
Crude oil to chemicals: light olefins from crude oil
A.
Corma
*a,
E.
Corresa
a,
Y.
Mathieu
a,
L.
Sauvanaud
a,
S.
Al-Bogami
b,
M. S.
Al-Ghrami
b and
A.
Bourane
b
aInstituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos s/n, 46022 Valencia, Spain. E-mail: acorma@itq.upv.es
bResearch and Development Center, Saudi Aramco, P.O. Box 62, Dhahran 31311, Saudi Arabia
First published on 26th September 2016
Abstract
The possibility to fulfill the increasing market demand and producers' needs in processing crude oil, a cheap and universally available feedstock, to produce petrochemicals appears to be a very attractive strategy. Indeed, many petrochemicals are produced as side streams during crude oil refining, which primary goal remains transportation fuel production. Availability of some critical feedstocks may then depend on local refining policy. In order to improve flexibility, it has been proposed to directly crack crude oil to produce petrochemicals, in particular light olefins (ethylene, propylene, butenes), using technologies derived from fluid catalytic cracking. This paper attempts to review the main research works done on the topic in the literature in the last five decades, focussing on process as well as catalyst technology, with a special interest for fluid catalytic cracking (FCC) based technology that can be used towards maximizing chemicals from crude oil. Factors investigated include use of severe cracking conditions, on-purpose additives (from ZSM5 to more exotic, metal doped additives), recycle streams and multiple riser systems.
1. Introduction
Crude oil refineries are generally oriented to the production of transportation fuels (gasoline, diesel and kerosene), with a minor but economically important side production of building blocks for the petrochemical industry, mainly light olefins (ethylene, propylene, butenes and butadiene) and BTX. These are the most common petrochemical feedstocks and their markets are expanding.1 They may be produced as side products of a fuel process (for example benzene from catalytic reforming). Alternatively, they can be produced from a cut with low value as fuel in a dedicated process, for example naphtha in steam crackers.2 Availability of these petrochemicals is dependent on the refining business. Therefore, it may be sound from the point of view of the petrochemical market to directly produce these basic intermediates from a universally available feedstock.Crude oil appears to be an ideal candidate, being cheaply available everywhere and compatible with a petrochemical business. While direct steam cracking of crude oil has been attempted, coils coking and limited product flexibility are major issues.3,4 Still, steam cracking processes with careful oil vaporization have been designed for this purpose5,6 and ExxonMobil has claimed to build a steam cracker using directly certain crude oils as feed. Several dedicated processes were also developed decades ago to directly crack crude oil using thermal processes with beds of different kind of particles, some of them inspired from fluid catalytic cracking technology, with reactor-solid circulation.7,8 Yet they were ethylene oriented, thus working at temperature generally over 700 °C, and did not compete well with naphtha steam cracking.
From the point of view of market demand, propylene production is creating new opportunities because:
• Propylene demand is growing faster than ethylene demand.
• Steam crackers have a limited ethylene to propylene ratio, usually not greater than 0.6.
• Many steam crackers are switching to cheap ethane, shrinking propylene production.
• On-purpose processes, such as propane dehydrogenation, metathesis, methanol to olefins, have their economics dictated by the feedstock price and availability, therefore limiting their applicability to niche markets.
Fluid catalytic cracking (FCC) has been the second major supplier of propylene after steam cracking, and has proven high flexibility in feedstock and product slate. Crude oil cracking in a FCC process may appear as an ideal candidate to fulfill petrochemical producer's needs. Fluid catalytic cracking units usually run on vacuum distillation products, namely vacuum gas oil (VGO) and vacuum residue (VR). Also, atmospheric residue (AR) can be used as a feedstock for FCC. In some small refineries it was shown that the FCC could substitute the main distillation unit, separating and converting the heavy part of the crude oil all in once.9 Problems associated with heavy material or metals in crude oil are readily addressed by Resid FCC (RFCC) technology (which treats, precisely, the heaviest part of the crude). Lighter fractions of the crude, especially the paraffinic naphtha, will crack to a lower extent under traditional FCC conditions.10 This problem has also been studied by most of the refiners with the aim of increasing propylene (and ethylene) yield in the FCC unit. All the technologies developed to enhance olefin yield in FCC are of high interest for converting crude to petrochemicals. Such a technology may probably be based on a conversion unit which can handle the heavy fractions of the crude oil, converting it partially to light olefins and reducing the amount of heavy products to a minimum. A modified fluid catalytic cracking process would be an ideal candidate. Others units may also be added to complement the conversion unit, such as steam cracking, to crack cleaner and lighter fractions into light olefins.
This review examines, in section 2, the technologies and refining strategies that may be used in a new kind of petrorefinery dedicated to enhance light olefins production from traditional refineries. After considering early thermal cracking intents of cracking crude oil in fluid beds (section 3), we will focus in section 4 on optimized and adapted conventional fluid catalytic cracking technology (process, catalyst, operation) and how it can be used as a petrochemicals oriented crude processing unit. Finally, a detailed overview of the commercialized FCC processes dedicated to maximize light olefins is given in section 5.
2. Refining strategy to maximize light olefins from crude oil
Direct processing of crude oil into small olefins was recognized early as an option to decrease costs in production of ethylene but also, be less dependent from refinery streams (for example naphtha) and general policy towards fuels. The main process to produce petrochemicals is the well-known steam cracking process that may be designed to handle feedstocks from ethane to naphthas or even gas oils. Attempts to process directly crude oil in steam crackers was however not successful due to fouling issues by coke. Thus, a number of processes or strategies have been proposed that suggest conditioning the feed (crude oil) by rejecting the heaviest part and contaminants of crude and upgrading the rest before feeding it to the steam cracker. The rejected part can be used as fuel to bring heat to the process or upgraded in a separate process.In the process described below in Fig. 1A, a raw separation of the crude oil is performed by partial vaporization at a temperature of 480–540 °C. The lighter vaporized fraction is then steam cracked under severe conditions (790–840 °C, 1450–1550 °F), while the heavier part remains liquid in the separation tank. This heavier part, if fed to the steam cracker, would produce significant amounts of coke on the walls of the radiant section (i.e. high temperature coil). A suitable device (distillation, packing, etc.) is used to knock out entrained liquid droplets. The liquid droplets are then contacted with steam introduced from the bottom part of the vaporizer at temperatures up to 700 °C (1300 °F) so that the heavy part of the crude oil can be mildly steam cracked. The coke formed during the process deposits on a packing and can be burned later. Packing may be used to enhance steam/liquid contact and/or distribution of oil across the reactor, and favours vaporization of the heavy crude oil fraction. The liquid falling through the packing finds increasingly hotter steam and increased steam to oil ratio. This favours vaporization, and the heavier parts that are more resistant to vaporization will finally be contacted with vapour at a temperature high enough to (mildly) crack the heavier fraction. Lighter products are then vaporized. Steam to oil ratio in the mild cracking section is preferably high. Globally, in the vaporization section, steam to oil ratio is 0.3/1 to 5/1, preferably 1/1. Steam enters the vaporization section at a temperature of 538–704 °C (1000–1300 °F). Contrary to prior practice, where the hydrocarbons are usually passed from the preheater to the hotter section as fast as possible, the vaporization reactor can be seen as a trap for the heavier components, which are eliminated through mild cracking, so that only light components with low coking tendency are fed to the radiant, high temperature zone. The non-vaporized material that formed coke on the packing of the vaporization reaction can be burned by conventional steam/air decoking performed during a normal furnace decoking cycle. Preheating the crude oil is also performed below 350 °C, a temperature notably lower than in a traditional steam cracker to avoid fouling, before loading to the vaporization section.
 | ||
| Fig. 1 Refining strategies to maximize light olefins from crude oil. (A) Process for steam cracking of crude oil with controlled vaporization.6 (B) Processing scheme combining pre-treatment of crude, steam cracking and catalytic cracking of the heavy fraction.21 (C) Steam cracking and bottoms upgrading process combination for petrochemical refinery.23 | ||
In addition to the packing, a catalyst bed may be disposed at the bottom of the vaporizer to enhance cracking, (Fig. 1), helping to remove metals such as Ni, Fe, V and trap non-vaporizable material such as asphaltenes. Materials such as alumina, silica–alumina, molecular sieves, and natural clays may be used. Catalyst may also be supported on random or structured supports (packing). Hydrogen can also be fed to the system in order to reduce coking/fouling.6 It has been also proposed to assist the mild cracking of the non-vaporized, heavy oil through controlled cavitation of a recycle pump at the bottom of the vaporizer. Implosion of cavitation bubbles provides for additional heat that helps vaporizing the remaining heavy fraction. As the heat is provided locally within the fluid and not through a hot wall, this minimizes coke formation on the wall. Claims of 97 wt% vaporization of a Sahara blend crude assisted with steam at 704 °C were reported.11 Some yields obtained with an Alaskan crude oil (1.2 CCR, API 39.2, 0.27 wt% sulfur) are reported in Table 1.12
| Yield (%) | ||
|---|---|---|
| SC temperature/°C | 829 | 843 |
| Hydrogen | 0.6 | 0.7 |
| Methane | 8.9 | 9.3 |
| Ethylene | 19.3 | 20.4 |
| Acetylene | 0.2 | 0.3 |
| Ethane | 2.6 | 2.4 |
| Propylene | 12.2 | 12.1 |
| Propane | 0.7 | 0.6 |
| Propadiene | 0.5 | 0.5 |
| Butadiene | 4.7 | 4.7 |
| Other C4 | 5.7 | 5.1 |
| C5+ | 44.6 | 43.9 |
Some of the non-vaporized heavy liquid may be withdrawn from the process and treated elsewhere.13 A gas condensate may also be used to dilute the crude oil and facilitate vaporization.14
Processing crude oil in steam cracking has been covered in research efforts of many companies.15 The crude is vaporized in two steps by passing into the convection zone of the cracking furnace, then separating vapors from liquids in a flash drum. Liquid droplets in vaporized crude oil, which may contain materials with high coking tendency, have to be carefully removed from the hydrocarbon/steam gas mixture.
Coalescence of these droplets is promoted using an expander or a series of expanders to reduce gas flow and subjecting the gas flow to a centrifugal force.16 The vaporized fraction is then fed to the steam cracker furnace.
The crude oil is then fractionated into naphtha (<220 °C AET) then gas oil (220–600 °C AET), which are cracked separately under optimized conditions. The non-vaporized residue is withdrawn from the process, avoiding coking issues. A recent announcement was made for the imminent start of a 1 Mtpy crude oil steam cracking unit at EXXONMOBIL Jurong Island Petrochemical complex in Singapore.
Shell also patented a similar technology.17 A crude oil steam mixture is preheated to at least 375 °C, more preferably 415 °C and the preheater wall is maintained wet to inhibit coking. A specially designed vapor/liquid separator that creates a swirl at the upper inlet is used to remove the non-vaporized part from the gas stream. The centrifuge effect created at the upper inlet forces the liquid droplets against the wall of the separator, and liquid further flows downwards in a thin film on the wall. This allows maintaining the gas stream hotter than in conventional flash drum and minimizes coking on the wall.
Saudi Aramco patented a number of configurations to pretreat the crude oil before steam cracking.18 The crude oil may be hydrotreated and/or solvent-deasphalted in order to produce a highly paraffinic, deasphalted and demetallized stream. Then, the upgraded stream can be further steam-cracked to produce C2–C4 olefins and BTX with an acceptable rate of coke formation. Both processes may be carried out under usual operating conditions with known technology, it being an advantage that the crude oil is easier to treat than heavier feeds such as atmospheric resid. Finally, the highly upgraded stream is steam cracked at a temperature of 400–900 °C, 0.3–2 steam/oil ratio and 0.05–2 s residence time.18c Depending on crude oil quality, the hydroprocessing step may be bypassed.18a The deasphalting step may be bypassed too if the heavier part of the crude oil is separated, feeding only the light fraction into hydroprocessing followed by steam cracking.19 The cut point for the separation can be, for example, 540 °C AET, so that the leftovers are compatible with residue fuel oil blend. Solvent deasphalting may also be carried out before the hydrotreating step.18b Heavy fuel oil from the pyrolysis (steam cracking) step may be blended with the asphalt from DAO.
In the scheme that considers splitting the crude oil at any point before or after pre-treatment, the heavy fraction can then be upgraded in a separate, dedicated process to yield more olefins. This process can be, for example, an FCC.20 Pyrolytic fuel oil (C10+) from the steam cracker may also be recycled for catalytic cracking. In this particular configuration, the catalytic cracker is run in a mode that favours light olefins and aromatics, that can be called high severity FCC, as represented in Fig. 1B. Derived from FCC technology, the processing temperature is higher, in the 590–620 °C range and catalyst circulation in the reactor has been inversed to a downflow, allowing the use of higher catalyst to oil ratio (8–20) as well as shorter gas residence time (0.2–0.7 s) than conventional risers.21,22 Different fractions can be fed at different points in the cracking reactor to optimize yields. ZSM-5 (or equivalent) additive amount range is indicated at 30–60 wt%, a level far above the traditional blending rate in FCC.
In a similar way, it has been proposed to combine a steam cracking process for the light fraction of a crude oil and a bottoms conversion process, in this case catalytic pyrolysis, to maximize the output of petrochemicals, as depicted in Fig. 1C.23
Catalytic pyrolysis processes will be detailed in the section dedicated to FCC technology. The crude oil may be hydroprocessed upstream to enhance the performance of the complex, as both steam cracker and catalytic converter will perform better towards olefins with more paraffinic feedstocks.
A further development would be processing the whole crude oil directly in a catalytic converter derived from FCC technology, simplifying the processing complex to a single unit. Catalytic cracking offers the possibility to process the crude oil without or with minimal pretreatment (which is a clear advantage of FCC process over steam cracking or hydroprocessing). Indeed, the FCC process has treated for decades the heavier part of the crude oil as main upgrading technology. Conversion of the lighter, paraffinic fraction of the crude oil may nevertheless require substantially different operating conditions than the heavier fraction, so it may be an advantage to split the crude in at least two fractions.24 This may be combined with the use of a specially designed process, for example two downer reactors in parallel with a common regenerator. A radical, one reactor new design was also proposed, taking advantage of the low coking tendency of light crude oils, so that longer catalyst residence time can be afforded as in older, fluid bed FCC units.25 A downwards moving bed is used, presenting a large temperature gradient, while oil is injected at the bottom and flows upwards, being cracked at increasing temperatures. Maintaining a temperature gradient aims at limited mixing of the catalyst bed, hence the concept of a moving bed rather than a fluid bed. Separate cracking sections may also be used. Steam is used to carry hydrocarbons through the system and maintain fluidization of the catalyst. The temperature profile is typically 350–750 °C, producing short olefins and C6–C8 aromatics in a 2–20 weight ratio.
3. Direct crude processing for olefins in dedicated equipment: an old history
3.1. Former thermal cracking attempts based on fluidized bed of particles
| Licensor | BASF | BASF | Chiyoda chemical | UBE | Lurgi | Gulf/S&W |
|---|---|---|---|---|---|---|
| Process/bed type | FB, 1 reactor | FB, reactor – regenerator | Fluid bed | Jet flow | Fluid bed | Fluid bed |
| Crude oil | Minas | Minas | Khafji | Minas | Irak | n/a |
| Heat supply | Crude partial combustion | Coke burning | Coke burning | Crude partial combustion | Coke burning | Coke burning |
| Particles in bed | Coke | Inorganic oxide | Coke | Inorganic oxide | Sand | Coke |
| Temperature/°C | 725 | 760 | 850 | 840 | 760 | 750 |
| Yields (wt%) | ||||||
| C2–C4 olefins | 41.5 | 41.5 | 37.6 | 47.8 | 41.6 | n/a |
| Ethylene | 23 | 25 | 26.8 | 28.1 | 23.1 | 22.5 |
| Propylene | 12.5 | 11.2 | 5.8 | 11.3 | 12.8 | 13.9 |
(a) Fluidized bed process. In this first implementation, coke particles were used as heat carrier. Heat generation and crude oil cracking were carried out in the same fluidized bed. In the lower part, oxygen was introduced to partially burn the coke. Combustion is a fast reaction, so all oxygen was consumed 1 to 2 meters above the injection grid. In the upper part of the fluid bed, crude oil as well as recycled heavy oil were injected with steam. Some catalytic material may be used in small amounts for controlling emissions, much like in the FCC regenerator. One problem of the process was to maintain the correct amount of coke particles with appropriate size and shape generated within the process, independently of the crude oil employed. So, careful control of coke combustion was needed. Fluidization ensured good temperature homogeneity in the bed between the upper (crude cracking) and lower part (combustion) despite the huge heat requirement. Yields up to 40 wt% of C2–C4 olefins were achieved with several crude oils at processing temperatures of 725–740 °C, with 20–24 wt% of ethylene (Table 3A). If necessary, coke excess could be recovered, as well as a naphthalene fraction from the light pyrolysis oil. A commercial unit producing 40
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tpy of ethylene was run for several years in Germany.
000 tpy of ethylene was run for several years in Germany.
| (A) BASF fluid bed process | ||||
|---|---|---|---|---|
| Feedstock | Crude oil | Heavy fuel oil | ||
| Minas | Bahia | Lybian | ||
| Cracking temperature/°C | 725 | 740 | 740 | 740 |
| Product yields (wt%) | ||||
| Ethylene | 23.0 | 23.5 | 20.6 | 15.3 |
| Propylene | 12.5 | 11.8 | 11.6 | 9.3 |
| Butenes + butadiene | 6.0 | 5.4 | 4.5 | 3.8 |
| Ethane | 4.5 | 4.6 | 4.6 | 4.7 |
| Pyrolysis light oil | 14 | 14.5 | 16.6 | 14.2 |
| (B) BASF fluid flow process | |||
|---|---|---|---|
| Feedstock | Crude oil | Heavy fuel oil | |
| Minas | Lybian | ||
| Cracking temperature/°C | 760 | 760 | 790 |
| Product yields (wt%) | |||
| Ethylene | 25.0 | 22.0 | 30.0 |
| Propylene | 11.2 | 10.5 | 9.8 |
| Butenes + butadiene | 5.3 | 5.8 | 5.1 |
| Ethane | 4.3 | 3.7 | 2.8 |
| Pyrolysis light oil | 17.5 | 22.5 | 23.0 |
(b) Fluid flow process. A second process was developed with a design similar to that of FCC. Instead of coke particles, an inorganic oxide material is used as heat carrier, and this one is in constant circulation between reactor and regenerator vessels. Riser technology was still to be developed, and the necessity of maintaining short gas residence time to avoid recracking led to the original reactor design. High gas velocity and a coarse heat carrier (0.3–2 mm) were used in a reactor with an unusual height to diameter ratio of less than one. Free space above the bed was minimized. An upper disengaging zone was placed above the reactor to minimize particle entrainment and rapidly quench the cracked gas. The coke and soot formed in the cracking process were then burnt in the regenerator, together with the recycled heavy oils, at temperatures above 900 °C. Solid recycle rate to crude oil input of 10–15 were reported. Yields were similar to the fluid bed process, with slightly higher ethylene yields due to a higher operating temperature (Table 3B). While the fluidized bed process was simpler in design (one reactor, no catalyst transfer), it required a larger reactor than the fluid flow process. The gaseous products were contaminated with CO and CO2 from the coke combustion. Also, it yielded more by-products, which may have been used to produce syngas to feed an ammonia plant. By the contrary, the Fluid Flow process could be run at higher temperature, maximizing ethylene yield.
:1 steam to oil ratio depending on the crude. More paraffinic crudes tend to give more light olefins. Propylene yields of 7–13 wt% were obtained with crudes at 800 °C, maximum yields are obtained at shortest gas residence time. Yield pattern with a Kuwait crude are summarized in Table 4.
