
Engineering of ZSM-5 zeolite crystals for enhanced lifetime in the production of light olefins via 2-methyl-2-butene cracking†
Sharon
Mitchell
,
Marilyne
Boltz
,
Jiaxu
Liu
and
Javier
Pérez-Ramírez
 *
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, CH-8093, Switzerland. E-mail: jpr@chem.ethz.ch
First published on 4th October 2016
Abstract
The selective cracking of low-value C5 olefins such as 2-methyl-2-butene (2M2B), a growing by-product of deep catalytic cracking, over ZSM-5 zeolites is an attractive route for the on-purpose production of ethylene and propylene. Yet, several aspects, including the deactivation behaviour and the effect of the crystal properties (i.e., the size, morphology, amount of defects, and mesoporosity) on the performance, still lack understanding. Here, the relative impact of these key crystal variables on the catalyst stability and selectivity is examined by preparing a series of tailored materials with equivalent acidic properties within the optimal range for this process. Specifically, zeolites with micron- and nanosized crystals of rounded-boat and coffin-shaped morphology and different defect concentrations are synthesised in hydroxide and fluoride media, whereas intracrystalline mesopores are subsequently introduced by their controlled desilication. In agreement with the comparable active site distributions, evaluation in 2M2B cracking evidences similar initial conversion and product distributions over all catalysts, with high propylene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylene ratios and high butylene yields compatible with a dimerisation–cracking mechanism. In contrast, major differences in the deactivation behaviour are demonstrated, clearly illustrating the dominant impact of the mesopore (external) surface area compared to the defect concentration or crystal morphology. Comparative evaluation in the widely studied conversion of methanol to olefins (MTO) reveals qualitatively similar trends. Quantitatively, however, a striking six-fold extension of the lifetime (347 versus 54 h) and a 3-fold higher light olefin space-time yield (6.67 versus 2.25 gC2–4= gzeolite−1 h−1) are observed over the best catalyst in 2M2B cracking with respect to MTO.
:ethylene ratios and high butylene yields compatible with a dimerisation–cracking mechanism. In contrast, major differences in the deactivation behaviour are demonstrated, clearly illustrating the dominant impact of the mesopore (external) surface area compared to the defect concentration or crystal morphology. Comparative evaluation in the widely studied conversion of methanol to olefins (MTO) reveals qualitatively similar trends. Quantitatively, however, a striking six-fold extension of the lifetime (347 versus 54 h) and a 3-fold higher light olefin space-time yield (6.67 versus 2.25 gC2–4= gzeolite−1 h−1) are observed over the best catalyst in 2M2B cracking with respect to MTO.
Introduction
Light olefins comprise some of the largest volume building blocks in the chemical industry with current demands of over 230 million tonnes per year.1 Traditional technologies for their production such as steam cracking of alkanes and fluid catalytic cracking cannot cover the rapidly expanding global market, particularly for propylene, which is obtained as a by-product in both processes.2,3 This generates a significant commercial incentive to identify efficient ‘on-purpose’ routes for light olefin production. Several catalytic processes have been developed to fill the supply and demand gap including variants on fluid catalytic cracking, methanol to olefins, methanol to propylene, propane dehydrogenation, olefin metathesis, and the cracking of light (C4+) olefin-rich refinery streams.2,3Converting low-value C5 olefins presents an attractive prospect.4,5 Currently, these streams are directly blended into fuels, but their use is increasingly limited by regulatory changes to reduce gasoline volatility.6 A particularly interesting substrate is 2-methyl-2-butene (2M2B), the main C5 olefinic component of selectively hydrogenated C5 cuts from steam cracking4 and a growing by-product of deep catalytic cracking (RIPP).7 The selective cracking of 2M2B to light olefins has been evaluated over various zeolites and zeotypes. Consistent with the majority of works on higher-olefin cracking,8 ZSM-5 (MFI-type) zeolites were found to yield superior stable and selective behaviour at high conversion compared to other framework types.4,9 The possible mechanisms of C4+ olefin cracking over zeolites have been extensively investigated.10,11 Different reactions, including monomolecular cracking and dimerisation–cracking pathways by β-scission, oligomerisation, isomerisation, and hydrogen transfer, can occur depending on the specific substrate, pore topology and acidic properties of the zeolite, and the temperature and weight hourly space velocity (WHSV) of the reaction. In the case of 2M2B, high light olefin selectivity and propylene:ethylene ratios are reportedly favoured over ZSM-5 zeolites with relatively low acid site densities (e.g., ≤100 mmol g−1), at temperatures above 723 K, and with short contact time across the catalytic bed (WHSV >10 h−1).4,11
Although these studies have provided important insights for the improved design of ZSM-5 catalysts for light olefin production from C5 olefins such as 2M2B, a number of factors require deeper understanding. Notably, there is a distinct lack of in-depth deactivation studies; almost all of the data analysed for C4+ olefin cracking was collected at times on stream of <3 h. Improved insight into the deactivation behaviour would benefit fixed-bed processes for C4–6 olefin conversions, such as PROPYLUR (Lurgi/Linde), one of the most successful technologies that has reached a large pilot scale to date.3 Furthermore, the impact of the crystal properties other than the amount and strength of acid sites on the performance of ZSM-5 zeolites has not been assessed.
In the well-known conversion of methanol to olefins (MTO), both hierarchical structuring of the porosity12 and reducing the concentration of internal defects13 have been linked to extended catalyst lifetimes. The effectiveness of the former, which has been achieved either by decreasing the crystal size or through the introduction of intracrystalline mesopores, has been linked to the improved accessibility of active sites within the zeolite. On the other hand, the benefits of reducing the defect concentration, which has been achieved by tailoring the synthesis or by post-synthetic modification, are typically attributed to the decreased retention of coke precursors. Albeit providing some hints for the design, the relative impact of each of these variables remains unclear, leaving insufficient knowledge to prepare the optimal material. Moreover, the lack of studies comparing the MTO reaction with other processes for light olefin production makes it difficult to predict the benefits of crystal engineering for C5 olefin cracking.
One of the main challenges of evaluating the effect of different crystal variables is the attainment of materials that only differ in a single property. In this context, we targeted the preparation of H-ZSM-5 catalysts with controlled crystal size, morphology, and defect concentration, but equivalent acidic properties. Specifically, micron- and nanosized crystals of rounded-boat and coffin-shaped morphology and with different defect concentrations are attained via controlled synthesis in alkaline or fluoride media, respectively. Intracrystalline mesopores are introduced by alkaline treatment of the purely microporous zeolites to attain hierarchical analogues. The catalysts are tested in the cracking of 2-methyl-2-butene under different conditions to quantify the impact of these properties on the catalyst performance. Evaluation of the same catalysts in the MTO reaction as a prototypical example for light olefin production highlights parallels and distinctions in terms of selectivity and deactivation behaviour compared to 2M2B cracking.
Experimental
Materials
Fumed silica (Aeroperl 300/3, Evonik), colloidal silica (LUDOX AS-40, 40 wt% aqueous suspension, Aldrich), tetraethyl orthosilicate (TEOS, 99%, Aldrich), sodium aluminate (AlNaO2, Sigma-Aldrich), aluminium sulphate (Al2(SO4)3·18H2O, 95%, Fluka), aluminium powder (99.9%, Acros organics), tetrapropylammonium bromide (TPABr, 98%, abcr), tetrapropylammonium hydroxide (TPAOH, 25 wt% aqueous solution, Alfa Aesar), ammonium hydroxide (NH4OH, 28 wt% aqueous solution, Sigma-Aldrich), ammonium fluoride (NH4F, 98%, Acros organics), sodium hydroxide (NaOH, 97%, Sigma-Aldrich), hydrochloric acid (HCl, 37 wt%, Sigma-Aldrich), hydrofluoric acid (HF, 40%, Sigma-Aldrich), and sulphuric acid (H2SO4, 97%, Sigma-Aldrich) were used for the zeolite syntheses.Zeolite synthesis
Micron- and nanosized ZSM-5 zeolites with a nominal Si/Al ratio of 50 were synthesised in OH− and F− media, adapting reported procedures.13b,14–16 All the hydrothermal syntheses were performed in static stainless steel Teflon-lined autoclaves (50 cm3). In OH− media, the micronsized ZSM-5 zeolite (coded OH-M) was prepared from a gel of the following molar composition: 100SiO2:1Al2O3:4(TPA)2O:3.4Na2O:2060H2O.14 Briefly, AlNaO2 was dissolved in distilled water for 10 min. Then, TPABr and NaOH (50 wt% aqueous solution) were added and the solution was stirred for an additional 10 min. Finally, colloidal SiO2 was slowly introduced under high mechanical stirring and the pH was adjusted to 12 using H2SO4 and NaOH (50 wt% aqueous solution). The gel was crystallised at 448 K for 24 h. The nanosized ZSM-5 zeolite (coded OH-N) was synthesised from a gel of molar ratio 100SiO2:1.5Al2O3:1.5(TPA)2O:60NH4OH:1500H2O.15 Al2(SO4)3·18H2O was dissolved in a solution of distilled water and NH3 under stirring prior to the addition of TPABr. After complete homogenisation, colloidal silica and the silicalite-1 seed suspension were carefully added into the mixture. The gel was crystallised under static conditions at 448 K for 24 h.
In F− medium, micronsized zeolite (coded F-M) was obtained from a gel of molar composition 100SiO2:1.5Al2O3:112NH4F:3.5(TPA)2O:7990H2O.16 AlNaO2 and TPABr were dissolved in distilled water to obtain a clear solution. Then, NH4F was added and finally the silica source was slowly dosed into the mixture under vigorous stirring. The pH was adjusted to 7 using a few drops of HF (40 wt%, Sigma-Aldrich) and the gel was aged for 2 h at room temperature. The hydrothermal synthesis was carried out at 448 K for 48 h. To prepare the nanosized zeolite (coded F-N), a silicalite-1 seed suspension was first synthesised using the clear solution method.13b The molar ratio of the gel was 100SiO2:18(TPA)2O:400EtOH:1720H2O, where the ethanol comes from the hydrolysis of TEOS. The reactants were mixed and stirred at room temperature overnight before the solution was crystallised at 373 K for 72 h. The resulting silicalite-1 suspension was directly used without any centrifugation or washing. The zeolite gel was then prepared having the following molar ratio: 100SiO2:1Al2O3:13.5TPAOH:14.2TPABr:62NH4F:2000H2O. The Al powder, NH4F, and TPABr were dissolved in water prior to the slow addition of fumed silica under vigorous stirring. The silicalite-1 seed (10 wt% silica content of the synthesis gel) was then added and the pH was decreased to 8 using HCl (37%, Sigma Aldrich). The resulting slurry was aged for 1 h and the hydrothermal synthesis was performed at 443 K for 4 h.
The solid products from the above syntheses were recovered by filtration, thoroughly washed with distilled water and dried at 338 K overnight. In the case of OH−-mediated routes, the zeolites were ion exchanged 3 times with ammonium nitrate (0.1 M, 100 cm3 gzeolite−1) for 8 h at room temperature. Finally, the samples were calcined in static air at 823 K (5 K min−1) for 5 h to obtain them in the protonic form. Hierarchical analogues were obtained by treatment of the micron- and nanosized zeolites (protonic form) in aqueous NaOH (0.2 M, 30 cm3 gzeolite−1) at 338 K for 30 min. The slurries were then quenched and the solids were collected by filtration, washed with distilled water, and dried for 10 h at 338 K. The resulting powders (coded with the suffix -H) were then ion exchanged with ammonium nitrate and calcined to obtain the protonic form as described above.
Characterisation
Powder X-ray diffraction (XRD) was measured in a PANalytical X'Pert Pro diffractometer using Ni-filtered CuKα radiation (λ = 0.1541 nm). Data were recorded in the 5–70° 2θ range with an angular step size of 0.05° and a counting time of 7 s per step. Argon sorption at 77 K was undertaken using a Micromeritics 3Flex instrument after sample evacuation at 573 K for 3 h. The content of Si and Al in the samples was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a Horiba Ultima 2 instrument equipped with a photomultiplier tube detector. Scanning electron microscopy (SEM) images were acquired with a Zeiss Gemini 1530 FEG microscope. Transmission electron microscopy (TEM) was performed using an FEI Tecnai F30 FEG microscope (300 kV). The zeolite powders were dispersed dry onto holey-carbon coated copper grids. Diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy was performed using a Bruker Optics Vertex 70 spectrometer equipped with a high-temperature cell (Harrick) and a liquid N2 cooled mercury cadmium telluride (MCT) detector. The samples were pre-dried at 673 K in N2 flow (100 cm3 min−1) for 3 h. Spectra were recorded in N2 at 473 K, in the range of 650–4000 cm−1, by co-addition of 320 scans and with a nominal resolution of 4 cm−1. Fourier transform infrared (FTIR) spectroscopy of adsorbed pyridine was conducted using a Bruker IFS 66 spectrometer. Self-supporting zeolite wafers (1 cm2) were first degassed at 10−3 mbar and 673 K for 4 h. After saturation with pyridine, weakly bound molecules were evacuated at room temperature for 15 min and subsequently at 473 K for 30 min. The concentrations of Brønsted and Lewis acid sites were determined using the reported extinction coefficients of 1.67 and 2.94 cm μmol−1, respectively.17 The coke content in the samples after the catalytic tests was quantified from the weight loss observed between 303 and 1173 K during thermogravimetric analysis in air using a Mettler Toledo TGA/DSC 1 system with a heating ramp of 10 K min−1.Catalytic tests
2-Methyl-2-butene (2M2B) cracking and the conversion of methanol (MeOH) to olefins were evaluated in a Microactivity-Reference unit (PID Eng&Tech) at ambient pressure and 673–773 K. The catalysts (0.03–0.3 g, 0.2–0.4 mm sieve fraction, mixed in a 1:30–1:3 mcat:mSiC ratio with SiC of 0.4–0.6 mm diameter) were loaded in a fixed-bed continuous-flow reactor (stainless steel, 13.5 mm i.d.) and heated for 1 h under a N2 flow at 773 K. 2M2B (Sigma-Aldrich, 95%) or MeOH (Sigma-Aldrich, 99.9%) was introduced via a Nexus 6000 high force infusion syringe pump into the preheated reactor using N2 as a carrier gas (48.9 cm3 min−1) to attain the desired weight hourly space velocity with respect to the zeolite content (WHSV = 12–40 g2M2B gzeolite−1 h−1 or 6 gMeOH gzeolite−1 h−1). The product mixture was analysed by online gas chromatography (GC 7890A, Agilent Technologies) using a HP PLOT Q capillary column and a flame ionisation detector. Individual products were identified and lumped together according to retention times established by comparison with pure standards, and carbon balances of between 96% and 99% were usually achieved. In the 2M2B cracking reaction, all 2M2B isomers were considered as reactant species when calculating the conversion, whereas in the methanol to olefin (MTO) reaction, methanol and dimethylether were lumped. In both reactions, the catalyst lifetime was determined as the time on stream during which the conversion was greater than 80%, which defines the length of a single catalytic cycle. Catalyst regeneration was accomplished using flowing air at 873 K for 3 h in a tubular furnace (Carbolite GHA).
Results and discussion
Synthesis and characterisation of the ZSM-5 catalysts
To decouple the impact of the distinct crystal variables, two sets of micron- (OH-M, F-M) and nanosized (OH-N, F-N) ZSM-5 zeolites were prepared by hydrothermal synthesis in OH− and F− media. Comparison of the X-ray diffraction patterns (Fig. 1a) reveals sharp reflections characteristic of a well-crystallised MFI-type structure in each of the resulting materials. The purely microporous nature of OH-M and F-M zeolites is confirmed by the virtually identical type I isotherms measured by Ar sorption at 77 K (Fig. 1b and c). As expected, OH-N and F-N zeolites exhibit increased uptake at higher relative pressures associated with the 2–3 times larger external surface areas (Smeso) compared with the micronsized samples (Table 1). Examination by scanning electron microscopy (Fig. 2) clearly evidences the distinct morphology and size of the resulting crystals. In particular, OH-M and OH-N zeolites prepared in OH− medium both comprise crystals of rounded-boat morphology with the longest dimension in the range of 5 and 0.4 μm, respectively. Comparatively, the F-N and F-M crystals synthesised in F− medium present a coffin-shaped morphology having well-developed facets, with average respective lengths of 0.6 and 24 μm. Independent of the synthesis medium, the nanosized samples exhibit a higher degree of twinning (intergrowth) visible on the (010) faces with respect to their micronsized analogues and are seen to be highly aggregated. Consistent with the close Si/Al ratios (50–60) attained (Table 1), infrared measurements of adsorbed pyridine confirm the comparable numbers of Brønsted (112–144 μmol g−1) and Lewis (11–23 μmol g−1) acid sites in the samples. This is a key accomplishment in order to quantify the effects of the other crystal properties.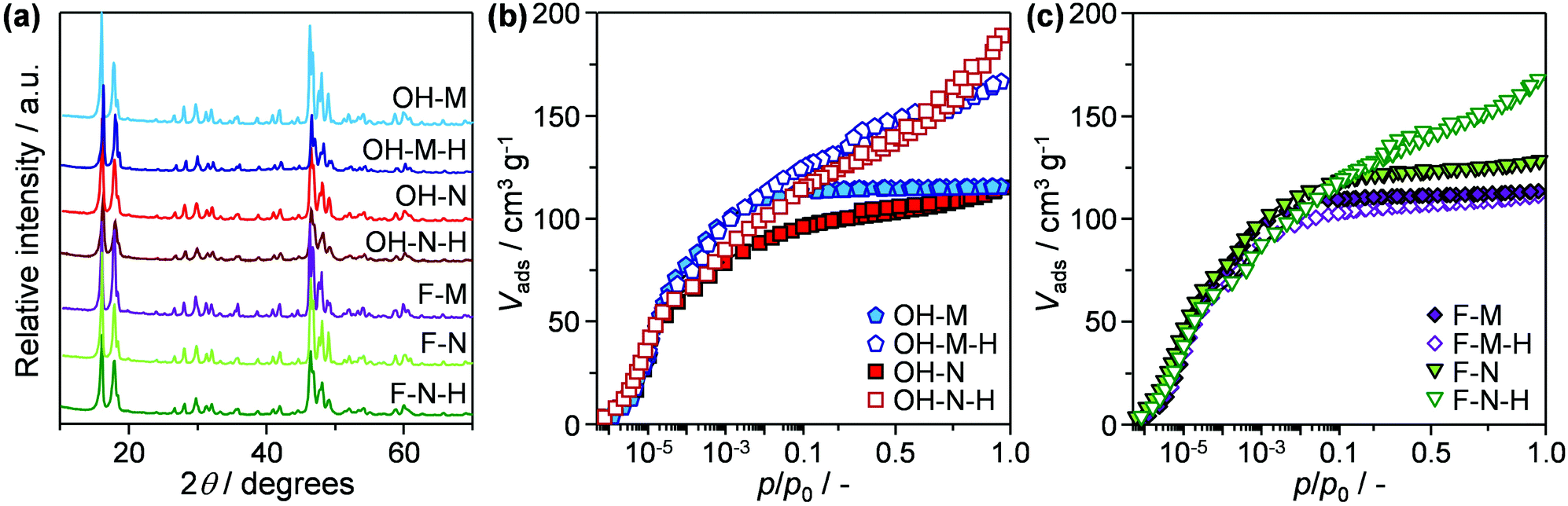 | ||
| Fig. 1 (a) X-ray diffraction patterns and (b and c) argon isotherms of the micron- and nanosized ZSM-5 catalysts and their desilicated analogues. | ||
| Sample | Si/Ala (mol mol−1) | L crystal b (μm) | V micro c (cm3 g−1) | V meso d (cm3 g−1) | S meso e (m2 g−1) | S BET f (m2 g−1) | c Brønsted g (μmol g−1) | c Lewis g (μmol g−1) | I int/Iexth |
|---|---|---|---|---|---|---|---|---|---|
| a ICP-OES. b Estimated by image analysis of 20 crystals by SEM. c NLDFT. d V meso = Vtotal − Vmicro. e t-Plot method. f BET method. g IR study of adsorbed pyridine. h Estimated from the intensity ratio of the bands at 3725 cm−1 (Iint) and 3745 cm−1 (Iext) in the DRIFT spectra. | |||||||||
| OH-M | 60 | 10 | 0.18 | 0.00 | 8 | 333 | 119 | 11 | 2.03 |
| OH-M-H | 48 | 10 | 0.17 | 0.10 | 57 | 343 | 108 | 56 | 1.04 |
| OH-N | 50 | 0.4 | 0.17 | 0.00 | 26 | 282 | 112 | 13 | 0.92 |
| OH-N-H | 42 | 0.4 | 0.17 | 0.10 | 103 | 364 | 102 | 31 | 0.78 |
| F-M | 50 | 24 | 0.17 | 0.00 | 9 | 310 | 144 | 15 | 1.81 |
| F-M-H | 35 | 24 | 0.16 | 0.01 | 18 | 308 | 92 | 30 | — |
| F-N | 60 | 0.6 | 0.19 | 0.01 | 20 | 356 | 146 | 23 | 0.96 |
| F-N-H | 53 | 0.6 | 0.16 | 0.08 | 93 | 375 | 128 | 40 | 0.80 |
 | ||
| Fig. 2 SEM micrographs of the ZSM-5 catalysts synthesised in OH− and F− media. The insets show zooms of 5 times the magnification of the main image. | ||
Complementary characterisation of the zeolite crystals was conducted by diffuse reflectance infrared Fourier transform spectroscopy in the hydroxyl-stretching region (Fig. 3). In line with the equivalent acidic properties, all of the samples exhibit a similarly intense band associated with the presence of Brønsted acid centres (3610 cm−1). In contrast, the relative intensity of bands assigned to other hydroxyl groups varies noticeably. These include terminal silanols at the external (3745 cm−1) or internal (3725 cm−1) surface of the zeolite, hydrogen-bonded silanol nests (broad band at ca. 3500 cm−1), and weaker bands at 3687 and 3656 cm−1, commonly assigned to extra-framework Al species. These bands all represent structural defects within the zeolites and their presence is expected to affect the catalyst performance.13 In particular, the presence of both silanol nests and internal silanols has been linked to the faster deactivation due to the increased retention of coke precursors.13 To quantify the defect concentrations in the zeolites prepared in this study we have evaluated the ratio of internal to external silanol groups as proposed by Barbera et al.13e (Table 1). In agreement with the expected defect-free nature of the micronsized zeolite synthesised in F− medium, the bands associated with terminal silanols appear much less prominently in F-M than in OH-M, and this sample exhibits a reduced presence of silanol nests. Due to their smaller crystal size, F-N and OH-N exhibit higher amounts of external silanols than F-M and OH-M, while F-N also exhibits increased amounts of internal silanols than F-M, which likely originate from the silicalite-1 seed applied during the synthesis.
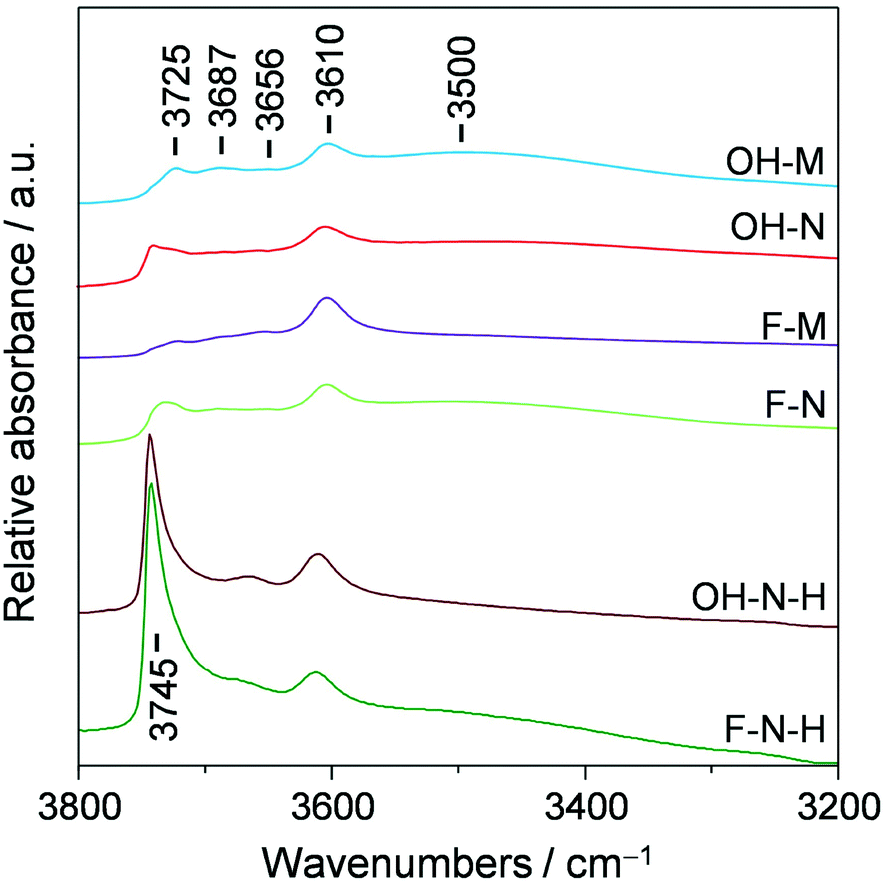 | ||
| Fig. 3 DRIFT spectra of the ZSM-5 catalysts synthesised in OH− and F− media and their desilicated analogues. | ||
Finally, to quantify the effectiveness of nanosizing vis-à-vis the development of intracrystalline mesopores, the purely microporous zeolites were also modified via desilication in alkaline media, yielding samples OH-M-H, OH-N-H, F-M-H, and F-N-H. As seen from the combined type I–IV behaviour observed in the Ar isotherms (Fig. 1b and c) and the derived porous properties (Table 1), this post-synthetic treatment successfully enhanced the mesoporosity (4–7-fold increase in Smeso) of all of the zeolites except in the case of F-M-H. Transmission electron microscopy clearly revealed the increased presence of intracrystalline mesopores in the desilicated F-N-H and OH-N-H samples than in the parent F-N and OH-N zeolites (Fig. 4). In the case of F-N-H, fragmentation of the initially coffin-shaped zeolite crystals is also noticeable. All of the desilicated samples preserved the crystalline MFI-type framework (Fig. 1a) and correspondingly a high micropore volume. The inefficiency of the alkaline treatment to generate mesopores in the case of F-M can be attributed to the quasi-absence of structural defects evidenced in this sample by the IR studies, which is believed to impede the hydrolysis of silicon from the framework.18 This is in agreement with an earlier study by Gil et al.,19 which showed that silanol nests are readily eliminated from the zeolite crystal upon desilication. However, the low concentrations of silanol nests detected in OH-N and F-N indicate that these are likely not the only defects where the hydrolysis of the framework initiates. The desilicated analogues exhibit slightly reduced Si/Al ratios, but almost fully preserved (ca. 10% drop) concentrations of Brønsted and higher numbers (ca. double) of Lewis acid sites compared to the corresponding parent zeolites (Table 1). These values are fully consistent with the general trends that have been observed in the acidic properties of hierarchical zeolites with increasing mesopore surface area.20 The significantly increased intensity associated with terminal silanols at the external surface evidenced by DRIFTS (Fig. 3) is also in agreement with previous observations.13b Due to the lack of mesopore development and the altered acidic properties, F-M-H was not considered for catalyst testing.
 | ||
| Fig. 4 TEM images of the nanosized ZSM-5 catalysts synthesised in OH− and F− media and their desilicated analogues. | ||
Evaluation in the cracking of 2-methyl-2-butene
Preliminary tests in 2M2B cracking evaluated the impact of the reaction temperature on the performance of the tailored zeolites (Fig. S1,† WHSV = 12 g2M2B gcatalyst h−1, 2 h on stream). All of the catalysts exhibit similar conversions, which are slightly increased at 773 K (X2M2B = 89–93%) than at 673 K (X2M2B = 82–85%). As expected due to the enhanced cracking activity, higher selectivity to light olefins (SC2–4=) is observed at 773 K (42–49 Cmol%) compared to 673 K (34–38 Cmol%), which is compensated for by a reduced selectivity to benzene, toluene, and xylene-range aromatics (SC6–8ar). Comparatively, the micronsized zeolites display slightly superior light olefin selectivity than the nanosized or desilicated samples at both temperatures. Subsequent variation of the weight hourly space velocity from 12 to 40 g2M2B gcatalyst h−1 at 773 K led to a drop in conversion of between 12% and 17% (Fig. S2†). Interestingly, increasing the space velocity (shorter contact time) enhanced the selectivity to light olefins over the nanosized zeolites leading to a convergence with the micronsized catalysts at ca. 50 Cmol%. Analysis of the propylene:ethylene ratio revealed a rising dependence (from 2.5 to 3.5) over the nanosized zeolites in this range. A similar falling tendency of the propylene selectivity with increasing conversion was previously reported in the conversion of methanol to hydrocarbons and was ascribed to the lower stability of propylene than ethylene towards further transformation.21 Comparatively, the product distribution over the micronsized zeolites showed less sensitivity to the contact time of the 2M2B with the catalyst bed, indicating that such secondary reactions are likely associated with the higher mesopore surface area of the nanosized and desilicated zeolites.
Stability tests in 2M2B cracking illustrate the relative variation in conversion and selectivity with time on stream (773 K, 12 g2M2B gcatalyst h−1). As expected, the catalysts, which initially exhibit very similar conversions, progressively deactivate due to coke deposition with a close to linear trend (Fig. 5a). To quantify the distinct behaviour observed, the deactivation rates were estimated based on the slopes of the conversion versus time on stream plots (Table 2). The widely varying values, ranging from −0.701 to −0.028% min−1, emphasise the major impact of the different crystal properties on the activity losses due to coke deposition. For comparative purposes, the catalyst lifetime has been defined as the time during which the conversion of 2M2B exceeded 80%, which also delimits a single catalyst cycle. Micronsized OH-M and F-M display modest catalyst lifetimes of around 15 h, while reasonable (2.6-fold) improvements are observed for the nanosized OH-N and F-N. Desilication of the parent zeolites yields more substantial lifetime extensions in all cases. Micronsized OH-M-H is stable over 4 times longer than OH-M, while OH-N-H and F-N-H display remarkably enhanced catalyst lifetimes of 347 and 190 h, respectively.
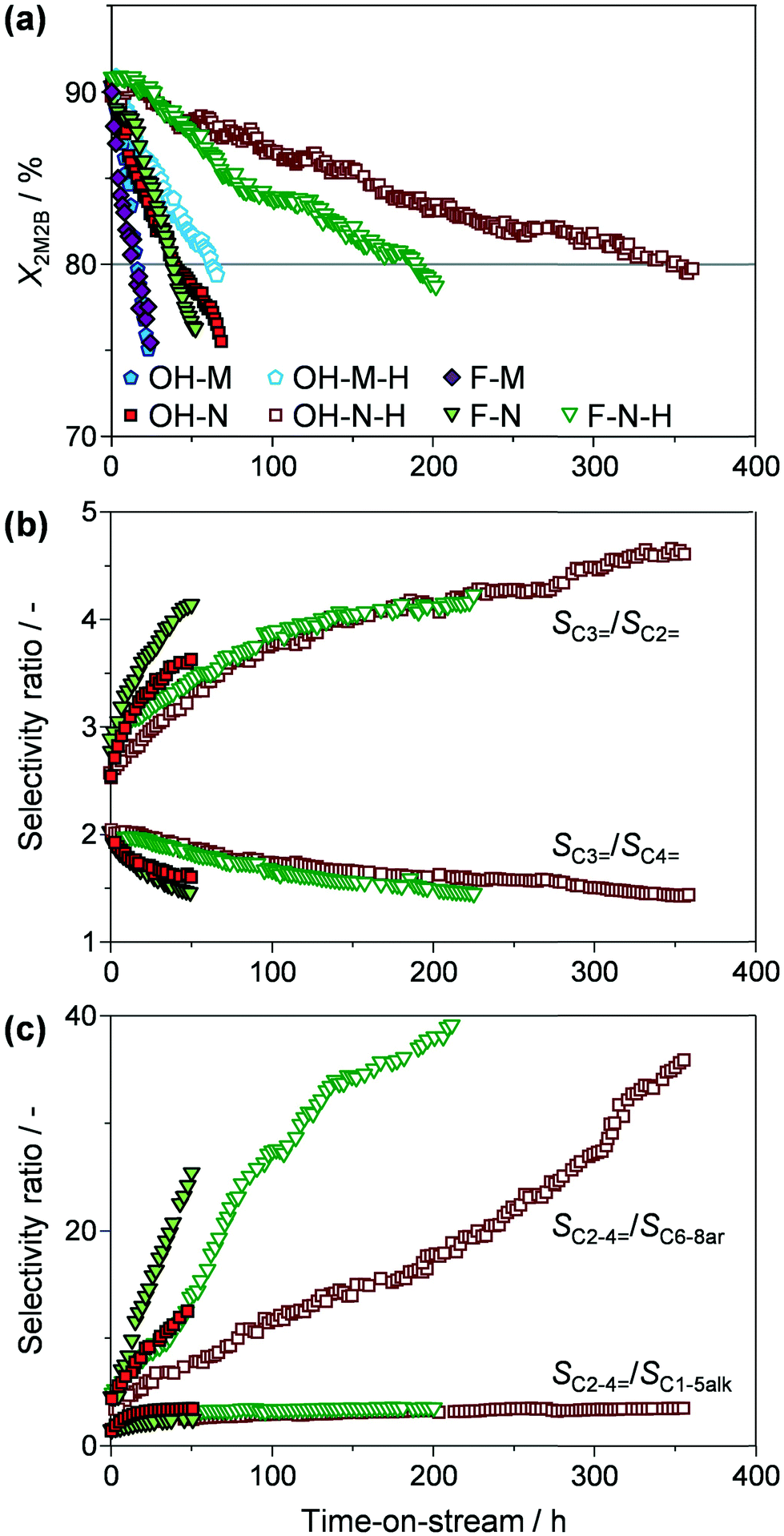 | ||
| Fig. 5 (a) Conversion of 2M2B and (b and c) selectivity ratios to individual or groups of products versus time on stream over the tailored ZSM-5 zeolites. The grey line in (a) indicates the degree of conversion at which a single catalytic cycle was defined. Conditions: T = 773 K, P = 1 bar, WHSV = 12 g2M2B gzeolite−1 h−1. | ||
| Sample | Lifetimea (h) | D b (% min−1) | S C2= c (Cmol%) | S C3= c (Cmol%) | S C4= c (Cmol%) | STYd (gC2–4= gzeolite−1 h−1) | S C1–3alk c (Cmol%) | S C4–7alk c (Cmol%) | S C6–8ar c (Cmol%) |
|---|---|---|---|---|---|---|---|---|---|
| a Time on stream during which X2M2B >80%. b Deactivation rate, estimated based on the slope of the conversion versus time on stream. c Selectivity to ethylene (SC2=), propylene (SC3=), butylenes (SC4=), light alkanes (SC1–3alk), gasoline-range alkanes (SC4–7alk), and to benzene, toluene, and xylene aromatics (SC6–8ar). d Space-time yield (STY) of C2–4 olefins. | |||||||||
| OH-M | 15 | −0.701 | 10 | 33 | 19 | 6.31 | 4 | 23 | 5 |
| OH-M-H | 63 | −0.166 | 10 | 34 | 20 | 6.50 | 4 | 23 | 5 |
| OH-N | 39 | −0.197 | 10 | 31 | 18 | 6.05 | 5 | 23 | 8 |
| OH-N-H | 347 | −0.028 | 9 | 35 | 20 | 6.67 | 4 | 22 | 6 |
| F-M | 15 | −0.508 | 9 | 31 | 18 | 5.75 | 5 | 22 | 9 |
| F-N | 39 | −0.287 | 9 | 31 | 19 | 6.14 | 4 | 21 | 6 |
| F-N-H | 190 | −0.060 | 9 | 34 | 19 | 6.29 | 3 | 22 | 4 |
Comparison of the average product distribution over a single catalytic cycle (Table 2) and of the selectivity evolution to different individual products and product groups (Fig. 5b and c and S3†) sheds further light on the performance of the zeolite catalysts. Similar selectivity trends are observed over all of the zeolites, resulting in comparable selectivity to different products per catalytic cycle (Table 2). Ethylene, propylene, and butylenes all formed in appreciable amounts, attaining very reasonable space-time yields to light olefins (5.75–6.67 gC2–4= gzeolite−1 h−1). However, the relative proportions varied with time on stream. Specifically, the propylene:ethylene ratio rose from 2.5 to 4, while the propylene:butylene ratio dropped from 2 to 1.5 over the lifetime of the catalyst. The overall increase in the selectivity to light olefins occurs at the expense of C6–8 aromatics (Fig. S3†), resulting in the sharp increase in the SC2–4=:SC6–8ar selectivity ratio (from 3 to 40) seen in Fig. 5c. At the same time, the value of the light olefin:C1–5 paraffin ratio (SC2–4=:SC6–8alk) varies less significantly (from 1.5 to 3).
The observed product compositions match well with those reported by Bortnovsky et al. over ZSM-5 zeolites.4 Interestingly, the selectivity variation seen during the catalytic cycle in this work reflects that previously demonstrated upon decreasing the concentration of Brønsted acid sites. The latter was linked to the preferred formation of oligomeric carbenium ion intermediates compared to the thermodynamic equilibrium ratios calculated for the conversion of 2M2B to light olefins via direct (protolytic) cracking or dimerisation–cracking routes (propylene:ethylene = 2.95, propylene:butylene = 0.83).4 The negligible role of the protolytic cracking mechanism under the conditions studied was further supported by the low amount of light gases (SC1–3alk) produced. In addition to oligomerisation and cracking, the observation of aromatic (primarily C6–8) and paraffinic (primarily C4–5) products confirms the occurrence of irreversible cyclisation and hydrogen-transfer reactions. However, as evidenced by the sharply increasing values of SC2–4=:SC6–8ar, the selectivity to aromatics drops rapidly with time on stream, forming only in minor amounts during the catalytic cycle.
Inspection of the relative trends (Fig. 5c and S3†) reveals that SC6–8ar decreases less rapidly with time on stream over the desilicated OH-N-H and F-N-H zeolites compared with their respective parent samples (i.e., OH-N and F-N). Reduced aromatic selectivity over ZSM-5 zeolites is typically attributed to the deactivation of stronger acidic sites due to coke deposition,22 implying that the presence of intracrystalline mesopores reduces the rate at which these sites are poisoned. Higher aromatic selectivity over hierarchical ZSM-5 has also been ascribed to both the improved diffusion of higher molecular weight products and an increased C4 hydrogen transfer index in the conversion of methanol to gasoline.23 However, the very similar initial values of SC6–8ar observed in 2M2B cracking suggest that in the absence of coke neither of these factors has a significant effect. Note that the decreasing selectivity to C6–8 aromatics likely drives the increased selectivity to light olefins rather than a significant change in the preferred cracking pathway. In contrast, the formation of paraffins from either reactant or product olefins remains significant during the whole catalytic cycle, suggesting that the hydrogen-transfer pathways leading to these products are independent of the processes which ultimately lead to aromatics. This is in line with the findings of an earlier study of the conversion of ethylene and propylene to higher hydrocarbons of ZSM-5 zeolites, which found that paraffin formation occurred at lower temperatures than that for aromatics formation.24
Thermogravimetric analysis of the catalysts isolated after the conversion had dropped to 80% (Fig. 6 and Table 3) revealed that similar amounts of coke had been deposited in the nanosized OH-N and F-N (5.8 and 6.6 wt%, respectively). In comparison, the desilicated analogues exhibited around 3 times higher coke contents (15.5 and 22.6 wt% for F-N-H and OH-N-H, respectively). The greater ability of the desilicated zeolites to accommodate coke without losing activity agrees with the findings of previous studies that have followed coke formation in the MTO reaction.25 By studying the location of coke, this has been related to the preferential deposition at the external surface due to the facilitated transport of coke precursors out of the micropores.25
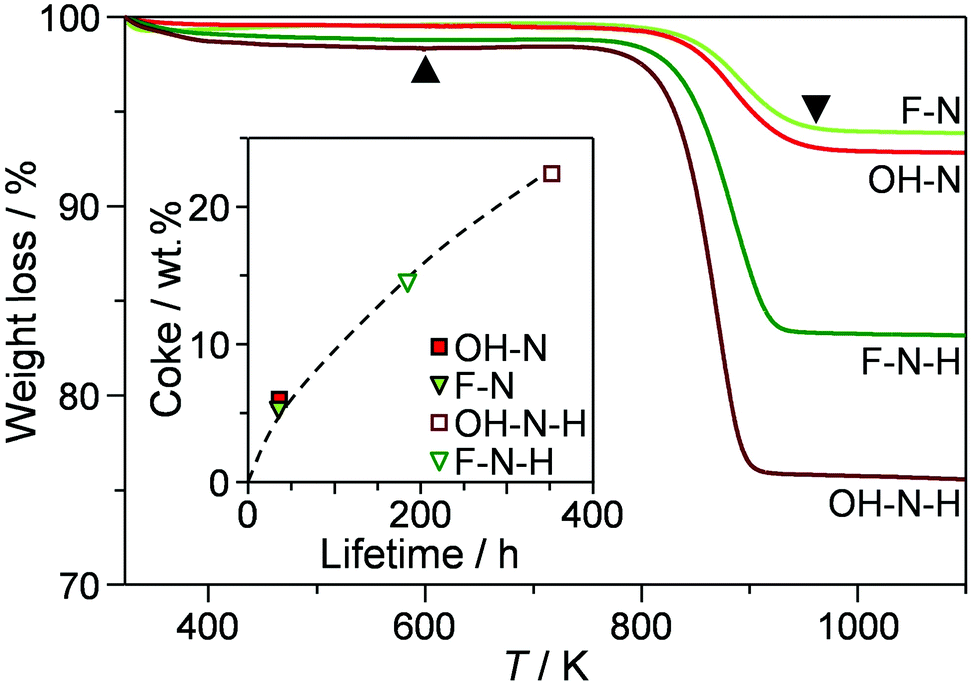 | ||
| Fig. 6 Thermogravimetric profiles of the nanocrystalline zeolites and their desilicated counterparts after deactivation to 80% conversion in the first catalytic cycle of 2M2B cracking. The amount of coke deposited (Table 3) was determined by the percentage weight loss in the range of 600–1000 K (indicated by the black triangles). The inset shows the relation between the coke content and the catalyst lifetime. | ||
| Sample | Cokea (wt%) | V micro b (cm3 g−1) | V meso c (cm3 g−1) | S meso d (m2 g−1) | c Brønsted e (μmol g−1) | c Lewis e (μmol g−1) |
|---|---|---|---|---|---|---|
| a Thermogravimetric analysis. b NLDFT. c V meso = Vpore − Vmicro. d t-Plot. e IR study of adsorbed pyridine. | ||||||
| OH-N | 6.6 | 0.16 | 0.01 | 24 | 85 | 17 |
| OH-N-H | 22.6 | 0.17 | 0.10 | 87 | 78 | 30 |
| F-N | 5.8 | 0.17 | 0.01 | 19 | 98 | 30 |
| F-N-H | 15.5 | 0.17 | 0.06 | 85 | 82 | 38 |
Hierarchical structuring can also alter the rate of coke formation. Considering the lifetimes of the catalysts in 2M2B cracking evidences an almost linear relation with the amount of coke deposited with the relative time on stream (Fig. 6, inset). This suggests that the coking rate was comparable in all catalysts, agreeing with the similar carbon balances observed. These results further support the conclusion that the crystal engineering did not impact the mechanism of 2M2B cracking and associated reaction paths leading to coke formation. Interestingly, these values are significantly lower than the amounts of coke previously reported after comparable deactivation of ZSM-5 zeolites of similar composition and mesopore surface area in the conversion of methanol to hydrocarbons with time on stream (e.g., 13 wt% after ca. 33 h).25 On the other hand, the single sharp weight loss centred around 873 K indicates that most of the carbonaceous species are deposited in the form of hard coke of comparable nature to that formed in the MTO reaction.25 Oxidative regeneration of the catalysts at 873 K almost completely restored the porosity (Table 3). Moderately reduced Brønsted acid site densities were observed with respect to the fresh catalysts. Dealumination is known to play a crucial role in the deactivation of ZSM-5 catalysts and has previously been evidenced in C4 olefin cracking reactions.26 Nonetheless, the acidic properties remain well within the optimal range of concentrations for this reaction.
Evaluation in the conversion of methanol to olefin (MTO)
As one of the most widely studied processes for the production of light olefins, the MTO reaction serves as a useful indicator of the performance of the tailored ZSM-5 zeolites. Targeting the efficient production of olefins,22a the catalysts were evaluated at 723 K and the weight hourly space velocity was maximised while ensuring an initial conversion of 100% over all catalysts. It is important to note that a slightly lower WHSV (longer contact time) than that used in the 2M2B cracking reaction had to be applied to reach full conversion. Fig. S4† presents the variation in conversion with time on stream and the corresponding selectivity to light olefins and to aromatics over the longest-lived catalyst (OH-N-H). The average product selectivity per catalytic cycle (also defined as the time for which XMeOH >80%) over each of the catalysts is reported in Table S1.†Quantitatively, very similar trends are observed in the catalyst stability compared with 2M2B cracking. The micronsized zeolites exhibit full conversion for ca. 15 h on stream before the onset of rapid deactivation, while activity losses are noticeably retarded over the nanosized zeolites, and even more significantly over the desilicated samples. Nonetheless, some differences are evident between the two reactions. One of the most obvious is the distinct shape of the conversion versus time-on-stream profiles. As opposed to the linear deactivation with different rates evidenced in the former (Fig. 5a), a characteristic plateau of varying duration at full conversion followed by a steep deactivation of a rather similar slope over all of the zeolites is seen in the MTO reaction. Moreover, despite the longer contact time, which is known to retard deactivation, significantly shorter catalyst lifetimes were evidenced (e.g., 54 h with respect to 347 h over the longest-lived OH-N-H catalyst). Several previous works have modelled the complex deactivation behaviour of ZSM-5 zeolites in the MTO reaction.27 It is known that a composition-dependent kinetic analysis is required to simulate the fixed-bed process with up to three product groups (oxygenates, aromatics, and light olefins) being considered to independently contribute to the coke deposition. While olefin cracking is undoubtedly important in both reactions, the significantly extended lifetimes evidenced in 2M2B cracking emphasise the highly detrimental role of oxygenates on the catalyst longevity.
Comparatively, slightly lower average selectivity to light olefins (SC2–4= = 38% over OH-N-H versus 49% in 2M2B cracking) and slightly higher selectivity to C6–8 aromatics and appreciably increased selectivity to C4–7 paraffins were evidenced in the MTO reaction compared to 2M2B cracking. The differences in product distribution can be attributed to the prevalent role of methanol in olefin methylation reactions, which are responsible for the formation of higher olefins and of substituted aromatic products that are subsequently cracked to propylene and ethylene via independent paths.28 Comparatively, this results in slightly lower space-time yields of light olefins compared to those achieved in 2M2B cracking under the conditions studied (e.g., 2.25 with respect to 6.67 gC2–4= gzeolite−1 h−1 over OH-N-H).
The trends observed over the tailored catalyst in 2M2B cracking and the MTO reaction provide direct insight into the relative impact of the distinct crystal properties. Consistent with previous observations, decreasing the crystal size benefits the catalyst longevity, which can be attributed to the increased external surface area. However, the crystal size is not a robust descriptor for the performance of the zeolite since it does not account for the presence of intracrystalline mesoporosity. The effects of both variables can be accounted for by the mesopore (external) surface area, which strongly correlates with the catalyst lifetime in these reactions (Fig. 7a). A similar dependence has previously been reported in the conversion of methanol to hydrocarbons.25 Comparatively, the small enhancements in the mesopore surface area (and corresponding catalyst lifetime) achieved upon significantly decreasing the crystal size vis-à-vis those evidenced in the desilicated samples demonstrate the significantly increased scope of developing intracrystalline mesopores for improving the coke tolerance with respect to nanosizing.
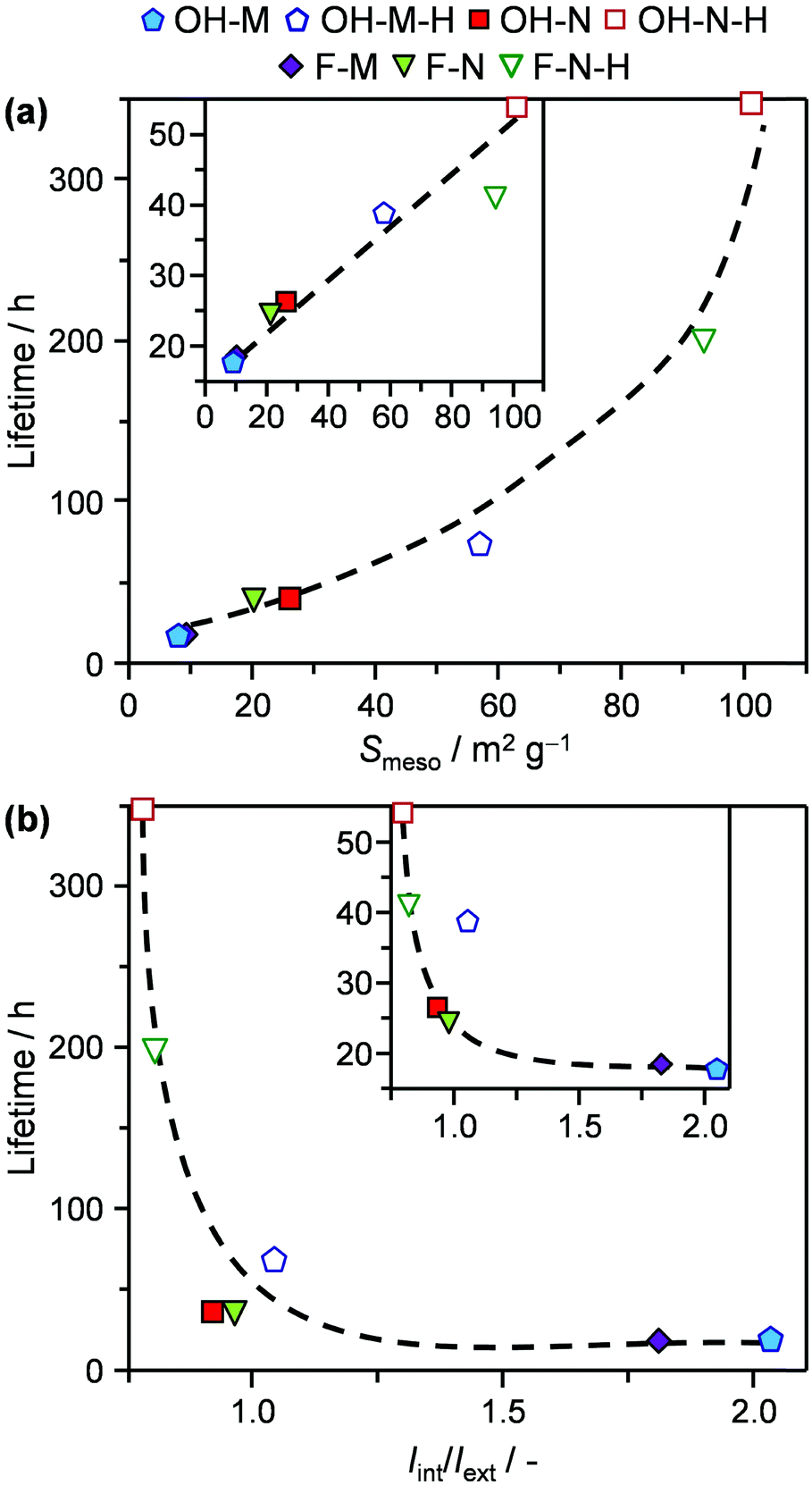 | ||
| Fig. 7 Correlations between the catalyst lifetime and (a) the mesopore surface area (Smeso) or (b) the amount of defects (Iint/Iext) in 2M2B cracking. The insets show the comparative trends in the MTO reaction. | ||
Concerning the concentration of defects, the situation becomes more complex. As mentioned, the deactivation rate of ZSM-5 zeolites in the conversion of methanol to hydrocarbons has previously been correlated with the intensity ratio of the IR bands for internal silanol and external silanol groups (Iint/Iext).13e Indeed, by plotting the measured catalyst lifetimes versus this parameter (Fig. 7b), similar conclusions can be drawn on comparison of pairs of purely microporous zeolites and their desilicated analogues. However, the relation is nonlinear. Furthermore, this descriptor fails to explain differences in the performance of purely microporous zeolites prepared by different routes (e.g., F-M and OH-M), which exhibit different fractions on internal and external silanols but equivalent lifetimes. Likewise, although comparable values of Iint/Iext were determined for OH-N-H and F-N-H, the former zeolite exhibited significantly extended lifetimes in both reactions. A limitation of this descriptor is that increasing the mesopore surface area can lead to a substantially higher Iext, without reducing Iint and thus low values of Iint/Iext can result even for materials with high internal defect concentrations. Apart from terminal silanols, the presence of Lewis acid sites was also recently linked with an increased turnover frequency in the cracking of n-pentane.29 Nonetheless, no direct link between the concentration of Lewis acid sites and the activity or deactivation behaviour of the catalyst could be established in either reaction studied. To the best of our knowledge, no clear relationship has previously been identified in the MTO reaction.
Finally, the virtually equivalent catalyst lifetimes evidenced for F-M and OH-M and for F-N and OH-N indicate that the crystal morphology has a negligible effect. Structure sensitivity deriving from the specific micropore orientation associated with the exposed crystallographic facets is one of the least addressed aspects in zeolite catalysis. This very likely stems from the complexity of the zeolite growth mechanism, meaning that the size and shape uniformity as well as the crystalline order of most of the catalysts studied to date remains insufficient to distinguish the impact of pore orientation. Nevertheless, advances in the development of ZSM-5 films have revealed that the separation performance is highly sensitive to the preferred orientation,30 and thus, the role of this crystal variable should not be completely discarded.
Conclusions
Light olefins can be obtained selectivity and in high productivity via 2-methyl-2-butene cracking over ZSM-5 zeolites. By synthesising tailored zeolites with equivalent acidic properties, we have quantitatively assessed the influence of crystal size, morphology, mesoporosity, and defect concentration on the catalyst performance. The similar initial activity and variations in product distribution observed over the catalysts with time on stream demonstrated both the absence of significant mass-transfer limitations and that the active site distribution was not significantly altered by tailoring the crystal properties. In contrast, major differences were observed in the deactivation behaviour between the catalysts. Enhancing the mesopore surface area by decreasing the crystal size and/or introducing intracrystalline mesopores was by far the most influential parameter. Up to 22-fold extensions in the catalyst lifetimes could be achieved by optimising this descriptor, which are attributed to the greater capacity to accommodate coke without blocking the zeolite micropores. The changes in selectivity related to the catalyst deactivation paralleled those expected upon decreasing the concentration of Brønsted acid sites in the sample. Comparatively, very similar deactivation behaviours were evidenced over zeolite crystals of coffin-shaped or rounded-boat morphology, but equivalent size. Furthermore, the comparison of zeolite crystals prepared with low concentrations of structural defects (terminal silanols or extra-framework aluminium species) in fluoride medium did not significantly alter the catalyst stability. Evaluation of the same set of catalysts in the conversion of methanol to olefins evidenced strongly curtailed lifetimes (e.g., from 347 h to just 54 h over the optimal catalyst) even when operating with longer contact times. Slightly reduced light olefin selectivity and propylene:ethylene ratios were also observed with respect to 2-methyl-2-butene cracking, highlighting the detrimental effect of olefin methylation in this reaction. Nonetheless, similar qualitative trends were evidenced in both reactions, demonstrating the robustness of the mesopore surface area as a descriptor for optimising the catalyst performance in the production of light olefins.
Acknowledgements
The Scientific Center for Optical and Electron Microscopy of ETH Zurich is thanked for the use of its facilities.References
- The IHS Chemical Economic Handbook online, https://www.ihs.com/products/propylene-chemical-economics-handbook.html (accessed May 2016) Search PubMed.
- (a) J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39 CrossRef CAS; (b) T. F. Degnan, Stud. Surf. Sci. Catal., 2007, 170, 54 CrossRef; (c) F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113 CrossRef CAS; (d) W. Vermeiren and J.-P. Gilson, Top. Catal., 2010, 52, 1131 CrossRef; (e) N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1 CrossRef CAS; (f) A. Farshi, F. Shaiyegh, S. H. Burogerdi and A. Dehgan, Pet. Sci. Technol., 2011, 29, 875 CrossRef CAS.
- G. Bellussi and P. Pollesel, Stud. Surf. Sci. Catal., 2005, 158, 1201 CrossRef.
- O. Bortnovsky, P. Sazama and B. Wichterlova, Appl. Catal., A, 2005, 287, 203 CrossRef CAS.
- (a) H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Industrial organic chemicals, John Wiley & Sons, New Jersey, 2012 Search PubMed; (b) J. Lee, U. G. Hong, S. Hwang, M. H. Youn and I. K. Song, Fuel Process. Technol., 2013, 109, 189 CrossRef CAS; (c) J. Lee, S. Park, U. G. Hong, J. O. Jun and I. K. Song, J. Nanosci. Nanotechnol., 2015, 15, 8311 CrossRef CAS PubMed.
- C. A. González-Rugerio, T. Keller, J. Pilarczyk, W. Sałacki and A. Górak, Fuel Process. Technol., 2012, 102, 1 CrossRef.
- (a) Z. Li, W. Shi, X. Wang and F. Jiang, Deep catalytic cracking process for light-olefins production, ACS Symposium Series, 2009, ch. 9, p. 33 Search PubMed; (b) Y.-K. Park, C. W. Lee, N. Y. Kang, W. C. Choi, S. Choi, S. H. Oh and D. S. Park, Catal. Surv. Asia, 2010, 14, 75 CrossRef CAS.
- X. Zhu, S. Liu, Y. Song and L. Xu, Appl. Catal., A, 2005, 288, 134 CrossRef CAS.
- A. Miyaji, Y. Sakamoto, Y. Iwase, T. Yashima, R. Koide, K. Motokura and T. Baba, J. Catal., 2013, 302, 101 CrossRef CAS.
- (a) J. Abbot and B. W. Wojciechowski, Can. J. Chem. Eng., 1985, 63, 462 CrossRef CAS; (b) J. S. Buchanan, J. G. Santiesteban and W. O. Haag, J. Catal., 1996, 158, 279 CrossRef CAS; (c) Y. V. Kissin, Catal. Rev.: Sci. Eng., 2001, 43, 85 CrossRef CAS; (d) P. Sazama, J. Dědeček, V. Gábová, B. Wichterlová, G. Spoto and S. Bordiga, J. Catal., 2008, 254, 180 CrossRef CAS; (e) L. Lin, C. Qiu, Z. Zhuo, D. Zhang, S. Zhao, H. Wu, Y. Liu and M. He, J. Catal., 2014, 309, 136 CrossRef CAS; (f) C.-J. Chen, S. Rangarajan, I. M. Hill and A. Bhan, ACS Catal., 2014, 4, 2319 CrossRef CAS; (g) P. Arudra, T. I. Bhuiyan, M. N. Akhtar, A. M. Aitani, S. S. Al-Khattaf and H. Hattori, ACS Catal., 2014, 4, 4205 CrossRef CAS.
- (a) L. F. Lin, S. F. Zhao, D. W. Zhang, H. Fan, Y. M. Liu and M. Y. He, ACS Catal., 2015, 5, 4048 CrossRef CAS; (b) M. Höchtl, A. Jentys and H. Vinek, Appl. Catal., A, 2001, 207, 397 CrossRef.
- (a) U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810 CrossRef CAS PubMed; (b) F. Mohammadparast, R. Halladj and S. Askari, Chem. Eng. Commun., 2014, 202, 542 CrossRef; (c) S. Mitchell, A. B. Pinar, J. Kenvin, P. Crivelli, J. Kärger and J. Pérez-Ramírez, Nat. Commun., 2015, 6, 8633 CrossRef CAS PubMed; (d) R. Khare, D. Millar and A. Bhan, J. Catal., 2015, 321, 23 CrossRef CAS; (e) M. Milina, S. Mitchell, D. Cooke, P. Crivelli and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 1591 CrossRef CAS PubMed.
- (a) S. Ivanova, C. Lebrun, E. Vanhaecke, C. Pham-Huu and B. Louis, J. Catal., 2009, 265, 1 CrossRef CAS; (b) Z. Qin, L. Lakiss, L. Tosheva, J.-P. Gilson, A. Vicente, C. Fernandez and V. Valtchev, Adv. Funct. Mater., 2014, 24, 257 CrossRef CAS; (c) M. S. Holm, S. Svelle, F. Joensen, P. Beato, C. H. Christensen, S. Bordiga and M. Bjørgen, Appl. Catal., A, 2009, 356, 23 CrossRef CAS; (d) I. Yarulina, J. Goetze, C. Gücüyener, L. van Thiel, A. Dikhtiarenko, J. Ruiz-Martinez, B. M. Weckhuysen, J. Gascon and F. Kapteijn, Catal. Sci. Technol., 2016, 6, 2663 RSC; (e) K. Barbera, F. Bonino, S. Bordiga, T. V. W. Janssens and P. Beato, J. Catal., 2011, 280, 196 CrossRef CAS.
- N. Danilina, F. Krumeich, S. A. Castelanelli and J. A. van Bokhoven, J. Phys. Chem. C, 2010, 114, 6640 CAS.
- T. Xue, Y. M. Wang and M.-Y. He, Microporous Mesoporous Mater., 2012, 156, 29 CrossRef CAS.
- F. L. Bleken, S. Chavan, U. Olsbye, M. Boltz, F. Ocampo and B. Louis, Appl. Catal., A, 2012, 447, 178 CrossRef.
- C. A. Emeis, J. Catal., 1993, 141, 347 CrossRef CAS.
- D. Fodor, A. Beloqui Redondo, F. Krumeich and J. A. van Bokhoven, J. Phys. Chem. C, 2015, 119, 5447 CAS.
- B. Gil, Ł. Mokrzycki, B. Sulikowski, Z. Olejniczak and S. Walas, Catal. Today, 2010, 152, 24 CrossRef CAS.
- S. Mitchell, M. Milina, R. Verel, M. Hernández-Rodríguez, A. B. Pinar, L. B. McCusker and J. Pérez-Ramírez, Chem. – Eur. J., 2015, 21, 14156 CrossRef CAS PubMed.
- F. C. Patcas, J. Catal., 2005, 231, 194 CrossRef CAS.
- (a) V. S. Nayak and V. R. Choudhary, Appl. Catal., 1984, 9, 251 CrossRef CAS; (b) N.-L. Michels, S. Mitchell and J. Pérez-Ramírez, ACS Catal., 2014, 4, 2409 CrossRef CAS; (c) E. Epelde, J. I. Santos, P. Florian, A. T. Aguayo, A. G. Gayubo, J. Bilbao and P. Castaño, Appl. Catal., A, 2015, 505, 105 CrossRef CAS.
- M. Bjørgen, F. Joensen, M. S. Holm, U. Olsbye, K.-P. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43 CrossRef.
- S. Bessel and D. Seddon, J. Catal., 1987, 105, 270 CrossRef.
- (a) J. Kim, M. Choi and R. Ryoo, J. Catal., 2010, 269, 219 CrossRef CAS; (b) M. Milina, S. Mitchell, P. Crivelli, D. Cooke and J. Pérez-Ramírez, Nat. Commun., 2014, 5, 3922 Search PubMed.
- G. Zhao, J. Teng, Z. Xie, W. Jin, W. Yang, Q. Chen and Y. Tang, J. Catal., 2007, 248, 29 CrossRef CAS.
- (a) P. L. Benito, A. G. Gayubo, A. T. Aguayo, M. Castilla and J. Bilbao, Ind. Eng. Chem. Res., 1996, 35, 81 CrossRef CAS; (b) T. V. W. Janssens, S. Svelle and U. Olsbye, J. Catal., 2013, 308, 122 CrossRef CAS.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770 CrossRef CAS PubMed.
- S. Schallmoser, T. Ikuno, M. F. Wagenhofer, R. Kolvenbach, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, J. Catal., 2014, 316, 93 CrossRef CAS.
- M. A. Snyder and M. Tsapatsis, Angew. Chem., Int. Ed., 2007, 46, 7560 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional catalytic data. See DOI: 10.1039/c6cy01009a |
| This journal is © The Royal Society of Chemistry 2017 |