
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective CO2 methanation on Ru/TiO2 catalysts: unravelling the decisive role of the TiO2 support crystal structure†
A.
Kim
ab,
C.
Sanchez
b,
G.
Patriarche
c,
O.
Ersen
d,
S.
Moldovan
d,
A.
Wisnet
e,
C.
Sassoye
*b and
D. P.
Debecker
 *a
*a
aInstitute of Condensed Matter and Nanoscience - Molecules, Solids and Reactivity (IMCN/MOST), Université catholique de Louvain, Croix du Sud 2 box L7.05.17, 1348 Louvain-La-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
bSorbonne Universités, UPMC Université Paris, CNRS, Collège de France, Laboratoire de Chimie de la Matière Condensée de Paris, 4 Place Jussieu, 75252 Paris Cedex, France. E-mail: capucine.sassoye@upmc.fr
cLaboratoire de Photonique et de Nanostructures (LPN), CNRS, Université Paris-Saclay, route de Nozay, F-91460 Marcoussis, France
dInstitut de Physique et Chimie des Matériaux de Strasbourg, UMR7504 CNRS-UNISTRA and NIE, 23 rue du Loess, B.P. 43, 67034 Strasbourg cedex 2, France
eDepartment of Chemistry and CeNS, Ludwig-Maximilians-University, Butenandtstrasse 11, 81377 Munich, Germany
First published on 21st September 2016
Abstract
The catalytic hydrogenation of CO2 is a relevant strategy for mitigating CO2 emissions and its applicability relies on our ability to prepare catalysts that are highly active under mild conditions. Understanding and improving these tailored catalysts requires innovative materials synthesis routes and advanced methods of characterization. In this study, mono-dispersed 2 nm RuO2 nanoparticles were prepared as a stable colloidal suspension and deposited onto different titania supports by impregnation. Supported RuO2 nanoparticles are homogeneously dispersed at the surface of the titania supports. Then, upon annealing and reduction, metallic Ru nanoparticles are obtained, which are active in the hydrogenation of CO2 to CH4. However, depending on the crystal structure of the different TiO2 supports (anatase, rutile, and a mixture of both), the catalysts exhibited drastically diverse catalytic performances. An array of characterization tools (N2-physisorption, H2-chemisorption, HR-TEM, STEM-HAADF, 3D tomographic analysis, XRD, and XPS) was used to unravel the origin of this support effect. It appeared that catalytic behaviour was related to profound morphological changes occurring during the annealing step. In particular, advanced electron microscopy techniques allow visualisation of the consequences of RuO2 nanoparticle mobility onto titania. It is shown that RuO2 sinters heavily on anatase TiO2, but spreads and forms epitaxial layers onto rutile TiO2. On anatase, large Ru chunks are finally obtained. On rutile, the formation of a particular “rutile-TiO2/RuO2/rutile-TiO2 sandwich structure” is demonstrated. These phenomena – along with the relative thermal instability of the supports – explain why the catalysts based on the commercial P25 titania support outperform those based on pure crystalline titania. The study opens new perspectives for the design of highly active CO2 methanation catalysts.
Introduction
Supported metal nanoparticles feature important structural properties that dictate their performance in heterogeneous catalysis.1–6 Indeed, a structure–performance relationship is found in many metal-catalysed reactions.7–13 Although the primary role of the support material is sometimes thought to be limited to serving as a physical carrier for intact metal nanoparticles, it has been recognized that the chemical nature of the support or the metal–support interactions can have a marked impact on catalytic activities and/or selectivity.14,15 Thus, the design of tailored supports for metal-based catalysts is a topical field of research which concerns the size, shape, texture, crystallinity, and redistribution process of the metal nanoparticles on their support.13,16,17CO2 hydrogenation to CH4 (CO2 methanation), also known as the Sabatier reaction, is an important catalytic process of fundamental academic interest with potential commercial application.18–20 In the current environmental context, this reaction not only reduces CO2 emissions but produces CH4, which can be directly transported through existing natural gas pipelines to be used as a fuel or as a chemical building block, simultaneously targeting both the valorisation of CO2 (reduction of greenhouse gases) and the vectorization of dihydrogen via CH4.
Although CO2 methanation has been a topical field of research for decades, the emphasis on the development in this area has only recently turned to catalytic performance with high selectivity at thermodynamically favourable conditions (i.e. low temperature and pressure) using different types of metals and supports.21
Reducible group VIII metal oxides have been widely used as support materials. Among them, TiO2 is best known for its high stability, high ultraviolet absorption, and semiconductor properties which allow its use in various applications in catalysis and photocatalysis.22 Furthermore, the existence of various crystalline phases of TiO2 contributes to the tuning of catalytic performance in heterogeneous catalysis through morphologic and electronic perturbations.12,16 For example, TiO2 is known as the most efficient support for noble metal catalysts including Pd, Ru and Rh in CO and CO2 methanation.21,23–25 Recently, TiO2 supported RuO2 nanoparticles have shown excellent oxidation capability as Deacon catalysts displaying different stabilities depending on the crystalline phases of TiO2.13,26
A study on the crystal phase effect of TiO2 on the structure and performance of Ru nanoparticles in CO2 hydrogenation has been recently reported.12 This study demonstrated the strong impact of the support crystal structure on Ru dispersion and therefore on methanation activity. In particular, rutile TiO2 was identified as the most promising support; thanks to the strong interaction it can develop with RuO2. However, the catalysts were prepared by a conventional impregnation method (IM), and this does not provide good control over the initial RuO2 dispersion, itself being a key parameter for the final Ru nanoparticle dispersion.
The impact of a structure–performance relationship in Ru-based catalysts has given rise to various preparation methods for well-dispersed and/or uniformly-sized nanoparticles of Ru and RuO2 for many catalytic applications in hydrogenation and oxidation reactions.27–30 Abe et al. have developed a dry technique for modifying the surfaces of powdery materials named as polygonal barrel-sputtering to prepare Ru nanoparticle loaded TiO2 with a narrow particle size distribution without the need of any heating which can cause nanoparticle sintering.31 Balaraju et al. have shown the superior catalytic performances of Ru/TiO2 catalysts prepared by a deposition–precipitation (DP) method compared with those prepared by a conventional impregnation (IM) method in the hydrogenolysis of glycerol, which was attributed to the presence of well-dispersed nano-sized Ru particles on TiO2.32 Also, Sassoye et al. have previously reported on the synthesis of mono-dispersed 2 nm RuO2 nanoparticles in an aqueous colloidal suspension and the preparation of Ru/TiO2 catalysts after reduction under H2.33 Such catalysts demonstrated superior catalytic performance in CO2 methanation compared to that prepared by an IM method.33
The successful preparation of a suspension of uniformly distributed 2 nm RuO2 nanoparticles and the suspected structure–performance relationship in Ru-catalysed CO2 methanation has prompted us to study the active phase–support interaction between pre-formed calibrated RuO2 nanoparticles and TiO2 supports with different crystalline phases. Indeed, starting from well-defined dispersed RuO2 nanoparticles, TiO2 supports with different crystalline phases might favour the stabilization of Ru in different states of dispersion, shape, structure.
In the present work, RuO2 nanoparticles of 2 nm were initially prepared by a colloidal method and supported on pure anatase, pure rutile, and P25 TiO2. The uniform size distribution of RuO2 nanoparticles facilitated a more accurate study of the influence of the TiO2 crystalline phases on RuO2 nanoparticles and the resulting catalytic behaviour in CO2 methanation. The morphological changes in the RuO2 nanoparticles were monitored after various thermal annealing temperatures and correlated with the CO2 methanation activity at low temperature (≤200 °C) and one standard atmospheric pressure.
Results and discussion
Basic characterization of supports and catalysts
XRD analyses confirmed the structure of the supports alone; mixed anatase and rutile phases for P25 TiO2, the pure anatase phase for the home-made anatase TiO2, and the pure rutile phase for the home-made rutile TiO2 (see the ESI,† SI-1).The 2D particle shapes of the three TiO2 supports, namely, P25, anatase, and rutile, were distinctive and well distinguishable from one another as shown by direct TEM observations in SI-2.† P25 TiO2 particles were rounded, rectangular-shaped, with anatase and rutile crystallites being impossible to distinguish; home-made anatase TiO2 particles were a mixture of oval and rhombus shapes; and home-made rutile TiO2 particles were needle-shaped. The particle sizes measured from TEM images were, on average, 25–30 nm for P25 TiO2, 6–7 nm for anatase TiO2, and 12 × 100 nm for rutile TiO2.
There was no difference in the shapes and sizes of the supports before and after the deposition of RuO2 nanoparticles with annealing at 150 °C (SI-3a–c†). The RuO2 nanoparticles have kept their initial ∼2 nm diameter on all supports and appeared globally well dispersed as proven by the 2D TEM micrographs. The 3D-STEM-HAADF tomography images (Fig. 1) provide the ultimate proof of good dispersion of RuO2 nanoparticles on the surface of P25 TiO2. To complement Fig. 1, a video showing the reconstructed volume with the catalyst particles deposited on the P25 TiO2 support under rotation is available in the ESI† (SI-4).
 | ||
| Fig. 1 Tomographic analysis of the RuO2/TiO2-P25 sample before calcination. (a) STEM-HAADF image extracted from the tilt series used to calculate the reconstruction of this aggregate. (b) 3D model of the studied aggregate, showing TiO2 in light grey, RuO2 nanoparticles in red, and gold nanoparticles deposited on the TEM membrane for alignment purposes in yellow. (c) Representative slices extracted from the reconstruction. The RuO2 nanoparticles are pointed by red arrows. A video showing the reconstructed volume with the catalyst particles deposited on the P25 TiO2 support under rotation is available in the ESI† (SI-4). | ||
ICP-AES elemental analysis of the catalysts (SI-5†) resulted in Ru contents of 2.35–2.60 wt%, indicating no Ru loss during the entire synthesis process (the small variation comes from the variation in the water content in the RuCl3·xH2O (x = 3–5) precursor). The specific surface areas obtained by the BET method (SBET) before and after the deposition of RuO2 followed by annealing at 150 °C were found to be consistent, as shown in Table 1.
| Annealing temperature (°C) | RuO2/TiO2-P25 (m2 g−1) | RuO2/TiO2-A (m2 g−1) | RuO2/TiO2-R (m2 g−1) |
|---|---|---|---|
| Pristine support | 50 | 139 | 71 |
| 150 | 52 | 132 | 64 |
| 250 | 45 | 118 | 60 |
| 350 | 50 | 64 | 41 |
| 450 | 46 | 60 | 32 |
In XPS, all 150 °C-annealed catalysts present Ru/Ti ratios that are higher than the 0.043 bulk Ru/Ti ratio (calculated based on the 2.5 wt% Ru in the final catalyst), confirming the good dispersion of Ru on all support surfaces (Table 2).
| References | Ru at% | Ti at% | Ru/Ti | |
|---|---|---|---|---|
| ox | met | |||
| RuO2/TiO2-P25-150 | 1.90 | 0.04 | 19.66 | 0.099 |
| RuO2/TiO2-P25-250 | 0.52 | 0.99 | 20.05 | 0.075 |
| RuO2/TiO2-P25-350 | 0.49 | 0.43 | 20.07 | 0.045 |
| RuO2/TiO2-P25-450 | 0.35 | 0.32 | 20.31 | 0.033 |
| RuO2/TiO2-A-150 | 0.85 | 0.07 | 20.38 | 0.045 |
| RuO2/TiO2-A-250 | 0.91 | 0.08 | 20.88 | 0.047 |
| RuO2/TiO2-A-350 | 0.87 | 1.06 | 19.54 | 0.098 |
| RuO2/TiO2-A-450 | 0.44 | 0.69 | 20.37 | 0.055 |
| RuO2/TiO2-R-150 | 0.96 | 0.17 | 21.15 | 0.053 |
| RuO2/TiO2-R-250 | 1.06 | 0.12 | 20.33 | 0.058 |
| RuO2/TiO2-R-350 | 0.66 | 0.04 | 20.39 | 0.034 |
| RuO2/TiO2-R-450 | 0.48 | 0.04 | 18.72 | 0.028 |
CO2 methanation activity
The RuO2/TiO2 catalysts were annealed at various temperatures, reduced in the reactor to obtain metallic Ru, and then tested in CO2 methanation at one atmospheric pressure and in the 50–200 °C temperature range. Under these conditions, as predicted by thermodynamic equilibrium calculations, selectivity to methane and water was always 100% for all three catalysts. The balance between converted CO2 and produced CH4 was always equilibrated. The specific activities of all catalysts with three supports at various annealing temperatures and reaction temperatures are provided in the ESI† (SI-6). The catalytic activities at 200 °C expressed in terms of the CH4 production rate (μmol methane produced per second and per gram of the catalyst) and in terms of CO2 conversion were compared for the three different TiO2 supports (Fig. 2). The CH4 production rate at a given reaction temperature increased with higher annealing temperatures for all three supports. | ||
| Fig. 2 Influence of annealing temperatures and TiO2 supports on CO2 methanation activity at 200 °C. Under the conditions used in this study, the highest activity level reported (2.57 μmolCH4 gcat−1 s−1) corresponds to a CO2 conversion of 27.4%. It is consistent with the activity levels reported by other groups (e.g. ∼7.8 μmolCH4 gcat−1 s−1 at 200 °C with a 5 wt% Ru/TiO2 catalyst reported by Lin et al.12). | ||
Strikingly, even if the Ru loading is strictly the same for the three studied supports, marked differences were observed in terms of activity depending on the nature of the support. The global trend (more evident as the annealing temperature increases) in terms of CH4 production rates is found to be P25 > anatase > rutile. This points to a decisive role of the TiO2 crystal structure in the catalytic behaviour of the final Ru/TiO2 catalysts. The activation energies of the catalysts annealed at 450 °C were obtained by scanning the catalytic activities from 100 to 200 °C, and were found to be 14.3, 14.3 and 15.4 kcal mol−1, for Ru/TiO2-P25-450, Ru/TiO2-A-450 and Ru/TiO2-R-450, respectively (SI-7†). The fact that similar values of activation energies are found suggests that the active species are the same in all the catalysts, i.e. the TiO2 crystal structure does not affect the reaction mechanism. The higher catalytic activity of P25 TiO2 supported catalyst compared to anatase or rutile TiO2 may therefore be attributed to a greater number of active sites. Thus the TiO2 crystal structure must play a role in the genesis and stabilization of the active phase.
Catalyst modifications upon annealing
Drastic morphology modifications are observed on all three catalysts after the thermal annealing, and these changes are more marked as the annealing temperature increases from 150 to 450 °C. The TEM images provided in Fig. SI-3† show that RuO2 evolves differently upon heating, depending on the crystal structure of the TiO2 support: hardly visible on P25, present as large crystals on anatase, and present as cracked thin layers on rutile. These general observations corroborate an earlier study on the growth of RuO2 on anatase and rutile TiO2.13 Importantly, while the size of the P25 particles remains constant upon heating, sintering of home-made rutile and anatase is observed by TEM (SI-8†). This is also confirmed by BET measurement (Table 1), as well as Scherrer calculation from the main diffraction peaks (SI-9†). The different sintering behavior is a factor influencing the catalytic properties. More in-depth observations at each temperature are discussed below sequentially for P25, anatase, and rutile-supported catalysts.P25
In P25, the anatase and rutile particles look alike, even in HR-TEM: similar shapes and numerous atomic planes presenting a d spacing below 3 Å (only the rutile 110 plane at 3.2 Å can be easily separated from the anatase 101 plane at 3.5 Å).As the calcination temperature increased from 150 °C (Fig. 4a) to 250, 350, and 450 °C, the RuO2 nanoparticles – initially clearly visible – became difficult to observe in HR-TEM (weaker contrast, disappearance of the RuO2 nanoparticles). After annealing at 250 °C, the TEM images revealed a tendency of RuO2 nanoparticles to aggregate in the form of layers (Fig. SI-10†). A darker layer around some TiO2 particles was observed (2–3 nm thick), corresponding to the detection of ruthenium by EDX spectroscopy, whereas Ru was absent elsewhere (EDX analysis spot is ∼100 nm). The interatomic spacing of the crystal lattice of this particle was measured to be around 3.2 Å, which is consistent with the (110) plane of rutile TiO2 (3.24 Å) as well as the (110) plane of RuO2 (3.18 Å). Thus, a RuO2 thin layer is seen on rutile TiO2 particles. This RuO2 redistribution phenomenon during thermal treatment or catalysis has also been proposed in the deacon process as well as total oxidation reactions.13,30
High-angle annular dark-field (HAADF) and bright field images were taken by scanning transmission electron microscopy (STEM) in order to obtain higher Z-contrast images of the 450 °C heated catalyst (Fig. 4b–f). Remarkably, the white spots and layers found in dark-field images were found only on rutile TiO2 particles. Energy dispersive X-ray spectroscopy (EDX) clearly identified those white spots as being Ru-rich. A higher atomic concentration of Ru was detected surrounding the rutile TiO2 particles, suggesting the presence of a RuO2 layer (Fig. 4c and d).The anatase TiO2 particles, which represent 80% of the P25 support, remained completely free of Ru (Fig. 4e and f).
Consistent with TEM observations, XRD analysis and Scherrer calculations showed that the crystallinity of P25 does not evolve with the increased annealing temperature of the catalyst (Fig. 3a and ESI† SI-9 and SI-11).