:1 steam to oil ratio28
| Feedstock | Cracking temperature | ||
|---|---|---|---|
| 750 °C | 800 °C | 850 °C | |
| Product yields (wt%) | |||
| Methane | 11.0 | 12.8 | 15.0 |
| Ethane | 3.3 | 2.7 | 2.0 |
| Ethylene | 19.7 | 24.7 | 26.8 |
| Propylene | 15.2 | 11.2 | 5.8 |
| Butenes | 4.8 | 2.7 | 1.8 |
| Butadiene | 5.8 | 4.9 | 3.2 |
| Aromatics | 15.7 | 13.0 | 21.8 |
| Other liquids | 12.5 | 13.3 | 11.4 |
| Coke | 7.4 | 7.7 | 8.9 |
Cracked gas from the reactor was quenched with water or gasoline. The quencher was a fluid bed of particles above the jet flow reactor, maintained at 350–400 °C. Coke formed on the solid particles in this section was burned in a regenerator at 800–900 °C, cooled down and returned to the quencher. Olefin production cost was estimated as 25–30% cheaper than with a naphtha steam cracker. Operating at very high temperatures, ethylene yields and total light olefins close to 30 wt% and 50 wt% respectively, could be obtained (Table 5).
| Product yields (wt%) | |
|---|---|
| Ethylene | 28.1 |
| Propylene | 11.3 |
| Butenes + butadiene | 8.4 |
| Pyrolysis gasoline | 12.2 |
| Heavy oil | 4.4 |
After heating at 350–400 °C and mixing with waste heat steam, hydrocarbons were contacted with sand at a temperature of 700–850 °C depending on the feedstock and the desired propylene to ethylene ratio. The fluidized bed was maintained at a narrow range temperature, and the gas residence time was set at 0.3–0.5 s. Cracked gas was then separated from sand by cyclones, and then cooled to 150 °C. Using feeds heavier than naphtha required some improvements: oil washes were performed in the heat recovery system to minimize fouling, while reactor effluent was quenched with gasoline to minimize secondary reactions that yields more aromatics at the expense of olefins. Sand was fed at a temperature 100–150 °C higher than the reactor temperature. The coke from the operation deposited on the sand as a film. Coke, together with recycled heavy fuel from the process was burned in a lift reactor acting as a regenerator. Sand was then stored in a hopper above the main reactor. Due to the very high temperature, all equipment was provided with refractory lining.
Circulation rate of the sand was approximately 20 times the amount of feed in the reactor. Other heat carriers than sand were tested such as corundum, which reduced by one order of magnitude the solid losses, but did not compensate for the higher cost of the material. Some tests were also carried out with undisclosed catalytic materials. While olefin yields remained nearly unchanged, this stimulated the production of hydrogen and CO. Operation at lower temperature was also conducted to boost propylene production, or dehydrogenate propane to reach high selectivity (80% at 50% conversion per pass). Typical yield pattern with light gasoline (ibp-160 °C) and a crude oil are reported in Table 6.
| Feed | Gasoline (40–160) | Iraqui crude | ||
|---|---|---|---|---|
| Trx/°C | 750 | 800 | 730 | 760 |
| Product yield (wt%) | ||||
| Ethylene | 25.9 | 31.6 | 19.6 | 23.1 |
| Propylene | 15.6 | 12.5 | 12.6 | 12.8 |
| Butenes | 6.4 | 2.6 | 4.7 | 2.0 |
| Butadiene | 3.8 | 3.7 | 3.4 | 3.7 |
| Total olefins | 51.7 | 50.4 | 40.3 | 41.6 |
| Methane | 14.1 | 15.7 | 9.6 | 10.8 |
| Ethane | 3.0 | 2.9 | 3.1 | 3.1 |
| Propane | 0.4 | 0.4 | 0.6 | 0.3 |
| Butanes | 0.5 | 0.4 | 0.5 | 0.2 |
| Total paraffins | 18.0 | 19.4 | 13.8 | 14.4 |
| CO2 + H2S | 0.3 | 0.2 | 0.9 | 0.4 |
| H2 | 0.8 | 1.0 | 0.6 | 0.8 |
| Co | 0.2 | 0.3 | 0.4 | 0.3 |
| Acetylene | 0.4 | 0.9 | 1.3 | 0.7 |
| Nitrogen | 0.2 | 0.2 | 0.2 | 0.2 |
| Total other gases | 1.9 | 2.6 | 3.4 | 2.4 |
| Gasoline (ibp-200) | 20.5 | 17.6 | 17.0 | 16.5 |
| Distillate (>200 °C) | 6.9 | 9.0 | 20.5 | 22.1 |
| Coke | 1.0 | 1.0 | 5.0 | 3.0 |
Using a paraffinic feed, yields of ethylene of 22.5 wt%, propylene of 13.9 wt% and liquids of 18.6 wt% were reported. While research was discontinued in the 1980s, further development using a catalytic material was carried out in the 1990s, and the process was renamed as Quick Contact. Claims of ethylene and propylene yields of 18–19 wt% were reported.
3.2. Other processes not using particulate materials
A number of other processes were also designed for the processing of crude oil or heavy oils to yield olefins, mainly ethylene, all having special features to tackle the coking problem. Some used superheated steam (above 1500 °C) for heat transfer or used molten salts to prevent coil walls coking. The following processes can be mentioned:
• Advanced Cracking Reactor, developed by Union Carbide and Kureha Chemical Industry Co. It uses steam heated in a burner at 2000 °C as heat transfer medium, cracking temperatures of 910–960 °C, very short contact time of 0.02 s and quenching with oil.
• Dow process, using superheated steam generated in a combustor.
• Cracking Oil by Steam and Molten Salts (COSMOS), where the crude oil or heavy oil is cracked in a tubular furnace at temperatures of 800 °C or above. Wall coking is prevented by spraying a mixture of LiCO2, Na2CO3 and K2CO3 in the furnace, forming a wet coating on the wall. This coating acts as a catalyst for coke and pitch steam reforming.
• Paccal cyclic thermal cracking, where atomized oil is sprayed downwards on checker bricks at 1200 °C in refractory lined tubular reactors, operating in pairs in a cyclic reaction/regeneration pattern.
3.3. Lessons from early technology
Fluid bed technology allowed obtaining yield patterns similar to that obtained through steam cracking, with a strong ability to cope efficiently with the contaminants present in a crude oil that impedes to feed it directly into a steam cracker. Yet due to the thermal nature of the process, the propylene to ethylene ratio was bound to remain low and relatively inflexible, while propylene demand increased faster than ethylene. Also, opportunity (cheap) crude oil tends to be significantly heavier than several decades ago, which implies that the process has to deal properly with a large fraction of heavy material. Introducing a catalyst is the obvious step to take forwards, leaving directly towards fluid catalytic cracking technology.4. FCC and FCC-related technology for enhancing propylene yields
4.1. Introduction
In the last few decades, a huge attention have been paid by refiners, researchers and catalysts manufacturers to optimize and redesign FCC unit processes and catalyst formulations in order to enhance the yield of light olefins produced via catalytic cracking of heavy hydrocarbons. The two major levers that can be used in this operation, used independently or complementary, are the optimization of the operating conditions of the unit, and the second one to the optimization of the FCC catalyst formulation.FCC process hardware and operation optimization will be reviewed in section 4.2, with focus on its impact on the yield of light olefins. The different sections will cover: reactor technology (i.e. riser vs. downer reactor), reaction temperature, feed atomization (i.e. pre-vaporization option), contact time, catalyst-to-oil ratio, hydrocarbons partial pressure, naphtha recycle, and the nature of the feed.
The two catalyst options that are concerned when it comes to enhancing light olefin yields in the FCC process are the addition of shape-selective ZSM-5 and the reformulation of the base cracking catalyst composed of Y zeolite and often an active matrix. Both of these methods have their advantages and drawbacks, and may be used synergetically to maximize light olefins yields. The catalytic system may also be tailored according to the process hardware and processed feedstock properties by optimizing the composition of a mixture of different zeolites and/or optimizing the properties of the Y zeolite. When processing heavy feedstocks, the pore size distribution of the matrix plays a crucial role. At short contact time, diffusion of large molecules may be a limitation, requiring the use of high accessibility materials. This last also helps in product diffusion, limiting hydrogen transfer reactions that otherwise reduces light olefins yield. Finally, the acidity of catalytic materials, as well as their strength and density can be adjusted to maximize light olefins in the product stream. All these issues will be discussed in section 4.3.
4.2. FCC process hardware and operation optimizations for maximum light olefins production
Because the FCC process involves successive reactions, the desired products such as olefins and gasoline are considered intermediate products. Suppression of back-mixing is the key to achieving maximum yield of these intermediates. A simplified reaction scheme about the competing reactions involved in light olefins production is shown in Fig. 2.35 Mainly the higher (linear) olefins are the reactants, which can be converted to light olefins. However, besides cracking and isomerization, these higher linear olefins can also undergo other reactions, such as hydrogen transfer and aromatization. Therefore, it can be concluded that the maximization of light olefins in the FCC is consistent with the objective of reducing the quantity of aromatics being produced and that the selection of an optimum and relatively short residence time allows the maximization of intermediate products such as gasoline and light olefins. A down-flow reactor would be then the optimal option to maximizing olefins yields.34
 | ||
| Fig. 2 FCC reaction model: production of aromatics vs. light olefins.35 | ||
It has to be taken into account that in a down-flow reactor, there is relatively little conversion in the entrance region since the solids are not held up there as much as they are in a riser.36 Thus, higher catalyst-to-oil ratio, reaction temperature, and/or catalyst activity will be necessary to compensate for lower catalyst holdup and lower catalyst residence time, thus maintaining an adequate conversion level. Fortunately, developments in highly accessible catalyst technologies make it possible to run down-flow systems under conventional FCC conditions without penalty in conversion.36,37
Characteristic improvements in LPG and light olefins yield by FCC down-flow operation compared to up-flow operation are given in Table 7. In addition, down-flow reactors allow working with higher catalyst-to-oil ratios because lifting of the catalyst by vaporized feeds and steam is no longer a limiting issued.38,39
| Yield (wt%) | ||||
|---|---|---|---|---|
| FCC process | C/O | Propylene | LPG | Bottoms |
| Riser | 7.8 | 4.8 | 18.0 | 11.4 |
| Downflow | 8.7 | 6.6 | 20.3 | 13.0 |
| Yield (wt%) | |||
|---|---|---|---|
| Process | Dry gas | Gasoline | Propylene |
| Base case | 2.6 | 45.0 | 4.2 |
| Higher temperature | 3.3 | 44.2 | 6.3 |
| ZSM-5 addition | 2.6 | 42.7 | 5.2 |
It has been observed that the synergy with the addition of ZSM-5 for boosting light olefins yield was limited. This is apparently due to the depletion of the precursor gasoline-range olefins, on which ZSM5 mainly acts.40
Both methods increase the cracking rate of gasoline olefins relative to the rate of hydrogen transfer, yielding short olefins rather than gasoline-range paraffins.
Almost all the conventional FCC industrial units use some type of device (atomizer) that combines the hydrocarbon feedstock with steam before being injected into the riser reactor. By means of especially designed nozzles, the potential energy of the feed and steam, along with the geometry of the nozzles, cause the feed to disperse into mist size droplets which allow a very fast feed vaporization when contacting with the hot catalyst. In order to improve further the initial contact between feed and catalyst, dedicated feedstock pre-vaporization systems, such as the PREVAP process designed by Petrobras S.A., were developed.42 In this system, feed is to a large extent vaporized before it makes contact with the hot cracking catalyst from the regenerator vessel. Heat exchange with the catalyst is then no longer necessary to vaporize the feed. As a result, coke deposits on the catalyst surface are reduced, gasoline and light olefins yields are improved, and the unit is able to operate under very short contact times (less than 0.5 s). One may visualize the PREVAP concept as follows: the feed is introduced into the riser in a “supercritical” state and instantaneously flashes over the feed nozzle, which acts as a “relief or restriction valve”. It is argued that PREVAP will be an important design innovation for maximum light olefins FCC technology aiming at extremely reduced contact times between catalyst and hydrocarbons, thus improving yield slate (Table 9).
| Yield (wt%) | ||||
|---|---|---|---|---|
| FCC process | Propylene | LPG | Gasoline | Bottoms |
| Base case | 4.5 | 16.3 | 40.3 | 15.7 |
| With PREVAP | 5.2 | 16.2 | 43.3 | 13.8 |
Downer reactors are ideally suited for this, while it may be difficult to lower residence time to the sub-second range in riser reactors, as the catalyst has to be pushed upwards. Catalyst backmixing does not help either. Proper downer design of downer reactors needs to ensure adequate mixing of the catalyst and feedstock at the reactor inlet and the fast separation of the catalyst and products at the reactor outlet.39 Downer reactors also allow working at higher catalyst-to-oil ratios, mitigating at the same time the lower conversion and bottom cracking implied by the use of relatively short residence times. Table 10 gives an example of the impact of the residence time on the dry gas and light olefins production in a conventional FCCU and illustrates the fact that there exists an optimum residence time which is dependent on the feedstock nature and the catalytic system used.43
| Yield (wt%) | |||
|---|---|---|---|
| Residence time/s | 2.5 | 1.5 | 0.7 |
| Dry gas (wt%) | 4.11 | 2.78 | 2.29 |
| Ethylene (wt%) | 6.02 | 6.88 | 5.96 |
| Propylene (wt%) | 19.36 | 22.30 | 21.64 |
| Butylene (wt%) | 12.56 | 13.41 | 14.12 |
Working at low residence time as depicted in the previous section usually results in lower conversion due to a decrease in bottom cracking. Raising the catalyst to oil ratio will then compensate the lower conversion due to short contact time directly increasing the catalytic cracking contribution.39 Circulating increased amounts of catalyst in a riser type unit may be problematic as larger amounts of steam may be used, reducing partial pressure and catalyst concentration. By the contrary, downer reactors are much less limited in these aspects.
An additional advantage for the production of light olefins is that the use of high catalyst-to-oil ratios decreases the temperature drop observed during the endothermic cracking reactions and facilitates the proper feed vaporization of the injected feedstock at the inlet of the FCCU reactor.
Finally, the use of high catalyst-to-oil ratios enhances the contribution of catalytic cracking over thermal cracking. Knowing that the thermal cracking favours ethylene production, the use of high catalyst-to-oil ratios favours catalytic cracking reactions and allows to decrease the ethylene-to-propylene ratio and to give the flexibility to adjust the light olefins production in function of the market, as shown in Table 11.34
| Yield (wt%) | |||
|---|---|---|---|
| Catalyst | Ethylene | Propylene | C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) /C3 /C3 |
| Quartz (thermal cracking) | 4.74 | 4.01 | 1.18 |
| Zeolite catalyst (mainly catalytic cracking) | 5.08 | 8.70 | 0.58 |
Regarding selectivity, it has been observed that the decrease of the hydrocarbon partial pressure in a FCCU favours LPG and in particular light olefins such as propylene and butylenes, whereas propane and butanes selectivity is lowered. This is illustrated in Fig. 3, where LPG olefinicity index (defined as the ratio of the total yield of LPG olefins to the total yield of LPG) is found to increase with the decrease of the hydrocarbon partial pressure, whatever the cracking severity represented here by different catalyst-to-oil ratios. Increased olefinicity is a direct consequence of hydrogen transfer reactions, which by being bimolecular, are more strongly affected by the change of the hydrocarbon partial pressure than unimolecular cracking reactions that mainly drives conversion.44,47
 | ||
| Fig. 3 Olefinicity index vs. hydrocarbon partial pressure.44 | ||
For the same level of conversion, less olefins are converted to paraffins when lowering the partial pressure.
Low hydrocarbon partial pressure may also directly affect residence time in the unit. As a consequence, thermal cracking may be proportionally lowered and dry gas yield decreased, improving selectivity towards more valuable products.
Steam is usually employed in the FCCU for carrying over the catalysts and dispersing the feed. At usual FCC temperature (520 °C) it was found acting essentially as a physical diluent with no noticeable influence on the chemistry of the cracking.48 In the case of processes such as steam catalytic cracking, working at substantially higher temperature, say above 600 °C, and in the presence of large amounts of steam, a noticeable (and reversible) effect on the reaction rate of small paraffins was found besides the evident hydrothermal deactivation. It was attributed to adsorption competition on the active site of the hydrocarbon with the water, which takes on more importance as the main cracking mechanism shifts from beta-scission to protolytic cracking at higher temperature.49
Nevertheless, it appeared that using this scheme, and even using a shape selective catalyst such as ZSM-5 zeolite, only a low conversion is achieved despite the high olefins content of the processed feedstock blend.10
This particular behaviour has been attributed to the competitive adsorption of gas oil molecules and particularly polynuclear aromatic products on the catalyst, hindering naphtha conversion. Therefore, other options to effectively crack and recycle FCC naphtha in the FCCU were explored as depicted in Fig. 4. Naphtha can be injected at several locations in the riser and stripper of the FCCU, offering a wide range of severity.53
 | ||
| Fig. 4 Different schemes for naphtha recycling in the FCC.52 | ||
FCC naphtha can be fed at the bottom of the riser before the gas oil injection zone (option A) where it may be cracked under higher severity conditions than in classical gas oil cracking; processed in a parallel riser or downer reactor sharing the same stripper and regenerator system than the main riser reactor and where it is possible to optimize the cracking conditions for maximum light olefins production from FCC naphtha (option B); or recycled at the end of the riser reactor or in the catalyst stripper, where it contacts a coked catalyst under mild temperature (option C).
As shown in Table 12, FCC naphtha reprocessing allows increasing light olefins and in particular propylene yields far beyond the values obtained when operating FCCU in a conventional way. Nevertheless, even if it is possible to achieve higher light olefins production through all the different proposed recycling schemes without disturbing to a large extent the coke selectivity and therefore the FCCU heat balance, the maximum light olefins is obtained when feeding the olefinic FCC naphtha at the bottom of the riser reactor (option A). Nevertheless, this option implies at the same time a much higher dry gas yield which can be a problem for the majority of the commercial FCCUs which are limited by compressor capacity.
| LCN recycle | None | Bottom of the riser | Parallel riser | Separator |
|---|---|---|---|---|
| Yields (wt%) | ||||
| Dry gas | 2.4 | 4.4 | 2.8 | 2.5 |
| LPG | 23.6 | 30.8 | 28.2 | 27.3 |
| Propylene | 8.6 | 12.5 | 10.6 | 10.0 |
| Butenes | 10.6 | 12.9 | 12.2 | 12.5 |
| Gasoline | 47.3 | 37.7 | 41.9 | 43.1 |
| Coke | 3.9 | 4.2 | 4.4 | 4.3 |
| Selectivity of LCN cracking | ||||
| Olefins LPG | 65 | 67 | 79 | |
| Propylene | 40 | 37 | 33 | |
| Gasoline composition (%) | ||||
| Paraffins | 33 | 35 | 41 | 40 |
| Olefins | 25 | 12 | 11 | 14 |
| Naphthenes | 10 | 9 | 10 | 10 |
| Aromatics | 32 | 44 | 39 | 37 |
On the contrary, reprocessing the FCC naphtha in the stripper zone (option C), even if less efficient, significantly increases the LPG olefins yields with the huge advantage to produce only small quantities of additional dry gas and coke yields and to lower in a large extent the olefin content of the cracked naphtha. In addition, the great advantage of option C is that the implementation of this strategy does not require major modifications of the FCCU and therefore a low capital investment compared to options A and B, which implies respectively a much higher compressor capability and the revamping of the FCCU to integrate the naphtha dedicated riser or downer reactor.
During the past three decades the FCC feed quality tended to be heavier, with an increasing tendency to incorporate resid. Also these feeds tended to be more aromatic, which difficult conversion towards light olefins. It was shown however that the use of resid is not always detrimental to olefins production, if the proper catalytic system is used, with high resistance to deactivation by coke and metals. While resid may contain large aromatic cores, yielding aromatics, it may also contain long aliphatic chains connecting these cores, which can be converted into olefins. Also, higher coke-on-catalyst tends to decrease hydrogen transfer reaction rate.54 Recently, tight oils also have entered the market and may be very interesting feedstocks for olefins production due to their paraffinic nature.55
Beyond traditional, oil based stocks, other stocks may also been interesting. Fischer–Tropsch waxes, produced either from ethane reforming or biomass gasification, would be ideal candidates, being highly paraffinic.56 There is no impediment in chemistry that ethanol be co-processed with other feeds, yielding ethylene by dehydration. Indeed, that would help reducing hydrocarbon partial pressure of the co-processed oil, but would put pressure on gas compressor if not designed upfront. Hydrotreated pyrolysis oils were found processable in FCC, with limited selectivity to small olefins.57 Vegetable oils were also proposed as feedstocks, as they possess long (linear) hydrocarbon chain to be cracked towards small olefins. Yet it seemed that propylene yield increase was very limited, being that these chains were more prone to intra cyclization, yielding gasoline components instead of small paraffins.58
4.3. FCC catalyst systems optimizations for maximum light olefins production
The introduction of rare earth (RE) in Y zeolite minimizes the framework dealumination under hydrothermal conditions and, as a consequence, increases activity per weight of zeolite and enhances the rate of hydrogen transfer.62 This improves both the activity and the hydrothermal stability of the catalyst. The degree of dealumination of the zeolitic component in the FCC catalysts has an important impact on the hydrogen transfer: the more dealuminated the zeolite, the less extended the reaction. On average, active sites of rare earth exchanged HY zeolites are weaker and in closer proximity to each other than those found in a more highly dealuminated catalyst characterized by lower unit cell size. As a result of the greater number of active sites, both the primary cracking and primary hydrogen transfer reactions that occur within the zeolite are enhanced. Primary cracking reactions involve the initial scission of the carbon–carbon bond to form higher valued liquid products in the gasoline pool range. Primary hydrogen transfer reactions are those that occur between cracked products to terminate the cracking reactions in the gasoline range, thus, reducing the overcracking of gasoline to C3's and C4's (Table 13).35
| Zeolite | Conversion | Gasoline | Propylene |
|---|---|---|---|
| Silica-alumina gel | 75.5 | 47.5 | 8.5 |
| REHY | 85.5 | 61.0 | 5.9 |
The hydrogen transfer reactions are thus greatly increased with the addition of rare earth to the USY zeolite, stabilizing at the same time the gasoline fraction selectivity. In addition, this reaction also promotes the hydrogenation of olefins, reducing gasoline octane number. Besides that, hydrogen transfer can also promote the formation of carbonaceous deposits on the catalyst and a correlation between coke formation and rare-earth exchange was found.
Hence, an optimum content of rare-earth element in Y zeolite based FCC catalyst should be searched according to the FCC unit production targets and the processed feedstock.63 The historical trend was towards the use of REUSY catalysts with high rare-earth levels as illustrated in Fig. 5, that accompanies the increasing trend in feed contaminants, especially metals.
 | ||
| Fig. 5 Rare-earth content and unit-cell size of equilibrium FCC catalysts.59 | ||
The recent surge in rare-earth price led to the development of RE-free catalysts with improved stability and gasoline selectivity, which may be of interest for the production of light olefins in combination with ZSM5. In addition, it has been observed that depending on the type of rare-earth cations introduced in the zeolite, different degrees of framework dealumination take place.64
Furthermore, a correlation between the decrease in the unit-cell size and the rare-earth ionic radius and its coordination to the oxygen framework was observed (Fig. 6). According to the Fichtner-Schmittler equation, the smaller the incorporated rare-earth cations, the higher will be the dealumination and the smaller the unit-cell size parameter.65
 | ||
| Fig. 6 Relationship between the Si/Al ratio (UCS) of the REHY zeolite and the ionic radius of the incorporated rare-earth cation.63 | ||
Therefore, knowing that the higher the FCC catalyst unit-cell size, the more intense the hydrogen transfer, it is clear that the use of low radius rare-earth cations to stabilize the HY zeolite is beneficial for the production of light olefins during FCC operations even if the activity and stability of the FCC catalyst will be negatively affected. Y zeolite used in FCC catalyst presents acid sites of Lewis and Brønsted types. The first type is mostly associated to non-framework aluminium species while the latter is related to acidic hydroxyl groups attached to the framework. It is universally recognized that the concentration, strength and distribution of acid sites play a key role in determining the overall activity and selectivity of the Y zeolite during FCC operation.64 While cracking reactions require the presence of strong acid sites, other reactions such as isomerization, cyclization and hydrogen transfer take place over weaker acid sites. Knowing that the incorporation of rare-earth elements in the Y zeolite limits the degree of framework dealumination, the SiO2/Al2O3 ratio is lower and therefore the unit-cell size of REHY is higher than that of an HY zeolite. It is quite interesting to note that rare-earth exchanged HSY zeolites equilibrate at a unit cell size of about 24.30 to 24.35 Å, while it is generally observed that HSY zeolites equilibrate at a unit cell size of about 24.25 ± 0.02 Å.66 The increase in the number of framework Al atoms in the rare-earth exchanged zeolitic compound of the FCC catalyst increases the probability to find paired acid sites which have a lower acidic strength than isolated acid sites (Fig. 7) but which are suitable for carrying out isomerization, cyclization and/or hydrogen transfer reactions.
 | ||
| Fig. 7 Acid site distribution in Y zeolites as a function of unit cell size.66 | ||
Cyclohexene cracking was used by Suarez et al. to characterize the ratio between isomerization (that requires only one site) and hydrogen transfer (that requires paired acid sites), and good correlation was found between this ratio and the ratio of total acid sites over paired acid sites.67 Below a unit cell size of 24.30 Å where most aluminium atoms are found isolated, isomerization to hydrogen transfer ratio increased sharply.
In summary, at lower unit cell sizes (below 24.30 Å) the activity of the zeolite is low due to the low concentration of active sites and at higher unit cell sizes hydrogen transfer reactions are favoured due to the rapid increase in the number of paired Al atoms. This explains the lower content of light olefins, olefinic compounds in the gasoline pool and the higher coke selectivity when processing gas oil over rare-earth exchanged zeolites.66 In addition to the increasing activity, the incorporation of rare-earth elements improves the olefins adsorption capacity of the Y zeolite based catalyst, thus further increasing the rate of hydrogen transfer reactions of rare-earth exchanged FCC catalysts.61
In addition, it appears that the ionic radius of the rare-earth element used to stabilize the Y zeolite has an influence on the impact of the rare-earth incorporation over the acidity of the zeolite, the density of the Brønsted acid sites being higher with larger ionic radius of the rare-earth atom (Fig. 8).68 Two possibilities are commonly presented: (i) the acidity related to the acid sites is increased due to a polarization mediated by the rare-earth cations and/or (ii) these new acid sites are stronger than those eliminated by ion exchange.
 | ||
| Fig. 8 Influence of the ionic radius of the rare-earth cation on the Brønsted acidity of Y zeolite.68 | ||
As an example, it was found that the Brønsted acid sites of CeHY zeolites are stronger than those of HY and the total number of acid sites decreases with the increasing cerium content.69
Finally, another important parameter which can help to tune the Y zeolite based FCC catalyst selectivity towards light olefins is the nature of the matrix (i.e. amorphous or active) and the zeolite-to-matrix ratio. Silica and/or alumina based materials are commonly used as binders (or matrices) and provide the dilution of active zeolite crystals, resistance to attrition and large pores for the access of feed molecules to active cracking sites. The binders may be designed to be acidic or non-acidic according to the different requirements. If coke reduction is required, a non-acidic binder such as silica gel can be used. On the other hand, an acidic binder is recommended if the FCC catalyst is used for residual feed cracking, since in this case large molecules in the residual feed must be cracked at the surface of matrix to smaller sizes that can diffuse into the pores of the zeolite to be further selectively cracked.70 It has been repeatedly reported that the use of active matrix allows to increase the gasoline yield and its octane number, the LPG yield and its olefinicity, together with the LCO yield due to the enhancement of the bottoms conversions (Table 14). However, the gasoline yield can increase and the coke make can increase due to some non-selective cracking reactions when using active matrices.71 Other materials were also studied as potential active matrix components: sepiolite (magnesium silicate), amorphous silico-alumino-phosphate (ASAPO) or other mixed oxides such as SiO2–ZrO2, or SiO2–Al2O3–MgO of a FCC catalyst and have shown that it is possible to change the overall FCC catalyst selectivity and in particular to increase even more the olefinicity of the LPG pool using new materials as matrices compared to commercial SiO2–Al2O3 based matrices.72
| Matrix SA/m2 g−1 | Product selectivity (%) | |
|---|---|---|
| Active matrix | Low SA inactive matrix | |
| 130 | 30 | |
| Propane | 1.4 | 1.3 |
| Propylene | 3.2 | 2.8 |
| Butanes | 6.0 | 5.7 |
| Butenes | 3.5 | 2.9 |
| C3= in C3 cut | 0.70 | 0.68 |
| C4= in C4 cut | 0.37 | 0.34 |
| Gasoline | 43.5 | 46.5 |
| Coke | 6.2 | 4.5 |
In commercial FCC catalysts having active matrices, the overall activity and selectivity is therefore determined by both zeolite and matrix and it is possible to adjust the catalyst activity and selectivity by changing the zeolite-to-matrix ratio. The optimum ratio generally depends on feedstock composition, process operating conditions and desired product slate, but the following general trends can be drawn. At high zeolite-to-matrix ratio, the selectivity pattern approaches that of pure zeolite cracking, while at low zeolite-to-matrix ratio the selectivity pattern is dominated by the matrix.
At constant catalyst activity, it is observed that a decrease in zeolite-to-matrix results in an increase of LCO, coke, dry gas yields, and a decrease in bottoms yields. The gasoline octane and olefin/paraffin ratio in LPG also increase under these conditions (Table 15).64
| USY | REUSY | REHY | REY | Active matrix | |
|---|---|---|---|---|---|
| Unit cell size | Low |
|
High | ||
| Framework Si/Al | High |
|
Low | ||
| RE content | Low |
|
High | ||
| Dry yield | Low | Low | Low | Low | High |
| C3/C4 yield | High | Moder. | Moder. | Low | High |
| C3/C4 olefins | High | Moder. | Moder. | Low | High |
| Coke/conversion | V. Low | V. Low | Low | Moder | High |
| Gasoline selectivity | Moder. | High | High | High | Low |
| Octane potential | High | Moder. | Low | Low | High |
| LCO selectivity | Moder. | Moder. | Low | Low | High |
| DO selectivity | Moder. | Moder. | High | High | Low |



Special attention should also be paid on diffusion issues with heavier feeds. Highly accessible catalysts will enhance bottoms cracking as well as diffusion of reaction products outside the catalyst, limiting hydrogen transfer reactions that reduce olefins yields.73 A catalyst with a high bottoms cracking ability will be in a short residence time process.74 The increase in mesopores surface area has been shown to increase resid cracking activity and lower coke yield.
In conclusion, the Y zeolite to be incorporated into a process dedicated to maximize light olefins should minimize hydrogen transfer reactions as much as possible while maintaining a good level of conversion. Rare-earth levels should be kept as low as possible while maintaining a good hydrothermal stability. USY type catalysts will be preferred, with reasonable amount of active matrix to maintain bottoms upgrading capacity. The right amount will depend on the targeted operating conditions, taking into account this will be some kind of high temperature, high catalyst-to-oil ratio operation. Thus, optimum composition may slightly differ than normal FCC operation, and catalysts with lower activity may be preferred as high temperature and CTO will boost catalytic activity.
This poses the problem of proper catalyst testing at the laboratory scale. Fixed bed, micro activity test, in spite of its well documented drawbacks, is still much used due to its simplicity, allowing a fast generation of preliminary performance data.47,75 It is well known that the long residence time of the catalyst, and the continuous deactivation during the test, tends to magnify hydrogen transfer reactions, yielding more aromatics and less olefins in the products, as well as more coke. This tendency is generally exacerbated with resids. A number of modifications of the original, ASTM-3907 protocol aimed at reducing these drawbacks, and usually features shorter reaction time, higher temperature, dilution of catalyst and feed.76,77 Catalytic fixed fluid bed (ACE) has emerged in the last years as a competitor of the MAT test as a standard catalytic cracking test. The main difference is that the catalyst bed is fluidized, which eliminates the formation of temperature and coke profiles along the bed.78 Nevertheless, the same time averaging effect occurs as with fixed bed. Y catalyst ranking have shown systematic deviations between the two protocols as well as with pilot plants.79,80 A number of equipments were designed to offer a more realistic simulation of riser cracking. The Riser Simulator is a fluid bed with gas recirculation working with a gas/catalyst contact time of 2–10 s.81 The Micro-riser features a transported bed reactor where very good piston flow was achieved.82 Residence time could be regulated from very short (50 ms) to several seconds by changing the numbers of coils of the reactor. Microdowner features a downer type transported bed reactor and is also capable to work at very short catalyst residence time (below 0.5 s).83 Injector and stripper design received special attention to ensure good feed dispersion and fast product separation, minimizing secondary reactions. Resids can also be handled. Its downer design allows working with very high catalyst to oil ratio and high reactor temperature, being a very good match for high severity processes simulation.
As in the case of Y zeolite, an improvement in accessibility of the catalyst will lower the occurrence of secondary reactions such as hydrogen transfer. ZSM5 with nanosized crystals (<100 nm) have shown improved activity, stability and slightly more selectivity to propylene.88 Mesoporosity can be introduced by alkaline treatment or secondary templating, improving selectivity to propylene.89
ZSM-5 based additives were firstly commercially introduced into the FCC process in the 1980s to improve gasoline octane numbers. Nevertheless, today the additive is principally used with the purpose of increasing C3–C5 olefins yields. The efficiency of the ZSM-5 based additives to increase gasoline octane and light olefins selectivity have been repeatedly proved since then.85,90,91
The addition of ZSM-5 additive is one of the most efficient methods for improving the yield of light olefins because it provides refiners with a high degree of flexibility to optimize the production output. Catalyst producers have developed methods to increase the concentration of ZSM-5 in their additives in order to avoid dilution effects at high levels of additive. The breakthrough technologies in this area involve the stabilization of the ZSM-5 to hydrothermal deactivation at higher concentrations in the additive.91 It is generally well accepted in the literature that the light olefins increase which occurs when using hydrothermally deactivated ZSM-5 combined with USY catalysts, is mainly due to the selective monomolecular cracking of gasoline pool olefins previously produced by the USY catalyst.44,86,92,93
Skeletal isomerization is energetically favoured with respect to cracking for lower carbon number carbenium ions, while higher carbon number carbenium ions are preferably cracked to C2–C5 olefins. Pentenes are simultaneously formed and consumed by ZSM-5 additives. Table 16 and Fig. 9 show some examples of changes of product selectivity and in particular of light olefins production which can be expected when adding 10 wt% of ZSM-5 additive to a FCC catalyst and processing different feedstocks.94
| Feedstock A | Feedstock B | Feedstock C | ||||
|---|---|---|---|---|---|---|
| Base USY | USY + 10 wt% ZSM-5* | Base USY | USY + 10 wt% ZSM-5* | Base USY | USY + 10 wt% ZSM-5* | |
| Conversion (wt%) | 60.0 | 60.0 | 60.0 | 60.0 | 60.0 | 60.0 |
| Gases yield (wt%) | 13.0 | +12.2 | 15.2 | +13.4 | 13.4 | +9.9 |
| Dry gases (wt%) | 1.4 | +0.7 | 2.5 | +0.8 | 2.1 | +0.6 |
| LPG (wt%) | 11.6 | +11.5 | 12.7 | +12.6 | 11.3 | +9.3 |
| Hydrogen yield (wt%) | 0.079 | −0.015 | 0.056 | −0.012 | 0.103 | −0.024 |
| Hydrogen transfer index | 1.63 | +0.56 | 2.90 | +0.88 | 2.01 | +0.09 |
| Propylene/i-butene ratio | 1.6 | +1.1 | 1.7 | +0.8 | 1.7 | +1.1 |
| i-C4/n-C4 ratio | 5.6 | −1.3 | 2.9 | −0.7 | 3.9 | +0.4 |
| i-C5/n-C5 ratio | 8.4 | −1.9 | 3.9 | −1.0 | 6.0 | +0.8 |
| Gasoline yield (wt%) | 43.8 | −12.5 | 37.9 | −11.4 | 39.1 | −10.6 |
| Light gasoline yield (wt%) | 29.9 | −8.7 | 27.9 | −6.9 | 27.1 | −7.8 |
| Heavy gasoline yield (wt%) | 13.9 | −3.8 | 10.0 | −4.5 | 12.0 | −2.8 |
| LCO (wt%) | 18.0 | +5.8 | 11.1 | −2.7 | 19.3 | −2.2 |
| HCO (wt%) | 22.0 | −5.8 | 28.9 | +2.7 | 20.7 | +2.2 |
| Coke yield (wt%) | 3.2 | +0.3 | 6.9 | +1.4 | 7.5 | +0.8 |
 | ||
| Fig. 9 Effect of the incorporation of 10 wt% ZSM-5 additive (2.5 wt% of pentasil crystal) to a USY FCC Ecat on light gases yield and olefinicity at a conversion level of 60 wt%.94 | ||
A complete and detailed review of ZSM-5 based catalyst modifications has been published by Rahimi et al.95
The optimization of the design of the H-ZSM-5 based catalyst for maximization of light alkenes should take into account the balance between the number/strength of acid sites and type/amount of metals incorporated in the modified catalyst and the necessary low coke ability and stability of the modified catalyst. Concerning acidity, it is well accepted that the global acidity of a given zeolite depends to a huge extent on the density and strength distribution of the Brønsted acid sites. The density of the Brønsted acid sites depends on the ion exchange capacity and is therefore directly proportional to the Al3+ content of the zeolite and is greatly affected by the Si/Al ratio and the dealumination stability of the material. On the other hand, as shown before, the strength of the Brønsted sites depends on the interaction between the proton and the zeolitic framework. For instance, a completely isolated Al tetrahedron will create the strongest type of Brønsted acid site. Therefore, the two main parameters governing the acid strength of the Brønsted sites are the structural characteristics of the zeolite and its chemical composition. The structural characteristic of the zeolite is related to the proton lability that depends on the angle formed between the two adjacent tetrahedral T at the oxygen carrying the proton.
Regarding the chemical composition factor, the acid strength depends on the number of Al atoms (aluminate tetrahedral) that are adjacent to a silanol (silicate tetrahedron) group.96 The strength of the Brønsted acid sites can also be affected by isomorphic substitution. For instance, it has been shown that gallium or iron zeolites exhibit dehydrogenation properties and are much less acidic than aluminum zeolites.97 Moreover boron-substituted for Si in pentasil ZSM-5 zeolites yield very weak acidity strength.98,99
The first usual and most widely used modification method applied for improving HZSM-5 zeolites performance is the incorporation of phosphorus in the zeolite framework.100 It is thought that phosphorus stabilizes the lattice aluminum ions by retarding aluminum from leaving the zeolite framework and hinders structural changes of zeolite that significantly results in enhancing the hydrothermal stability of the catalyst.101–105 Therefore, even if the incorporation of phosphorus has a negative impact on the initial acidity of the zeolite, phosphorus modified ZSM-5 zeolite retains a larger fraction of the acidity compared to H-ZSM-5 zeolite when the catalyst is subjected to severe hydrothermal conditions.90 It has been reported that the incorporated phosphorus species are responsible for both the enhanced acid stability and the significantly improved catalytic performance in the cracking of C4 olefins and naphtha light olefins (i.e., ethylene and propylene).103,104 An increasing propylene and butenes selectivity is also claimed when using phosphorus modified HZSM-5 in the catalytic cracking of n-decane.105
Altogether, the modification of HZSM-5 zeolite with phosphorus decreases Brønsted acidity; and/or converts strong Brønsted acid sites of HZSM-5 into weak Brønsted acid sites that increases the density of the weak Brønsted acid sites without changing the overall acid–base properties. Reducing the Brønsted acidity of phosphorous modified HZSM-5 provides a pathway for reaction to occur with enhanced light olefin production.103,105Table 17 gives an example of acidity modification when incorporating phosphorus to a HZSM-5 zeolite.
| Zeolite | Amount of acid sites (μmol g−1) (200 °C) | Amount of acid sites (μmol g−1) (350 °C) | ||||||
|---|---|---|---|---|---|---|---|---|
| B | L | B + L | L/B | B | L | B + L | L/B | |
| HZSM-5 | 524.4 | 49.4 | 574.8 | 0.09 | 477.8 | 28.7 | 506.5 | 0.06 |
| 0.1P/HZSM-5 | 501.6 | 89.7 | 591.3 | 0.18 | 506.3 | 25.3 | 531.5 | 0.05 |
| 0.5P/HZSM-5 | 534.9 | 52.9 | 587.8 | 0.10 | 477.8 | 27.6 | 505.4 | 0.06 |
| 1P/HZSM-5 | 296.8 | 43.7 | 340.5 | 0.15 | 255.6 | 19.5 | 275.1 | 0.08 |
On the other hand, it appears that the incorporation of low electronegative charge-balancing cations such as alkali metals and/or alkali earth metals allows modifying the ZSM-5 acid character by weakening acid strength or reducing acid sites and is even able to make the zeolite basic. It is observed that the quantity of the stronger acid sites reduces with the alkaline earth modification by increasing the ionic radius. Barium as the strongest Lewis base in alkaline earths series resulted in the highest yield of light olefins.106 Moreover, the base strength of the ZSM-5 catalyst rises with increasing the aluminium content of the framework and reduces with decreasing the ionic radius of the exchanged cation (e.g. Li < Na < K < Rb < Cs). It is also influenced by the exchange of Al and/or Si ions in the zeolite framework for Ga and/or Ge, respectively. It is thought that the modification of ZSM-5 zeolite based catalysts with the elements enhancing the surface basicity decreases the readsorption of the basic compounds of the cracking products, such as ethylene, propylene and butenes, and this is apparently the major cause of the lower aromatics and higher light olefins formation. In addition, the alkaline-earth metal treatment of HZSM-5 zeolite is able to limit hydrogen transfer reactions and stimulate the dehydrogenative cracking by controlling the HZSM-5 acid character and dehydrogenation activity, leading to an improvement of light olefins yield.101,107,108
Nevertheless, it has been reported that the thermal and hydrothermal stabilities of the alkali-treated ZSM-5 zeolites are slightly deteriorated because of the introduction of mesopores caused by the desilication. Weak alkali solutions dissolve a small amount of silica and alumina, and so create mesopores by enlarging the micropores of zeolites, while strong alkali solutions destroy their crystal structure; therefore, converting them to amorphous materials with macropores and extremely small acid sites.109,110Fig. 10 gives an example of the decrease of the ZSM-5 zeolite activity when incorporating alkali metals and/or alkaline-earth metals to a HZSM-5 zeolite.
 | ||
| Fig. 10 Activity of La, Al and Ba ion-exchanged ZSM-5 zeolites in the n-hexane cracking reaction.103 | ||
The possibility to incorporate transition-metal elements to a HZSM-5 zeolite has also been studied by various authors. Metals are generally associated to detrimental effects in the FCCU (primarily Ni, V, Fe) as they tend to promote dehydrogenation reactions leading to coke, making their use unlikely in heavy feed cracking. Yet it may be of high interest in the case of very low coking feeds such as naphtha, cracked in dedicated processes as described in section 5.2.
Due to their electron configuration, transition metals are able to form chemical bonds with neutral molecules and it has been observed that the incorporation of transition metals creates new Lewis acid sites in HZSM-5 zeolites, leading to an increase of the acid activity of zeolites and influencing directly the selectivity of the catalytic cracking products.
Therefore, the incorporation of transition-metal ions into zeolites leads to interesting bifunctional catalysts in which metal and acid centers can act simultaneously.111 The transition metals, such as Ni, Co, Zn, Cu, Cr, and Mn, introduced into pentasil type zeolites are generally used as active components for hydrogenation or dehydrogenation,112,113 or aromatization reactions.114,115 The reported results show that Co, Cu, Zn, and Ag loaded sites on HZSM-5 zeolites accelerate the dehydrogenation cracking and cyclo-dehydrogenation reactions.101,107,108
Altogether, the combination of Lewis sites for the dehydrogenation of the paraffinic feedstock with Brønsted acid sites for the cracking of subsequent higher olefins to light olefins, could enhance the yield of light olefins.116
It has been reported that iron or chromium incorporation in HZSM-5 leads to increasing the selectivity to aromatic hydrocarbon and decreasing the total olefins, because of improving further oligomerization and cyclization reactions.117,118
In addition, the use of Cu-exchanged HZSM-5 with an optimum balance between dehydrogenation activity of the metal and acid function of the shape selective HZSM-5 zeolite seems promising to improve the yield of light olefins. Indeed, it has been observed that a small amount of copper ions hinders framework dealumination during hydrothermal treatment compared to HZSM-5 or the Ga- and Cr-modified samples. The total number of acid sites of the Cu/HZSM-5 sample seems, at the same time, to be higher than the rest of the metal transition modified ZSM-5 zeolites.119 Meanwhile, addition of Zn by chemical vapor deposition (CVD) seems to cause a decrease of the Brønsted acidity intensity of ZSM-5 zeolite, without increasing the density of Lewis acid sites, unlike Ga/HZSM-5 catalysts.120
Finally, the influence of rare-earth modification on the catalytic performance of the ZSM-5 zeolite has not been completely elucidated and is still controversial. Nevertheless, it has been reported that the introduction of a rare-earth metal ion into HZSM-5 zeolites could have a great impact on its acidic properties, modifying the density of acid sites and at the same time changing the acid nature (i.e., the Lewis/Brønsted ratio).121,122 However, if some researchers have observed that the incorporation of lanthanum in HZSM-5 zeolite lowers the number of strong acid sites and increases the number of weak acid sites,121 other authors claim that the total amount of acid sites of rare earth-modified HZSM-5 zeolites increases and estimate that the amount of weak, strong, and total acid is respectively impacted as follows:122
• Sm > Nd > Pr > Eu > Ce > La > HZSM-5 > Gd for weak acidity.
• Nd > Eu > La > Sm > Ce > Pr > Gd > HZSM-5; for strong acidity.
• Nd > Sm > Eu > Pr > Ce > La > HZSM-5 > Gd for total acidity.
On the other hand, the introduction of rare-earth elements also modifies the basicity of ZSM-5 zeolites as rare-earth oxides usually possess some basic character. Indeed, considerable changes of the basicity have been observed when loading an increasing amount of La to a HZSM-5 zeolite. It has been therefore suggested that the La-loading of the HZSM-5 zeolite implies a generation of basic sites on the surface of the latter.101 In addition, as the amount of La loading increases, the olefin adsorption over La/HZSM-5 zeolites decreases.107 Therefore, the main reason for the higher yield of ethylene and propylene is due to the decrease of the rate of the bimolecular reactions which is negatively affected by the decrease of readsorption of the basic compounds of the cracking products, such as ethylene, propylene and butenes. Yields of ethylene plus propylene close to 60 wt% at 650 °C were reported.101
For the cracking of naphtha rich in small paraffins, which are difficult to crack under typical FCC conditions, specialty catalysts beyond Y and ZSM5 may be used. Petrobras developed special additives based on a mixture of ZSM-5 and ferrierite, which are said to increase propylene yield over traditional ZSM-5 addition.128 Ferrierite, a small pore zeolite (4–5.5 Å pore size) was found to suppress hydrogen transfer and cyclization reactions that lead to higher small paraffins and aromatic yield in products.129,130 Propane yield was found stable while increasing propylene yield upon ferrierite addition to the catalytic system, while a substantial increase is observed upon addition of ZSM-5. However, ferrierite has a lower activity than ZSM-5, thus a mixture of the two additives was found optimal. Optimal mixture is claimed as at least 0.5:1 FER:MFI weight ratio. High silica to alumina ratio is preferred, and ratios as high as 200–400 for FER and 600 to 1600 for MFI has been claimed.131 Traditional stabilization by phosphorus of pentasil-type catalyst was also claimed.132
In processes using very high temperature as well as high amount of steam such as the proposed steam catalytic cracking, it was proposed to replace ZSM5 by IM5, which presented a higher activity. As with ZSM5, IM5 hydrothermal resistance could be largely improved by incorporating phosphorus, leading to a catalyst with improved overall activity, and improved selectivity towards ethylene and aromatics while preserving propylene selectivity.133
5. Commercial FCC and FCC-related processes designed for maximizing light olefins
Increasing the yield of the valuable light olefins, especially propylene, remains a major challenge for many integrated refineries, not only for economic and environmental reasons but also because an increasing reliance on deep upgrading of crude oil. In this landscape, deep catalytic conversion processes, such as FCC, will play an increasing role.This section reviews the different catalytic cracking technologies that have been significantly investigated and/or commercialized aimed at maximizing petrochemicals (C2–C4 olefins and BTX). A list of the processes described below can be found in Table 18. We have segregated processes depending on the philosophy applied for boosting olefins.
| Process name | Developer/licensor | Trx (°C) (1st riser) (2nd riser if applicable) | C2 yield |
C3 yield |
Status/remarks |
|---|---|---|---|---|---|
| VGO/resid cracking + naphtha recycle | |||||
| Maxofin | ExxonMobil/KBR | 510–540 VGO | 4 | 18 | Naphtha recycle, commercial |
| 550–600 naphtha | |||||
| Indmax | Indian Oil Co./ABB Lummus | 530–600 | 3–7 | 17–25 | Commercial (1 unit), naphtha recycle optional |
| Selective component cracking (SCC) | ABB Lummus | 500–600 | n/a | 24 | Naphtha recycle |
| PetroFCC | UOP LLC | 510–620 | 6 | 22 | RxCat, naphtha recycle |
| High olefins catalytic cracking | Petrobras | n/a | 3–9 | 8–14 | Downer, naphtha recycle |
| Industrial scale test | |||||
| Petroriser | Total/Axens | 510–540 resid | >3 | >12 | R2R implementation |
| 550–600 naphtha | 127000 bpd unit commissioned |
||||
| TMP (propylene/middle distillate) | China Petrochemical University/Petrochina | 500–520 | <4 | 20 | Multiple recycles, dual risers, |
| 520–540 | |||||
| 40000 bpd unit revamp |
|||||
| Middle distillate and light olefins selective process (MILOS) | SHELL | 500–540 | 2–3 | 10–17 | Naphtha recycle in dense bed. Pilot plant studies |
| 565–620 | |||||
| High severity VGO/resid cracking | |||||
|---|---|---|---|---|---|
| Deep catalytic cracking (I & II) | RIPP-SINOPEC/Stone &Webster | 510–575 | 2–6 | 14–21 | Commercial |
| Catalytic pyrolysis process (CPP) | RIPP-SINOPEC/Stone &Webster | 560–700 | 8–19 | 17–23 | Commercial trials |
| High severity FCC (HS-FCC) | KFUPM/JCCP/Saudi Aramco/Axens | 580–630 | 2–3 | 17–25 | Downer. 3000 bpd semi-commercial unit (Japan) |
| NeXCC | Neste Oy/Fortum | 520–650 | n/a | 16 | Multi port cyclones compact design. Pilot studies |
| Naphtha cracking dedicated process | |||||
|---|---|---|---|---|---|
| Superflex | ExxonMobil/KBR | 600–650 | 20 | 40 | C4–C10 olefinic feeds, |
| 1 com. operation (SASOL 2006) | |||||
| Propylene catalytic cracking | ExxonMobil | 510–630 | 2–7 | 6–17 | 1.5 bpd pilot plant unit |
| Cracked naphtha feed | |||||
| Advanced cracking olefins | ExxonMobil/KBR | 650–750 | 25–30 | 30–35 | Small paraffins feed |
First, the traditional FCC process is boosted raising reactor temperature and adding (more) ZSM5 to the catalyst inventory, within certain limits as the process flow remains very close to a traditional FCC. Catalyst circulation rate and compressor capacity may be limited, which in turn limit the increase of temperature, usually in the 530–560 °C range. In addition, an additional riser is often added to crack recycled naphtha stream in optimized conditions. This approach requires the minimum investment.
In order to increase further olefins production, dedicated, high temperature high catalyst to oil ratio were developed. They often rely on downer design to reach short time on stream so that extreme temperature (above 600 °C) can be reached while dry gas and coke yields remain under control. Reactor temperature implies catalyst to oil ratio in the 15–40 range, significantly above traditional riser designs.
The main problem for optimal naphtha recracking is the need to use the same catalyst as for VGO cracking, which implies a higher rate of hydrogen transfer than desirable due to the presence of some form of Y zeolite and matrix as base cracking catalyst for heavy fraction. Hence, some processes were developed based on FCC technology but fed with a range of naphtha instead. Special catalyst formulations are then allowed, such as all ZSM5 or metal-containing materials, allowing higher selectivity to olefins or improved cracking activity for difficult materials such as small paraffins.
5.1. Boosting olefins yields from heavy feeds cracking
| Process change | Pros | Cons |
|---|---|---|
| Temperature increase (contact time) | Low-cost route to increased olefins | Excess dry gas/coke make |
| Cracked naphtha reprocessing | Product selectivity optimization | Increased dry gas, large loss of gasoline |
| Spent catalyst recycle | Increased catalyst-to-oil ratio | High circulation rate, poor selectivity |
High severity operation relies mainly on higher cracking temperature, which also entails higher catalyst-to-oil ratio. The goal is to over-crack distillates (beyond maximum gasoline mode), producing increased quantities of light olefins. This “brute force” method is the simplest way of increasing light olefins production, but comes with major drawbacks such as an increased production of dry gas that may overload the gas plant (compressor and separator) and higher coke make, especially at higher residence time. Careful control of residence time will help controlling the formation of undesired products. While downers are ideally suited to work at low residence time, enabling high temperature and catalyst to oil ratios, this implies a major revamping of the unit, with uncertain economics in spite of better product slate. Grassroots downers units are more likely to be built.
Re-processing of the olefin-rich cracked naphtha can yield incremental light olefins. Visbreaker or coker naphthas may also be reprocessed. This operation can be carried out in the same or a separated reactor, or even a separate process if a custom catalyst is used. Recycling cracked naphtha will nevertheless entail an increase in dry gas yield and maximal gasoline product loss.
In the partial-catalyst-recycle approach, part of the spent catalyst is recycled back to the reaction section to increase the catalyst to oil ratio, achieving lower mixing temperature (less thermal cracking), and lower hydrogen transfer because of partial catalyst deactivation. Nevertheless, catalyst circulation needs to be increased largely, and selectivity is not always optimal.
Besides these modifications, nearly all designs also feature highly efficient atomizing nozzles to improve feed vaporization, as well as riser termination devices and efficient strippers to reduce recracking, minimizing dry gas and coke yield.
In conjunction with appropriate process modifications, optimized catalysts will be used, featuring low hydrogen transfer and high accessibility. The basic catalyst formulation relies on Y zeolite with low rare earth, highly accessible matrix to boost bottoms cracking when short contact time are used, together with large amounts of shape-selective ZSM5 catalyst. Beyond basics, proportions of the different components will be adapted to each design.
• High olefins catalytic cracking developed by Petrobras.
• Maxofin process by UOP.
• Petroriser from Axens.
• TMP from Sinopec.
• MILOS from SHELL.
Parallel risers have been arranged in different ways depending on the base FCC proprietary technology, featuring all the advances already applied by each licensor for cracking VGO at high severity. The fraction most commonly recycled is the light cracked naphtha, in particular the C5 to C7 fraction which has the highest content of olefins.
Several papers cite a Petrobras High Olefins FCC process running on naphtha recycle. A downer technology was also developed, with at least a pilot plant in use.135 A special additive formulation is used, comprising a mixture of ZSM5 and ferrierite zeolites, detailed in section 4.3.3.
An example of Maxofin process operation and yields is compared with FCC in Table 20.136 While VGO cracking operation is maintained at the same temperature as the traditional process (538 °C), ZSM-5 is added to the catalysts inventory and naphtha is recycled and cracked at 593 °C, with a catalyst to oil ratio of 25 (riser outlet temperature).
| Operating mode | Max C3 |
Interm. | Max. fuels |
|---|---|---|---|
| Recycle | Yes | No | No |
| ZSM-5 | Yes | Yes | No |
| Riser temperature/°C | 538/593 | 538 | 538 |
| Product yield (wt%) | |||
| C2 minus | 7.6 | 2.3 | 2.2 |
| Ethylene | 4.3 | 2.0 | 0.9 |
| Propylene | 18.4 | 14.4 | 6.2 |
| Butenes | 12.9 | 12.3 | 7.3 |
| Gasoline | 18.8 | 35.5 | 49.8 |
| Coke | 8.3 | 6.4 | 5.9 |
Compared with the base case with ZSM-5, propylene yield is boosted 4 wt%, ethylene and butenes adds another 3 wt%, with gasoline decreasing 17 wt%. The naphtha resulting from this operation is very rich in aromatics, with BTX making up to 60 wt% of C5–C10 naphtha fraction. The Maxofin technology is simply derived from the robust, well known Orthoflow® technology and adapted with dual riser technology. The dual riser operation, sharing a common catalyst, ensures that enough coke will be produced in the process to maintain heat balance.
Besides recycling naphtha from the primary cracking, it has also been proposed to incorporate other streams to the second riser, for example other olefinic streams, such as coker naphtha or C4 raffinate, increasing further the amount of light olefins produced.
Paraffinic naphtha may also be fed to Maxofin unit, but will convert to a lesser extent into small olefins due to the low reactivity of small paraffins at temperatures below 650 °C and relatively short contact time.
Petroriser is an adaptation of R2R technology from IFP/S&W with a second riser dedicated to recycling light naphtha.137 Besides increasing propylene (and ethylene) as well as reducing naphtha, there is a synergy between naphtha recycle and resid cracking, the first acting as a “chemical” cooler due to the very low coke make during naphtha cracking. Thus, it helps maintaining the heat balance, compensating the high coke from the resid operation. Axens announced that Petroriser™ technology had been licensed in Abu Dhabi (Ruwais refinery) for a very large RFCC unit (127000 bpd), using an atmospheric resid (API 21°, 4–6 CCR). Propylene yields above 12 wt% are expected. TMP process developed by State Key of Heavy Oil processing/Petrochina is another variant of the dual riser operation, aimed at optimizing propylene yield as well as maintaining reasonable distillate quality and reducing olefins in gasoline.138,139 It builds on a series of previous studies on dual riser/multiple injection points technology for example.140–142
Atmospheric resid (AR) is cracked in a first riser, with riser outlet temperature in the range of 500–520 °C. A C4 fraction may be injected upstream the AR to be cracked at very high temperature and cool down the catalyst before contacting the resid. In a second riser, naphtha is recycled to be cracked and increase olefins yields. Heavy cracked oil may be injected upstream naphtha, in order to crack this recycled feed under more severe conditions and reduce it as much as possible. The second riser outlet temperature is 520–540 °C. Some yields obtained by processing a Daqing AR (d20 = 900 kg m−3, CCR 4.5 wt%) are summarized in Table 21. Residence times in both risers are reported to be below 2 s.
A naphtha recycle process was also investigated by Shell: middle distillate and light olefins selective process (MILOS). Besides improving light olefins yields, its second objective was also to preserve or improve diesel cut, which is important for the European market. The second reactor is not a riser but a dense bed run at 565–620 °C, a temperature high enough to achieve reasonable naphtha conversion.143–145 The dense bed is fed with fresh catalyst from the regenerator.
Spent catalyst from this dense bed is mixed with regenerated catalyst and fed to the primary riser were the main cracking reaction takes place. Heavy oil/unconverted oil may be recycled to the reactor to maximize conversion. Catalyst used for maximum middle distillate mode may have moderate activity and a zeolite/matrix ratio between 1 and 2. ZSM-5 up to 10 wt% may be used to increase propylene yield.
With the appropriate feed and increasing VGO cracking severity, propylene yields of 23 wt% are claimed. Running in middle distillate mode may allow propylene yield as high as 17 wt% as compared with DCC process in Table 22. In both examples the feed has around 12.4 wt% hydrogen.
| Yield (wt%) | ||
|---|---|---|
| DCC | MILOS | |
| Dry gas | 10.3 | 5.4 |
| C2 minus (excl C2) |
4.9 | 2.3 |
| Ethylene | 5.4 | 3.1 |
| LPG | 36.3 | 37.6 |
| Propane | 3.0 | 2.1 |
| Propylene | 17.0 | 17.1 |
| Butanes | 4.9 | 5.8 |
| Butylenes | 11.4 | 12.6 |
| Gasoline | 29.5 | 28.2 |
| LCO | 10.4 | 15.8 |
| HCO | 5.8 | 8.0 |
| Coke | 6.6 | 5.0 |
5.1.3.1. Indmax (I-FCC, I-Max). This process was developed by Indian Oil Corporation and licensed by ABB Lummus Technology Inc.146,147 The range of operating conditions proposed for the process is the typical for maximizing olefins production: a reactor temperature in the 530–600 °C range, allowing a CTO between 12 and 20, a dilution steam of 3 to 50 wt% and a regeneration temperature of 650 to 750 °C. Feedstocks are the same as VGO, including residue, to be cracked in a riser type reactor.
In order to boost further the light olefins production, part of the products is recycled to the riser and injected above (downstream) the VGO inlet. Recycle stream can include up to 40 wt% of the naphtha, the diesel and the unconverted liquid products. In this configuration, LPG olefins yields of 20 to 40% were obtained, with a selectivity up to 80% of olefins in LPG fraction.
The catalyst employed in the Indmax process is claimed to be composed of 1–6 wt% of ultra stable Y-zeolite, 8–25 wt% of pentasil zeolite, 0–8 wt% of bottom selective active material, 0–1 wt% of rare-earth components and 91–60 wt% of non-acidic component and binder. A catalyst composition is adapted depending on the nature of the processed feedstock. The Indmax catalyst formulation is also highly tolerant of metals and can operate with high vanadium concentration on the equilibrium catalyst. This ability to accept feeds with high metal levels is very important for residue operations.109
The first commercial unit to utilize the Indmax process was commissioned in 2003 at the Guwahati Refinery (Assam, India). The plant performance test run data is presented in Table 23. The unit operates on a heavy feed with CCR of 3.75 wt%.
| Feed | |
|---|---|
| Feed composition | Atm. residue + delayed coker liquids |
| Feed density (ATB + CCHGO) | 0.9456 |
| Coker naphtha | 0.7164 |
| CCR (ATB + CHGO) | 3.75 wt% |
| Yield (wt%) (fresh feed basis) | |
|---|---|
| Dry gas/ethylene | 7.4/3.3 |
| LPG/propylene | 36.3/17.2 |
| Gasoline (C5 – 200 °C) | 34.7 |
| Conversion, wt% (up to 200 °C D86) | 86 |
| Gasoline octane (RON) | 99 |
5.1.3.2. Selective component cracking, SCC (ABB Lummus)148,149. The cracked products to be recycled to the process covers a range of materials such as higher carbon number olefins or straight run products from other conversion units. These components are injected separately, downstream from the fresh feed injection point through a set of injectors in the riser reactor system situated at a point where the conditions are ideal for cracking these components due to the high activity as well as the high temperature that the catalyst presents at this point. A unique feature applied in this invention that helps to preserve the yield of light olefins formed in the riser reaction zone is the fact that the cyclone situated at the reactor outlet is operated at a lower pressure than the interior of the vessel in the separation zone. This provides for a complete separation of the reacting hydrocarbons from the catalyst so as to quickly terminate secondary chain reactions. With an adequate paraffinic feed, yields of 24% propylene were reported (Table 24).149
| Feed API | 25.9 |
| Riser outlet, °C | 570 |
| Product yield (wt%) | |
| Dry gas | 7.2 |
| Propane | 2.8 |
| Propylene | 23.8 |
| Butane + butenes | 20.4 |
| Gasoline | 25.5 |
| LCO | 9.6 |
| Bottoms | 3.2 |
| Coke | 7.5 |
The combination with short contact time cracking allowed designing a reactor with two different contact time zones in the same riser reactor: the first one with higher temperature and very short contact time, followed by a second one with cooler reaction temperature and longer contact time.150–152 Reactions products are extracted between the first and second zone. This allows treating two feeds under significantly different reaction conditions: for example a lighter feed with lower reactivity (for example naphtha) in the first zone under more severe conditions, and a second, heavier feed in the second zone under milder conditions.
Two versions were proposed: in the first one, the feed is injected together with regenerated catalyst into a disengaging vessel, where it mixes with recycled spent catalyst. Hydrocarbons products are evacuated from the disengaging vessel through the cyclone, while the catalyst mixture is fed to the second, riser-type reaction zone.151 In the second version, regenerated catalyst and spent catalyst are readily mixed in the bottom Y section of the riser, and the mixture is fed to a riser reactor.152 In a first injection point a first feed is injected. At a short distance, a separation device allows separating 60–90% of the vapor from this first section as well as part of the catalyst, while the rest of the catalyst and vapor flow their path to the riser reactor. Downstream the separation device, a second feed is injected as shown in Fig. 11A.
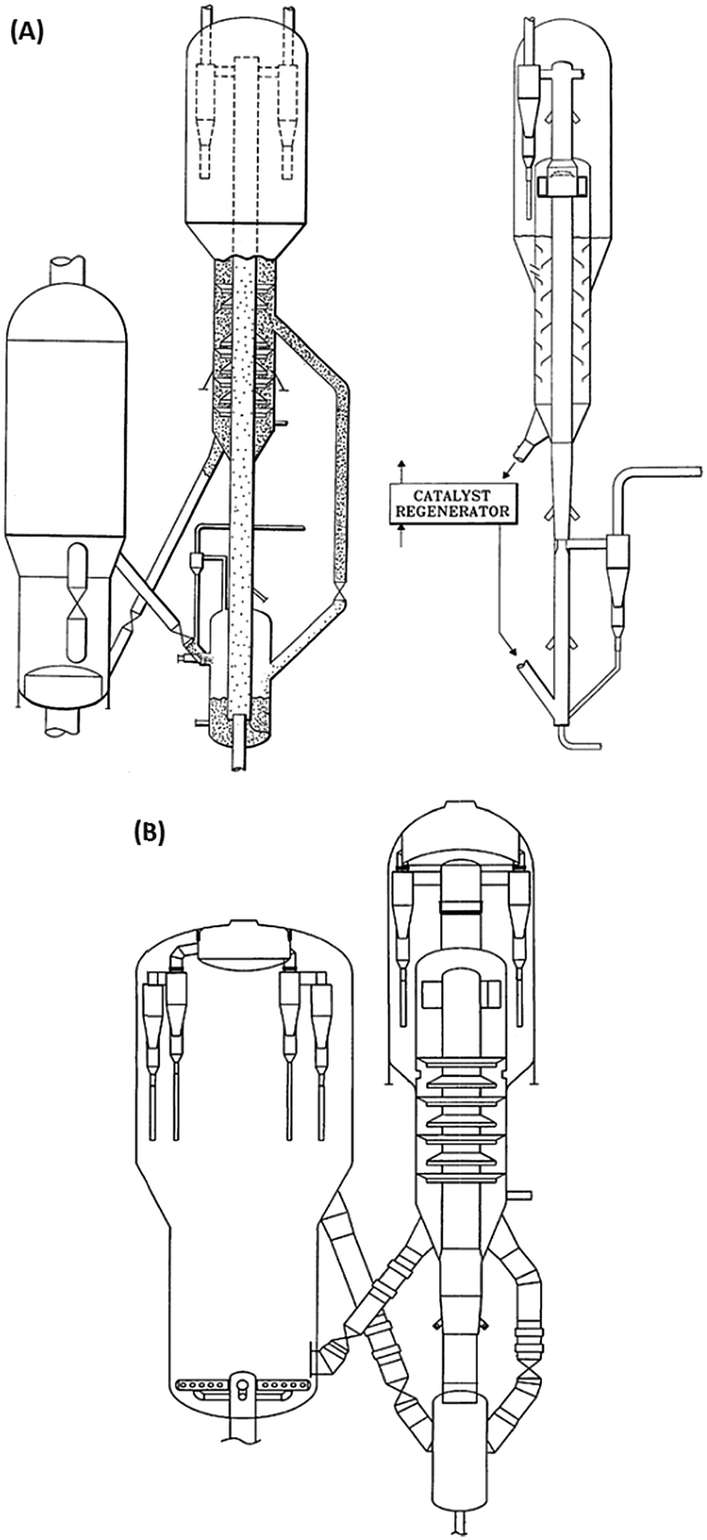 | ||
| Fig. 11 (A) RxCat reactor configurations. Reprinted from Lomas et al.151,152 (B) PetroFCC process schematics.153 | ||
This concept then allows producing higher yields of olefins processing for example low boiling range feedstocks fractions (i.e. naphtha for example) in the first zone and heavy boiling hydrocarbons in the riser zone. One limitation is the need to use the same catalyst compositions for each cracking zone.
The composition of the catalyst used in this configuration includes amorphous clay type catalysts with a 30% or more ZSM-5 zeolite dispersed in a porous inorganic carrier such as silica, alumina or zirconia. Circulating of a certain amount of inert material may present an advantage, decreasing the average coke-on-solid ratio of the solid entering the regenerator without affecting the solid to oil ratio on the reactor side of the process. Suitable inert solids are any refractory material with low coke production such as alpha alumina, fused alumina and low surface area clays.
The combination of RxCat technology and severe cracking operation conditions in the riser are the basis of the PetroFCC process licensed by UOP.153 A scheme of the unit is presented in Fig. 11B, where the RxCat mixing vessel is clearly visible. This process gives high yields of propylene, light olefins and aromatics from feedstocks which can include conventional FCC feeds as well as higher boiling or residual feeds.
The catalyst used in the petroFCC process has two components: a first component comprising a large pore molecular sieve (X zeolite or Y zeolite) with a limited content of rare-earth elements (≤1 wt%) and a second component comprising at least 1 wt% of a zeolite with no greater than medium pore size (for instance ZSM-5 or ST-5) (RxCat technology). The high concentration of medium or small pore zeolite in the second component of the catalyst composition improves selectivity to light olefins by further cracking the lighter naphtha range molecules.
However, at the same time, the resulting smaller concentration of the first catalyst composition still exhibits sufficient activity to maintain conversion of the heavier feed molecules to a reasonably high level. Regenerated catalyst is blended with coked catalyst, with a ratio of the coked to the regenerated catalyst in a range of 0.3 to 3.0 weight bases, to make the blended catalyst which is introduced in the riser reactor. The blended catalyst composition will contain at least 0.1 wt% coke before contacting the feed.
The recycling of the coked catalyst along with regenerated catalyst allows increasing the catalyst to feed ratio within the riser, avoiding the heat balance limitations of a conventional FCCU. UOP claims that blends of coked and regenerated catalyst (up to 50 wt%) have comparable activity to that of regenerated catalyst.152
Moreover, authors argue that recycling coked catalyst including large pore zeolite and/or an active amorphous material and a zeolite with no greater than medium average pore size and blending it with regenerated catalyst improve the yield of light olefins and the overall conversion. Additionally, according to them, the lower temperature of the catalyst resulting from blending hot regenerated catalyst on cooler recycled catalyst improves olefins selectivity.
The following operation conditions are typically used in the PetroFCC process in order to maximize the olefins yield and selectivity: a reactor temperature in the 510–620 °C range, a catalyst to oil ratio of at least 10:1 and preferably between 15 to 25 g g−1 and a steam diluent flow rate between 10 to 55 wt% of the feed flow rate in order to minimize the hydrocarbon partial pressure and achieve a contact time no greater than 2 seconds. Yields patterns from traditional FCC unit and a PetroFCC unit are compared in Table 25.
| Yield (wt%) | ||
|---|---|---|
| FCC | PetroRiser | |
| H2S, H2, C1, C2 | 2.0 | 3.0 |
| Ethylene | 1.0 | 6.0 |
| Propane | 1.8 | 2.0 |
| Propylene | 4.7 | 22.0 |
| Butanes | 4.5 | 5.0 |
| Butenes | 6.5 | 14.0 |
| Gasoline | 53.5 | 28.0 |
| LCO | 14.0 | 9.5 |
| Bottoms | 7.0 | 5.0 |
| Coke | 5.0 | 5.5 |
5.2. High severity, dedicated process for light olefins from heavy feeds
Yields of up to 15 wt% propylene and 15 wt% butylenes were claimed using a reactor temperature in the 500–650 °C range, catalyst to oil ratio in the 2–12 w/w range, steam to feedstock ratio of 0.01–2 by weight, WHSV in the range of 0.2 to 20 h−1 and a reaction pressure from 1.5 to 3 bar in a fluidized or dense phase transfer reactor. Further patents were taken on optimized conditions to enhance further the production of light olefins, as well as high octane gasoline.156,157
Two types of DCC operating modes are usually presented in the literature depending on the product slate desired. DCC type I is focused on propylene maximization and is carried out at higher severity reaction conditions while DCC type II works at lower severity operation conditions with the aim of increasing iso-olefins yields. Process yields for each DCC type are compared in Table 26 with conventional FCC yields and typical steam cracking yields obtained with a light gas oil.2,35,158 Reactor temperatures in DCC modes are substantially higher than in FCC, but remain largely below that of steam cracking (SC). Steam usage is also higher than for FCC, but, again, remains considerably lower than for SC. DCC catalyst circulation rates are higher than FCC operations.
| Type I | Type II | FCC | SCa | |
|---|---|---|---|---|
| a Typical yields from a vacuum distillate, low severity steam cracking. | ||||
| T/°C | 530–575 | 505–555 | 500–550 | 750–870 |
| Cat/oil (w/w) | 8–15 | 7–12 | 5–10 | — |
| Steam (feed wt%) | 20–30 | 10–15 | 1–6 | 30–80 |
| Type of cracking | Riser & bed | Riser | Riser | Batch |
| Typical yields (wt%) | ||||
| Dry gases | 11.9 | 5.6 | 3.5 | 28.8 |
| LPG | 42.2 | 34.5 | 17.6 | 27.3 |
| Ethylene | 6.1 | 2.3 | 0.8 | 19.4 |
| Propylene | 21.0 | 14.3 | 4.9 | 13.9 |
| Butylenes | 14.3 | 14.7 | 8.1 | 7.0 |
| Gasoline | 26.6 | 39.0 | 54.8 | 18.9 |
| Diesel | 6.6 | 9.8 | 10.2 | 25 |
| HCO | 6.1 | 5.8 | 9.3 | |
| Coke | 6.0 | 4.3 | 4.3 | — |
While paraffinic VGO feedstocks gives the highest propylene and isobutylene yields, naphthenic and aromatic feeds can also be processed, albeit a lower olefins yield is found due to the lower hydrogen content of the feed.110
The optimal conditions for olefins production imply an increase in dry gases yield meanwhile gasoline yield is reduced. This new yield product distribution will be economically affordable depending on the marked demand.
First patents claimed the use of acidic solid catalyst, formed by USY zeolite, a pentasil zeolite or a mixture of them supported on Kaolinite.159 Pentasil-type materials are abundantly used to promote propylene yield, while Y zeolite content was lowered as high temperature and residence time increased activity.
Each mode of operation may use an optimized catalyst. Formulations may contain varying amounts of high silica zeolite having pentasil type structure and Y zeolite. The catalyst named CRP-1 was developed for use in the DCC maximum propylene operation and presents a relatively low activity to ensure high olefin selectivity, while CS-1 and CZ-1 were developed to produce high isobutylene and isoamylene selectivity as well as propylene selectivity.154 All these catalysts, which can be modified/stabilized using well known FCC techniques such as adding rare earth and/or phosphorus, incorporate a high matrix activity for the primary cracking of the heavy hydrocarbons, with a modified mesoporous pentasil zeolite structure to enhance the secondary cracking of gasoline.160,161 These modifications are aimed at improving skeletal isomerization activity, lowering hydrogen transfer, maintaining coke selectivity and hydrothermal stability as well as high metal tolerance.
In order to increase further the yield of light olefins, a segregated feed pattern was implemented.162 A first cracking reaction is operated at relatively high weight hourly space velocity (>50 h−1), to convert approximately 35–60 wt% of the hydrocarbons feedstock at a temperature between 500 to 730 °C. A second cracking zone is operated at relatively low weight hourly space velocity (<30 h−1) to complete conversion, at a temperature between 480 and 680 °C, and a dilution steam of 20 wt% based on the weight of the feedstock. The riser features a first narrower and a second wider reactor section. Regeneration is carried out in two steps. In a first step, 40–80% of the coke deposited on catalyst is burned, while the remainder is burned at temperatures above 700 °C, to achieve complete removal of the carbon with excess oxygen. This configuration was claimed to produce more propylene and butenes, and less ethylene.
The DCC process can be integrated with other units to improve further olefins yields or change olefins distribution. Steam cracking may be used to crack efficiently small paraffins recycled from DCC output.23,163 In addition, integration with a disproportion unit would allow increasing propylene yield at the expense of ethylene and butenes. With a DCC unit producing 42–48 wt% of C2–C4 olefins, it was proposed to flexibilize propylene output from the complex between 17 and 33 wt%.
An example of operating conditions and resulting yields is given in Table 27A. The treated VGOs have a content of hydrogen of 12.8 wt%, which corresponds to an H/C ratio of approximately 1.76, close to that of paraffins.
| (A) | Operating mode | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| a Including 0.6 to 1.0% losses. | |||
| Riser outlet/°C | 576 | 610 | 640 |
| Regenerator/°C | 720 | 725 | 760 |
| Catalyst to oil ratio | 14.5 | 16.9 | 21.1 |
| Steam to oil ratio | 0.30 | 0.37 | 0.51 |
| Yields (wt%)a | |||
| Dry gas | 17.6 | 26.3 | 37.1 |
| LPG | 43.7 | 36.6 | 28.5 |
| Gasoline | 17.8 | 17.6 | 14.8 |
| LCO + HCO | 11.8 | 9.0 | 7.9 |
| Coke | 8.4 | 9.7 | 10.7 |
| Olefins yield (wt%) | |||
| Ethylene | 9.8 | 13.7 | 20.4 |
| Propylene | 24.6 | 21.5 | 18.2 |
| Butenes | 13.2 | 11.3 | 7.5 |
| (B) | Feed | |||
|---|---|---|---|---|
| VGO | AR | AR | VR | |
| H/C mol ratio | 1.89 | 1.82 | 1.79 | 1.76 |
| Aromatic carbon (wt%) | 6.8 | 10.9 | 13.0 | 13.8 |
| Yields (wt%) | ||||
| Dry gas | 26.3 | 24.1 | 28.2 | 27.4 |
| LPG | 41.7 | 42.2 | 37.1 | 35.2 |
| Gasoline | 16.3 | 16.9 | 14.9 | 16.5 |
| LCO + HCO | 4.5 | 5.1 | 5.4 | 6.2 |
| Coke | 11.2 | 11.18 | 14.3 | 14.8 |
| Olefins yield (wt%) | ||||
| Ethylene | 13.5 | 13.8 | 12.2 | 12.1 |
| Propylene | 22.6 | 22.6 | 19.3 | 19.9 |
| Butenes | 11.9 | 10.7 | 10.4 | 8.4 |
Increasing temperature from 580 to 640 °C, the propylene to ethylene ratio could be shifted from 2.25 to 0.9. In this example, and probably due to larger residence time, maximum propylene production was observed at 580 °C, while ethylene yield increased continuously with temperature. Residence time was not reported but the space velocities mentioned are in the range of 60–90 s, and seems that a fluidized bed, not riser cracking, is used for this operation. Gas residence time is not reported.
