
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systems biocatalysis: para-alkenylation of unprotected phenols†
Eduardo
Busto‡
a,
Michaela
Gerstmann
b,
Felix
Tobola
b,
Edmund
Dittmann
b,
Birgit
Wiltschi
*b and
Wolfgang
Kroutil
 *a
*a
aDepartment of Chemistry, NAWI Graz, BioTechMed Graz, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: wolfgang.kroutil@uni-graz.at; Fax: +43 316 380 9840
bAustrian Centre of Industrial Biotechnology (ACIB), Petersgasse 14, 8010 Graz, Austria. E-mail: birgit.wiltschi@acib.at
First published on 13th October 2016
Abstract
Commercially available phenol derivatives were transformed with pyruvate to form a new C–C bond leading to the corresponding para-coumaric acids and only one molecule of water as an innocent side product in buffer. The reaction was catalysed by a biocatalytic system consisting of two enzymatic steps, which were run simultaneously: (i) in the first step catalysed by a tyrosine phenol lyase the C–C coupling of phenol derivatives with pyruvate and ammonia yielded L-tyrosine derivatives, (ii) which were transformed in the second step to the final product via ammonia elimination catalysed by a tyrosine ammonia lyase. The reactions proceeded with exquisite regio- and stereoselectivity yielding just the para-products with the (E)-configuration. The method represents an efficient approach for the direct alkenylation of phenols on a preparative scale (up to 0.6 mmol) affording (E)-para-coumaric acids in excellent isolated yields without requiring chromatographic purification. Co-expression of the involved enzymes in a single host gave access to single catalyst preparation.
1. Introduction
α,β-unsaturated carboxylic acids are central compounds in organic chemistry which are not only widely present in a broad range of biologically active molecules but also exhibit outstanding potential1 to be used as building blocks for organic synthesis.2 Among them, coumaric acid represents a privileged scaffold being part of a broad range of pharmacologically active compounds widely distributed in the plant kingdom. For instance, caffeic acid and specially its phenethyl ester have demonstrated “in vitro” activities such as anti-inflammatory and anti-tumour effects or provided protection against UV light.3 Furthermore, p-coumaric acid, the most abundant isomer of coumaric acid, is known to reduce the risk of stomach cancer by reducing the formation of carcinogenic nitrosamines.4 Related compounds such as ferulic acid have shown remarkable activities against breast and liver cancers.5 Additionally, functionalized derivatives such as chlorogenic acid and cichoric acid have been shown to slow the release of glucose to the bloodstream and inhibit the HIV integrase, respectively.6Traditional methods to prepare p-coumaric acid derivatives are generally based on Knoevenagel7 or Heck8 reactions, thus requiring a functional group (aldehyde or halogen, respectively) in the para-position to the phenolic hydroxyl. On the other hand, biocatalytic approaches are limited to the carboxylation of para-vinylphenol derivatives using carboxylases,9 which require the corresponding p-hydroxy styrene precursors to be prepared using Wittig reactions of the corresponding aldehydes.10 Direct C–H alkenylations of phenols have been reported with (i) Ru-catalysts,11 (ii) Pd-catalysis to access ortho-coumaric acid derivatives,12 and (iii) Fe-containing mesoporous aluminosilicates.13 All direct alkenylation methods lead to ortho products.
2. Results and discussion
We have recently reported a biocatalytic system for the para-vinylation of phenols (Scheme 1a).14 In the present work, the biocatalytic system was adapted for the preparation of p-coumaric acid derivatives from unsubstituted phenols using only pyruvate as a stoichiometric reagent (Scheme 1b), which represents a para-selective alkenylation. This contribution represents the first example of direct para-selective alkenylation, without requiring an auxiliary group at the p-position (Br, CHO, etc.) with respect to the phenolic OH moiety. | ||
| Scheme 1 a) Vinylation of phenols. b) C–H para-alkenylation of phenols leading to p-coumaric acid derivatives. | ||
The cascade15 investigated was designed by combining two enzymatic reaction steps. In the first step of the cascade, the C–C coupling between pyruvate, ammonia and different phenol derivatives 1 should give access to substituted L-tyrosine derivatives 3 (Scheme 2).16 Subsequent elimination of ammonia by a tyrosine ammonia lyase (TAL)17 should give the (E)-p-coumaric acids 4 in the second step. The overall reaction requires only pyruvate and phenols as stoichiometric reagents since ammonia is internally recycled. For the first step the M379V variant of the recombinant tyrosine phenol lyase from Citrobacter freundii was used as a cell-free extract due to its broad scope towards non-natural substrates.16 For the elimination step the recombinant TAL from Rhodobacter sphaeroides gave the best results when employed as an E. coli whole cell catalyst.14
 | ||
| Scheme 2 Two-step cascade for the biocatalytic alkenylation of phenols. TAL: recombinant tyrosine ammonia lyase from Rhodobacter sphaeroides employed as an E. coli whole cell catalyst; TPL: recombinant tyrosine phenol lyase from Citrobacter freundii M379V used as a cell-free extract. | ||
To identify the optimal pH value for the overall biotransformation, the biocatalytic alkenylation of phenol 1a was studied at pH values between pH 8–10 (Fig. 1) since the TPL as well as the TAL gave the highest conversions at alkaline pHs.14 At pH 8 the formation of (E)-4a was observed albeit at a low rate due to the accumulation of L-3a in the reaction (see the ESI† for a detailed profile). This is consistent with the lower activity of the TAL. The highest concentration of (E)-4a was obtained at pH 10 where TPL and TAL also gave the highest conversions in the individual reactions. This is in contrast to the vinylation cascade (Scheme 1A),14 where pH 8 turned out to be optimum due to a compromise with the last additional step, the decarboxylation, which proceeded best at pH 6.
 | ||
| Fig. 1 Conversion of 1a to p-coumaric acid (E)-4a at different pH values. Reaction conditions: 1a (23 mM), pyruvate (46 mM), NH4Cl (180 mM), TPL M379V (5 mg cell-free extract, 1.5 U), TAL (20 mg freeze-dried E. coli/TAL, 0.18 U), buffer (50 mM, potassium phosphate, pH 8, CHES, pH 9–10), PLP (0.04 mM), Et2O (5% v/v), 30 °C, 850 rpm. | ||
Monitoring the alkenylation of 1a over time at pH 10 and 30 °C revealed that under the conditions employed the two steps were well coupled since the concentration of L-3a never exceeded 15% of the total amount of products (Fig. 2). Accumulation of 3a would indicate that elimination is the rate-limiting step of the cascade. The formation of (E)-4a followed a linear profile for the first two hours, then slowed down reaching completion after 24 h when the starting materials and intermediates were below the detection limit. Although all reactions are reversible, the high conversion resulted from the thermodynamic preference for the formation of (E)-4a (−24.0 kcal mol−1 for the transformation of the L-tyrosine binding state in the enzyme to (E)-coumarate + H2N–MIO).17a
 | ||
| Fig. 2 Time course for the alkenylation of 2-fluorophenol (1a) into 3-fluoro-4-hydroxycinnamic acid (E)-4a. Reaction conditions: 1a (23 mM), pyruvate (46 mM), NH4Cl (180 mM), buffer (50 mM, CHES, pH 10), PLP (0.04 mM), TPL M379V (5 mg cell-free extract, 1.5 U in 1 mL total volume), TAL (20 mg freeze-dried E. coli/TAL, 0.18 U in 1 mL total volume), Et2O (5% v/v), 30 °C, 850 rpm. | ||
Next, the scope of the cascade was investigated using a set of 2- and 3-substituted phenols. Phenols bearing halogens (fluoro, chloro and bromo, 1a–c) in the 2-position were efficiently transformed with >99% conversion and perfect selectivity towards the E-isomer (Table 1, entries 1–4). 2-Methylphenol (1d) was also transformed achieving in this case good conversion of the alkenylated product (entry 5). Higher amounts of (E)-4d were formed at longer reaction times (48 h, entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1a–g | 1 [mM] | 1 [%]b | 3 [%]b | 4 [%]b,c |
| a Reaction conditions: phenol 1, pyruvate (2 equiv.), NH4Cl (180 mM), TPL (50 mg cell-free extract, 15 U), TAL (200 mg freeze-dried E. coli/TAL, 1.8 U), CHES buffer (pH 10, 10 mL, 50 mM), PLP (0.04 mM), Et2O (5% v/v), 30 °C, 120 rpm, 24 h. b Determined by reverse-phase HPLC analysis. c Isolated yields are shown in brackets. d Reactions performed on an analytical scale only. e Double amounts of the two biocatalysts were used. f Reaction time: 48 h. | |||||
| 1 | a | 23 | <1 | <1 | >95 |
| 2 | a | 46 | <1 | <1 | >95(96) |
| 3 | b | 23 | <1 | <1 | >95(85) |
| 4 | c | 23 | <1 | <1 | >95(96) |
| 5 | d | 23 | 10 | 20 | 71d |
| 6 | d | 23 | 2 | 15 | 83d |
| 7 | e | 23 | <1 | <1 | >95(88) |
| 8 | f | 23 | 26 | 5 | 69d |
| 9 | g | 23 | <1 | <1 | >95(80) |

The bioalkenylation protocol was also successfully applied for 3-substituted phenols (entries 7–8), observing quantitative conversion of 3-fluorophenol (1e) and high conversion of 3-chlorophenol (1f). Interestingly, a 2,3-disubstituted phenol (1g) was also efficiently transformed into the corresponding p-hydroxycinnamic acid (entry 9). However, phenols bearing polar ortho groups such as in catechol or o-aminophenol were not transformed at all.
Subsequently, para-alkenylation was successfully shown on a semi-preparative scale (0.6 mmol, 46 mM substrate concentration), isolating for instance the fluorinated derivative (E)-4a as a single isomer in 96% isolated yield after a simple extraction protocol without requiring chromatographic purification (entry 2). Semi-preparative biotransformations were also performed for 2-substituted phenols (1b–c), the 3-substituted phenol (1e) as well as for the 2,3-substituted phenol (1g), demonstrating the potential of the present methodology for the preparation of the corresponding alkenylated products in excellent isolated yields (80–96%) and complete stereoselectivity without requiring chromatographic purification steps.
Since the cascade requires two enzymes, the next step was to co-express both enzymes in a single host, which simplifies enzyme production and handling of the catalysts. Co-expression of enzymes in a single cell enables the generation of efficient whole-cell catalysts.18 To identify the most suitable construct for the expression of the two enzymes in E. coli, two bicistronic constructs were designed, wherein in one case the TPL was first in line followed by TAL (construct 1), while in the other case the order is reversed, thus the TAL enzyme comes first (construct 2) (Fig. 3). The TPL and TAL genes were assembled under the control of a strong inducible promoter (T7/lacO). The translation of TAL was driven by the RBS of pET28a(+) and the gene carried an N-terminal hexahistidine-tag. TPL was driven by the RBS of the pET22b(+) vector. The constructs were generated using Gibson isothermal assembly.
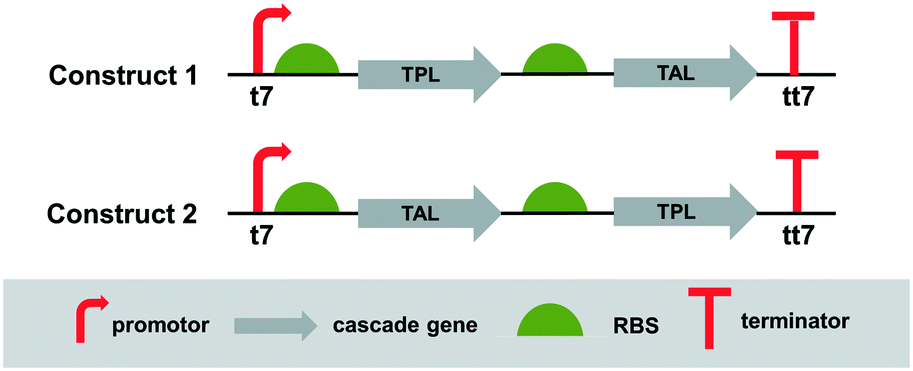 | ||
| Fig. 3 Design of constructs for co-expression of TPL and TAL. t7: IPTG inducible T7/lacO promoter; tt7: T7 terminator. | ||
Initial rates for the overall cascade were measured for freeze-dried cells of E. coli containing the overexpressed enzymes encoded by constructs 1–2. A higher activity was observed for the transformation of 1a to (E)-4a with E. coli(TPL–TAL) (1.2 mU mg−1) compared to E. coli(TAL–TPL) (0.23 mU mg−1). The TAL activities for the single step transformation of 3a into 4a were 1.35 mU mg−1 for E. coli(TPL–TAL) and 0.31 mU mg−1 for E. coli(TAL–TPL). Consequently, the transformation of 1a into (E)-4a was monitored over time for the best performing catalyst, that is, E. coli(TPL–TAL) (Fig. 4). The reaction reached completion after about 19 hours. This may indicate a positive influence of the co-expression in a single cell, because the TAL activity of the E. coli(TPL–TAL) catalyst was limiting (intermediate 3a reached up to 23%). However, only 22% of the units used in the experiment were employed for the profile in Fig. 2 and the reaction only took roughly double the amount of time. Nevertheless, the expression level may be improved for efficient catalyst preparation.
 | ||
| Fig. 4 Time course for the alkenylation of 2-fluorophenol (1a) into 3-fluoro-4-hydroxycinnamic acid (E)-4a employing E. coli(TPL–TAL). Reaction conditions: total volume: 1 mL, 1a (23 mM), pyruvate (46 mM), NH4Cl (180 mM), buffer (50 mM, CHES, pH 10), PLP (0.04 mM), E. coli(TPL–TAL) (30 mg freeze-dried cells, 36 mU), Et2O (5% v/v), 30 °C, 850 rpm. | ||
3. Conclusions
In conclusion, phenols have been efficiently para-alkenylated using a biocatalytic system involving two steps, namely C–C bond formation19 and NH3-elimination. Only pyruvate is required as the stoichiometric reagent. Since only one molecule of water is released in the transformation, the overall reaction possesses an excellent atom economy (82% for (E)-4a–e, 83% for (E)-4b, 86% for (E)-4c and 83% for (E)-4g). p-Alkenylation was successfully performed on a semi-preparative scale at substrate concentrations of 23–46 mM obtaining the alkenylated p-coumaric acid products as a single (E)-isomer in excellent isolated yields (80–96%) without requiring chromatographic purification. By co-expression of the two enzymes in a single host, only a single catalyst preparation may be required for the reaction.4. Experimental section
4.1 General
Chemical reagents were purchased from different commercial sources and used without further purification. Melting points were measured on samples in an open capillary tube and were uncorrected. 1H and 13C-NMR spectra were obtained using a Bruker spectrometer (1H, 300.13 MHz; 13C 75.5 MHz). The chemical shifts are given in delta (δ) and the coupling constants in Hertz (Hz). Mass spectra experiments (MS) were carried out by ESI+ using an Agilent HPLC coupled to a MS.4.2 Single enzyme preparations
Single biocatalysts were prepared as previously described.14 Tyrosine phenol lyase M379V (TPL) from Citrobacter freundii (0.30 U mg−1) was used as a freeze-dried cell-free extract and prepared as previously described.14 Tyrosine ammonia lyase (TAL) from Rhodobacter sphaeroides (9.2 mU mg−1) was used as a whole-cell freeze-dried catalyst.144.3 Co-expression of TPL and TAL
The coding sequences for tyrosine phenol lyase (TPL) from Citrobacter freundii M379V and tyrosine ammonia lyase (TAL) from Rhodobacter sphaeroides were PCR amplified from pET22b(+)-TPL and pET28a(+)-TAL14 using the following primers: TPL with pBP856 (5′-gataacaattccggaattgtgagcggataacaattccggaattcaaggagatatacatatgatgaactatccggc-3′) and pBP857 (5′-gctgcccatggtatatctccttatttaaatttattagatataatcgaagcgcgcggtaaaaaaac-3′) for construct 1; with pBP939 (5′-gctgcaaca gagtccggtttaagctagcaaggagatatacatatgatgaactatccggc-3′) and pBP943 (5′-ggggttatgctagggatcggatccttattagatata atcgaagcgcgcggtaaaaaaac-3′) for construct 2. TAL was amplified with pBP858 (5′-cgcgcgcttcgattatat ctaataaatttaaataaggagatataccatgggcagcagcc-3′) and pBP866 (5′-caaggggttatgctagggatcggatccttaaaccggactctgttgcagcaga tg-3′) for construct 1; and with pBP937 (5′-gataacaattccggaattgtgagcggataacaattccggaattcaaggagatataccatgggcagcagccatc-3′) and pBP938 (5′-atagttcatcatatgtatatctccttgctagcttaaaccggactctgttgcagcagatg-3′) for construct 2. The PCR fragments included the RBSs of pET28a(+) (TAL) and pET22b(+) (TPL) and TAL carried an N-terminal hexahistidine-tag.14 pCAS1 (ref. 18c) was linearized with EcoRI and BamHI and dephosphorylated using alkaline phosphatase (Thermo Fisher Scientific, Waltham, MA). All PCR and DNA fragments were gel purified (Promega, Fitchburg, WI) before the TPL and TAL genes were inserted into pCAS1 by Gibson isothermal assembly.20 The resulting constructs 1 and 2 were sequence verified.For the co-expression of the two cascade enzymes, E. coli BL21-Gold(DE3) (Agilent Technologies, Santa Clara, CA) was transformed with constructs 1 and 2. The cells were grown in LB medium (Carl Roth GmbH, Karlsruhe, Germany) until they reached a density of OD600 ∼ 0.6–0.8. The expression of the cascade enzymes was induced with 1 mM IPTG (Sigma-Aldrich, St. Louis, MO) and the expression was performed at 28 °C overnight. The cells were then harvested by low speed centrifugation (2800 × g, 4 °C, 20 min), washed once in PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4) and the pellets were stored at −20 °C until lyophilisation in 15 mL of PBS overnight.
4.4 Overall activities for E. coli(TPL–TAL) and E. coli(TAL–TPL)
For better comparison of constructs 1 and 2, the activity for the transformation of the corresponding phenol 1a into the cinnamic acid derivative (E)-4a was determined by measuring the initial rate by HPLC. One unit of activity was defined as the amount of catalyst prepared that catalysed the formation of 1 μmol of 3-fluoro-4-hydroxycinnamic acid (4a) per minute under the following conditions: 30 °C, 850 rpm, pH 10. The assay mixture contained 2-fluorophenol (1a, 23 mM), pyruvate (46 mM), NH4Cl (180 mM) and E. coli whole cells overexpressing TPL and TAL (30 mg). The reaction activities were determined corresponding to 1.2 mU mg−1E. coli whole cells for construct 1 and 0.23 mU mg−1E. coli whole cells for construct 2.4.5 Biotransformations on analytical scale
Recombinant TPL M379V from C. freundii (1.5 U, 5 mg freeze-dried cell-free extract) and TAL from R. sphaeroides (0.18 U, 20 mg freeze-dried E. coli/TAL cells) were rehydrated in a CHES buffer (50 mM, 180 mM NH4Cl, 0.04 mM PLP, pH 1, 1 mL) for 10 min at 30 °C and 120 rpm. The corresponding phenols 1a–g (0.023 mmol, 23 mM) and pyruvate (0.046 mmol, 46 mM; 11.5 μL of 4 M stock solution in CHES buffer pH 10) were added to the mixture and the reaction was incubated at 21 °C and 800 rpm for 24 h. Reactions were quenched with a MeCN/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution containing 0.1% TFA (1 mL), centrifuged to remove the cells and the supernatant was injected in the HPLC system for conversion measurement.
:1 solution containing 0.1% TFA (1 mL), centrifuged to remove the cells and the supernatant was injected in the HPLC system for conversion measurement.
4.6 Biotransformations on preparative scale
Recombinant TPL M379V from C. freundii (15 U, 50 mg freeze-dried cell-free extract) and TAL from R. sphaeroides (3.68 U, 400 mg freeze-dried E. coli/TAL cells) were rehydrated in a CHES buffer (50 mM, 180 mM NH4Cl, 0.04 mM PLP, pH 10, 10 mL) for 10 min at 30 °C and 120 rpm. Then the phenol (0.23 mmol, 23 mM) and pyruvate (101.2 mg, 0.46 mmol, 46 mM) were added to the mixture and the reaction was incubated for 24 h at 21 °C and 170 rpm. The reaction was stopped with an aqueous saturated NH4Cl solution (5 mL), acidified with 2 M HCl (1 mL) and extracted with EtOAc (3 × 15 mL). The combined organic phases were dried (Na2SO4) and the solvent was evaporated under reduced pressure. The crude product was washed with n-heptane (3 × 5 mL) obtaining the corresponding p-hydroxycinnamic acids as white solids (80–96%).4.7 Reaction scale and isolated yields
1a: (51 mg, 0.46 mmol, 46 mM); isolated yield: (E)-4a: 80.6 mg, 96%. 1b: (30 mg, 0.23 mmol, 23 mM); isolated yield: (E)-4b: (39 mg, 85%). 1c: (40 mg, 0.23 mmol, 23 mM); isolated yield: (E)-4c: (54 mg, 96%). 1e: (26 mg, 0.23 mmol, 23 mM); isolated yield: (E)-4e: (37 mg, 88%). 1g: (30 mg, 0.23 mmol, 0.23 mM); (E)-4g: isolated yield: (36 mg, 80%).4.8 Analytics
Conversions for substrates were determined with an Agilent chromatograph UV detector at different wavelengths using a Luna C18 column (25 cm × 4.6 mm I.D.). Sample preparation: a MeCN/H2O solution (1 mL, 1:1) containing 0.1% TFA was added to an aliquot of the reaction solution (1 mL). The protein was removed by centrifugation and the solution was filtered through VIVAspin membrane polyethersulfone filters. The resulting solution was analysed by HPLC under the conditions stated below (see the ESI† for retention times). Column: Luna C18 5 μm; flow: 1 mL min−1; temperature: 30 °C; gradient: from 100% H2O (0.1% TFA) to 100% MeCN (0.1% TFA) in 22 min. Wavelength: 280 nm. The relative amount of the species was determined by correcting the value with the response factor of each compound.
Acknowledgements
E. B. received funding from the European Commission through a Marie Curie Actions Intra European Fellowship (IEF) as part of the “BIOCASCADE” project (FP7-PEOPLE-2011-PIEF-GA-2011-298030). M. G., B. W. and W. K. have been supported by the Austrian BMWFJ, BMVIT, FFG, SFG, Standortagentur Tirol and ZIT through the Austrian FFG-COMET-Funding Program. COST Action CM1303 “Systems Biocatalysis” is acknowledged.Notes and references
- (a) H. Röller, K. H. Dahm, C. C. Sweely and B. M. Trost, Angew. Chem., Int. Ed. Engl., 1967, 6, 179–180 CrossRef; (b) A. T. Fuller, G. Mellows, M. Woolford, G. T. Banks, K. D. Barrow and E. B. Chain, Nature, 1971, 234, 416–417 CrossRef CAS PubMed.
- (a) G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491–3495 CrossRef CAS PubMed; (b) X.-Y. Chen, Z.-H. Gao, C.-Y. Song, C.-L. Zhang, Z.-X. Wang and S. Ye, Angew. Chem., Int. Ed., 2014, 53, 11611–11615 CrossRef CAS PubMed.
- (a) M. Demestre, S. M. Messerli, N. Celli, M. Shahossini, L. Kluwe, V. Mautner and H. Maruta, Phytother. Res., 2009, 23, 226–230 CrossRef CAS PubMed; (b) Z. Orban, M. Mitsiades, T. R. Burke Jr., M. Tsokos and G. P. Chouros, NeuroImmunoModulation, 2000, 7, 99–105 CrossRef CAS PubMed; (c) T.-W. Chung, S.-K. Moon, Y.-C. Chang, J.-H. Ko, Y.-C. Lee, G. Cho, S.-H. Kim, J.-G. Kim and C.-H. Kim, FASEB J., 2004, 18, 1670–1681 CrossRef CAS PubMed; (d) V. Staniforth, L.-T. Chung and N.-S. Yang, Carcinogenesis, 2006, 27, 1803–1811 CrossRef CAS PubMed; (e) K. Natajaran, S. Singh, T. R. Burke Jr., D. Bruberger and B. B. Aggarwal, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9090–9095 CrossRef.
- L. R. Ferguson, S.-T. Zhu and P. J. Harris, Mol. Nutr. Food Res., 2005, 49, 585–593 CAS.
- (a) Y. S. Lee, Arch. Pharmacal Res., 2005, 28, 1183–1189 CrossRef CAS; (b) M. Kampa, V. I. Alexaki, G. Notas, A. P. Nifli, A. Nistikaki, A. Hatzoglou, E. Bakogeorgou, E. Kowimtzoglou, G. Blekces and D. Boskou, Breast Cancer Res., 2004, 6, 63–74 CrossRef PubMed.
- (a) K. L. Johnston, M. N. Clifford and L. M. Morgan, Am. J. Clin. Nutr., 2003, 78, 728–733 CAS; (b) J. Y. Lee, K. J. Yoon and Y. S. Lee, Bioorg. Med. Chem. Lett., 2003, 13, 4331–4334 CrossRef CAS PubMed.
- (a) N. Sharma, U. K. Sharma, R. Kumar, N. Katoch, R. Kumar and A. K. Sinha, Adv. Synth. Catal., 2011, 353, 871–878 CrossRef CAS; (b) V. Percec, M. Peterca, M. J. Sienkowska, M. A. Ilies, E. Aqad, J. Smidkral and P. A. Heiney, J. Am. Chem. Soc., 2006, 128, 3324–3334 CrossRef CAS PubMed.
- A. Shard, N. Sharma, B. Bharti, S. Dadhwal, R. Kumar and A. K. Sinha, Angew. Chem., Int. Ed., 2012, 51, 12250–12253 CrossRef CAS PubMed.
- (a) C. Wuensch, T. Pavkov-Keller, G. Steinkellner, J. Gross, M. Fuchs, A. Hromic, A. Lyskowski, K. Fauland, K. Gruber, S. M. Glueck and K. Faber, Adv. Synth. Catal., 2015, 357, 1909–1918 CrossRef CAS PubMed; (b) C. Wuensch, S. M. Glueck, J. Gross, D. Koszelewski, M. Schober and K. Faber, Org. Lett., 2012, 14, 1974–1977 CrossRef CAS PubMed.
- C. Wuensch, J. Gross, G. Steinkellner, K. Gruber, S. M. Glueck and K. Faber, Angew. Chem., Int. Ed., 2013, 52, 2293–2297 CrossRef CAS PubMed.
- (a) D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325–7328 CrossRef CAS PubMed; (b) M. C. Reddy and M. Jeganmohan, Eur. J. Org. Chem., 2013, 1150–1157 CrossRef CAS.
- X.-S. Zhang, Z.-W. Li and Z.-J. Shi, Org. Chem. Front., 2014, 1, 44–49 RSC.
- S. Haldar and S. Koner, Beilstein J. Org. Chem., 2013, 9, 49–55 CrossRef CAS PubMed.
- E. Busto, R. C. Simon and W. Kroutil, Angew. Chem., Int. Ed., 2015, 54, 10899–10902 CrossRef CAS PubMed.
- (a) W.-D. Fessner, New Biotechnol., 2015, 32, 658–664 CrossRef CAS PubMed; (b) R. Sigrist, B. Z. d. Costa, A. J. Marsaioli and L. G. de Oliveira, Biotechnol. Adv., 2015, 33, 394–411 CrossRef CAS PubMed; (c) V. Köhler and N. J. Turner, Chem. Commun., 2015, 51, 450–464 RSC; (d) J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC; (e) J.-K. Guterl and V. Sieber, Eng. Life Sci., 2013, 13, 4–18 CrossRef CAS; (f) E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS; (g) J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed.
- (a) B. Seisser, R. Zinkl, K. Gruber, F. Kaufmann, A. Hafner and W. Kroutil, Adv. Synth. Catal., 2010, 352, 731–736 CrossRef CAS; (b) K. Kim and P. A. Code, Bioorg. Med. Chem. Lett., 1999, 9, 1205–1208 CrossRef CAS PubMed; (c) R. Stephen-Phillips, K. Ravichandran and R. L. Von Tersch, Enzyme Microb. Technol., 1989, 11, 80–83 CrossRef.
- (a) S. Pilbák, O. Farkas and L. Poppe, Chem. – Eur. J., 2012, 18, 7793–7802 CrossRef PubMed; (b) J. A. Kyndt, T. E. Meyer, M. A. Cusanovic and J. J. Von Beeumen, FEBS Lett., 2002, 512, 240–244 CrossRef CAS PubMed.
- (a) P. Both, H. Busch, P. P. Kelly, F. G. Mutti, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2016, 55, 1511–1513 CrossRef CAS PubMed; (b) Z. Xu, Z. Xu, B. Tang, S. Li, J. Gao, B. Chi and H. Xu, Biochem. Eng. J., 2016, 109, 28–34 CrossRef CAS; (c) G. Gourinchas, E. Busto, M. Killinger, N. Richter, B. Wiltschi and W. Kroutil, Chem. Commun., 2015, 51, 2828–2831 RSC; (d) S.-Y. Chen, C.-X. Yang, J.-P. Wu, G. Xu and L.-R. Yang, Adv. Synth. Catal., 2013, 355, 3179–3190 CrossRef CAS; (e) E. Lorenz, S. Klatte and V. F. Wendisch, J. Biotechnol., 2013, 168, 289–294 CrossRef CAS PubMed; (f) M. Schrewe, N. Ladkau, B. Bühler and A. Schmid, Adv. Synth. Catal., 2013, 355, 1693–1697 CrossRef CAS.
- (a) For selected reviews on biocatalytic C–C bond formation see: N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS PubMed; (b) Science of Synthesis: Biocatalysis in Organic Synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, ch. 2.1 and 2.2, pp. 1–212 Search PubMed; (c) Y. Miao, M. Rahimi, E. M. Geertsema and G. J. Poelarends, Curr. Opin. Chem. Biol., 2015, 25, 115–123 CrossRef CAS PubMed; (d) B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed; (e) M. Müller, Adv. Synth. Catal., 2014, 354, 3161–3174 CrossRef; (f) V. Resch, J. H. Schrittwieser, E. Siirola and W. Kroutil, Curr. Opin. Biotechnol., 2011, 22, 793–799 CrossRef PubMed; (g) M. Pohl and A. Liese, Industrial Processes Using Lyases for C-C, C-N and C-O Bond Formation, in Biocatalysis in the Pharmaceutical and Biotechnology Industry, ed. R. N. Patel, CRC Press, 2007, pp. 661–676 Search PubMed.
- D. G. Gibson, L. Young, R.-Y. Chuang, J. C. Venter, C. A. Hutchison III and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy01947a |
| ‡ Present address: Department of Organic Chemistry I, Universidad Complutense de Madrid, 28040 Madrid (Spain). |
| This journal is © The Royal Society of Chemistry 2016 |