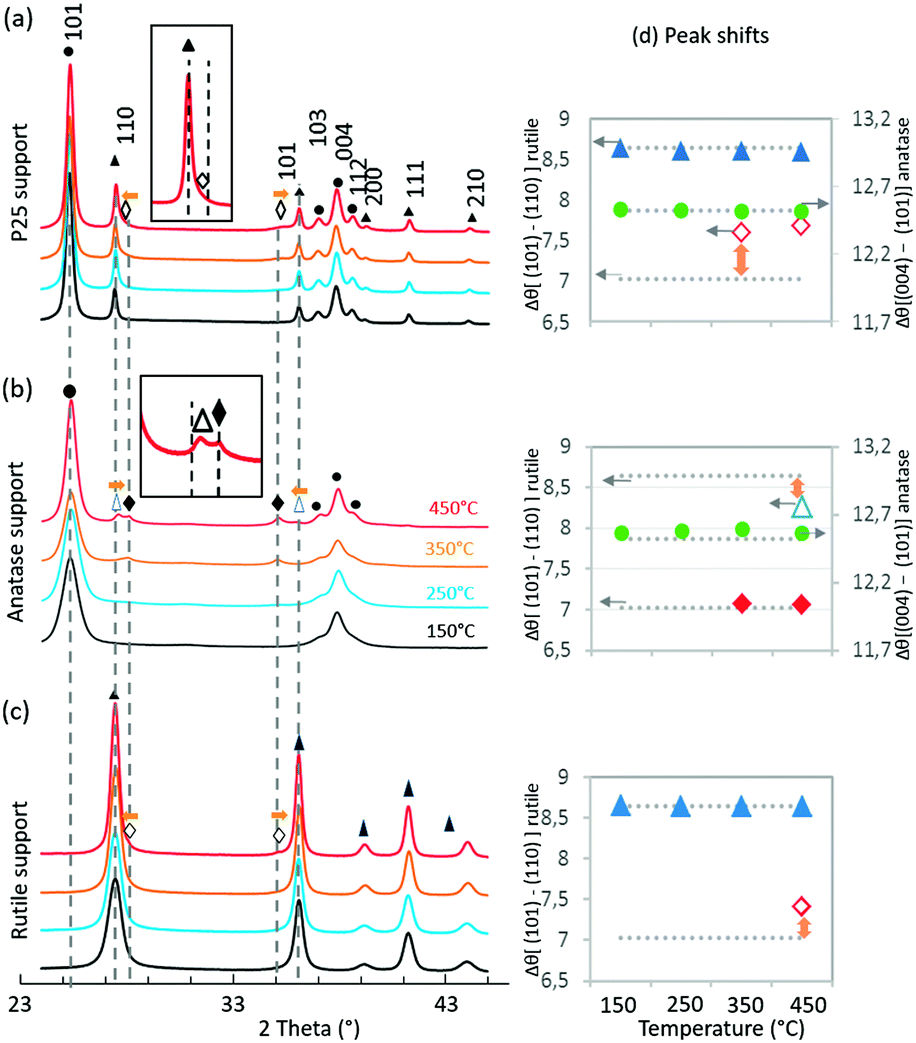 | ||
| Fig. 3 XRD patterns of the catalysts annealed at different temperatures. P25 support (a), anatase support (b) and rutile support (c). The vertical dotted grey lines represent the expected peaks based on ICDD (021-1276 TiO2 rutile, 021-1272 TiO2 anatase and 043-1027 RuO2); experimental peaks are assigned to TiO2 anatase (●), TiO2 rutile (▲), shifted TiO2 rutile (△), RuO2(♦) and shifted RuO2 (◇). For each phase, the difference has been calculated between the 2θ positions of the 2 main peaks. (d) The horizontal dotted grey lines represent the corresponding calculated values from ICDD. | ||
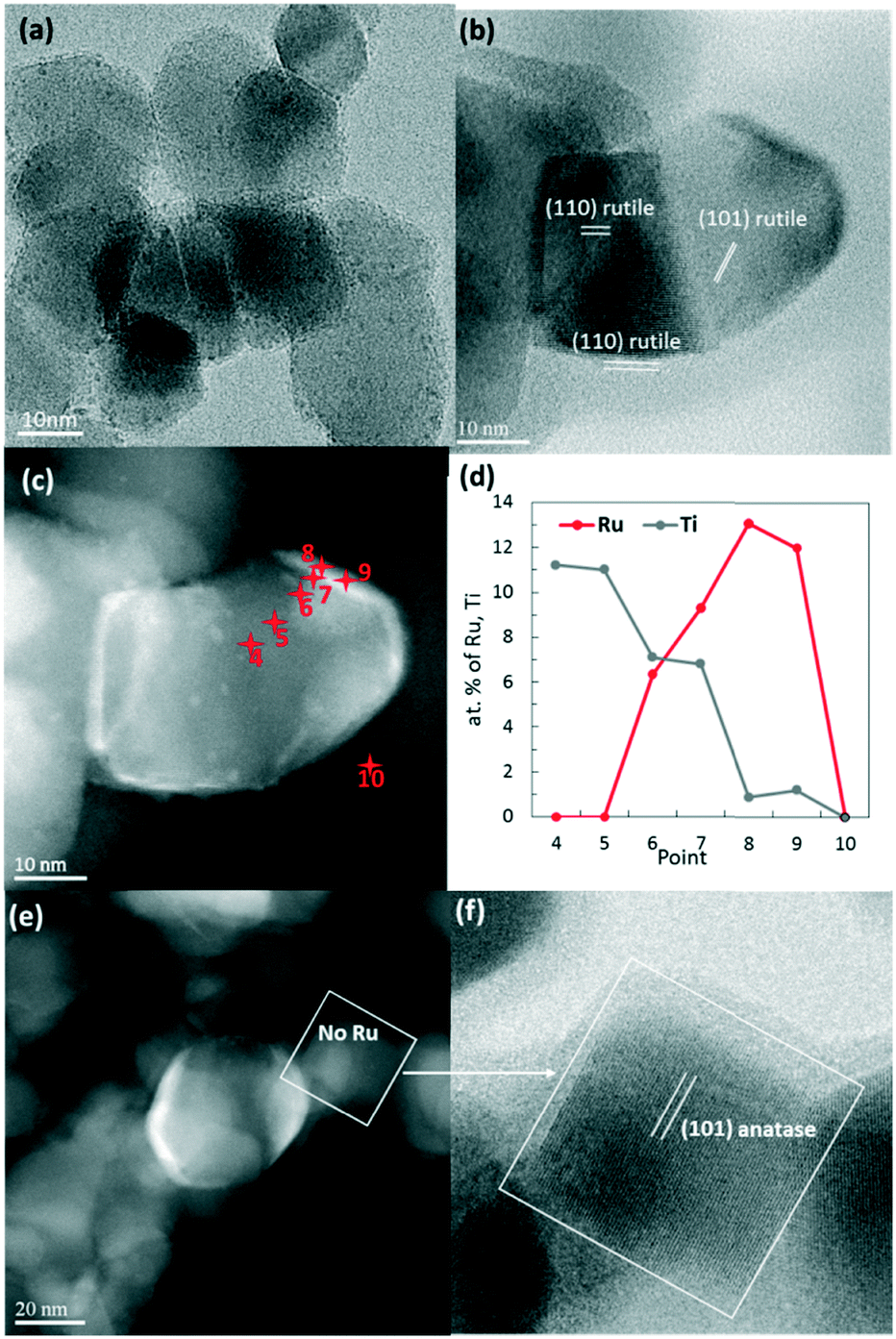 | ||
| Fig. 4 RuO2/TiO2-P25 heated at 150 °C (a) and 450 °C (b–f). STEM-BF (bright field) micrograph (b) of an identified rutile P25 particle and the corresponding STEM-HAADF image (c) revealing RuO2 as white layers and particles. EDX analysis (d) was performed on 10 chosen spots (as indicated by the red points) to verify that the Ru concentration is high on the white areas. A HAADF image (e) showing a naked particle and a zoomed-in image corresponding to the bright field image (f) identifying this particle as anatase. | ||
The TiO2 diffraction peaks (both anatase and rutile phases) do not shift in position at all annealed temperatures. Deconvolution of RuO2 peaks, however, showed that, with increased annealing temperature, the (110) RuO2 and (101) RuO2 peaks shift in opposite directions, both towards the associated (110) and (101) rutile TiO2 peaks. This is particularly clear in Fig. 3d where Δθ[(101)–(110)] is higher than the expected value from ICDD for RuO2. This observation is a clear indication of epitaxial interactions between RuO2 and rutile TiO2 and is supported by the fact that RuO2 and rutile TiO2 present the same rutile crystal structure with very close lattice parameters (for rutile TiO2, a = 4.5933 Å, c = 2.9592 Å – 021-1276; and for RuO2, a = 4.4994 Å, c = 3.1071 Å – 043-1027). Indeed, the degree of mismatch between the pure reference RuO2 and the pure rutile reference TiO2 stands at 1.8% for the (110) surface and 2.7% for the (101) surface, values that are largely below the reported 5% limit allowing epitaxial layer growth. As discussed in detail in the ESI† (SI-12), the shift in RuO2 peak positions towards rutile TiO2 peak positions has consequences on the RuO2 structure. The epitaxial growth of RuO2 on rutile TiO2 implies that the RuO6 octahedra are less distorted compared to RuO6 octahedra from the RuO2 crystal alone.
XPS analysis showed a decrease in the atomic ratio of Ru/Ti with the increase in annealing temperatures (Table 2). This corroborates the sintering or concentration of RuO2 on rutile TiO2 particles as previously discussed based on the TEM observations.
Anatase
The SBET of the RuO2/TiO2-A catalyst decreased drastically by annealing from 150 to 450 °C, indicating the occurrence of support sintering. This trend was well correlated with TEM observations.RuO2 appeared relatively well dispersed at 150 °C (Fig. 5a). TEM and EDX on various regions show that this remained the case at 250 °C (SI-13a†). After annealing at 350 °C or 450 °C, some large dark crystals were found among the smaller TiO2 anatase particles (Fig. 5b). EDX confirmed that a large part of the TiO2 surface was free of Ru and that the large dark crystals were RuO2 (SI-13b†). Interestingly, rod-shaped crystals were also observed and identified as TiO2 rutile rods (Fig. 5b and SI-13c†). Crystallization of TiO2 rutile has been triggered by the rutile RuO2 structure, leading to anatase-to-rutile transformation at temperatures significantly lower than the usual ∼600 °C.34,35 Contrary to the case of the rutile phase of P25, RuO2 does not migrate toward the newly grown rutile TiO2 crystal.
 | ||
| Fig. 5 TEM images of the RuO2/TiO2-A catalyst after annealing at 150 °C (a), 350 °C (b) and 450 °C (c); at 150 °C, RuO2 particles are relatively well dispersed (a) whereas large RuO2 nanocrystals crystallize separately from TiO2 anatase at 350 °C and 450 °C (b and c, respectively). TiO2 rutile also crystallizes from RuO2 crystals. The RuO2 phase is pointed by red arrows or lines. The white circles show the areas where EDX has been performed to confirm the identification of the crystals (SI-13†). | ||
From XRD analysis (Fig. 3b), the pure anatase TiO2 peaks became narrower with increased annealing temperature consistent with the increase in crystal size (SI-9†). Deconvolution of the RuO2 peaks was only possible from 350 °C (SI-11†). RuO2 peaks appeared at the expected position for rutile RuO2 (Fig. 3d). This is in relation with TEM images where large RuO2 crystallites were observed to be clearly separated from the anatase TiO2 support. At 350 °C and 450 °C, (110) and (101) rutile TiO2 peaks were observed, with a satisfactory deconvolution only possible at 450 °C. Interestingly, these rutile TiO2 peaks were identified as (110) and (101) TiO2 rutile peaks shifted, respectively, towards (110) and (101) RuO2 peaks. This observation indicates the occurrence of rutile TiO2 crystallization starting at 350 °C through the epitaxial lattice matching mechanism from RuO2 to TiO2.
XPS analysis showed that the Ru/Ti ratios remained similar for different annealing temperatures with the exception of the 350 °C annealed catalyst. Due to the sintering of both support and Ru species on the pure anatase support, establishment of a reliable trend at various annealing temperatures becomes critical.
Rutile
TEM observations showed a gradual evolution of RuO2/TiO2-R catalysts with increasing annealing temperatures (ESI† SI-14 and Fig. 6).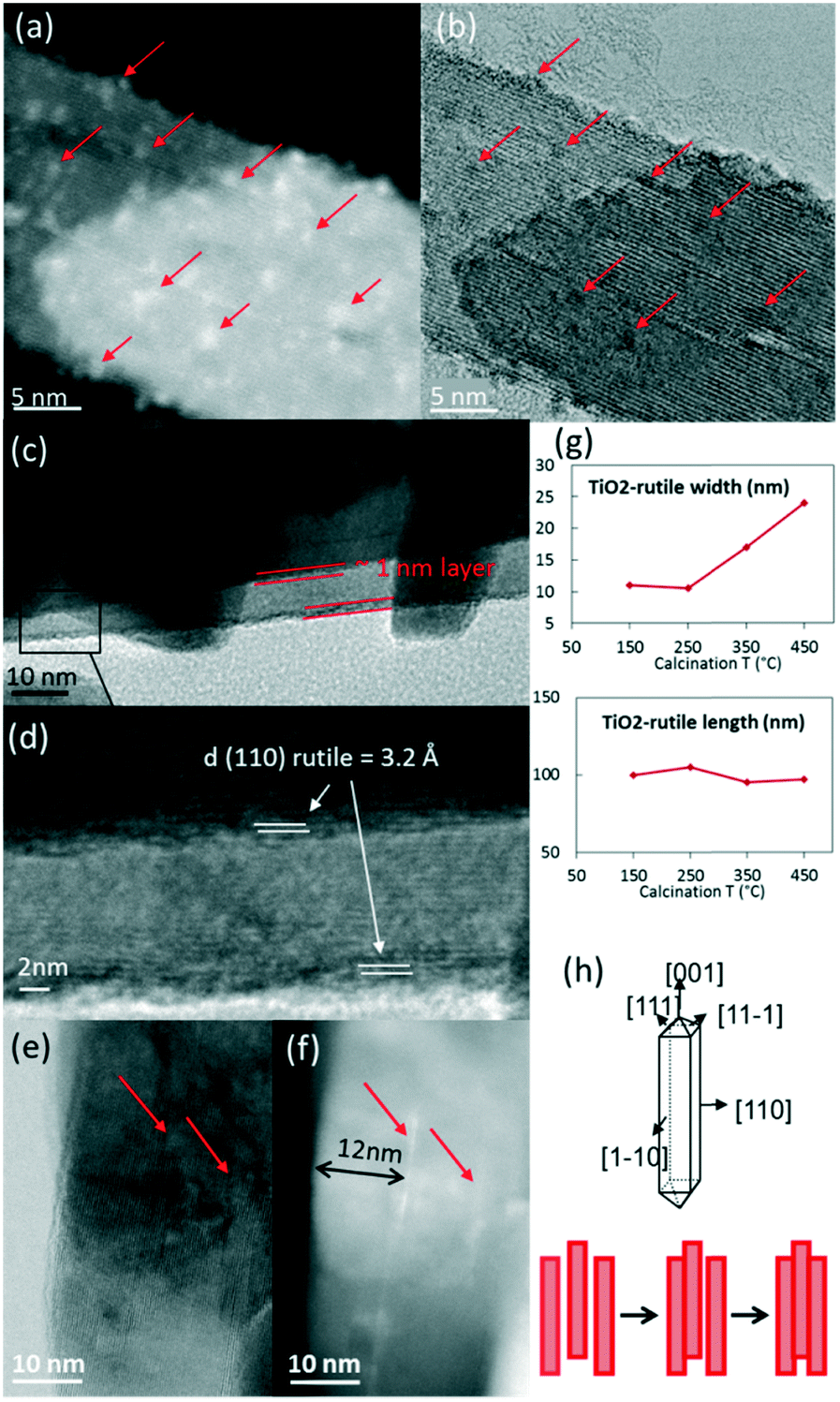 | ||
| Fig. 6 STEM-HAADF (a) and STEM-BF images (b) of RuO2/TiO2-R heated at 150 °C. RuO2/TiO2-R heated at 450 °C (c–f) showing the 110 RuO2 and TiO2 rutile planes (c and d) as well as the thin RuO2 layers in the bright field image (e) and the corresponding HAADF image (f). TiO2 rutile particle size evolution with temperature (g) allows the development of a schematic drawing (h) of TiO2-rutile growth in the direction of the (110) facet. The red arrows point at RuO2. | ||
After annealing at 150 °C, the RuO2 nanoparticles were well distributed on the intact rod-shaped rutile TiO2 particles (Fig. 6a and b).
Starting from 250 °C, the nanoparticles started to evolve into thin RuO2 layers. With increasing annealing temperature, the ∼1 nm thick layer around the rutile TiO2 rods became better defined. The thin layer was verified to have a d-spacing of 3.2 Å, which corresponds to the (110) plane of rutile RuO2 (3.18 Å) (Fig. 6c and d). This is an indication that the (110) RuO2 plane is oriented parallel to the (110) rutile TiO2 plane of the support, forming the so-called epitaxial layer, similar to that on the rutile particles of P25.
The support itself was clearly affected by the thermal treatment (Fig. 3c and SI-8†). The width of the support particles was found to be approximately doubled from 150 °C to 450 °C (also confirmed by Scherrer calculations, SI-9†), while the length remained constant. It suggests stacking of the rod-shaped rutile TiO2 particles in the direction of the (110) facet, as depicted in Fig. 6g and h. The STEM-BF images and their corresponding HAADF images revealed the presence of white lines of RuO2 layers every ∼12 nm on average, which corresponds to the initial width of the rutile TiO2 rod (Fig. 6e and f). This is evidence that RuO2 epitaxial layers on the (110) facet of rutile TiO2 act as “glue” between the 12 nm-wide TiO2 rutile rods, resulting in the “sandwiching” of the RuO2 layers between the TiO2 rods.
Discussion on the RuO2 nanoparticle migration
The redistribution process of RuO2 during heat treatment from anatase TiO2 particles to rutile TiO2 particles appears to play a major role in catalyst activation. This phenomenon only occurs for small RuO2 particles (2 nm or smaller).13 In this size range, surface tension dominates most physicochemical properties of nanomaterials, especially the interface behaviour and surface stability. RuO2 arrival on rutile TiO2 is clearly driven by epitaxy stabilization. The departure of the ruthenium atoms from the anatase TiO2 surface is more controversial. Two possible pathways are proposed: RuO2 local volatilization (RuO3 and RuO4) followed by redeposition36–38 and RuO2 nanoparticle diffusion.39,40 As discussed in detail in the ESI† (SI-20), it is difficult to totally exclude one of the two mechanisms. The fact that the Ru loading remains constant from RuO2 deposition until after the catalytic test indicates that volatilization is unlikely. This is also further supported by thermodynamic calculations.41 In any case, if volatilization occurs, it has to remain local (volatilization followed by immediate re-deposition). We rather propose that small RuO2 nanoparticles diffuse at the surface of the TiO2 particles, driven by the Oswaltd ripening mechanism, as it is well documented in the literature.42–44 Subsequently, the diffusion of RuO2 nanoparticles leads to two different phenomena in terms of sintering: isotropic growth of RuO2 crystals on the pure anatase TiO2 support or epitaxial growth of the RuO2 layer on rutile TiO2 (P25 and pure rutile TiO2 supports).Catalyst modifications upon reduction
A close observation of the morphology of the RuO2 phase at different annealing temperatures indicated unambiguously that the nature of the support dictates the behaviour of the RuO2 nanoparticles, and this was correlated with different catalytic performances. Yet, the actual active species in the methanation reaction are the reduced states of Ru over TiO2 obtained after an in situ thermal treatment under H2 at 200 °C.The three most active catalysts for each support, annealed at 450 °C, were analysed by conducting TPR (SI-15†). The H2 consumption profiles for the three catalysts were not overlapping, thus confirming the strong impact of the support structure. For the P25 supported catalyst, two close reduction events were observed, as often reported for titania-supported RuO2.45 The origin of the two reduction peaks is sometimes attributed to an inhibiting effect of residual water on the reduction kinetics.46 The complex reduction profile is often attributed to a heterogeneous distribution of RuO2 particles47 or to the presence of different RuO2 species which develop different types of interactions with the respective TiO2 supports.45 Here, the oxidized Ru species on the different TiO2 supports show very different reduction patterns, confirming that Ru species are different from the respective catalysts, probably in terms of size, accessibility, or support interaction. While reduction occurs below 200 °C on P25, for anatase and rutile, the two reduction peaks are shifted towards higher temperatures. Interestingly, by looking at the relative intensities of the reduction peaks, TPR shows that RuO2 in Ru/TiO2-R-450 is less reducible, corresponding to trapped RuO2 layers.
In Ru/TiO2-P25-450 post methanation, as RuO2 is reduced into metallic Ru, the epitaxial lattice matching over rutile TiO2 is suppressed. Additionally, the electronic density is higher in metallic ruthenium than in RuO2. As a result, the TEM image contrast between metallic Ru and TiO2 becomes higher than that between RuO2 and rutile TiO2. This allows one to clearly distinguish metallic Ru patches that are non-homogeneously dispersed on TiO2 P25 particles. Most TiO2 particles remain “naked” (Ru-free) and a few particles are covered by Ru nanoparticles (Fig. 7a). The HR-TEM images clearly showed (110) TiO2 rutile planes underneath Ru particles (SI-16a and b†) and 101 anatase planes on the naked TiO2 particles. 3D-TEM tomographic analysis (Fig. 8) of Ru/TiO2-P25-450 after reduction and methanation unambiguously confirms the 2D TEM observations by showing the presence of Ru particles (mean size of 2.8 ± 1.0 nm) localized preferentially on the surface of rutile TiO2 particles, leaving the TiO2 anatase particles naked. A media file corresponding to the tomography analysis is uploaded in the ESI† (SI-17, after test-mix.avi). This unambiguously shows that the Ru particles that accumulated specifically onto the rutile TiO2 particles in the form of RuO2 epitaxial layers during annealing remain localized over the same TiO2 rutile particles upon reduction. As observed previously during the thermal annealing treatment, the mean particle size of TiO2 P25 has not been affected by the catalytic test and remained at 25 ± 14 nm.
 | ||
| Fig. 7 TEM images of the catalysts post reduction and methanation: (a) Ru/TiO2-P25-450 post methanation showing Ru-free anatase TiO2 and Ru-covered rutile TiO2 particles; (b) Ru/TiO2-A-450 post methanation showing agglomerated Ru particles (indicated by white circles); bright field (c and d) and HAADF (e and f) images of Ru/TiO2-R-450 post methanation. The white arrows show the metallic Ru, and the red arrows show the trapped RuO2. | ||
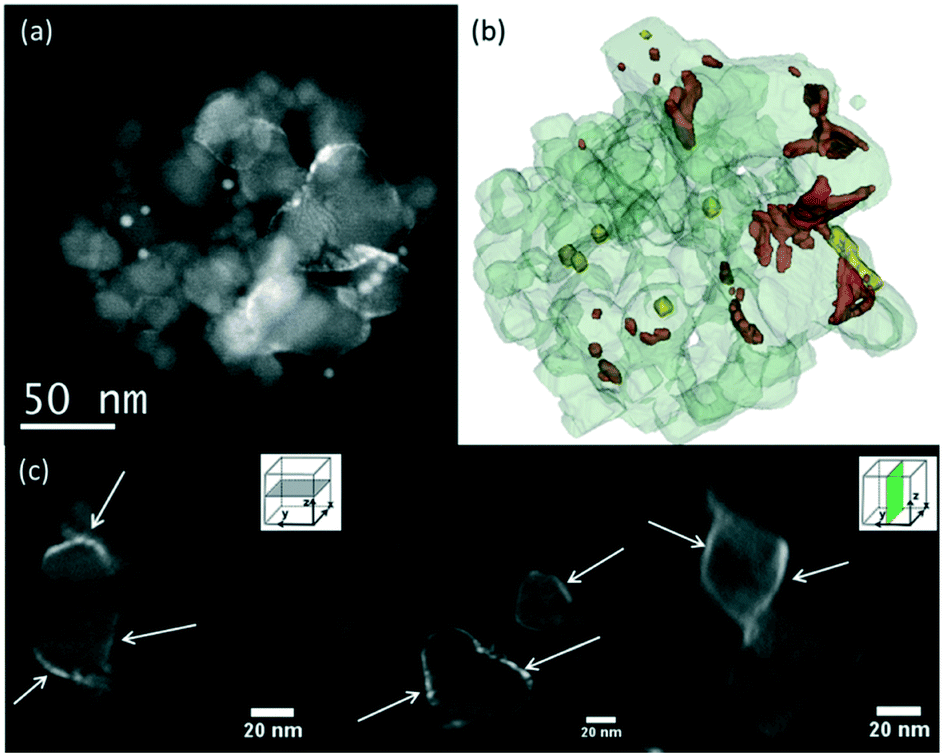 | ||
| Fig. 8 3D-TEM tomographic analysis of Ru/TiO2-P25-450 after reduction and the catalytic test showing the presence of Ru particles on the surface of rutile TiO2 particles. (a) STEM-HAADF image showing the particle aggregate used for 3D reconstruction, (b) volume reconstruction of the studied aggregate, with Ru nanoparticles in red, TiO2 in light grey and Au nanoparticles used for tomogram alignment in yellow. (c) Representative slices extracted from the reconstruction. Ru particles are pointed by the white arrows. | ||
On the anatase support, metallic Ru particles appear as agglomerates of smaller particles, as shown in Fig. 7b. The approximate sizes of these agglomerates are in the same range as that of the RuO2 crystal prior to reduction and the catalytic test (50 to 100 nm). It is suggested that RuO2 has undergone fragmentation during reduction, but no significant re-dispersion, leaving most of the anatase particles uncovered. The Ru particles appear crystalline (100, 002 and 101 planes of metallic Ru) (SI-16c and d†), with a mean size of 3.8 ± 1.0 nm. As observed on the non-reduced sample, a few rutile TiO2 crystals could be seen, systematically decorated with Ru particles (SI-16c†), again attesting the absence of Ru mobility during reduction and methanation. The size of anatase TiO2 particles remains unchanged after reduction and methanation (11.5 ± 2.6 nm).
On pure rutile TiO2, metallic crystalline Ru particles can be easily seen after methanation (Ru planes observed on SI-16e and f†); they are mainly localized close to the tip of the TiO2 rutile needles (Fig. 7c and e). Their mean size is 3.5 ± 1.0 nm. The TiO2 needles have not sintered during reduction or reaction (length of 96 ± 17 nm and width of 24 ± 5 nm). The Ru particles, however, appear scarce on the numerous images that were obtained in comparison with the P25 and anatase supports. Instead, the STEM-HAADF images show trapped Ru-containing layers in between the rutile TiO2 crystals (Fig. 7c–f), indicating that the “sandwiching” of RuO2 is maintained upon reduction.
XPS analysis after methanation revealed that the Ru/Ti ratios remained similar to the values obtained before methanation for all three supports (Table 3), indicating that the catalyst dispersion was barely modified after reduction and methanation. For all three supports, the proportion of surface metallic Ru increased after reduction and methanation (SI-18†). It is, however, important to note that the metallic Ru proportion on the rutile support was significantly lower compared to the P25 and anatase supports. This is explained by the “sandwiched” RuO2 layers described above that are protected from H2 exposure and thus reduction.
| Support | Ru/Ti (XPS) | |
|---|---|---|
| Before reduction | After methanation | |
| P25 | 0.033 | 0.036 |
| Anatase | 0.055 | 0.068 |
| Rutile | 0.028 | 0.031 |
On the P25 support, the proportion of oxidized and metallic Ru in the spent catalysts was always around 40% oxidized and 60% metallic Ru species (after being exposed to air) regardless of the annealing temperature (ESI,† SI-18a and b). This indicates that the beneficial effect of a high annealing temperature is exerted through the morphological changes observed for the active phase and not through a change in reducibility of the RuO2 phase.
H2 chemisorption resulted in the Ru dispersion values of 24%, <5%, and 13% for Ru/TiO2-P25-450, Ru/TiO2-A-450, and Ru/TiO2-R-450, respectively. A lower Ru dispersion suggests sintering of Ru nanoparticles. Based on the fact that the P25 and rutile supports follow the same pattern of RuO2 modification through epitaxial layer formation, the lower Ru dispersion value of Ru/TiO2-R-450 compared to Ru/TiO2-P25-450 is clearly attributed to the loss of Ru in between the rutile TiO2 particles. On the other hand, the lowest Ru dispersion value of Ru/TiO2-A-450 is due to the high degree of sintering of RuO2 resulting in highly agglomerated Ru after reduction. In conclusion, dispersion is strongly affected by the modifications that occur during annealing as described above.
Decisive factors dictating the methanation activity
The catalysts prepared on different crystalline TiO2 supports show distinctive behaviors in the catalytic CO2 methanation. Although the selectivity to CH4 was 100% for all the catalysts, the level of activity was markedly affected by the crystallinity of the TiO2 support used. As a general trend, the P25 supported catalysts showed the best CH4 production rate, followed by anatase- and then rutile-supported catalysts.Taking into account the dispersion data (H2 chemisorption), an apparent turnover frequency (TOF) can be calculated on the basis of the amount of Ru available at the catalyst surface. At 200 °C, the TOF (expressed as mole of CH4 produced per mole of surface-accessible Ru per second) reached ∼7 s−1 for both Ru/TiO2-P25-450 and Ru/TiO2-R-450. This indicates that the specific activity of P25 and rutile-supported catalysts is directly related to the dispersion of the Ru phase. The TOF is found to be higher for Ru/TiO2-A-450 (about 43 s−1). This suggests that different Ru species exist on the anatase TiO2 support, exhibiting a higher density of active sites compared to those stabilized onto P25 or rutile TiO2 supports. Indeed, methanation is thought to be a structure-sensitive reaction, since different physical states of active species have been reported to have different intrinsic activities. The importance of size and size distribution for the active nanoparticles has been proven in the case of ammonia synthesis (Ru particles) and CO2 methanation (Rh particles) under mild conditions.48–50 Moreover, the role of the exposed active face, defects, steps or terraces on the catalyst surface has been widely discussed for Ru/TiO2 catalysts.51
In both commonly proposed pathways for CO2 methanation (i.e. via formate intermediates or by direct CO2 dissociation into CO(ads) and O(ads)), the dissociation of CO(ads) is generally recognized to be the rate-determining reaction and is expected to proceed at different rates on different Ru surface species.21,52,53
As recently confirmed by near ambient pressure X-ray photoelectron spectroscopy, the active state of ruthenium is the metallic one.54 Yet the level of performance is dictated by the state of the catalyst after annealing. Detailed TEM observations of the catalyst after various annealing temperatures provide a link between the morphology and the catalytic behaviour (Fig. 9). In summary, on anatase and rutile TiO2 phases, two main phenomena occur during annealing, both driven towards the stabilization of the system: intrinsic growth of the particles and RuO2–TiO2 epitaxy. Concerning anatase, the weak interaction between RuO2 and anatase TiO2 results in the growth of large rutile RuO2 crystals, as well as separate TiO2 anatase sintering. After reduction, the resulting large metallic Ru aggregates have low proportions of surface Ru (detected by H2 chemisorption) which translates into modest levels of specific methanation activity, despite a higher TOF. Concerning the rutile support, the strong interaction between RuO2 and rutile TiO2 promotes the formation of continuous epitaxial layers, dominantly over the (110) facet. A “sandwiching” phenomenon is also observed, concomitantly with TiO2 rutile sintering (increase in particle width). This results in the loss of Ru species, embedded between TiO2 rods. The low activity of the rutile supported catalyst is explained by this embedding of the active phase. The P25 particles do not sinter and the stabilization occurs through RuO2–rutile TiO2 epitaxial interactions. RuO2 is thus spread on the TiO2 rutile surface, with anatase particles acting as the diluent and preventing the TiO2 rutile–RuO2–TiO2 rutile stacking. With a high amount of Ru available at the surface, as compared to the catalyst supported on pure rutile, Ru/TiO2-P25-450 presents the highest specific activity.
 | ||
| Fig. 9 Graphical illustration of the shape evolution of the RuO2/TiO2 catalysts; after RuO2 nanoparticle deposition, after thermal annealing at 450 °C, and after reduction and methanation. Red indicates RuO2, pink indicates thin RuO2 layers, white indicates Ru depleted areas, and black indicates metallic Ru. | ||
Conclusions
Ru/TiO2 methanation catalysts were studied systematically, starting from calibrated RuO2 nanoparticles as the precursor for active Ru species, which are subsequently supported on anatase, rutile, and a mixture of the two (P25) crystal structures of TiO2. Different crystalline TiO2 as supports for RuO2 nanoparticles are shown to have a marked impact on catalytic performance. This is rationalized by studying how the physico-chemical properties of the catalyst’s active phase depend on the crystalline structure of the support.We show that the annealing step provokes intense modifications of the catalyst properties. These modifications are strongly dependent on the crystal structure of the support. The weak interactions between RuO2 and anatase TiO2 cause sintering and growth of RuO2 crystals, whereas the strong interactions (i.e. lattice-matched interfacial structure) between the RuO2 and rutile TiO2 lead to a transformation of nanoparticles into epitaxial RuO2 layers sandwiched between rutile TiO2 rods. In both cases, the specific activity is decreased, either because the amount of surface accessible Ru drops significantly. On the surface of P25 TiO2, the mixing of anatase and rutile particles gives rise to a more favourable situation where Ru does interact strongly with rutile but remains fully accessible.
These results provide an insight into the design of supported catalysts taking into account the possibility of balancing RuO2–TiO2 interactions in a favourable way. The present study should prompt further work on the tuning of the anatase/rutile ratio in search of higher catalytic activity not only in CO2 methanation but also in various catalytic reactions.
Experimental section
Catalyst preparation
Pure anatase TiO2 particles were prepared by microwave hydrothermal treatment at 200 °C for 2 h of an aqueous solution of TiCl4 with the acidity adjusted to pH 6. The resultant precipitates were collected by centrifugation and then washed with water and nitric acid.55Pure rutile TiO2 particles were obtained by refluxing at 120 °C for 3 days an aqueous solution of TiCl4 in 1 M HCl followed by washing the resultant precipitates with water and nitric acid.56
A highly stable colloidal suspension of monodispersed RuO2 nanoparticles was obtained by dropwise addition of 15% v/v H2O2 diluted in H2O into 0.011 M RuCl3·xH2O (x = 3–5) dissolved in H2O so that the final concentration is [Ru] ≈ 0.007 M. The solution was heated at 95 °C for 2 h. Once cooled to room temperature, an appropriate amount of TiO2 powder (P25 from Degussa, home-made pure anatase or home-made pure rutile) was added to the colloidal suspension of RuO2 nanoparticles to yield 2.5 wt% of Ru in the final catalyst. The mixture was put in an oven at 50 °C overnight and the excess water was removed by rotary evaporation. The resulting powder was then annealed/calcined at 150, 250, 350, or 450 °C for 16 h in static air and washed 3 times with water. The catalysts are denoted as RuO2/TiO2-P25, RuO2/TiO2-A, and RuO2/TiO2-R for P25, pure anatase, and pure rutile TiO2 supported RuO2, respectively. After reduction under continuous flow of H2 at 200 °C (see section 2.3), the catalysts were denoted as Ru/TiO2-P25, Ru/TiO2-A, and Ru/TiO2-R, respectively. Different calcination temperatures are indicated with extended numerical notations, e.g. RuO2/TiO2-P25-150.
Catalyst characterization
Transmission electron microscopy (TEM) images were obtained using a FEI Tecnai 120 Twin microscope operating at 120 kV and equipped with a Gatan Orius CCD numeric camera. The samples were prepared by ultrasonic dispersion of the powders in water and a droplet of the dispersion was then placed onto a carbon-coated copper grid.High-resolution analysis (HR-TEM) images were obtained by using a JEOL JEM 2010 microscope operating at 200 kV and equipped with a Gatan camera. The sample preparation was the same as in TEM sample preparation.
High angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images were obtained using a Jeol 2200FS microscope equipped with a spherical aberration corrector on the probe and an EDX system from Jeol. The convergence semi-angle of the probe was 30 mrad and the current was 150 pA. The inner and the outer semiangles for the dark-field detector (upper DF detector) were 100 and 170 mrad, respectively.
3D TEM tomography data were acquired using a JEOL 2100F electron microscope. The acquisition of bright field (BF) and dark field (DF) tilt series was carried out simultaneously in scanning mode (STEM). A camera length of 10 cm was chosen for this experiment. It corresponds to inner and outer semiangles of 60 and 160 mrad, respectively, for the HAADF detector. A 100 μm condenser aperture was employed, allowing one to reach a probe diameter of about 0.12 nm with a current density of 0.5 pA Å−2. Under these conditions, the tomography series were acquired using Digital Micrograph software (tomography plugin), giving access to an automatic increment of the tilt angles and sharp control of the specimen drift and defocusing. A high tilt specimen holder from Gatan was employed for a tilting range of −65° to 65°, with an equal angular step of 2.5°.
The precision of these nanoscale 3D analyses greatly benefits from the DART reconstruction method that minimizes the artefacts due to the missing wedge. Indeed, after the fine alignment of all projections, the 3D volume was calculated using the discrete algebraic reconstruction technique (DART). For this purpose, a preliminary simultaneous iterative reconstruction technique (SIRT) reconstruction was performed. By constraining the reconstruction volume with a mask which roughly equals the particle shapes, reliable material densities can be deduced. Subsequently, the density of the gold particles is used to perform a DART reconstruction, which is discrete in terms of grey values.57 This means that each voxel (unit fragment of the volume) is attributed to either vacuum or gold. Consequently, DART is superior to SIRT when exact particle boundaries are to be determined.58
X-ray diffraction (XRD) measurements were performed with Cu Kα radiation using a Bruker D8 Advance diffractometer equipped with a Lynx eye detector. The 2θ diffractograms were recorded between 24–50° with a step size of 0.04° and a steep time of 20 s per step. The ICDD-PDF2 database was used to identify the crystalline phases. When possible, TiO2 and RuO2 XRD peaks were deconvoluted, using WinPLOTR.59 The Scherrer equation was used to calculate the crystallite size of particles (SI-19†).60
X-ray photoelectron spectroscopic (XPS) analyses were performed using an SSX 100/206 photoelectron spectrometer from Surface Science Instruments (USA) equipped with a monochromatised micro focused Al X-ray Kα source (powered at 20 mA and 10 kV), a 30° solid angle acceptance lens, a hemispherical analyser and a position-sensitive detector. The samples were pressed in small stainless steel troughs of 4 mm diameter and placed on a multi-specimen holder. The pressure in the analysis chamber was around 10–6 Pa. The angle between the surface normal and the axis of the analyser lens was 55°. The analysed area was approximately 1.4 mm2 and the pass energy was set at 150 eV. Atomic concentration ratios were calculated by normalizing surface area ratios with sensitivity factors based on Scofield cross-sections. In addition, all binding energies were calculated taking as reference the C–(C, H) component of the C 1s adventitious carbon peak fixed at 284.8 eV. Peak decomposition was performed using the CasaXPS program (Casa Software Ltd., UK) with a Gaussian/Lorentzian (85/15) product function and a Shirley non-linear sigmoid-type baseline. The following peaks were used for the quantitative analysis: O 1s, C 1s, Ti 2p, and Ru 3d. The 3d Ru peak was decomposed into 3 doublets assigned to Ru0, Ru4+ and the related RuO2 plasmon species, respectively.61 The positions of these species have been imposed at 280.0 ± 0.1 eV, 291.0 ± 0.1 eV and 282.8 ± 0.1 eV, respectively. The binding energy difference between the 3/2 and 5/2 contributions of each doublet was fixed to 4.17 eV. Besides, the Ru3d3/2/Ru3d5/2 ratio was fixed to 0.667. The FWHM of each component was limited at 2.5 eV.
TPR experiments were performed using 100 mg of each catalyst after in situ purging under an inert gas (Ar) at 140 °C for one hour. The analysis was carried out under 2.5 v/v% H2 diluted in inert gases (2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :82.5:15 v/v% H2:He:Ar) in a stream of 20 mL min−1 from 20 °C to 500 °C using a 5 °C min−1 temperature ramp. H2 consumption and H2O production were measured simultaneously via a QMC 311 quadrupole mass spectrometer (Balzers) coupled in line with the reactor.
:82.5:15 v/v% H2:He:Ar) in a stream of 20 mL min−1 from 20 °C to 500 °C using a 5 °C min−1 temperature ramp. H2 consumption and H2O production were measured simultaneously via a QMC 311 quadrupole mass spectrometer (Balzers) coupled in line with the reactor.
The weight percentages of Ru and Ti inside the catalysts were measured by Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES) using an ICAP 6500 from Thermo Scientific. The materials were dried at 105 °C before the measurement.
The specific surface area of the catalysts was obtained by means of a nitrogen adsorption–desorption isotherm collected at −196 °C using a BELSORB-mini II (BEL Japan, Inc.). The samples were outgassed overnight at 140 °C prior to analysis. SBET was calculated by the Brunauer–Emmet–Teller (BET) method at the N2 relative pressure range of 0.05 < P/P0 < 0.30.
H2 chemisorption at 100 °C was used to measure the exposed Ru atoms using an ASAP 2010C apparatus from Micromeritics. A catalyst with a weight between 150–200 mg was loaded into a Pyrex tube, and subsequently degassed under He at 150 °C for 30 min. After evacuation, the sample was reduced under pure H2 at 200 °C for 2 h (same as in situ reduction for methanation, see section 2.3) followed by purging with He at 100 °C for 1 h and adsorption of H2. Two isotherms were measured in the range of 0.08–95 kPa. The first accounts for reversible and irreversible chemisorption. The sample was evacuated to desorb reversibly adsorbed H2. The second isotherm was then measured which accounts only for the reversibly adsorbed H2. The subtraction of the linear part of the two isotherms gave the total amount of irreversibly adsorbed (chemisorbed) H2. The amount of surface Ru atoms was calculated from the amount of chemisorbed H2, assuming that the chemisorption stoichiometry is H:Ru = 1.62,63 Dispersion is defined as surface Ru atoms divided by total Ru atoms in the catalyst.
Methanation of carbon dioxide
200 mg of a catalyst with a particle size between 100 and 315 μm was loaded in a continuous flow fixed bed reactor and reduced in situ at 200 °C for 2 h under 30 ml min−1 of H2 prior to the catalytic reaction. The reaction was carried out at 1 atm in the temperature range of 50 to 200 °C under a 20 ml min−1 flow of a reaction mixture (CO2 (10 vol%), H2 (40 vol%) diluted in He). Each temperature was maintained for 52 min (3 GC injections). The exit gases were quantified using a gas chromatograph (Varian CP3800) equipped with Hayesep Q, Molsieve 5A, and CP-Sil-5CB columns. The separated gases were detected by a flame ionization detector (CH4) and a thermal conductivity detector (CO and CO2). The analysis parameters were set so as to allow analysis every 19 min and to obtain measurements accurate within about 1% (relative) for the methane production rate (mole of methane produced per gram of catalyst per second). All transfer lines were maintained at 110 °C to avoid water condensation.Acknowledgements
A. K. thanks the European doctoral school IDS FunMat for the PhD fellowship. The authors acknowledge Wallonie-Bruxelles International, the Ministère Français des Affaires étrangères et européennes and the Ministère de l'Enseignement supérieur et de la Recherche for their financial support in the framework of the Hubert Curien partnership (Tournesol), as well as the METSA network for 3D STEM tomography. The authors also acknowledge the Fondation Collège de France. AK and DPD thank Dr. François Devred for the technical and logistical support in the project.References
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- B. R. Cuenya, Thin Solid Films, 2010, 518, 3127–3150 CrossRef CAS.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS PubMed.
- C. T. Campbell, Acc. Chem. Res., 2013, 46, 1712–1719 CrossRef CAS PubMed.
- G. Prieto, J. Zečević, H. Friedrich, K. P. de Jong and P. E. de Jongh, Nat. Mater., 2012, 12, 34–39 CrossRef PubMed.
- E. V. Kondratenko, A. P. Amrute, M.-M. Pohl, N. Steinfeldt, C. Mondelli and J. Perez-Ramirez, Catal. Sci. Technol., 2013, 3, 2555–2558 CAS.
- S. Tada, R. Kikuchi, A. Takagaki, T. Sugawara, S. T. Oyama, K. Urasaki and S. Satokawa, Appl. Catal., B, 2013, 140–141, 258–264 CrossRef CAS.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- J. Martins, N. Batail, S. Silva, S. Rafik-Clement, A. Karelovic, D. P. Debecker, A. Chaumonnot and D. Uzio, Catal. Commun., 2015, 58, 11–15 CrossRef CAS.
- N. Mahata, K. V. Raghavan, V. Vishwanathan, C. Park and M. A. Keane, Phys. Chem. Chem. Phys., 2001, 3, 2712–2719 RSC.
- Q. Lin, X. Y. Liu, Y. Jiang, Y. Wang, Y. Huang and T. Zhang, Catal. Sci. Technol., 2014, 4, 2058 CAS.
- G. Xiang, X. Shi, Y. Wu, J. Zhuang and X. Wang, Sci. Rep., 2012, 2, 801 Search PubMed.
- M. S. Chen and D. W. Goodman, Catal. Today, 2006, 111, 22–33 CrossRef CAS.
- R. A. Van Santen, Acc. Chem. Res., 2009, 42, 57–66 CrossRef CAS PubMed.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- Y. Zhang and T. Ren, Chem. Commun., 2012, 48, 11005–11007 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- K. R. Thampi, J. Kiwi and M. Graetzel, Nature, 1987, 327, 506–508 CrossRef CAS.
- P. J. Lunde and F. L. Kester, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 27–33 CAS.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141–153 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- M. A. Henderson, S. D. Worely and S. D. Worley, J. Phys. Chem., 1985, 89, 1417–1423 CrossRef CAS.
- K. Urasaki, K.-I. Endo, T. Takahiro, R. Kikuchi, T. Kojima and S. Satokawa, Top. Catal., 2010, 53, 707–711 CrossRef CAS.
- A. Erdohelyi, M. Pasztor and F. Solymosi, J. Catal., 1986, 98, 166–177 CrossRef CAS.
- D. Crihan, M. Knapp, S. Zweidinger, E. Lundgren, C. J. Weststrate, J. N. Andersen, A. P. Seitsonen and H. Over, Angew. Chem., Int. Ed., 2008, 47, 2131–2134 CrossRef CAS PubMed.
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- Q. Lin, Y. Huang, Y. Wang, L. Li, X. Y. Liu, F. Lv, A. Wang, W.-C. Li and T. Zhang, J. Mater. Chem. A, 2014, 2, 5178–5181 CAS.
- K. Seki, Catal. Surv. Asia, 2010, 14, 168–175 CrossRef CAS.
- D. P. Debecker, B. Farin, E. M. Gaigneaux, C. Sanchez, C. Sassoye, B. Farin, E. M. Gaigneaux, C. Sanchez and C. Sassoye, Appl. Catal., A, 2014, 481, 11–18 CrossRef CAS.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 CAS.
- M. Balaraju, V. Rekha, B. L. A. Prabhavathi Devi, R. B. N. Prasad, P. S. Sai Prasad and N. Lingaiah, Appl. Catal., A, 2010, 384, 107–114 CrossRef CAS.
- C. Sassoye, G. Muller, D. P. Debecker, A. Karelovic, S. Cassaignon, C. Pizarro, P. Ruiz and C. C. Sanchez, Green Chem., 2011, 13, 3230–3237 RSC.
- S. C. Pillai, P. Periyat, R. George, D. E. McCormack, M. K. Seery, H. Hayden, J. Colreavy, D. Corr and S. J. Hinder, J. Phys. Chem. C, 2007, 111, 1605–1611 CAS.
- N. Wetchakun, B. Incessungvorn, K. Wetchakun and S. Phanichphant, Mater. Lett., 2012, 82, 195–198 CrossRef CAS.
- L. Ji, J. Lin and H. C. Zeng, Chem. Mater., 2001, 13, 2403–2412 CrossRef CAS.
- W. E. Bell and M. Tagami, J. Phys. Chem., 1963, 67, 2432–2436 CrossRef CAS.
- H. Schaefer, A. Tebben, W. Gerhardt, V. Harald and A. Tebben, Z. Anorg. Allg. Chem., 1963, 321, 41–55 CrossRef CAS.
- X. Paquez, G. Amiard, G. de Combarieu, C. Boissiere and D. Grosso, Chem. Mater., 2015, 27, 2711–2717 CrossRef CAS.
- T. W. Hansen, A. T. De La Riva, S. R. Challa, A. K. Datye, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- F. Garisto, AECL-9552, Whiteshell Nucl. Res. Establ., 1988 Search PubMed.
- S. B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested and S. Helveg, J. Am. Chem. Soc., 2010, 132, 7968–7975 CrossRef CAS PubMed.
- K. Yoshida, A. Bright and N. Tanaka, J. Electron Microsc., 2012, 61, 99–103 CrossRef CAS PubMed.
- A. D. Benavidez, L. Kovarik, A. Genc, N. Agrawal, E. M. Larsson, T. W. Hansen, A. M. Karim and A. K. Datye, ACS Catal., 2012, 2, 2349–2356 CrossRef CAS.
- M. A. G. Hevia, A. P. Amrute, T. Schmidt and J. Pérez-Ramírez, J. Catal., 2010, 276, 141–151 CrossRef CAS.
- H. Madhavaram, H. Idriss, S. Wendt, Y. Kim, M. Knapp, H. Over, J. Aßmann, E. Löffler and M. Muhler, J. Catal., 2001, 202, 296–307 CrossRef CAS.
- I. Balint, A. Miyazaki and K. Aika, Society, 2001, 932–938 CAS.
- C. Fernández, C. Sassoye, D. P. Debecker, C. C. Sanchez, P. Ruiz, C. Fernandez, C. Sassoye, D. P. Debecker, C. C. Sanchez and P. Ruiz, Appl. Catal., A, 2014, 474, 194–202 CrossRef.
- C. Fernández, C. Sassoye, N. Flores, N. Escalona, E. M. Gaigneaux, C. Sanchez and P. Ruiz, Appl. Catal., A, 2015, 502, 48–56 CrossRef.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS.
- C. Chiu, A. Genest, A. Borgna, N. Rösch and N. Rosch, Phys. Chem. Chem. Phys., 2015, 17, 15324–15330 RSC.
- F. Solymosi and A. Erdohelyi, Stud. Surf. Sci. Catal., 1981, 7, 1448–1449 CrossRef CAS.
- I. A. Fisher and A. T. Bell, J. Catal., 1996, 162, 54–65 CrossRef CAS.
- S. Carenco, C. Sassoye, M. Faustini, P. Eloy, D. P. Debecker, H. Bluhm and M. B. Salmeron, J. Phys. Chem. C, 2016, 120, 15354–15361 CAS.
- F. Dufour, S. Cassaignon, O. Durupthy, C. Colbeau-Justin and C. Chaneac, Eur. J. Inorg. Chem., 2012, 2012, 2707–2715 CrossRef CAS.
- C. Magne, F. Dufour, F. Labat, G. Lancel, O. Durupthy, S. Cassaignon and T. Pauporté, J. Photochem. Photobiol., A, 2012, 232, 22–31 CrossRef CAS.
- K. J. Batenburg, S. Bals, J. Sijbers, C. Kuebel, P. A. Midgley, J. C. Hernandez, U. Kaiser, E. R. Encina, E. A. Coronado and G. Van Tendeloo, Ultramicroscopy, 2009, 109, 730–740 CrossRef CAS PubMed.
- A. Zuerner, M. Doeblinger, V. Cauda, R. Wei and T. Bein, Ultramicroscopy, 2012, 115, 41–49 CrossRef CAS PubMed.
- J. Rodriguez-Carvajal and T. Roisnel, WinPLOTR, a graphic tool for powder diffraction, Institut Laue Langevin, 2016 Search PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- H. Over, A. P. Seitsonen, E. Lundgren, M. Smedh and J. N. Andersen, Surf. Sci., 2002, 504, L196–L200 CrossRef CAS.
- J. G. Goodwin Jr., J. Catal., 1981, 68, 227–232 CrossRef.
- J. Okal, M. Zawadzki, L. Kepinski, L. Krajczyk and W. Tylus, Appl. Catal., A, 2007, 319, 202–209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy01677d |
| This journal is © The Royal Society of Chemistry 2016 |