Laboratory results conducted on a fixed fluidized bed and rather short gas residence time (1–4 s) showed that propylene yield passes through a maximum in the range of 630–660 °C, independently of the feed used, while ethylene yield increased continuously with temperature.164–166 Residence time, catalyst to oil ratio and temperature were varied to shift product yield, although temperature changes had the largest effects (Table 28).
| Reaction temperature (°C) | |||||||
|---|---|---|---|---|---|---|---|
| 600 | 620 | 640 | 660 | 680 | 700 | 716 | |
| Yield (wt%) | |||||||
| Dry gas | 12.8 | 16.1 | 19.6 | 23.1 | 27.7 | 32.4 | 35.6 |
| LPG | 45.7 | 46.6 | 45.8 | 43.2 | 39.3 | 33.9 | 30.1 |
| Gasoline | 24.2 | 20.0 | 18.3 | 16.9 | 15.1 | 15.5 | 14.1 |
| LCO + HCO | 8.3 | 7.9 | 6.7 | 6.1 | 6.3 | 6.2 | 5.7 |
| Coke | 9.0 | 9.5 | 9.6 | 10.8 | 11.6 | 12.0 | 14.4 |
| Olefins yield (wt%) | |||||||
| Ethylene | 8.7 | 10.8 | 12.4 | 13.8 | 15.6 | 17.3 | 18.3 |
| Propylene | 18.2 | 21.3 | 22.5 | 22.6 | 22.1 | 19.9 | 18.0 |
| Butenes | 10.8 | 11.2 | 11.2 | 10.7 | 9.7 | 8.2 | 6.8 |
The feedstock has the second largest influence on yields. Increasing aromatics content of the feed, measured by the weight percentage of hydrogen in fed or KUOP factor, will decrease small olefins yields. The feed tested had an H/C molar ratio between 1.76 and 1.89, which corresponds to hydrogen content in the feed between 12.8 and 13.6 wt%. Increasing steam to oil ratio from 0.3 to 1.6 was also reported as very beneficial, increasing small olefins yield (Table 27B).
Also, the increase of conversion with increased steam to oil ratio (i.e. decreasing oil partial pressure) was explained by lower catalyst deactivation, although coke yield barely changed with steam to oil ratio (decreasing only 10 wt% at higher steam to oil ratio). The presence of a significant amount of aromatic rings, and especially dealkylated aromatic rings, may be the source of a significant deactivation that gets lowered (diluted) with increasing steam.
While thermal cracking contribution is often obviated in fluid catalytic cracking (but for dry gas yield), its contribution may become significant in light olefins production owing the much higher processing temperatures. It was shown that thermal pyrolysis could reach yields of light olefins close to those of catalytic cracking when the temperature reached 700 °C (Fig. 12). Nevertheless, olefins distribution is much displaced toward ethylene, in thermal cracking, yielding lower P/E ratio, generally below 1.
 | ||
| Fig. 12 Light olefin yield with or without catalyst, against process temperature.167 | ||
Recycling of distillate streams such as naphtha and diesel is a common technique to increase light olefins yields. Nevertheless, in the case of catalytic pyrolysis, it showed little effects.167,168 These streams are already largely aromatic, with little paraffin remaining. The source for small olefins is then mostly alkyl groups on aromatics, yielding around 10 wt% light olefins and increasing selectivity towards rings or ring systems with small alkyl groups (BTX, naphthalenes, phenanthrenes, etc.).
A large number of catalyst acronyms have been used by RIPP/Sinopec, yet formulation is rarely disclosed. As for any catalyst system aimed at maximizing light olefins, the matrix and first cracking component (for example Y zeolite) will be tailored with an easily accessible pore system to enhance the conversion of large molecules with rapid diffusion of intermediate products, that are further cracked into small olefins on the second cracking component. Desirable properties of the second cracking component, based on ZSM-5 type structure, are enhanced hydrothermal stability, and high acidity and/or some dehydrogenation ability to promote small paraffin cracking into olefins.
Hydrothermal resistance of the zeolitic components may be stabilized by phosphorus (MFI) and La (FAU) as detailed in section 4.3.
A preparation of ZSM-5 structure incorporating rare-earth ions, based on a seeding method in which an REY zeolite is dispersed in a gel containing Si-, Al-, Na-sources and water and the mixture converted into a MFI type zeolite, was also claimed to give enhanced hydrothermal stability.160 Resulting material, further modified under hydrothermal conditions, was denoted as ZRP. Phosphorus incorporation during synthesis resulted in the development of CEP-1 catalyst.23
Treatment with alkaline ions was found to enhance further the selectivity to ethylene by modifying acid site distribution and strength (PMZ materials).
Metals were also added to increase even further selectivity to small olefins. Phosphorus exchanged ZSM-5 was further impregnated with Mg and Ni, Zn or Cu and yielded more propylene and ethylene (and slightly more coke) at the expense of other components. Total C2–C4 olefins increased from 48 to 53 wt%.169 Claimed compositions for the modified ZSM-5 were SiO2/Al2O3 ratio between 15 and 60, 2–8 wt% P, 0.5–3 wt% alkaline earth (Mg or Ca), 0.5 to 3 wt% metal (Cr, Fe, Co, Ni, Cu, Zn), all based on corresponding oxide weight. Zn-bearing ZSM-5 was found most active, followed by Ni. Coke production was slightly higher with Ni. Silver-promoted ZSM-5 was also said to promote ethylene and propylene yields in thermal cracking of gas oil at 650 °C.23 An early patent from Pop et al. also reported enhanced activity and selectivity for Cu, Ag, Co-exchanged mordenite.170 Cracking of a C9–C14 paraffin cut was performed with high steam to hydrocarbon ratio (2–3), yielding 67 wt% C2–C4 olefins at 725 °C.
CEP catalyst is often compared with LCM materials, which are claimed as oxide based material designed for heavy oil cracking (HCC process). It is based on silica-alumina, which may be doped with alkaline or alkaline earth or transition-metal oxides. It may also carry some molecular sieve catalyst.171
Several models of increasing complexity with detailed light olefins composition were developed for heavy-oil cracking pyrolysis and fitted from laboratory results.172–174 Deactivation was modelled by an exponential of time or coke on catalyst, with largely different values. Deactivation was found to be small by Liu et al., but significant by Meng. Differences in catalyst type and process temperature may have explained the divergent findings. Thermal and catalytic contributions were not separated, so that intermediate activation energies were found, in the 100–200 kJ mol−1 range. Dry gas and ethylene formation presented higher activation energies probably because of a significant thermal contribution.
The CPP process may be used in combination with a steam cracker (SC, on the lighter fraction of the feed) to yield maximum short olefins from full cut crude oil, as detailed in section 2. Paraffins yield much better results than naphthenes or aromatics (aromatics rings will hardly crack) so that paraffinic crudes are much preferred for increasing small olefins yields. Hydrotreatment of non-paraffinic crudes may be carried out, but this brings a number of problems related to metals and CCR in the crude oil. Naphthas from steam cracking or CPP are also very rich in BTX. Economic evaluation showed that CPP plants integrated with steam crackers will have a very fast payout.23
Spent catalyst can also be recycled to the reactor to increase the catalyst to oil ratio in the same way as PetroFCC design does. The use of baffled separation devices for both reactor and regenerator circulating beds allowed to design a very compact unit as shown in Fig. 13, reducing catalyst inventory and improving heat transfer efficiency.176,177
 | ||
| Fig. 13 A simplified schema of the NEXCC process reaction system.175 | ||
It also permits maintaining a higher solid density in the circulating beds, allowing a higher conversion rate per volume.
As solid catalyst, NEXCC process can use conventional cracking catalyst based on natural and synthetic aluminum silicates, zeolites, clays, Y and ZSM-5 zeolite, stabilized by rare-earth and phosphorus. Short contact time and efficient separation and stripping enables the unit to use also improved cracking catalyst with higher loadings of zeolite. An increase of 10 points of propylene + butenes yields could be obtained while maintaining bottoms conversion (Table 29). It was also claimed that the flexibility of the design allowed extending operation to other reactions that may take advantage of short contact time, high temperature and efficient heat transfer between reactor and regenerator. For example, it was proposed to carry out thermal cracking of crude oil or heavy oil at short contact time of 0.2–0.5 s, high reactor temperature between 650 and 950 °C with an inner particulate heat carrier to yield light olefins and aromatics.
| FCC | NEXCC pilot plant | |
|---|---|---|
| Dry gases | Base | +1.6 |
| Propylene | Base | +5.0 |
| i-Butane | Base | −0.8 |
| i-Butene | Base | +2.3 |
| C4 alkanes | Base | −1.3 |
| C4 olefins | Base | +4.9 |
| Total LPG | Base | +8.4 |
| Gasoline | Base | −10.0 |
| LCO + HCO | Base | 0.0 |
Dehydrogenation of small paraffins was also proposed as a process with high heat requirements. Ethane to butane feedstocks could be processed at temperature ranging from 650 to 750 °C, 0.5–4 s contact time, with typical Cr–alumina or vanadium based dehydrogenation catalysts, yielding ethylene, propylene or butenes depending on the feed. Other reactions such as oxidative coupling of methane, or gasification of a variety of feedstocks ranging from methane to biomass, coal or bottoms residues were also proposed.
To accomplish this objective, the HS-FCC process works at high reaction temperature (550–700 °C), short contact time (0.1–3 s) and high catalyst to oil ratio (15–50 wt/wt) in a downer type reactor. After reaction, the mixture of the products, unreacted materials and catalyst is forwarded into a separation zone (initially a box-type or a U-bent type), where at least 90% of the catalyst is separated before the catalyst is precisely removed from the mixture in a cyclone separation zone. The separation system was later improved by the use of a baffled separation system. After stripping, the catalyst is lifted by the circulating flow regenerator to a hopper from which it is fed back to the downer reactor. Feedstocks are the usually used in fluid catalytic cracking.
A good management of temperature in the reactor and reactor inlet is more critical than in conventional FCC due to the higher mean temperature and shorter contact time involved.180 Increasing reactor temperature for higher olefins yield and bottoms cracking implies raising sharply inlet temperature, leading to adverse effects such as increased thermal cracking, thus dry gas yield, and coke yield.
In order to mitigate this effect, it was proposed to have two or more catalyst inlets in the downer reactor. A first part of the regenerated catalyst, between 20 and 95% of the total catalyst flow to reactor, is fed at the reaction zone inlet where it contacts with hydrocarbon, while the rest (5–80 wt%) is fed downstream at one or several locations before the separation device. A mixture of regenerated catalyst and incompletely regenerated catalyst can also be fed at the reaction zone inlet which allows decreasing catalyst inlet temperature. The raw oil is then cracked under mild conditions, restraining the generation of unsuitable by-products such as dry gas and coke. In another improvement, residual oil is recycled to the process and serves as quench oil at the inlet of the separation device.
While it is common to quench the reaction mixture at reactor outlet, it becomes less effective at higher catalyst to oil ratio due to the larger thermal mass. Also, the higher reactor temperature would call for a higher temperature drop during the quench for being effective, increasing quench flow rate to unsustainable levels. It was found however that the use of residual oil, which is rich in heavy aromatics, was effective as quench media although temperature drop was minimal after quench.
Catalysts used in these inventions contain the ultrastable Y-zeolite (2–60 wt%), a matrix and a crystalline aluminosilicate zeolite or SAPO each having smaller pores than ultrastable Y-type zeolite. It may also be preferable to use a Y zeolite with a low rare-earth content, for example lower than 0.5 wt%, in order to minimize hydrogen transfer reactions.181 At the same time, larger amounts (5–40 wt%) of shape selective zeolite, more preferably ZSM-5 zeolite, are recommended to take full advantage of the less saturated products yielded by low RE cracking catalysts, maximizing light olefins content.
Based on the intrinsic features of the HS-FCC, maximum light olefin yield can be obtained by the combination of optimized catalyst system and operation conditions. The yields of light olefins, LPG, gasoline, LCO, HCO and coke are presented in Table 30 for conventional FCC and HS-FCC.
| FCC | HS-FCC | |||
|---|---|---|---|---|
| Base | +10 wt% ZSM-5 | Base | +10 wt% ZSM-5 | |
| Dry gases | 5.3 | 6.4 | 4.6 | 5.5 |
| LPG | 20.7 | 29.3 | 30.9 | 40.5 |
| Propylene | 7.5 | 13.0 | 10.7 | 18.4 |
| Butenes | 8.8 | 13.6 | 16.1 | 17.8 |
| Gasoline | 43.4 | 34.1 | 45.4 | 34.0 |
| LCO | 15.0 | 15.4 | 9.4 | 9.3 |
| HCO | 13.3 | 12.7 | 6.6 | 7.1 |
| Coke | 2.3 | 2.1 | 3.1 | 3.5 |
In conventional FCC, the base catalyst yielded about 16 wt% light olefins and 43 wt% gasoline compared to 29 wt% light olefins and 45 wt% gasoline in HS-FCC. In the case of ZSM-5 addition, the yield of light olefins increased to more than 37 wt%, particularly propylene which showed an increase of 72 wt%. The results showed that, in both cases, the rise in light olefins was accompanied with a drop in gasoline yield since the addition of ZSM-5 accelerates the cracking of gasoline to lighter products.182
Millisecond catalytic cracking process was commercialized by UOP and CEPOC.183 In this process a stream of hydrocarbons is sprayed perpendicularly into a downwards flow of catalyst (Fig. 14). The mixture is then fed directly into a separation device located in front of the feed inlet, ensuring very low residence time of catalyst and feed in the reactor. In the MSCC process catalyst used is composed of at least 40 wt% of zeolitic component and contact times are less than 0.5 s. It was observed that the bottoms conversion was limited, hence the very high zeolite content in the catalyst formulation.
 | ||
| Fig. 14 MSCC reactor – enlarged view of the horizontal contactor.183 | ||
The solid downflow allows increasing significantly the catalyst to oil ratio as well as access to very short contact time, which allows in turn increasing the reactor temperature, while thermal cracking is maintained controlled. Although this is not its primary objective, this process arrangement is ideal to promote formation of light olefins when combined with the adequate catalyst.
In 1994, Cepoc revamped its conventional FCCU to a short-contact-time system known as the MSCC process and some of the benefits realized were:184
• Increased resid processing capability and improved yield structure through enhanced catalytic reactions.
• Increased overall liquid-volume yield and higher hydrogen content in the unconverted products.
• Higher LPG olefinicity, gasoline octane and light-cycle oil (LCO) cetane. Reduced dry gas yield.
• Reduced dehydrogenation by contaminant metals even though metals levels increased.
• Reduced catalyst addition rate from 15 to 25 tons per day to 8–10 tons per day.
The TCR technology developed at the beginning of the 1980s by Stone & Webster was further extended, replacing the inert solid carrier by a catalyst.185,186 While initially designed for olefins production, it was promoted later as a short contact time process with the advantages inherent to this type of process for the processing of VGO: reduced dry gas, higher gasoline yield, lower coke yield.187
The flow scheme is similar to the HS-FCC process. Its main feature is a downer reactor, with contact times claimed to be as short as 200 ms.188 The catalyst distributor, feed injection and downer outlet separator were carefully designed to ensure a good contact between hydrocarbons and catalyst as well as an efficient separation to take full advantage of short contact time. The solid is fed to the reactor from a hopper situated above the injection zone. Hydrocarbon is injected in the annular area surrounding the solid inlet, through angled openings to limit erosion of the wall.
A mixing plug creates several discrete mixing zones of reduced volume, ensuring more homogeneous distribution of solid and gas across the reactor section. If reaction time has to be kept minimal, solid separation must also be fast. Such a system is reported to separate more than 98% of the solid within 30 ms. The solid from the reactor impinges the lower wall of the separator, accumulating at the bottom and flowing by gravity to the stripping section. Gas is forced to turn upwards and enters a cyclone where the remains of solid are separated back to stripper.
The stripped catalyst is then contacted with air in an upwards transport bed, being regenerated and brought back to the catalyst hopper.187 Operating parameters were claimed as 540–650 °C reactor temperature, 5–15 catalyst to oil ratio, steam to hydrocarbon ratio 0.2–0.4, residence time in reactor from 0.02 to 5 s.
5.3. Dedicated catalytic cracking processes for light olefins: boosting small olefins yields through naphtha cracking
• Superflex, as an extension of Maxofin process (KBR).
• Propylene catalytic cracker (ExxonMobil).
Superflex® is based on technology developed by ARCO Chemical Company (now LyondellBasell, patent owner) and is licensed by KBR with the same Orthoflow® reactor technology found for KBR FCC processes.136 Its preferred feed is C4–C10 streams with high content of olefins, and may be sourced from C4–C5 fractions from steam cracker, light cracked naphtha from FCC, coker naphtha or even olefinic stream from the Fischer–Tropsch process. Non-aromatic, C4–C6 fractions are recycled to extinction in the process to maximize small olefins and BTX output. Operating conditions may be in the range of 630–650 °C reactor temperature, with typically 10 wt% steam added to the hydrocarbon. Depending on feedstock, propylene yield averages 40 wt% while total ethylene plus propylene yield averages 60 wt%. This process was proposed as a good synergy with a steam cracker based on naphtha or distillate, which generates a substantial amount of olefins in pyrolytic naphtha.189 One unit is in operation in SASOL, running on an olefinic naphtha stream derived from FT operation. Yields of 10–15 wt% fuel gas, 15–20 wt% ethylene, 35–40 wt% propylene, 5–7 wt% propane and 20–25 wt% gasoline can be attained recycling C4–C6 fractions to extinction. The naphtha stream produced from the process is highly aromatic, and thus may be used as a high octane, low olefin blend component for motor gasoline, or separated to valorise BTX.
Maintaining heat balance may be difficult with naphtha feed and low-coke making catalysts such as ZSM-5 or similar. Thus, additional heat has to be generated. This can be made directly by injecting a fuel, for example fuel oil recycled from the process, into the regenerator.190
Dual riser may also be used as in Maxofin™ process, in order to process paraffinic and olefinic stream under separate conditions, and where an amount of coke precursor, such as diolefins, is added to one of the feeds to provide enough coke for the process to be autothermal.191 It has also been proposed to use the technology with a Ga-doped ZSM-5 catalyst to obtain BTX from small paraffins, for example propane.192
The propylene catalytic cracker process from ExxonMobil is also based on a reactor regenerator system derived from Exxon FCC technology, and using ZSM-5 as main catalyst. Butylenes, C5 and/or light cracked naphtha are fed to the reactor. Temperature is in a range that allows high conversion of olefins but that is not high enough to obtain high conversion of small paraffins, so that a fraction of naphtha is conserved.193 Carefully designed riser reactor provides narrow residence time, that allow producing C2 and C3 streams with very high olefin content. Chemical grade (95%) propylene stream may be produced directly from the unit, avoiding costly fractionation.
Other technologies may be used, for example fixed bed technology (Propylur process) or oligomerization/cracking technology (Flexene process).
The Flexene process licensed by AXENS is an integration between FCC and PolyNaphtha™ (Oligomerization process) unit to improve either propylene yield or middle distillate or gasoline (Fig. 15). For maximum propylene mode, C4–C9 olefins are recovered, oligomerized to higher C8 and C12 olefins and recycled back to the FCC unit to be cracked to propylene. Based on the process conditions, about 3–6 wt% of propylene can be added to the product stream.180,194–196 Processing oligomerized olefins showed better selectivity for propylene production than recycling light cracked naphtha (LCN) alone. The selectivity of the two options compared to the base case where direct C4 is cracked is illustrated in Fig. 16. The relative propylene selectivity when cracking oligomerized C4 stream is about 2.3 compared to 1.9 for cracking LCN.
 | ||
| Fig. 15 Integration of PolyNaphthaTM with FCC technology for propylene maximization.196 | ||
 | ||
| Fig. 16 Enhancement of propylene selectivity with flexene vs. recycle.194 | ||
This process was developed to produce short olefins from streams rich in small alkanes such as light straight run naphtha. It has the advantage over steam cracking to produce propylene and ethylene at a ratio of 1:1 or higher, whereas naphtha steam cracking is usually limited to 0.5:1.
Another advantage is a reduction in dienes, and particularly acetylene, which facilitates further purification of small olefins to polymer grade.197
K-COT™ process has similar or slightly lower ethylene yield than a steam cracker (Fig. 17) but with twice the yield of propylene. The total yield of ethylene and propylene can reach 60–65 wt%, similar or higher than a steam cracker.
 | ||
| Fig. 17 Yield pattern in ACO process compared with thermal cracking for LSR naphtha processing.172 | ||
It uses a robust, zeolite catalyst, most likely based on pentasil type, at a temperature between 600 and 700 °C, with the injection of steam. The catalyst is specially formulated to show high performance with paraffinic feeds, and high hydrothermal resistance.
Compact Orthoflow® design is used in the K-COT™ process. Typical features of high performance FCC, such as closed cyclones, are also used. As happens in cracking, the process is endothermic, yet the very low coke make from naphtha requires external heat to be supplied.
As Superflex process, the process may uses dual risers with the purpose of building coke and generating heat in the regenerator without the need of firing supplemental fuels. Propylene to ethylene ratio decreases with temperature, from 1.1 to 0.7. Tail gas is lower than in thermal crackers.
Also, the C4–C6 fraction can be recycled to the reactor without hydrogenation (but benzene). Gasoline is also richer in BTX than pyrolysis gasoline from steam cracker.
A commercial trial was programmed for 2010 at Ulsan refinery in partnership with SK Energy. The operating conditions of this process are similar to that of catalytic pyrolysis process described in an earlier section, but with the advantage of using a specialty catalyst designed for naphtha cracking.
As a kind of catalytically assisted steam cracking, it also has the advantage to run at substantially lower temperature, 650–750 °C compared with typically 850 °C for naphtha steam cracking.
As coke is formed on the catalyst, it is not affected by wall coking, as the catalyst is constantly regenerated. Also, the in situ heat generation by coke combustion avoids the contact with extremely hot walls that accelerates the coke formation in steam crackers.
6. Summary and outlook
Today crude oil refining is centered on the production of fuel as main product, with collateral production of minor (but valuable) tonnage of sub-products such as petrochemicals. In certain areas, due to an increasing olefins market demand coupled with a limited supply from the refinery, new transformation complexes focused on petrochemicals production directly from crude oil may appear, with fuels regarded as indirect secondary products. A central part of such complex could be a catalytic conversion unit such as an FCC like process optimized for light olefins.The very limited flexibility of thermal processes, such as steam cracking, and in particular the low and relatively inflexible propylene to ethylene ratio, together with the difficulties to use low grade crude as feedstock due to excessive wall coking, indicates that future technologies based on catalytic cracking will be better positioned to fulfill impending market demand. The technology already existing in FCC field to boost light olefins production could be extended to process the whole crude oil fraction, propelling the direct petrochemicals production in the 30–50% yield range, which is far beyond the current levels in refinery. Process optimization for light olefins in FCC process were largely detailed in this review and, generally, features cracking temperature significantly above FCC range. As a consequence, short contact time and high catalyst-to-oil ratio will be used, making downer reactors an ideal reactor configuration. The cracking component based on Y zeolite is tailored to have as low hydrogen transfer activity as possible. That means low rare-earth composition, and high accessibility to favour bottoms conversion while limiting hydrogen transfer, yielding a maximum of material to be transformed further into light olefins by a shape selective catalyst. Active material content, including active matrix, in inventory may not require to be high since high CTO and temperature will bolster catalyst activity. This is even more true if one considers that coke make needs is reduced to minimum and, for instance, active ZSM-5 with optimized Si/Al ratio in the range of the hundreds and high accessibility may be used, to limit hydrogen transfer while keeping high activity.
The conversion of the lighter, naphtha range part of the crude oil, usually very paraffinic, may prove however more challenging using the same operating conditions and the same catalyst than for the heavier part of the crude oil. This stable light fraction may be cracked under conditions (higher temperature, very active catalysts), that would not be adequate with heavier feeds due to excessive coke and dry gas yields. It will remain challenging to find a catalyst formulation with an activity fully adapted to the whole range of the crude oil, from very stable, low molecular weight paraffins to high molecular weight, high coke make asphaltenes. Some may prefer to process the light fraction in a separate reactor. This is often carried out with the cracked naphtha, which is reprocessed in a reactor (riser or downer) in parallel to the main conversion reactor to crack the abundant stock of gasoline range olefins and increase further light olefins yields. Improving further the light olefins yield of this light fraction may be carried out in a separate process with a dedicated catalyst of high cracking activity, eventually improved with metals not allowed in traditional FCC for their coke make. Until a better catalyst is developed, multi-reactors units may remain an essential feature of such processes.
As an alternative strategy, a number of units may come to support the catalytic cracking converter, with the objective of converting lighter fractions that may not crack efficiently towards olefins or aromatics in the catalytic cracker. A steam cracker may be used in its traditional way and a catalytic reformer may be used if more aromatics are sought. Metathesis units may be used to flexibilize further the ratio between light olefins, transforming ethylene and butenes into more propylene. Finally, an important issue in the near future that may impact an olefins oriented catalytic cracker will be the incorporation of increasing amounts of unconventional feedstocks with diverse compositions such as extra heavy crude oils, light tight oils and/or biocrudes derived from biomass resources. The intrinsic properties of these feeds, will call for additional research to obtain optimized processes and catalytic systems adapted to each of these emerging feedstocks.199 The high average molecular weight, low hydrogen to carbon ratio and high contaminants content for extra heavy crude oils will call for catalysts with better resistence to contaminants (metals) and ultra low coke make. Active cracking component amount may be reduced, and a significant part of the cracking performed thermally. By contrast, low molecular, paraffinic tight oils may benefit from highly active materials, as small paraffins are difficult to crack. Also, increased resistance to Ca/Na/Fe was recommended. ZSM5 catalysts may be used for the direct conversion of acidic, high oxygen content and low thermal stability biocrudes to obtain preferentially aromatics or chemicals.200,201 Meanwhile, hydrotreated, triglyceride-based oils yielding feedstocks similar to a paraffinic crude oil may be preferred to obtain light olefins.
Acknowledgements
The authors thank Saudi Aramco for material and financial support. Financial support by the Spanish Government-MINECO through program “Severo Ochoa” (SEV 2012-0267), Consolider Ingenio (2010-Multicat, CSD-2009-0050), MAT2012-31657, CTQ2015-70126-R (MINECO/FEDER), by the European Union through ERC-AdG-2014-671093–SynCatMatch and by the Generalitat Valenciana through the Prometeo program (PROMETEOII/2013/011) is also acknowledged.Notes and references
-
Kirk-Othmer Encyclopedia of Chemical Technology, 4th edn, 1991, vol. 9, p. 902 Search PubMed
.
-
A. Chauvel and G. Lefebvre, in Petrochemical processes, Editions Technip, Paris, 1989, ch. 2.1, pp. 117–164 Search PubMed
- Y. C. Hu, Hydrocarbon Process., 1982, 61(11), 109–116 CAS
- Y. C. Hu, Hydrocarbon Process., 1983, 62(5), 88–96 CAS
-
D. H. Powers, US Pat., 6743961, 2004 (Equistar Chemicals, LP) Search PubMed
-
D. H. Powers, US Pat., 7019187, 2006 (Equistar Chemicals, LP) Search PubMed
- P. Schmalfeld, Hydrocarbon Process. Pet. Refin., 1963, 42(7), 145–148 CAS
- D. Kunii, Chem. Eng. Sci., 1980, 35, 1887–1910 CrossRef CAS
-
W. D. Fitzharris, S. J. Ringle and K. S. Nicholes, US Pat., 4859310, 1989 (Amoco Corporation) Search PubMed
- A. Corma, F. V. Melo, L. Sauvanaud and F. J. Ortega, Appl. Catal., A, 2004, 265, 195–206 CrossRef CAS
-
D. H. Powers, US Pat., 6979757, 2005 (Equistar Chemicals, LP) Search PubMed
-
R. S. Bridges, R. B. Halsey and D. H. Powers, US Pat., 2001016673, 2001 (LyondellBasell Industries) Search PubMed
-
D. H. Powers, US Pat., 2008093261, 2008 (Lyondell Chemical Company) Search PubMed
-
D. H. Powers, US Pat., 2009048475, 2009 (Lyondell Chemical Company) Search PubMed
-
G. B. Wirth and C. E. Jahnig, US Pat., 3617493, 1971 (Esso Research and Engineering Company) Search PubMed
-
R. C. Stell, J. L. Bancroft, A. R. Dinicoloantonio and G. Stephens, US Pat., 7097758, 2004 (ExxonMobil Chemical Patents Inc.) Search PubMed
-
D. Yuk-Kwan Ngan, P. Chan and A. J. Baumgartner, US Pat., 6632351, 2003 (Shell Oil Company) Search PubMed
-
A. Bourane, R. Shafi, E. Sayed, I. A. Abba and A. Akhras, WO Pat., 2013112966, 2013 (Saudi Arabian Oil Company) Search PubMed
-
R. Shafi, A. Bourane, I. A. Abba and A. Akhras, WO Pat., 2013112970, 2013 (Saudi Arabian Oil Company) Search PubMed
-
I. A. Abba, R. Shafi, A. Bourane, E. Sayed and A. Akhras, WO Pat., 2013142609, 2013 (Saudi Arabian Oil Company) Search PubMed
-
T. Ino, T. Okuhara, M. Abul-Hamayel, A. Aitani and A. Maghrabi, US Pat., 6656346, 2003 (King Fahd University of Petroleum and Minerals; Petroleum Energy Center) Search PubMed
- T. Ino, Y. Fujiyama, H. Redhwi, A. Aitani and M. R. Saeed, Davison Catagram, 2004, vol. 94, pp. 45–49 Search PubMed.
-
W. Xieqing, X. Chaogang, L. Zaiting and Z. Genquan, in Practical advances in Petroleum Processing, ed. C. S. Hsu and P. Robinson, Spinger-Verlag, New York, 2006, ch. 5, vol. 1, pp. 149–168 Search PubMed
-
I. A. Abba, R. Shafi, A. Bourane and E. Sayed, WO Pat., 2013142563, 2014 (Saudi Arabian Oil Company) Search PubMed
-
W. Xu and I. Abba, WO Pat., 2013188729, 2013 (Saudi Arabian Oil Company) Search PubMed
- A. Steinhofer, Hydrocarbon Process., 1965, 44(8), 134–142 CAS
- T. Kunugi, D. Kunii, H. Tominaga, T. Sakai, S. Mabuchi and K. Takeshige, J. Jpn. Pet. Inst., 1973, 16(3), 232–248 CrossRef CAS
-
T. Ueda, M. Ohta and S. Sugano, US Pat., 4259177, 1981 Search PubMed
- M. Matsunami, Y. Suxukawa and G. Nakawa, Hydrocarbon Process., 1970, 49(11), 121–126 Search PubMed
- P. Schmalfeld, Erdoel Kohle, Erdgas, Petrochem., 1963, 16(6-I), 534–537 CAS
- P. Schmalfeld, Erdoel Kohle, Erdgas, Petrochem., 1961, 14, 537–541 CAS
- P. Schmalfeld, Chim. Ind., 1961, 86, 231–242 CAS
-
H. N. Woebcke, A. H. Bhojwani, R. J. Gartside and A. R. Johnson, US Pat., 4318800, 1982 (Stone & Webster Engineering Corp.) Search PubMed
- A. Ma'adhah, M. Abul-Hamayel, A. Aitani, T. Ino and T. Okuhara, Oil Gas J., 2000, 98(33), 66–70 Search PubMed
- P. O'Connor, A. Hakuli and P. Imhof, Stud. Surf. Sci. Catal., 2004, 149, 305–321 CrossRef
- C. R. Schlosser, A. De Rezende Pinho, P. O'Connor, R. D. Baptista, E. F. Sandes and M. A. Torem, Akzo Nobel Catalysts Symposium, ed. S. Docter and Akzo Nobel, The Netherlands, June 2001 Search PubMed.
- A. De Rezende Pinho, E. Morgado and K. Maeshiro, Prepr. - Am. Chem. Soc., Div. Pet. Chem., 2003, 48(3), 167–169 CAS
- A. Aitani, T. Yoshikawa and T. Ino, Catal. Today, 2000, 60, 111–117 CrossRef CAS
- A. Farshi, Pet. Sci. Technol., 2011, 29, 2113–2122 CrossRef CAS
- J. S. Buchanan and Y. G. Adewuyi, Appl. Catal., A, 1996, 134, 247–262 CrossRef CAS
-
R. Sadeghbeigi, in Fluid catalytic cracking handbook – Design, operation and troubleshooting of FCC facilities, Gulf Publishing Company, Houston, 2nd edn, 2000, ch. 6, pp. 182–205 Search PubMed
-
C. R. Schlosser, A. De Rezende Pinho and C. Guerra Pereira, WO Pat., 0039250, 2000 (Petrobras S.A.) Search PubMed
-
R. M. Pittman and L. L. Upson, US Pat., 6538169, 2003 (UOP LLC.) Search PubMed
- A. Corma, O. Bermúdez, C. Martinez and F. J. Ortega, Appl. Catal., A, 2002, 230, 111–125 CrossRef CAS
- Y. X. Zhao and B. W. Wojciechowski, J. Catal., 1996, 163, 365–373 CrossRef CAS
- M. Forissier, M. Formenti and J. R. Bernard, Catal. Today, 1991, 11, 73–83 CrossRef CAS
- U. A. Sedran, Catal. Rev.: Sci. Eng., 1994, 36(3), 405–431 CAS
- A. Corma and F. J. Ortega, J. Catal., 2002, 233(2), 257–265 CrossRef
- A. Corma, J. Mengual and P. J. Miguel, Appl. Catal., A, 2012, 417–418, 220–235 CrossRef CAS
-
L. L. Upson, US Pat., 6113776, 2000 (UOP LLC.) Search PubMed
-
J. Herbst, H. Owen and P. Shipper, US Pat., US5053204, 1991 (Mobil Oil Corporation) Search PubMed
- A. Corma, F. V. Melo, L. Sauvanaud and F. J. Ortega, Catal. Today, 2005, 107–108, 699–706 CrossRef CAS
- J. Knight and R. Mehlberg, Hydrocarbon Process., 2011, 90(9), 91–95 CAS
-
(a) A. Devard, G. De la Puente, F. Passamonti and U. Sedran, Appl. Catal., A, 2009, 353, 223–227 CrossRef CAS
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC
-
(a) D. Kubicka and R. Cerny, Ind. Eng. Chem. Res., 2012, 51(26), 8849–8857 CrossRef CAS
- G. Fogassy, N. Thegarid, Y. Schuurman and C. Mirodatos, Green Chem., 2012, 14, 1367–1371 RSC
- X. Dupain, D. J. Costa, C. J. Shaverien, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2007, 72, 44–61 CrossRef CAS
- D. Wallenstein and R. H. Harding, Appl. Catal., A, 2001, 214, 11–29 CrossRef CAS
- A. Corma, F. Mocholí, V. Orchillés, G. S. Koermer and R. J. Madon, Appl. Catal., 1991, 67, 307–324 CrossRef CAS
- A. Corma, V. Faraldos and A. Mifsud, Appl. Catal., 1989, 47, 125–133 CrossRef CAS
- G. M. Woltermann, J. S. Magee and S. D. Griffith, Stud. Surf. Sci. Catal., 1993, 76, 105–144 CrossRef CAS
- E. F. Sousa-Aguiar, F. E. Trigueiro and F. M. Z. Zotin, Catal. Today, 2013, 218–219, 115–122 CrossRef CAS
- J. Scherzer, Stud. Surf. Sci. Catal., 1993, 76, 145–182 CrossRef CAS
- H. Fichtner-Schmittler, U. Lohse, G. Engelhardt and V. Patzelova, Cryst. Res. Technol., 1984, 19(1), K1–K3 CrossRef CAS
- L. A. Pine, P. J. Maher and W. A. Wachter, J. Catal., 1984, 85, 466–476 CrossRef CAS
- W. Suarez, W. C. Cheng, K. Rajagopalan and A. W. Peters, Chem. Eng. Sci., 1990, 45(8), 2581–2588 CrossRef CAS
- J. W. Roelofsen, H. Mathies, R. L. De Groot, P. C. M. Van Woerkom and H. Angad Gaur, Stud. Surf. Sci. Catal., 1986, 28, 337–344 CrossRef CAS
- F. Lemos, F. Ramoa Ribeiro, M. Kern, G. Giannetto and M. Guisnet, Appl. Catal., 1987, 29, 43–54 CrossRef CAS
- J. Shen and A. Auroux, Stud. Surf. Sci. Catal., 2004, 149, 35–70 CrossRef CAS
- J. E. Otterstedt, Y. M. Zhu and J. Sterte, Appl. Catal., 1988, 38, 143–155 CrossRef CAS
- A. Corma, C. Martinez and L. Sauvanaud, Catal. Today, 2007, 127, 3–16 CrossRef CAS
- P Imhof, E. Rautiainen and J. Gonzalez, Hydrocarbon Process., 2005, 84(9), 109–114 CAS
-
P. O'Connor, F. P. Olthof, R. Smeink and J. Coopmans, in Fluid Cracking Catalysts, ed. M. L. Occelli and P. O'Connor, Marcel Dekker, 1998, vol. 74, pp. 85–98 Search PubMed
- A. Corma and L. Sauvanaud, Catal. Today, 2013, 218–219, 107–114 CrossRef CAS
- D. Wallenstein, R. H. Harding, J. Witzler and X. Zhao, Appl. Catal., A, 1998, 167, 141–155 CrossRef CAS
- P. Imhof, M. Baas and J. A. Gonzales, Catal. Rev.: Sci. Eng., 2004, 46, 151–161 Search PubMed
-
J. C. Kayser, US Pat., 6069012, 1998 (Kayser Technology Inc) Search PubMed
-
A. V. Sapre and T. M. Leib, in Fluid Catalytic Cracking II, ACS Symp. series, ed. M. L. Occelli, 1991, vol. 452, pp. 144–164 Search PubMed
- C. P. Kelkar, M. Xu and R. J. Madon, Ind. Eng. Chem. Res., 2003, 42, 426–433 CrossRef CAS
- D. W. Kraemer, U. A. Sedra and H. I. DeLasa, Chem. Eng. Sci., 1990, 45, 2447 CrossRef CAS
- M. P. Helmsing, M. Makkee and J. A. Moulijn, Chem. Eng. Sci., 1996, 51, 3039–3044 CrossRef CAS
- A. Corma, C. Martinez, F. V. Melo, L. Sauvanaud and J. Y. Carriat, Appl. Catal., A, 2002, 232, 247–263 CrossRef CAS
- J. Buchanan, Appl. Catal., 1991, 74, 83–94 CrossRef CAS
- J. Biswas and I. E. Maxwell, Appl. Catal., 1990, 58, 19–27 CrossRef CAS
- Y. G. Adewuyi, D. J. Klocke and J. S. Buchanan, Appl. Catal., 1995, 131, 121–133 CrossRef CAS
- S. Mandal, D. Bhattacharyya, V. B. Shende, A. K. Das and S. Ghosh, ACS Symp. Ser., 1994, 571, 335–348 CrossRef CAS
- H. Konno, T. Okamura, T. Kawahara, Y. Nakasaka, T. Tago and T. Masuda, Chem. Eng. J., 2012, 207–208, 490–496 CrossRef CAS
- M. A. Bari Siddiqui, A. M. Aitani, M. R. Saeed and S. Al-Khattaf, Top. Catal., 2010, 53, 1387–1393 CrossRef CAS
- T. F. Degnan, G. K. Chitnis and P. H. Schipper, Microporous Mesoporous Mater., 2000, 35–36, 245–252 CrossRef CAS
- R. H. Harding, A. W. Peters and J. R. D. Nee, Appl. Catal., A, 2001, 221, 389–396 CrossRef CAS
- C. S. Triantafillidis, N. P. Evmiridis, L. Nalbandian and I. A. Vasalos, Ind. Eng. Chem. Res., 1999, 38, 916–927 CrossRef CAS
- R. J. Madon, J. Catal., 1991, 129, 275–287 CrossRef CAS
- Y. Mathieu, A. Corma, M. Echard and M. Bories, Appl. Catal., A, 2012, 439–440, 57–73 CrossRef CAS
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS
-
C. Marcilly, Acido-Basic Catalysis: Application to Refining and Petrochemistry, Editions Technip, Paris, 2005 Search PubMed
- D. H. Lin, G. Coudurier and J. C. Vedrine, Stud. Surf. Sci. Catal., 1989, 49, 1431–1448 CrossRef
- I. E. Maxwell and W. H. J. Stork, Stud. Surf. Sci. Catal., 2001, 137, 747–819 CrossRef CAS
- C. T. W. Chu, G. H. Kuehl, R. M. Lago and C. D. Chang, J. Catal., 1985, 93, 451–458 CrossRef CAS
- G. Jiang, L. Zhang, Z. Zhao, X. Zhou, A. Duan, C. Xu and J. Gao, Appl. Catal., A, 2008, 340, 176–182 CrossRef CAS
- Y. Yoshimura, N. Kijima, T. Hayakawa, K. Murata, K. Suzuki, F. Mizukami, K. Matano, T. Konishi, T. Oikawa, M. Saito, T. Shiojima, K. Shiozawa, K. Wakui, G. Sawada, K. Sato, S. Matsuo and N. Yamaoka, Catal. Surv. Jpn., 2000, 4(2), 157–167 CrossRef CAS
- Y. Wei, Z. Liu, G. Wang, Y. Qi, L. Xu, P. Xie and Y. He, Stud. Surf. Sci. Catal., 2005, 158, 1223–1230 CrossRef
- N. Xue, X. Chen, L. Nie, X. Guo, W. Ding, Y. Chen, M. Gu and Z. Xie, J. Catal., 2007, 248, 20–28 CrossRef CAS
- D. Liu, W. C. Choi, C. W. Lee, N. Y. Kang, Y. J. Lee, C. H. Shin and Y. K. Park, Catal. Today, 2010, 164, 154–157 CrossRef
- T. Blasco, A. Corma and J. Martínez-Triguero, J. Catal., 2006, 237, 267–277 CrossRef CAS
- P. Tynjälä and T. Pakkanen, J. Mol. Catal. A: Chem., 1996, 110, 153–161 CrossRef
- K. Wakui, K. Satoh, G. Sawada, K. Shiozawa, K. Matano, K. Suzuki, T. Hayakawa, Y. Yoshimura, K. Murata and F. Mizukami, Catal. Lett., 2002, 81, 83–88 CrossRef CAS
- K. Wakui, K. Satoh, G. Sawada, K. Shiozawa, K. Matano, K. Suzuki, T. Hayakawa, Y. Yoshimura, K. Murata and F. Mizukami, Catal. Lett., 2002, 84, 259–264 CrossRef CAS
- J. S. Jung, J. W. Park and G. Seo, Appl. Catal., A, 2005, 288, 149–157 CrossRef CAS
- M. Ogura, S. Y. Shinomiya, J. Tateno, Y. Nara, M. Nomura, E. Kikuchi and M. Matsukata, Appl. Catal., A, 2001, 219, 33–43 CrossRef CAS
- A. J. Maia, B. Louis, Y. L. Lam and M. M. Pereira, J. Catal., 2010, 269, 103–109 CrossRef CAS
- T. K. Katranas, K. S. Triantafyllidis, A. G. Vlessidis and N. P. Evmiridis, Stud. Surf. Sci. Catal., 2005, 155, 347–354 CrossRef CAS
- V. A. Tsiatouras, T. K. Katranas, C. S. Triantafillidis, A. G. Vlessidis, E. G. Paulidou and N. P. Evmiridis, Stud. Surf. Sci. Catal., 2002, 142, 839–846 CrossRef
- T. Mole, J. R. Anderson and G. Creer, Appl. Catal., 1985, 17, 141–154 CrossRef CAS
- D. B. Lukyanov, Stud. Surf. Sci. Catal., 1997, 105, 1301–1308 CrossRef
- N. Rane, M. Kersbulck, R. A. Van Santen and E. J. M. Hensen, Microporous Mesoporous Mater., 2008, 110, 279–291 CrossRef CAS
- J. Lu, Z. Zhao, C. Xu, A. Duan and P. Zhang, Catal. Lett., 2006, 109, 65–70 CrossRef CAS
- J. Lu, Z. Zhao, C. Xu, P. Zhang and A. Duan, Catal. Commun., 2006, 7, 199–203 CrossRef CAS
- A. A. Lappas, C. S. Triantafillidis, Z. A. Tsagrasouli, V. A. Tsiatouras, I. A. Vasalos and N. P. Evmiridis, Stud. Surf. Sci. Catal., 2002, 142, 807–814 CrossRef
- B. S. Kwak and W. M. H. Sachtler, Korean J. Chem. Eng., 1996, 13(4), 356–363 CrossRef CAS
- R. W. Hartford, M. Kojima and C. T. O'Connor, Ind. Eng. Chem. Res., 1989, 28, 1748–1752 CrossRef CAS
- W. Xiaoning, Z. Zhen, X. Chunming, D. Aijun, Z. Li and J. Guiyuan, J. Rare Earths, 2007, 27, 321–328 CrossRef
- L. Bonetto, M. A. Camblor, A. Corma and J. Pérez-Pariente, Appl. Catal., 1992, 82, 37–50 CrossRef CAS
- K. Tarach, K. Góra-Marek, J. Tekla, K. Brylewska, J. Datka, K. Mlekodaj, W. Makowski, M. C. Igualada López, J. Martínez Triguero and F. Rey, J. Catal., 2014, 312, 46–57 CrossRef CAS
- A. Corma, J. Martínez-Triguero and C. Martínez, J. Catal., 2001, 197, 151–159 CrossRef CAS
- A. Corma and J. Martínez-Triguero, J. Catal., 1997, 165, 102–120 CrossRef CAS
- A. Corma, M. J. Díaz-Cabañas, J. Martínez-Triguero, F. Rey and J. Rius, Nature, 2002, 418, 514–517 CrossRef CAS PubMed
-
R. Bastiani, A. de Rezende Pinho, R. Wasserman and I. B. do Espirito Santo, WO Pat., 2013126974, 2013 (Petrobras) Search PubMed
- R. Bastiani, Y. L. Lam, C. Assumpção Henriques and V. Teixeira da Silva, Fuel, 2013, 107, 680–687 CrossRef CAS
-
H. Abrevaya, S. F. Abdo and R. Lyle Patton, US Pat., 6867341, 2005 (UOP LLC) Search PubMed
-
G. P. Towler and H. Abrevaya, US Pat., 8137533, 2012 (UOP LLC) Search PubMed
-
M. de Almeida, A. Costa, Y. Lam and E. Mattos, Eur. Pat., 2047905, 2008 (Petrobras) Search PubMed
- A. Corma, J. Mengual and P. J. Miguel, Appl. Catal., A, 2013, 460–461, 106–115 CrossRef CAS
- D. Soni, P. J. Angevine, R. Rao, G. Saidulu, D. Bhattacharyya and V. Krishnana, 20th Ethylene Producers Conference, AIChE Spring Meeting, New Orleans, 2008, 575–583 Search PubMed.
- A. Pinho, K. Maeshiro, W. K. Huziwara, E. Morgado Jr., M. B. B. Almeida, M. Silva and N. Patricio Jr., Proc. World Pet. Congr., 2002, 17(3), 523–530 Search PubMed
- P. K. Niccum, R. B. Miller, A. M. Claude, M. A. Silverman, N. A. Bhore, G. K. Chitnis, S. J. McCarthy and K. Liu, NPRA Annual Meeting, San Francisco, California, USA, 1998, paper AM-98-18 Search PubMed.
- J. P. Margotin, paper presented at Petrofed Leadership seminar on Innovations in Gas & Oil sectors, October 19th, 2011, New Delhi.
- C. Yang, First Saudi-Chinese Oil Refining Forum, October 28–29th, 2013.
- C. Yang, X. Chen, J. Zhang, C. Li and H. Shan, Appl. Petrochem. Res., 2014, 4(4), 435–439 CrossRef PubMed
- J. Zhang, H. Shan, X. Chen, C. Li and C. Yang, Ind. Eng. Chem. Res., 2013, 52, 658–668 CrossRef CAS
- C. Li, C. Yang and H. Shan, Ind. Eng. Chem. Res., 2007, 46, 4914–4920 CrossRef CAS
- G. Wang, G. Yang, C. Xu and J. Gao, Appl. Catal., A, 2008, 341, 98–105 CrossRef CAS
- C. N. Eng, S. C. Kang and Y. K. Park, Paper presented at the 2007 Spring National Meeting - Houston, Texas.
- M. Nieskens, paper AM-08-54, presented at NPRA – Annual Meeting, San Diego, CA, March 9–11, 2008.
-
W. Mo, G. A. Hadjigeorge and F. H. H. Khouw, US Pat., 7632977, 2009 (Shell Oil Company) Search PubMed
-
S. Mandal, et al., US Pat., 5846402, 1998 (Indian Oil Corporation Ltd.) Search PubMed
- M. R. Rao, D. Bhattacharya, S. Mandal, A. K. Das, V. Krishnan, S. Makhija and S. Ghosh, Prepr. - Am. Chem. Soc., Div. Pet. Chem., 2003, 48(3), 166 CAS
-
S. Dalip and L. F. Castagnos, WO Pat., 2005/073347, 2005 (ABB Lummus Global, Inc.) Search PubMed
- S. Dalip, Davison Catalagram, 2004, 94, 51–56 Search PubMed
-
K. A. Couch, K. D. Seibert and P. V. Opdorp, NPRA Annual Meeting, Texas, March 23–25, 2003, Paper No. AM-04-45; Search PubMed
-
D. A. Lomas, US Pat., 5965012, 1999 (UOP LLC) Search PubMed
-
D. A. Lomas, US Pat., 6010618, 2000 (UOP LLC) Search PubMed
-
R. M. Pittman and L. L. Upson, US Pat., 6538169, 2003 (UOP LLC) Search PubMed
-
W. S. Letzsch, in Handbook of Petroleum Refining Processes, ed. R. A. Meyers, 3rd edn, 2004, ch. 3.2, pp. 3.35–3.45 Search PubMed
- Z. Li, S. Liu and X. Ge, ACS Symp. Series, FCC III, ed. M. Occelli, 1994, ch. 4, vol. 571, pp. 33–42 Search PubMed.
-
Y. Huo, Z. Wang, Y. Wang and Y. Lu, US Pat., 5326465, 1994 (China Petro-Chemical Corporation and Research Institute of Petroleum Processing) Search PubMed
-
Z. Li, C. Xie, W. Shi, F. Jiang, S. Liu, R. Pan and S. Li, US Pat., 5670037, 1997 (China Petro-Chemical Corporation and Research Institute of Petroleum Processing, SINOPEC) Search PubMed
- D. Dharia, W. Letzsch, H. Kim, D. McCue and L. Chapin, Hydrocarbon Process., 2004, 83(4), 61–66 CAS
-
Z. Li, S. Liu and X. Ge, US Pat., 4980053, 1990 (Research Institute of Petroleum Processing, SINOPEC) Search PubMed
-
X. Shu, W. Fu, M. He, M. Zhou, Z. Shi and S. Zhang, US Pat., 5232675, 1993 (China Petro-Chemical Corporation and Research Institute of Petroleum Processing) Search PubMed
-
Z. Shi, W. Shu, Y. Ye, X. Ge, P. Cao, S. Liun, C. Xie, Z. Li, X. Shu, X. Yang, W. Fu, M. Zhou and M. He, US Pat., 5380690, 1995 (China Petro-Chemical Corporation) Search PubMed
-
W. S. Letzsch, US Pat., 6905591, 2003 (Stone & Webster Process Technology Inc.) Search PubMed
-
J. N. Rubin, US Pat., 5523502, 1996 (Stone & Webster Engineering Corp.) Search PubMed
- X. Meng, C. Xu, J. Gao and L. Li, Appl. Catal., A, 2005, 294, 168–176 CrossRef CAS
- X. Meng, C. Xu, L. Li, J. Gao and Z. Liu, Energy Fuels, 2009, 23, 65–69 CrossRef CAS
- Z. Genquan and X. Chaogang, China Pet. Process. Petrochem. Technol., 2013, 15(3), 7–12 Search PubMed
- Z. Liu, X. Meng, C. Xu and J. Gao, Chin. J. Chem. Eng., 2007, 15(3), 309–314 CrossRef CAS
- X. Meng, C. Xu, L. Li and J. Gao, Energy Fuels, 2011, 25(8), 3382–3388 CrossRef CAS
-
F. Zhang, X. Shu, Z. Shi, W. Wang, F. Qin and X. Wang, US Pat., 6080698, 2000 (RIPP & China Chemical Corporation) Search PubMed
-
G. Pop, G. Ivanus, S. Boteanu, P. Tomi and E. Pop, US Pat., 4172816, 1979 (Institutul de Inginerie Tehnologica) Search PubMed
-
Y. Sha, Z. Cui, G. Wang and M. Wang, US Pat., 6420621, 2002 (China PetroChemical Corporation & Luoyang Petrochemical Engineering Corporation SINOPEC) Search PubMed
- X. Meng, C. Xu, L. Li and J. Gao, Ind. Eng. Chem. Res., 2003, 42, 6012–6019 CrossRef CAS
- X. Meng, C. Xu, L. Li and J. Gao, Appl. Catal., A, 2006, 301, 32–38 CrossRef CAS
- Y. Liu, X. Chen, H. Zhao and C. Yang, Chin. J. Chem. Eng., 2009, 17(1), 78–82 CrossRef CAS
-
J. Hiltunen, K. Fagerstolt, O. Krause, K. Kääriäinen, S. Ruottu and A. Halme, WO Pat., 9527019, 1995 (Neste Oy) Search PubMed
-
S. Ruottu, K. Kääriäinen and J. Hiltunen, US Pat., 6045688, 2000 (Neste Oy) Search PubMed
- J. Hiltunen, V. M. Niemi, K. Lipiäinen, I. Eilos, P. Hagelberg, P. Knuuttila, K. Jääskeläinen, J. Majander and J. Röppänen, Stud. Surf. Sci. Catal., 2001, 134, 111–132 CrossRef CAS
- A. Ma'adhah, Y. Fujiyama, H. Redhwi, M. Abul-Hamayel, A. Aitani, M. Saeed and C. Dean, Arabian J. Sci. Eng., 2008, 33(1B), 17 Search PubMed
- Y. Fujiyama, M. H. Al-Tayyar, C. F. Dean, A. Aitani and H. H. Redhwi, Stud. Surf. Sci. Catal., 2007, 166, 1–12 CrossRef CAS
-
Y. Fujiyama, US Pat., 5904837, 1999 (Nippon Oil Co., Ltd. and Petroleum Energy Center) Search PubMed
-
T. Ino and S. Ikeda, US Pat., 5951850, 1999 (Nippon Oil Co., Ltd. and Petroleum Energy Center) Search PubMed
-
Y. Fujiyama, M. Adachi, T. Okuhara and S. Yamamoto, US Pat., 6045690, 2000 (Nippon Oil Co., Ltd.) Search PubMed
-
D. B. Bartholic, US Pat., 4985136, 1991 Search PubMed
- M. W. Schnaith, P. A. Sexson, D. R. True, D. B. Bartholic, Y. K. Lee, I. S. Yoo and H. S. Kang, Oil Gas J., 1998, 96(25), 53–58 CAS
-
R. J. Gartside and H. N. Woebcke, US Pat., 4338187, 1982 (Stone & Webster Engineering Corp.) Search PubMed
-
R. J. Gartside, A. R. Johnson, J. L. Ross and D. Duncan, US Pat., 4663019, 1987 (Stone & Webster Engineering Corp.) Search PubMed
-
A. R. Johnson, R. J. Gartside, J. L. Ross and D. Duncan, US Pat., 5976355, 1999 (Stone & Webster Engineering Corp.) Search PubMed
-
R. J. Gartside, in Fluidization VI, ed. J. R. Grace and M. A. Bergougnou, Engineering Foundation, New York, 1989, pp. 25–32 Search PubMed
-
R. B. Miller, C. Santner and M. J. Tallman, US Pat., 7128827, 2006 (Kellogg, Brown & Root LLC) Search PubMed
-
R. B. Peterson, C. Santner and M. J. Tallman, US Pat., 7153479, 2006 (Kellogg, Brown & Root LLC) Search PubMed
-
C. N. Eng and R. B. Miller, US Pat., 7491315, 2009 (Kellogg, Brown & Root LLC) Search PubMed
-
P. K. Niccum and E. A. Gbordzoe, WO Pat., 2008159696, 2008 (Kellogg, Brown & Root LLC) Search PubMed
-
P. K. Ladwig, J. E. Asplin, G. F. Stuntz, W. A. Wachter and B. E. Henry, US Pat., 6069287, 2000 (Exxon Research & Engineering
Co) Search PubMed
- C. Dupraz, (R)FCC Product Flexibility with FlexEneTM, at: Global Technology Forum – Asian Refining Technology Conference - GTF ARTC 2012 – Bangkok.
- FlexEne, The flexibility offered by FlexEne can be illustrated by the following modes of operation, Available at: www.axens.net/product/technology-licensing/11001/flexene.html.
- M. Gagniere, A. Pucci and E. Rousseau, Petroleum Technology Quarterly (ptq), Q2, 2013 Search PubMed.
- M. J. Tallman, C. N. Eng, S. Choi and D. S. Park, AIChE Spring Meet. 7th Global Congr. Process Saf., Conf. Proc., Chicago, IL, United States, Mar. 13–17, 2011 Search PubMed.
- M. Tallman, Hydrocarbon Process., 2010, 89(4), 57–59 CAS
- P. O'Connor, Stud. Surf. Sci. Catal., 2006, 166, 227–251 CrossRef
- A. Corma, G. W. Huber, L. Sauvanaud and P. O'Connor, J. Catal., 2007, 247, 307–327 CrossRef CAS
- P. S. Rezaei, H. Shafaghat and M. A. W. W. Daud, Appl. Catal., A, 2014, 469, 490–511 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |