, 49, 79–82 CrossRef CAS PubMed.
- M. Liu, Z. J. Lim, Y. Y. Gwee, A. Levina and P. A. Lay, Angew. Chem., Int. Ed., 2010, 49, 1521–3773 Search PubMed.
- I. Bratsos, T. Gianferrara, E. Alessio, C. G. Hartinger, M. A. Jakupec and B. K. Keppler, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, 2011, p. 151 RSC; M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, J. Anal. At. Spectrom., 2005, 20, 856–863 RSC; A. K. Bytzek, K. Boeck, G. Hermann, S. Hann, B. K. Keppler, C. G. Hartinger and G. Koellensperger, Metallomics, 2011, 3, 1049–1055 RSC.
- A. Levina, J. B. Aitken, Y. Y. Gwee, Z. J. Lim, M. Liu, A. M. Singharay, P. F. Wong and P. A. Lay, Chem. – Eur. J., 2013, 19, 3609–3619 CrossRef CAS PubMed.
- M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46–51 RSC.
- M. J. Clarke, Coord. Chem. Rev., 2002, 232, 69–93 CrossRef CAS; M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46–51 RSC.
- J. J. Soldevila-Barreda and P. J. Sadler, Curr. Opin. Chem. Biol., 2015, 25, 172–183 CrossRef CAS PubMed; T. Sriskandakumar, S. Behyan, A. Habtemariam, P. J. Sadler, P. Kennepohl, A. A. Nazarov, C. H. Hartinger and P. J. Dyson, J. Organomet. Chem., 2014, 751, 251–260 CrossRef.
- P. Tomšík, D. Muthná, M. Řezáčová, St. Mičuda, J. Ćmielová, M. Hroch, R. Endlicher, Z. Červiková, E. Rudolf, St. Hann, D. Stíbal, B. Therrien and G. Süss-Fink, J. Organomet. Chem., 2015, 782, 42–51 CrossRef.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem. – Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed; Z. Adhireksan, G. E. Davey, P. R. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Dröge, U. Roethlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 3462 Search PubMed.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. Nazarov, G. Wagnières, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed; A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Int. J. Oncol., 2008, 33, 1281–1289 Search PubMed.
- J. M. L. Ebos, C. R. Lee, W. Cruz-Munoz, G. A. Bjarnason, J. G. Christensen and R. S. Kerbel, Cancer Cell, 2009, 15, 232–239 CrossRef CAS PubMed; J. M. L. Ebos and R. S. Kerbel, Nat. Rev. Clin. Oncol., 2011, 8, 210–221 CrossRef PubMed.
- R. F. S. Lee, S. Escrig, M. Croisier, S. Clerc-Rosset, G. W. Knott, A. Meibom, C. A. Davey, K. Johnsson and P. J. Dyson, Chem. Commun., 2015, 51, 16486–16489 RSC.
- A. Weiss, X. Ding, J. R. van Beijnum, I. Wong, T. J. Wong, R. H. Berndsen, O. Dormond, M. Dallinga, L. Shen, R. O. Schlingemann, R. Pili, C.-M. Ho, P. J. Dyson, H. van den Bergh, A. W. Griffioen and P. Nowak-Sliwinska, Angiogenesis, 2015, 18, 233–244 CrossRef CAS PubMed.
- X.-J. Meng, M. L. Leyva, M. Jenny, I. Gross, S. Benosman, B. Fricker, S. Harlepp, P. Hébraud, A. Boos, P. Wlosik, P. Bischoff, C. Sirlin, M. Pfeffer, J.-P. Loeffler and C. Gaiddon, Cancer Res., 2009, 69, 5458–5466 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed; A. Bergamo, A. Masi, A. F. A. Peacock, A. Habtemariam, P. J. Sadler and G. Sava, J. Inorg. Biochem., 2010, 104, 79–86 CrossRef PubMed.
- N. J. Farrer and P. J. Sadler, Medicinal inorganic chemistry: state of the art, new trends, and a vision of the future, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH, Weinheim, 2011 Search PubMed.
- D. C. Ware, B. D. Palmer, W. R. Wilson and W. A. Denny, J. Med. Chem., 1993, 36, 1839–1846 CrossRef CAS PubMed.
- G. O. Ahn, K. J. Botting, A. V. Patterson, D. C. Ware, M. Tercel and W. R. Wilson, Biochem. Pharmacol., 2006, 71, 1683–1694 CrossRef CAS PubMed.
- L. L. Parker, S. M. Lacy, L. J. Farrugia, C. Evans, D. J. Robins, C. C. O'Hare, J. A. Hartley, M. Jaffar and I. J. Stratford, J. Med. Chem., 2004, 47, 5683–5689 CrossRef CAS PubMed.
