
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation of cinnamyl alcohol using bimetallic Au–Pd/TiO2 catalysts: a deactivation study in a continuous flow packed bed microreactor
Gaowei
Wu
a,
Gemma L.
Brett
b,
Enhong
Cao
a,
Achilleas
Constantinou†
a,
Peter
Ellis
c,
Simon
Kuhn
d,
Graham J.
Hutchings
b,
Donald
Bethell
e and
Asterios
Gavriilidis
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK. E-mail: a.gavriilidis@ucl.ac.uk
bSchool of Chemistry, Cardiff University, CF10 3AT, UK
cJohnson Matthey Technology Centre, Blounts Court, Sonning Common, Reading RG4 9NH, UK
dDepartment of Chemical Engineering, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
eDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK
First published on 26th May 2016
Abstract
The stability of a bimetallic Au–Pd/TiO2 catalyst was examined in a packed bed microreactor for the oxidation of cinnamyl alcohol dissolved in toluene. The catalyst was prepared by co-impregnation with a Au–Pd weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19. Experiments were performed at 80–120 °C, oxygen concentration 0–100% and total pressure 4 bara. Principal products observed were cinnamaldehyde, 3-phenyl-1-propanol and trans-β-methylstyrene. Although the same catalyst was shown to possess good stability in the oxidation of benzyl alcohol, it deactivated during the oxidation of cinnamyl alcohol, particularly at elevated reaction temperatures. Higher concentration of oxygen used for the reaction led to improved cinnamaldehyde selectivity but lower conversion and higher deactivation rates. Treatment with hydrogen recovered only a fraction of the activity. Deactivation was attributed to Pd leaching and a complex effect of oxygen.
:19. Experiments were performed at 80–120 °C, oxygen concentration 0–100% and total pressure 4 bara. Principal products observed were cinnamaldehyde, 3-phenyl-1-propanol and trans-β-methylstyrene. Although the same catalyst was shown to possess good stability in the oxidation of benzyl alcohol, it deactivated during the oxidation of cinnamyl alcohol, particularly at elevated reaction temperatures. Higher concentration of oxygen used for the reaction led to improved cinnamaldehyde selectivity but lower conversion and higher deactivation rates. Treatment with hydrogen recovered only a fraction of the activity. Deactivation was attributed to Pd leaching and a complex effect of oxygen.
1. Introduction
Selective oxidation of alcohols is important in organic chemistry and often performed with stoichiometric inorganic reagents.1,2 In recent years, there has been a growing demand for the development of heterogeneous catalysts for oxidation of alcohols with molecular oxygen, since it is a green and efficient process.3,4 However, large-scale applications of aerobic alcohol oxidation are still limited to few catalysts with sufficient reactivity and stability.5 Among these heterogeneously catalysed oxidation reactions, oxidation of cinnamyl alcohol is one of the most studied, since cinnamyl alcohol is a simple aromatic allylic alcohol.4,6–10 The target product, cinnamaldehyde, is also an important chemical intermediate in organic transformations and has wide applications in the food and perfume industries.11–13 Up to now, studies of cinnamyl alcohol oxidation have mainly focused on monometallic catalysts, and platinum-group metals (particularly Pd) have been intensively investigated.10,12,14–16 Due to the co-existence of reactive hydroxyl and alkenyl functional groups in cinnamyl alcohol, complex reaction pathways are possible in this reaction (Fig. 1).17–20 Apart from cinnamaldehyde arising from dehydrogenation of cinnamyl alcohol, 3-phenyl-1-propanol and trans-β-methylstyrene can also be produced through hydrogenation and hydrogenolysis reactions, respectively. These three products could potentially further lead to 1-phenylpropane, styrene, 3-phenyl-1-propanal, ethylbenzene and benzaldehyde.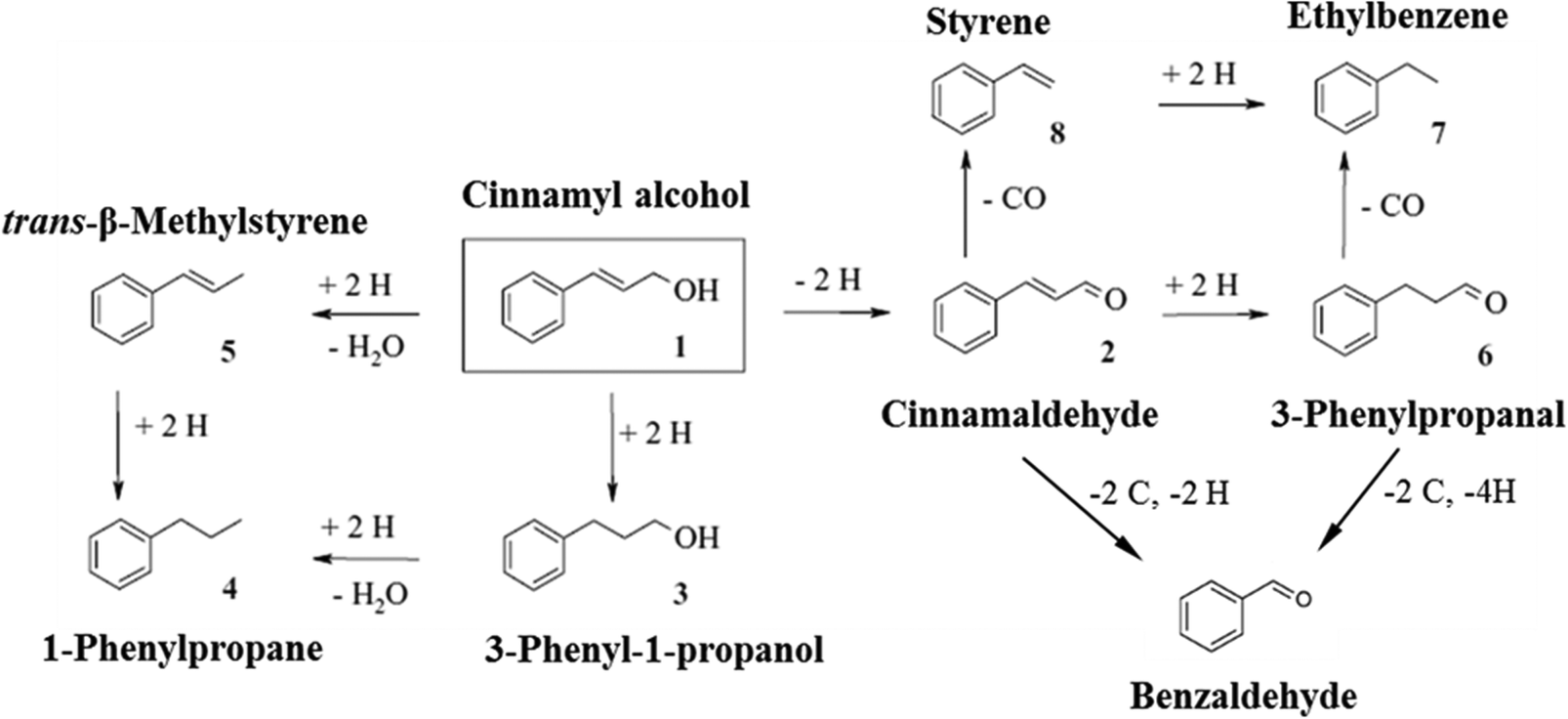 | ||
| Fig. 1 Reaction network proposed from Baiker and co-workers.17,20 | ||
Marked catalyst deactivation has been observed by comparison of the reaction rates at different reaction times.12,17 However, the active phase for the reaction, as well as the reason for the deactivation, is still a matter of debate. Baiker and co-workers17 demonstrated that metallic Pd was much more active than oxidic Pd in cinnamyl alcohol oxidation, which is consistent with the well-known dehydrogenation mechanism of selective alcohol oxidation to aldehydes on platinum-group metals catalysts.21 Deactivation caused either by the products from decarbonylation reactions poisoning the catalyst or by over-oxidation of the metal was suggested. The major role of oxygen was thought to rapidly and continuously remove the strongly adsorbed CO and thus ensure free Pd sites for the dehydrogenation reaction.17 On the other hand, Lee and co-workers22,23 found Pd-based catalyst activity directly correlated with the proportion of exposed surface palladium oxide. The rapid reduction of PdO to Pd led to catalyst deactivation under static oxygen.12 Flowing oxygen at ambient pressure contributed to suppressing the reduction of palladium oxide to metal, which ensured the maintenance of a selective oxidation activity.
Compared with monometallic catalysts, bimetallic catalysts provide an attractive approach for catalyst development. Preparation of bimetallic catalysts and their use for the transformation of bio-renewable substrates, including selective oxidations, has been reviewed recently by Sankar et al.24 Enache et al.25 showed that the addition of Au to Pd/TiO2 improves selectivity for benzyl alcohol oxidation. Good reusability of the catalyst was demonstrated, while no metals were detected in the filtered reaction mixtures. Dimitratos et al.6 found that Au-promoted Pd/C catalysts showed a significantly enhanced activity in the selective oxidation of cinnamyl and benzyl alcohols, when compared to monometallic catalysts. However, no information about catalyst reusability was provided. Generally, heterogeneous oxidation of cinnamyl alcohol has been carried out in batch reactors, due to their simplicity and flexibility. However, catalyst deactivation cannot be easily identified and monitored through a single run in batch reactors. To check the stability, catalysts need to be filtered and re-used in another run.10 Even with such experiments though, transient leaching of supported metal may not be detected easily, since the leached metal could redeposit onto the support.26
Over the past decade, microreactors have emerged as an excellent alternative for chemical synthesis, due to high mass and heat transfer coefficients and high-resolution reaction time control.27,28 The small volumes of microreactors also make it possible to use smaller amount of both reagents and catalyst for the rapid assessment of catalysts. Liquid phase catalytic oxidation chemistry in continuous-flow microreactors has recently been summarized from both technological and chemical perspective by Gemoets et al.29 In this study, the stability of a 1% bimetallic Au–Pd (5:95)/TiO2 catalyst is examined in a packed bed capillary microreactor for oxidation of cinnamyl alcohol. Various reaction conditions are investigated, in an attempt to explore the possible contributions to catalyst deactivation.
2. Experimental section
2.1 Catalyst preparation
1 wt% bimetallic Au–Pd/TiO2 catalysts were prepared by co-impregnation, similar to our previous work.25 An aqueous solution of HAuCl4·3H2O (Johnson Matthey) and PdCl2 (Johnson Matthey) with Au-to-Pd weight ratio equivalent to 1:19 was added to TiO2 (Degussa, P25) to form a slurry. The resultant slurry was spray-dried (nozzle temperature 220 °C), and then calcined in static air at 400 °C for 3 h. The powder was pelletized, crushed, and sieved to the desired particle size range.
2.2 Capillary microreactor setup
The schematic of the capillary microreactor setup is shown in Fig. 2. The gas flow was controlled with a mass flow controller (MFC, Brooks GF40) and the liquid feed was pumped with an HPLC pump (Knauer P 2.1S). Benzyl alcohol (Sigma-Aldrich, ≥99%) and 0.5 M cinnamyl alcohol (Sigma-Aldrich, 98%) dissolved in toluene (Sigma-Aldrich, ≥99.5%) were used as received. The gas and liquid flows were combined in a T-mixer (Upchurch) to generate a gas–liquid slug flow and fed into the reactor. The capillary microreactor was a 30 cm long PTFE tube (Upchurch, 0.8 mm I.D.) packed with the Au–Pd/TiO2 catalyst particles (particle size 53–63 μm). Unless otherwise specified, each experiment was performed with fresh catalyst. The microreactor was submerged in a stirred oil bath for isothermal operation and the temperature of the oil bath was controlled and measured by a hot plate fitted with a thermocouple (Stuart). The catalyst was kept under flowing oxygen until the oil bath reached the desired reaction temperature and then the liquid was pumped into the reactor to start the reaction. A sample of the product mixture was collected in a 2 mL vial every 30 min, and a 50 mL vessel was connected in parallel to minimize the pressure drop after taking a sample. The system pressure was sustained by a back pressure regulator (BPR, Swagelok) at the outlet and measured with two pressure sensors (Zaiput). | ||
| Fig. 2 Schematic of the capillary microreactor set-up used in this work. | ||
The collected sample was quantitatively analysed by a gas chromatograph (Agilent 7820A) fitted with a HP-INNOWax capillary column and a flame ionization detector. Alcohol conversion (Con.) and selectivity to each product were calculated according to the following equations.
 | (1) |
 | (2) |
2.3 Catalyst characterization
X-ray photoelectron spectra (XPS) was carried out using a Thermo Scientific K-alpha photoelectron spectrometer with monochromatic AlKα radiation. Spectra were collected in steps of 0.15 eV and plotted with the CasaXPS software. Inductively coupled plasma (ICP) measurements were carried out on an Agilent 7900 ICP-MS equipped with a micromist nebuliser in organic phase mode. Quantification was carried out by comparison with calibration curves of Au and Pd.3. Results and discussion
3.1 Oxidation of benzyl alcohol as benchmarking experiment
The co-impregnation method employed in this study is one of the easiest ways to synthesize bimetallic catalysts.24,25 It is also considered to be ideal for the discovery of active catalyst phase, since a wide variation in nanostructures is available in the prepared catalyst.30 Oxidation of benzyl alcohol is often used as a model reaction for oxidation of alcohols. In order to verify the reactivity of the prepared Au–Pd/TiO2 catalyst and the performance of the capillary microreactor system, oxidation of benzyl alcohol was initially carried out for benchmarking.Conversion and selectivity of the oxidation of benzyl alcohol as a function of time are shown in Fig. 3. It can be observed that the conversion of benzyl alcohol (88%) stabilized after 1 h and then stayed constant over the whole 30 h. The selectivities to benzaldehyde and toluene, the two main products, also showed stable trends. Hence, the Au–Pd/TiO2 catalyst exhibited high stability in this particular oxidation reaction. This is consistent with previous work.25,31 Furthermore, it has been indicated that benzaldehyde can be formed by both the oxidation and disproportionation of benzyl alcohol, while toluene is produced only from the disproportionation.32 Thus, the higher selectivity to benzaldehyde (67%) than that to toluene (25%) shows the good activity of Au–Pd/TiO2 catalyst in the oxidation of benzyl alcohol. These results obtained in the packed bed capillary microreactor were similar to those in a silicon-glass packed bed microreactor.31
 | ||
| Fig. 3 Solvent-free oxidation of benzyl alcohol as a function of reaction time. Reaction conditions: Au–Pd/TiO2 catalyst, 20 mg; oxygen flow, 0.8 mL min−1 at standard temperature and pressure (STP, 0 °C, 1 bara); benzyl alcohol flow, 0.008 mL min−1; reaction temperature, 120 °C; reaction pressure, 4 bara. Con., conversion of benzyl alcohol; SB, selectivity to benzaldehyde; ST, selectivity to toluene. | ||
3.2 Effect of reaction temperature on oxidation of cinnamyl alcohol
For oxidation of cinnamyl alcohol, a blank experiment was first conducted at 100 °C with TiO2 support and pure oxygen. The conversion was observed to be 9%, with a 22% selectivity to cinnamaldehyde. Then, the prepared Au–Pd/TiO2 catalyst was studied for oxidation of cinnamyl alcohol at different reaction temperatures. Cinnamaldehyde, 3-phenyl-1-propanol, and trans-β-methylstyrene were observed to be the main products. There were still 3 unidentified by-products in the GC chromatograms, even though all the potential products shown in Fig. 1 had been tested. The carbon loss could be up to 20% at high conversion. Since, this study is focused on the stability of the Au–Pd/TiO2 catalyst, the conversion of cinnamyl alcohol and the selectivity to cinnamaldehyde were the main criteria to evaluate catalyst performance.From Fig. 4, the conversion of cinnamyl alcohol was observed to be rather low (9%) at 80 °C, although it was stable within the investigated duration. The selectivity to cinnamaldehyde fluctuated around 54%. 3-Phenyl-1-propanol was also detected with a selectivity of 6% while no trans-β-methylstyrene was produced. When the reaction was performed at 100 °C, the conversion initially started from a high value (41%) and gradually decreased to 19% after 7 h. The selectivity to 3-phenyl-1-propanol also presented a similar trend, dropping from 20% to 7% within 3 h and then remaining stable. Since 3-phenyl-1-propanol is formed through the hydrogenation of cinnamyl alcohol, this might indicate that the hydrogenation reactivity of the catalyst was suppressed as reaction progressed, which caused the decrease in the total conversion. The selectivity to cinnamaldehyde increased from 54% at 80 °C to 64% at 100 °C. trans-β-Methylstyrene was detected only at low concentrations (both <3%). When the reaction was conducted at 120 °C, the initial conversion was further enhanced (58%), but it still decreased to 33% at 7 h. The selectivity to cinnamaldehyde was about 3% lower than that at 100 °C and similar behaviour was also observed for the selectivities to 3-phenyl-1-propanol and trans-β-methylstyrene. The initial cinnamaldehyde yield was 35%, which corresponded to 2.8 gCD gcat−1 h−1 cinnamaldehyde produced.
 | ||
| Fig. 4 Oxidation of cinnamyl alcohol as a function of reaction time at different reaction temperatures. a) 80 °C; b) 100 °C; c) 120 °C. Reaction conditions: Au–Pd/TiO2 catalyst, 10 mg; oxygen flow, 2.0 mL min−1 at STP; 0.5 M cinnamyl alcohol in toluene, 0.020 mL min−1; reaction pressure, 4 bara. Con., conversion of cinnamyl alcohol; SCD, selectivity to cinnamaldehyde; SPP, selectivity to 3-phenyl-1-propanol; STR, selectivity to trans-β-methylstyrene; dotted line, average of the selectivity to cinnamaldehyde. | ||
3.3 Effect of catalyst reduction on oxidation of cinnamyl alcohol
The active phase for cinnamyl alcohol oxidation is still under debate, but the pre-reduction of Pd-based catalysts with alcohol or hydrogen before reaction has been reported to enhance the reaction rate.17 In an attempt to address the issue of catalyst deactivation, Au–Pd/TiO2 was first pre-reduced in situ at the reaction temperature (100 °C) with 0.5 M cinnamyl alcohol in toluene for 0.5 h, followed by introducing oxygen to start the reaction. The reaction results are shown in Fig. 5a. The initial conversion of cinnamyl alcohol was higher as compared to the non-reduced catalysts (Fig. 4b), but the activity drop to a similar level after 6 h. To ensure that the catalyst had been satisfactorily reduced, the fresh catalyst was also pre-reduced with hydrogen at 100 °C in another experiment and then purged with helium before reaction. From Fig. 5b, the pre-reduced catalyst with hydrogen showed similar results to the pre-reduced catalyst with cinnamyl alcohol. The hydrogen reduction process was carried out again and subsequently the reaction was continued (shown in Fig. 5b). Some recovery of catalyst activity was observed, indicating that a small part of the deactivation is reversible. | ||
| Fig. 5 Oxidation of cinnamyl alcohol as a function of reaction time under different pre-reduction conditions. Catalyst pre-reduced with a) 0.5 M cinnamyl alcohol; b) 100% hydrogen (re-reduced with hydrogen during the reaction). Reaction conditions: Au–Pd/TiO2 catalyst, 10 mg; oxygen flow, 2.0 mL min−1 at STP; 0.5 M cinnamyl alcohol in toluene, 0.020 mL min−1; reaction temperature, 100 °C; reaction pressure, 4 bara. Con., conversion of cinnamyl alcohol; SCD, selectivity to cinnamaldehyde; SPP, selectivity to 3-phenyl-1-propanol; STR, selectivity to trans-β-methylstyrene; dotted line, average of the selectivity to cinnamaldehyde. | ||
The palladium oxidation state of fresh and spent catalysts was investigated using XPS. From Fig. 6a and b, it can be observed that PdO was the main phase in the fresh catalyst without any pre-reduction while more Pd existed in the used catalyst. This shows that PdO could be reduced by cinnamyl alcohol during the reaction (corresponding reaction data are in Fig. 4b), due to the relatively high reaction temperature.33 After pre-reduction with hydrogen, most of the PdO was converted into Pd (Fig. 6c). Notably, Pd was still the main phase after the reaction. At first glance, it seems that these results agree with Lee and co-authors' contention that Pd/C and Pd/SBA-15 catalyst deactivation was caused by the reduction of PdO to Pd.12,22 If so, the pre-reduced catalyst should present lower reactivity from the beginning, given that most of the PdO had been reduced before reaction. Actually, the opposite is observed, i.e. the pre-reduced catalyst shows higher initial conversion than the non-reduced (corresponding reaction data are in Fig. 4b and 5). So, the reduction of PdO to Pd does not seem to be the reason for the deactivation in this case. As for adsorbed hydrocarbons, they were difficult to be identified on the catalyst surface through C1s scanning (shown in Fig. 6), due to adventitious carbon contamination.
 | ||
| Fig. 6 XPS of Pd 3d (left) and C 1s (right). Catalysts without any pre-reduction a) fresh and b) used catalysts after 7 h reaction at 100 °C with 100% oxygen. Catalysts after pre-reduction with hydrogen c) fresh and d) used catalysts after 7 h reaction at 100 °C with 100% oxygen. | ||
To further explore the cause of catalyst deactivation, the effluent was collected from the experiment in Fig. 4b over 7 h and then analysed by ICP to check for metal leaching. The analysis revealed no Au but 2.4 ppm of Pd from catalyst leaching, which corresponded to 24% of total Pd loaded on the fresh catalyst. Thus, catalyst deactivation could be correlated to metal leaching. Indeed, metal leaching is often reported to be responsible for catalyst deactivation in oxidation of alcohols, though the cause of metal leaching is rarely specified.4 In palladium-catalysed coupling reactions, metal leaching has also been detected during the oxidative addition and accordingly a (quasi)homogeneous mechanism has been suggested.26 Alloying of Pd with Au has been observed to greatly limit the Pd leaching.34 A “release and catch” catalytic system, which combines the benefits of homogeneous and heterogeneous catalysis, has been proposed as a potential solution.35 It is noteworthy that the degree of Pd loss in our study was lower, compared with the conversion decreasing from 41% to 19% (see Fig. 4b). This suggests additional factors contributing to catalyst deactivation, such as selective Pd leaching (i.e. depending on particle size and/or composition) that would also alter the local Au/Pd ratio, which is known to influence catalytic activity.
3.4 Effect of oxygen concentration on oxidation of cinnamyl alcohol
For selective oxidation reactions such as that of cinnamyl alcohol, the reaction conditions have been suggested to maintain the Pd constituent in a metallic state with an optimal availability of oxygen to avoid catalyst poisoning by alcohol degradation or over-oxidation of the metal.33,36 In batch reactors, Baiker and co-workers15,17 reduced the stirring speed and the gas flow rate to make oxygen transport from the gas phase to the Pd surface the rate-limiting step. In the packed bed capillary microreactor, different oxygen concentrations were used to investigate the role of oxygen in the cinnamyl alcohol oxidation. This was achieved using air or nitrogen, while keeping the same gas-to-liquid ratio.From Fig. 7a, it can be observed that the Au–Pd/TiO2 catalyst still showed deactivation in terms of conversion, when using air as the oxidant gas (21 vol% oxygen). The average selectivity to cinnamaldehyde (59%) was lower than that with pure oxygen. Furthermore, relatively high and stable selectivity to 3-phenyl-1-propanol (12%) was maintained during the reaction. This indicates the hydrogenation of cinnamyl alcohol was favoured when less oxygen was present.
 | ||
| Fig. 7 Oxidation of cinnamyl alcohol as a function of reaction time under different oxygen concentrations. a) 21 vol%; b) 0 vol%. Reaction conditions: Au–Pd/TiO2 catalyst, 10 mg; gas flow, 2.0 mL min−1 at STP; 0.5 M cinnamyl alcohol in toluene, 0.020 mL min−1; reaction temperature, 100 °C; reaction pressure, 4 bara. Con., conversion of cinnamyl alcohol; SCD, selectivity to cinnamaldehyde; SPP, selectivity to 3-phenyl-1-propanol; STR, selectivity to trans-β-methylstyrene; dotted line, average of the selectivity to cinnamaldehyde. | ||
Since the catalyst still deactivated even at low oxygen concentration, the reaction was carried out under pure nitrogen conditions (oxygen-free conditions). Surprisingly, a relatively high steady-state conversion of cinnamyl alcohol was obtained with average selectivity to cinnamaldehyde of 44% (Fig. 7b). An upward trend was observed for the selectivity to trans-β-methylstyrene (from 16% to 25%), while the selectivity to 3-phenyl-1-propanol dropped from 22% to 15%.
These results are consistent with the dehydrogenation mechanism of alcohol oxidation. The reaction on the metal surface should involve a first dehydrogenation of alcohol to produce adsorbed metal alkoxide and metal hydride.4,21 The metal alkoxide is subsequently dehydrogenated into the corresponding aldehyde through β-hydride elimination. Last, the metal hydride reacts with dissociatively adsorbed oxygen to regenerate the metal surface. In this case, hydrogen is also required during the formation of 3-phenyl-1-propanol and trans-β-methylstyrene (shown in Fig. 1). Interestingly, the average selectivity to cinnamaldehyde (44%) is almost equal to the sum of 3-phenyl-1-propanol and trans-β-methylstyrene (40%). This indicates the reactant itself acts as hydrogen acceptor in the absence of oxygen. This phenomenon was also observed in the oxygen-free oxidation of benzyl alcohol, where equal amounts of toluene and benzaldehyde were produced.32,37
The effluent collected from the reaction under oxygen-free conditions over 7 h (Fig. 7b) was analysed with ICP to check for catalyst leaching. 1.6 ppm of Pd was found, which corresponded to 16% Pd loss. This explains the slight drop in the cinnamyl alcohol conversion under nitrogen conditions from 70% to 60% (as shown in Fig. 7b). Pd leaching might be caused by the formation of complexes through the interaction of the products with the catalyst. The Pd loss is lower than that under oxygen-rich conditions (24%); this may be due to accelerated leaching when the metal is oxidised.38
To better illustrate the effect of oxygen concentration at the reactor inlet, these results are summarized in Fig. 8. Clearly, there is a strong correlation between the oxygen concentration and the reactivity in terms of conversion and selectivity. With decreasing oxygen concentration the conversion was enhanced and the deactivation rate decreased. On the other hand, comparatively low selectivity to cinnamaldehyde was obtained under low oxygen concentration. These contribute to the comparable yield to cinnamaldehyde at some points (shown in Fig. 8c). On the basis of initial dehydrogenation of cinnamyl alcohol followed by secondary hydrogenation to form 3-phenyl-1-propanol and trans-β-methylstyrene, the cinnamaldehyde yield should be independent of oxygen concentration in the feed. Using the data of the initial conversion (to minimise the effects of catalyst deactivation) and the average selectivity, the aldehyde yields are 26% (100% O2), 33% (21% O2) and 31% (0% O2), i.e. roughly independent of oxygen and confirming the correctness of the assumptions. Hardacre et al.36 reported a higher alcohol conversion and lower selectivity to cinnamaldehyde in the absence of oxygen compared with experiments in the presence of oxygen when Pd/Al2O3 catalyst was used in a rotating disc reactor at 80 °C. Baiker and co-workers observed low conversion on Pd/Al2O3 catalyst under argon atmosphere at 40 °C, while in air, higher conversion was obtained due to oxidative removal of CO and other degradation products. However, they argue that an optimal level of oxygen is required to keep the catalyst surface clean but not overoxidise the catalyst.17 With our catalyst, we believe that the presence of Au can alter the electronic and geometrical properties of the synthesized particles.24,25
 | ||
| Fig. 8 Comparison of catalyst performance and deactivation under different oxygen concentrations during reaction. a) Conversion of cinnamyl alcohol; b) selectivity to cinnamaldehyde; c) yield to cinnamaldehyde. Reaction conditions: Au–Pd/TiO2 catalyst, 10 mg; gas flow, 2.0 mL min−1 at STP; 0.5 M cinnamyl alcohol in toluene, 0.020 mL min−1; reaction temperature, 100 °C; reaction pressure, 4 bara; dotted line, average of the selectivity to cinnamaldehyde. | ||
Since the rate of catalytic dehydrogenation appears to be independent of oxygen concentration but different percentage decrease in cinnamyl alcohol conversion was observed under different oxygen concentrations, yet with similar percentage loss of Pd, poisoning of the surface by adsorbed oxygen might be also responsible for the catalyst deactivation.1,21,39 In the absence of oxygen, the metal hydride is consumed in rapidly reducing cinnamyl alcohol to either 3-phenyl-1-propanol or trans-β-methylstyrene. When oxygen is present, adsorbed oxygen on the metal surface competes with adsorbed cinnamyl alcohol for metal hydride. Therefore, the selectivities to 3-phenyl-1-propanol and trans-β-methylstyrene go down and so does the cinnamyl alcohol conversion. Activity was only partially recovered when re-reducing the catalyst with hydrogen (Fig. 5b). Hence, the irreversible deactivation might be caused by Pd leaching (possibly accelerated in the presence of oxygen) and strong adsorption of poisoning species formed under oxygen conditions.15,17
To further explore the effect of oxygen on catalyst reactivity and deactivation, an oxygen/nitrogen switching experiment was performed with the same catalyst packing. From Fig. 9, deactivation as expected was observed within 3 h under pure oxygen conditions. Then, a distinct rise of conversion was obtained when changing the gas from oxygen to nitrogen. The selectivity to cinnamaldehyde also decreased simultaneously, together with the increase of selectivities to 3-phenyl-1-propanol and trans-β-methylstyrene. The trend of conversion and selectivities under pure nitrogen conditions seems to be slightly different from that in Fig. 7b, which might be caused by changes of surface properties or the presence of poisoning species from the previous reaction stage with oxygen. Nevertheless, these results clearly indicate an inhibiting effect of adsorbed oxygen on the cinnamyl alcohol conversion.
 | ||
| Fig. 9 Oxidation of cinnamyl alcohol as a function of reaction time under different gas conditions. Reaction conditions: Au–Pd/TiO2 catalyst, 10 mg; gas flow, 2.0 mL min−1 at STP; 0.5 M cinnamyl alcohol in toluene, 0.020 mL min−1; reaction temperature, 100 °C; reaction pressure, 4 bara. Con., conversion of cinnamyl alcohol; SCD, selectivity to cinnamaldehyde; SPP, selectivity to 3-phenyl-1-propanol; STR, selectivity to trans-β-methylstyrene. | ||
Switching experiments between oxidation of cinnamyl alcohol and benzyl alcohol were also carried out with the same catalyst packing, since different catalyst stabilities were observed in these two reactions. As shown in Fig. 10, catalyst deactivation was first observed in cinnamyl alcohol oxidation. When the reaction was changed to benzyl alcohol oxidation, the catalyst still presented good stability in benzyl alcohol oxidation over the following 20 h. When it was changed back to cinnamyl alcohol oxidation, the trend of catalyst deactivation continued. This suggests that the catalyst deactivation in cinnamyl alcohol oxidation due to metal leaching and presence of oxygen does not apply to benzyl alcohol oxidation, and this may be due to the different catalytic sites for the two reactants, different reaction mechanism as well as the different products.
 | ||
| Fig. 10 Oxidation of cinnamyl alcohol and benzyl alcohol as a function of reaction time. Reaction conditions: Au–Pd/TiO2 catalyst, 10 mg; oxygen flow, 2.0 mL min−1 at STP; 0.5 M cinnamyl alcohol in toluene, 0.020 mL min−1; neat benzyl alcohol, 0.010 mL min−1; reaction temperature, 100 °C; reaction pressure, 4 bara. CA, cinnamyl alcohol; BnOH, benzyl alcohol; Con., conversion of cinnamyl alcohol or benzyl alcohol; SCD, selectivity to cinnamaldehyde; SPP, selectivity to 3-phenyl-1-propanol; STR, selectivity to trans-β-methylstyrene; SB, selectivity to benzaldehyde; ST, selectivity to toluene. | ||
4. Conclusions
A bimetallic Au–Pd/TiO2 catalyst was studied for oxidation of cinnamyl alcohol in a packed bed microreactor. The catalyst showed stable conversion and selectivity for the oxidation of benzyl alcohol to benzaldehyde. For cinnamyl alcohol oxidation, catalyst deactivation was observed which was more severe at high reaction temperature and high oxygen concentration. Catalyst pre-reduction with cinnamyl alcohol or hydrogen before reaction, improved initial activity but did not affect catalyst deactivation. Higher oxygen concentration did not affect the rate of cinnamaldehyde formation but led to lower initial alcohol conversion by suppressing the formation of the by-products, 3-phenylpropanol and trans-β-methylstyrene, leading to higher cinnamaldehyde selectivity. Pd was detected in the reactor effluent during cinnamyl alcohol oxidation. Thus, Pd leaching and the presence of oxygen seem to be causes for catalyst deactivation. The higher deactivation rates and amount of Pd leached at higher oxygen concentration, along with partial recovery of activity after hydrogen treatment, indicate that the role of oxygen is complex, affecting deactivation in several ways (possibly including competing with alcohol on the catalyst, removing by-products from surface, accelerating Pd leaching). Switching substrates between benzyl alcohol and cinnamyl alcohol showed deactivation during cinnamyl alcohol oxidation but steady performance during benzyl alcohol oxidation, demonstrating different deactivation characteristics for the two reactions.Acknowledgements
Financial support by EPSRC, UK (grant EP/L003279/1) is gratefully acknowledged. G. W. acknowledges the Chinese Council Scholarship (CSC) and UCL for his studentship. We thank Simon Waller for help with the ICP experiments.References
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- D. S. Mannel, S. S. Stahl and T. W. Root, Org. Process Res. Dev., 2014, 18, 1503–1508 CrossRef CAS PubMed.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS.
- C. M. A. Parlett, P. Keshwalla, S. G. Wainwright, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, ACS Catal., 2013, 3, 2122–2129 CrossRef CAS.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- M. Kotani, T. Koike, K. Yamaguchi and N. Mizuno, Green Chem., 2006, 8, 735–741 RSC.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS PubMed.
- J. Cocchiara, C. S. Letizia, J. Lalko, A. Lapczynski and A. M. Api, Food Chem. Toxicol., 2005, 43, 867–923 CrossRef CAS PubMed.
- C. M. A. Parlett, L. J. Durndell, A. Machado, G. Cibin, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, Catal. Today, 2014, 229, 46–55 CrossRef CAS.
- A. Buonerba, A. Noschese and A. Grassi, Chem. – Eur. J., 2014, 20, 5478–5486 CrossRef CAS PubMed.
- S. Mori, M. Takubo, K. Makida, T. Yanase, S. Aoyagi, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. Commun., 2009, 5159–5161 RSC.
- T. Mallat, Z. Bodnar, P. Hug and A. Baiker, J. Catal., 1995, 153, 131–143 CrossRef CAS.
- L. J. Durndell, C. M. A. Parlett, N. S. Hondow, K. Wilson and A. F. Lee, Nanoscale, 2013, 5, 5412–5419 RSC.
- C. Keresszegi, T. Bürgi, T. Mallat and A. Baiker, J. Catal., 2002, 211, 244–251 CrossRef CAS.
- J. D. Grunwaldt, C. Keresszegi, T. Mallat and A. Baiker, J. Catal., 2003, 213, 291–295 CrossRef CAS.
- B. Qi, Y. Wang, L.-L. Lou, L. Huang, Y. Yang and S. Liu, J. Mol. Catal. A: Chem., 2013, 370, 95–103 CrossRef CAS.
- M. Caravati, D. M. Meier, J. D. Grunwaldt and A. Baiker, J. Catal., 2006, 240, 126–136 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef CAS.
- A. F. Lee and K. Wilson, Green Chem., 2004, 6, 37–42 RSC.
- A. F. Lee, S. F. J. Hackett, J. S. J. Hargreaves and K. Wilson, Green Chem., 2006, 8, 549 RSC.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- D. Cantillo and C. O. Kappe, ChemCatChem, 2014, 6, 3286–3305 CrossRef CAS.
- K. S. Elvira, X. C. i. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- J.-I. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noel, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- G. J. Hutchings and C. J. Kiely, Acc. Chem. Res., 2013, 46, 1759–1772 CrossRef CAS PubMed.
- M. Morad, M. Sankar, E. Cao, E. Nowicka, T. E. Davies, P. J. Miedziak, D. J. Morgan, D. W. Knight, D. Bethell, A. Gavriilidis and G. J. Hutchings, Catal. Sci. Technol., 2014, 4, 3120–3128 CAS.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem. – Eur. J., 2011, 17, 6524–6532 CrossRef CAS PubMed.
- J.-D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 25586–25589 CrossRef CAS PubMed.
- A. Villa, D. Wang, N. Dimitratos, D. Su, V. Trevisan and L. Prati, Catal. Today, 2010, 150, 8–15 CrossRef CAS.
- M. Gruttadauria, F. Giacalone and R. Noto, Green Chem., 2013, 15, 2608–2618 RSC.
- C. Hardacre, E. A. Mullan, D. W. Rooney and J. M. Thompson, J. Catal., 2005, 232, 355–365 CrossRef CAS.
- E. H. Cao, M. Sankar, S. Firth, K. F. Lam, D. Bethell, D. K. Knight, G. J. Hutchings, P. F. McMillan and A. Gavriilidis, Chem. Eng. J., 2011, 167, 734–743 CrossRef CAS.
- J. H. Vleeming, B. F. M. Kuster and G. B. Marin, Catal. Lett., 1997, 46, 187–194 CrossRef CAS.
- Z. Gogová and J. Hanika, Chem. Eng. J., 2009, 150, 223–230 CrossRef.
Footnote |
| † Current address: Division of Chemical and Petroleum Engineering, School of Engineering, London South Bank University, London SE1 0AA, UK. |
| This journal is © The Royal Society of Chemistry 2016 |