- A. K. Renfrew, N. S. Bryce and T. W. Hambley, Chem. Sci., 2013, 4, 3731–3739 RSC.
- S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC.
- R. Pettinari, F. Marchetti, F. Condello, C. Pettinari, G. Lupidi, R. Scopelliti, S. Mukhopadhyay, T. Riedel and P. J. Dyson, Organometallics, 2014, 33, 3709–3715 CrossRef CAS.
- M. A. Weiss, D. Bonvin, R. H. Berndsen, E. Scherrer, T. J. Wong, P. J. Dyson, A. W. Griffioen and P. Nowak-Sliwinska, Sci. Rep., 2015, 5, 8990 CrossRef PubMed.
- M. L. Fishel, M. P. Gamcsik, S. M. Delaney, E. G. Zuhowski, V. M. Maher, T. Karrison, R. C. Moschel, M. J. Egorin and M. E. Dolan, Cancer Chemother. Pharmacol., 2005, 55, 333–342 CrossRef CAS PubMed.
- E. E. Ramsay and P. J. Dilda, Front. Pharmacol., 2014, 5, 1–16 CAS.
- C. K. Mirabelli, R. K. Johnson, C. M. Sung, L. Faucette, K. Muirhead and S. T. Crooke, Cancer Res., 1985, 45, 32–39 CAS.
- J. Zhu, Z. Chen, V. Lallemand-Breitenbach and H. de Thé, Nat. Rev. Cancer, 2002, 2, 705–713 CrossRef CAS PubMed; Z. X. Shen, Z. Z. Shi, J. Fang, B. W. Gu, J. M. Li, Y. M. Zhu, J. Y. Shi, P. Z. Zheng, H. Yan, Y. F. Liu, Y. Chen, Y. Shen, W. Wu, W. Tang, S. Waxman, H. De Thé, Z. Y. Wang, S. J. Chen and Z. Chen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5328–5235 CrossRef PubMed; G. McCollum, P. C. Keng, J. C. States and M. J. McCabe, J. Pharmacol. Exp. Ther., 2005, 313, 877–887 CrossRef PubMed.
- A. M. Tsimberidou, L. H. Camacho, S. Verstovsek, C. Ng, D. S. Hong, C. K. Uehara, C. Gutierrez, S. Daring, J. Stevens, P. B. Komarnitsky, B. Schwartz and R. Kurzrock, Clin. Cancer Res., 2009, 15, 4769–4776 CrossRef CAS PubMed; L. Horsley, J. Cummings, M. Middleton, T. Ward, A. Backen, A. Clamp, M. Dawson, H. Farmer, N. Fisher, G. Halbert, S. Halford, A. Harris, J. Hasan, P. Hogg, G. Kumaran, R. Little, G. J. M. Parker, P. Potter, M. Saunders, C. Roberts, D. Shaw, N. Smith, J. Smythe, A. Taylor, H. Turner, Y. Watson, C. Dive and G. C. Jayson, Pharmacol., 2013, 72, 1343–1352 Search PubMed.
- P. J. Hosein, M. D. Craig, M. S. Tallman, R. V. Boccia, B. L. Hamilton, J. J. Lewis and I. S. Lossos, Am. J. Hematol., 2012, 87, 111–114 CrossRef CAS PubMed.
- J. R. Berenson, S. Jaganath, D. Reece, R. Boccia, R. Soebel, A. Belch, B. Schwartz, R. P. Gale and M. Hussein, J. Clin. Oncol., 2007, 25, 8109 Search PubMed.
- J. Wu, C. Henderson, L. Feun, P. Van Veldhuizen, P. Gold, H. Zheng, T. Ryan, L. S. Blaszkowsky, H. B. Chen, M. Costa, B. Rosenzweig, M. L. Nierodzik, H. Hochster, F. Muggia, G. Abbadessa, J. Lewis and A. X. Zhu, Invest. New Drugs, 2010, 28, 670–676 CrossRef CAS PubMed.
- S. L. Soignet, P. Maslak, Z. G. Wang, S. Jhanwar, E. Calleja, L. J. Dardashti, D. Corso, A. DeBlasio, J. Gabrilove, D. A. Scheinberg, P. P. Pandolfi and R. P. Warrell Jr., N. Engl. J. Med., 1998, 339, 1341–1348 CrossRef CAS PubMed.
- N. Garnier, G. G. Redstone, M. S. Dahabieh, J. N. Nichol, S. V. Del Rincon, Y. Gu, D. S. Bohle, Y. Sun, D. S. Conklin, K. K. Mann and W. H. Miller Jr., Mol. Pharmacol., 2014, 85, 576–585 CrossRef PubMed.
- E. E. Ramsay, S. Decollogne, S. Joshi, A. Corti, M. Apte, A. Pompella, P. J. Hogg and P. J. Dilda, Mol. Pharm., 2014, 11, 1500–1511 CrossRef CAS PubMed.
- R. Huang, N. Southall, Y. Wang, A. Yasgar, P. Shinn, A. Jadhav, D.-T. Nguyen and C. P. Austin, Sci. Transl. Med., 2011, 3, 80ps16 Search PubMed.
- S. H. Park, J. H. Lee, J. S. Berek and M. C. Hu, Int. J. Oncol., 2014, 45, 1691–1698 CAS.
- C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and M. P. Rigobello, Free Radicals Biol. Med., 2007, 42, 872–881 CrossRef CAS PubMed.
- V. Milacic, D. Fregona and Q. P. Dou, Histol. Histopathol., 2008, 23, 101–108 Search PubMed; T. Gamberi, F. Magherini, T. Fiaschi, I. Landini, L. Massai, E. Valocchia, L. Bianchi, L. Bini, C. Gabbiani, S. Nobili, E. Mini, L. Messori and A. Modesti, Mol. BioSyst., 2015, 6, 1653–1667 RSC.
- S. L. McGovern, E. Caselli, N. Grigorieff and B. K. Shoichet, J. Med. Chem., 2002, 45, 1712–1722 CrossRef CAS PubMed.
- H. Schmidbaur, Chem. Soc. Rev., 1995, 24, 391–400 RSC.
- C. S. Allardyce, R. Bays and N. Thévenaz, Chimia, 2015, 69, 10–16 CrossRef CAS PubMed.
- S. M. Chiu, L. Y. Xue, M. Lam, M. E. Rodriguez, P. Zhang, M. E. Kenney, A. L. Nieminen and N. L. Oleinick, Photochem. Photobiol., 2010, 86, 1161–1173 CrossRef CAS PubMed.
- I. Ashur, R. Goldschmidt, I. Pinkas, Y. Salomon, G. Szewczyk, T. Sarna and A. Scherz, J. Phys. Chem. A, 2009, 113, 8027–8037 CrossRef CAS PubMed.
- D. Kessel, M. Castelli and J. J. Reiners Jr., Photochem. Photobiol., 2002, 76, 314–319 CrossRef CAS PubMed.
- A. E. OConnor, M. M. McGee, Y. Likar, V. Ponomarev, J. J. Callanan, D. F. OShea, A. T. Byrne and W. M. Gallagher, Int. J. Cancer, 2012, 130, 705–715 CrossRef CAS PubMed.
- C. Lottner, R. Knuechel, G. Bernhardt and H. Brunner, Cancer Lett., 2004, 215, 167–177 CrossRef CAS PubMed; C. Lottner, R. Knuechel, G. Bernhardt and H. Brunner, Cancer Lett., 2004, 213, 171–180 CrossRef; J. Mao, Y. Zhang, J. Zhu, C. Zhang and Z. Guo, Chem. Commun., 2009, 908–910 RSC.
- M. Pernot, T. Bastogne, N. P. E. Barry, B. Therrien, G. Koellensperger, S. Hann, V. Reshetov and M. Barberi-Heyob, J. Photochem. Photobiol., B, 2012, 117, 80–89 CrossRef CAS PubMed.
- C. Ceresa, A. Bravin, G. Cavaletti, M. Pellei and C. Santini, Curr. Med. Chem., 2014, 21, 2237–2265 CrossRef CAS PubMed.
- R. W. Orrell, Curr. Opin. Invest. Drugs, 2006, 7, 70–80 CAS.
- M. Patel and B. J. Day, Trends Pharm. Sci., 1999, 20, 359–364 CrossRef CAS PubMed; P. D. Harvey, S. Tasan, C. P. Gros, C. H. Devillers, P. Richard, P. Le Gendre and E. Bodio, Organometallics, 2015, 34, 1218–1227 CrossRef.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS PubMed.
- C. M. Crew, Chem. Biol., 2010, 17, 551–555 CrossRef CAS PubMed; A. K. Arakaki, W. Tian and J. Skolnick, BMC Genomics, 2006, 7, 315 CrossRef PubMed; G. L. Verdine and L. D. Walensky, Clin. Cancer Res., 2007, 13, 7264–7270 CrossRef PubMed.
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drug, 2015, 33, 201–214 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |