
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light photoredox catalysis using a macromolecular ruthenium complex: reactivity and recovery by size-exclusion nanofiltration in continuous flow†
Javier
Guerra
 ab,
David
Cantillo
a and
C. Oliver
Kappe
*a
ab,
David
Cantillo
a and
C. Oliver
Kappe
*a
aInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at
bCrystal Pharma, Gadea Pharmaceutical Group, a division of AMRI, Parque Tecnológico de Boecillo, Valladolid, 47151, Spain
First published on 4th February 2016
Abstract
A novel macromolecular photoredox catalyst based on [Ru(bpy)3]2+ anchored to a 2nd-generation PAMAM dendrimer has been developed. Its catalytic activity under visible light irradiation and recyclability using organic solvent nanofiltration with a size-exclusion membrane have been explored under continuous flow conditions.
Visible-light photoredox catalysis has recently emerged as a very powerful tool for organic synthesis.1 These methods rely on the redox properties of so-called photoredox catalysts upon absorption of low energy visible light. The single-electron transfer processes triggered by catalyst irradiation permits the construction of bonds (especially C–C bonds) difficult to achieve by conventional means, and in many instances avoids the use of stoichiometric amounts of oxidants or reductants.1 The use of visible light instead of UV prevents deterioration of substrates and products in the reaction mixture, and constitutes a greener method of activation. Notably, this synthetic method has been shown as a valuable tool in the preparation of complex molecules and natural products such as heitziamide A2 and aplyviolene.3 The most commonly used photoredox catalysts are ruthenium and iridium polypyridyl complexes. Tris(2,2′-bipyridine) ruthenium(II) (Ru(bpy)32+) (Fig. 1) is probably the most popular catalyst and a plethora of transformation using this additive have been reported.1 Ruthenium and iridium are scarce elements and the cost of these metal complexes can be considered a major drawback of photoredox catalysis. Moreover, the use of potentially toxic metals is an important concern in API synthesis, and meticulous purification procedures are typically required to meet the specification limits for metal residues. Several organic photosensitizers have been reported to substitute the use of ruthenium or iridium compounds in some instances.4 Yet, most transformations being reported still rely on the photoredox catalysis properties of Ru(bpy)32+.1
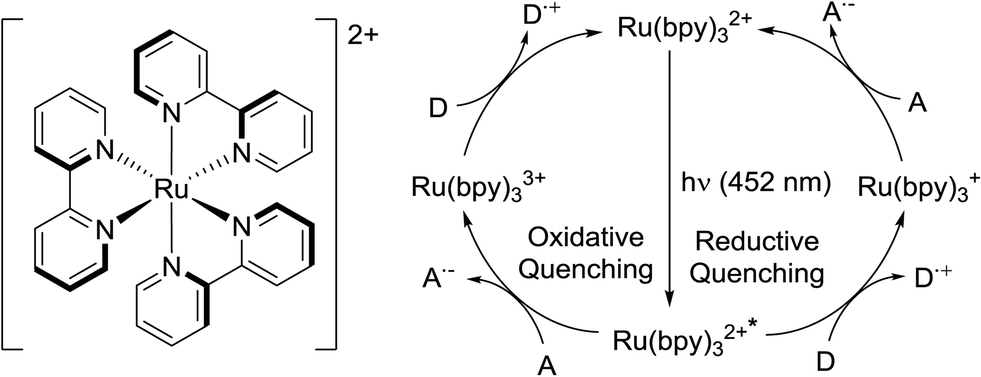 | ||
| Fig. 1 Structure of Ru(bpy)32+ and its oxidative and reductive quenching cycles. | ||
Photochemistry in general and photoredox catalysis in particular have benefited from technological advances such as the use of continuous flow reactors.5 The relatively low reactor volume and in particular the high surface to volume ratio of capillary flow reactors assure intense and uniform irradiation of the reaction mixture. Important reaction enhancements including reduced reaction times and catalyst loadings have been described for Ru(bpy)32+ and related systems.5 However, catalyst loadings of 0.5–1 mol% are still typically required.
There is therefore little doubt that development of catalyst recovery/recycling technology for metal-based photoredox catalysts is highly desirable. In the context of continuous flow chemistry several recycling techniques for metal catalysts have been reported in the literature.6 These include catalyst immobilization, liquid–liquid biphasic conditions, or organic solvent nanofiltration. Solvent nanofiltration typically requires “enlargement” of the catalyst.6 Different soluble platforms such as dendrimers, polymers and polyhedral silesquioxanes have been used to anchor metal centers, in most cases palladium catalysts for cross-coupling chemistry. An example of continuous recovery/recycling of a ruthenium Grubbs–Hoveyda type catalyst anchored on a polyhedral oligomeric silsesquioxane for ring metathesis was described by Grela.7 Bergbreiter and coworkers prepared a modified recyclable Ru(bpy)32+ based photocatalyst decorated with multiple polyisobutene chains, that could be separated by selective precipitation from the reaction mixture.8 More recently, the group of Reiser has described a continuous flow procedure for the use and recycling of a polyisobutene immobilized fac-Ir(ppy)3 complex.9 Constant conversions with only minor losses of catalyst could be achieved using a biphasic system.9 Recycling of a fluorinated photocatalyst using a biphasic system with supercritical CO2 has also been reported.10 However, to the best of our knowledge, merger of organic solvent nanofiltration and photoredox catalysis using a macromolecular metal catalyst has never been described.
On the basis of all the above, we envisaged a strategy for the continuous flow utilization and in-line recycling of a ruthenium bipyridyl-based macromolecular photoredox catalyst (Fig. 2). To achieve this goal, we synthesized a second-generation polyamidoamine (PAMAM) dendrimer11 decorated with bipyridine pendant ligands to which Ru(bpy)22+ was attached. The resulting macromolecular Ru(bpy)32+ catalyst was used for a set of model photoredox catalysis reactions under continuous flow conditions, using a photoreactor equiped with blue (450 nm) LEDs. After the reactions, the catalyst could be retrieved from the reaction mixture and recycled by continuous solvent nanofiltration with a size-exclusion membrane. Herein we report on the synthesis of the dendrimer immobilized ruthenium catalyst, its properties as photoredox catalyst, and the recovery/recycling of the dendrimer using organic solvent nanofiltration.
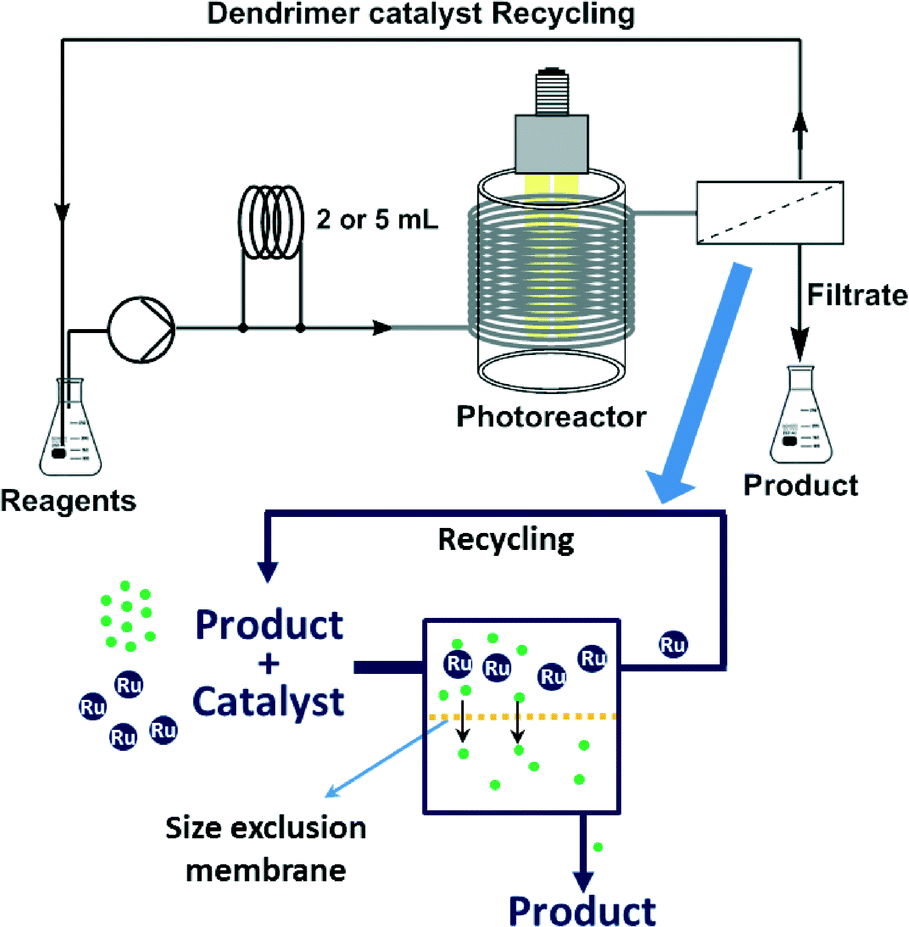 | ||
| Fig. 2 Proposed setup for macromolecular photoredox catalyst use and recycling, and schematics of the continuous organic solvent nanofiltration using a size-exclusion membrane. | ||
As platform for the construction of our macromolecular Ru(bpy)32+ catalyst a second-generation polyamidoamine (PAMAM) dendrimer was chosen.11,12 We used the 16 primary amino groups located at the periphery of the dendrimer to introduce the [Ru(bpy)3]2+ moieties, following an analogous synthetic route to that reported by Abruña et al.12 and Velders et al.13 (Scheme 1). The bipyridine pendant ligand 4 was constructed following a methodology previously reported from 4,4′-dimethyl-2,2′-dipyridyl 1 and bromo dioxolane 3.12,13 Attachment of the bipyridine to the dendrimer surface was carried out by imination of the dendrimer amino groups with 4 followed by reduction (Scheme 1).14 Purification of the bypiridine dendrimer derivative was performed by nanofiltration using a size-exclusion membrane (NF080105, Solsep BV, The Netherlands) installed in a modified commercial continuous liquid–liquid separator (Zaiput) (Fig. S2†). This procedure facilitates the removal of reactants or byproducts with a molecular weight lower than 1 kDa. A Kaiser test was performed after the bipyridine coupling and purification to rule out the presence of free primary amines (Fig. S1†). Chelation of the Ru was performed following previous methodologies.12,13 Purification of the resulting G2-PAMAM-Ru dendrimer was again carried out by continuous nanofiltration in methanol. The final G2-PAMAM-Ru catalyst was characterized by means of NMR, MALDI-TOF, ICP-MS as well as UV-visible spectroscopy measurements (see ESI† for details). Notably, ICP-MS analysis revealed that the catalyst contained an average of 14.7 Ru atoms per dendrimer unit. Measurement of the absorptivity coefficient by UV-visible spectroscopy agreed with this value (see ESI† for details).
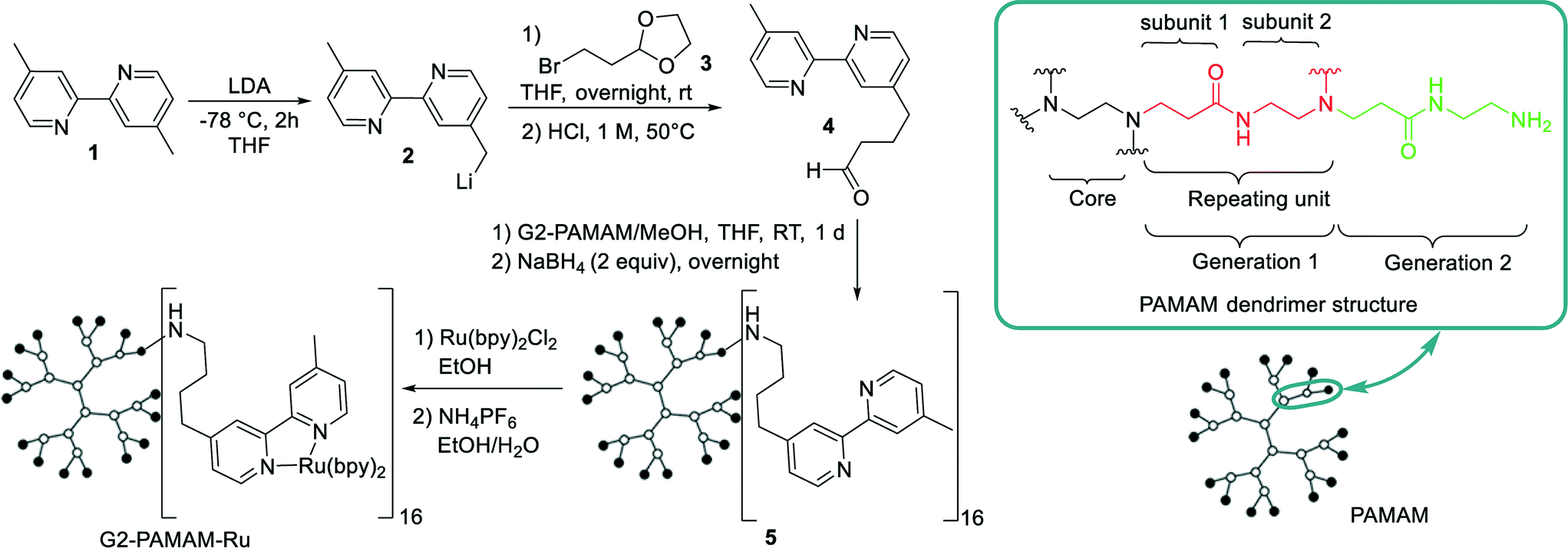 | ||
| Scheme 1 Synthetic sequence for the preparation of the G2-PAMAM-Ru dendrimer catalyst and detail on the PAMAM structure. | ||
The catalytic efficiency of our macromolecular catalyst was tested for three model photochemical reactions: the phosphine-free Appel reaction, reductive opening of chalcone epoxide and azide reduction. These reactions have been previously used as model for benchmarking studies with Ru(bpy)32+ under continuous flow.15 The continuous flow photochemical reactions were performed in a Vapourtec EasyMedChem instrument with a UV150 photochemical reactor (FEP tubing, 1.0 mm id, 10 mL volume) equipped with a 450 nm blue LED (Fig. S5†). The reaction mixtures were introduced into the reactor using sample loops after the system had been stabilized pumping solvent through the system at the desired flow rate.
The first transformation attempted with our catalytic system was the phosphine-free Appel reaction.16 In this reaction an alcohol 6 (Table 1) is converted into the corresponding bromide 7 using CBr4 as bromine source. The classical reaction conditions require stoichiometric amounts of triphenylphosphine as reducing agent. The phosphine-free photochemical version requires the use of DMF as solvent or co-solvent as it plays a role in the reaction mechanism.17 Apart from the G2-PAMAM(Ru)16(PF6)32 catalysts, a modified version with chloride as counteranion was also employed. The solubility of the hexafluorophosphate salt of G2-PAMAM-Ru in DMF was very poor and therefore a mixture of acetone/DMF was used (Table 1). Gratifyingly, excellent results were obtained for primary, secondary, and tertiary alcohols with both the hexafluorophosphate and chloride catalysts. Full or near to full conversion to the desired bromine derivative 7 was obtained in all cases, in analogy with the results obtained with the typical monomeric ruthenium catalysts (Table 1, entries 1, 2 and 5).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Catalyst | Solvent | Conv.b (%) |
| a Conditions: 0.2 M substrate, 2 equiv. CBr4, 2 mL reaction mixture. b Determined by GC-MS. c 0.35% [Ru] was used. | ||||
| 1 | 1-Nonanol | Ru(bpy)3Cl2 | DMF | 99 |
| 2 | 1-Nonanol | Ru(bipy)2(Me2bpy)(PF6)2 | Ac/DMF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
99 |
| 3c | 1-Nonanol | G2-PAMAM(Ru)16(PF6)32 | Ac/DMF 1:6 |
99 |
| 4 | 1-Nonanol | G2-PAMAM(Ru)16Cl32 | DMF | 99 |
| 5 | PhCH2CH2OH | Ru(bipy)2(Me2bpy)(PF6)2 | Ac/DMF 12:1 |
94 |
| 6 | PhCH2CH2OH | G2-PAMAM(Ru)16(PF6)32 | Ac/DMF 1:2 |
99 |
| 7 | PhCH2OH | G2-PAMAM(Ru)16(PF6)32 | Ac/DMF 12:1 |
97 |
| 8 | Ph2CHOH | G2-PAMAM(Ru)16(PF6)32 | Ac/DMF 12:1 |
99 |
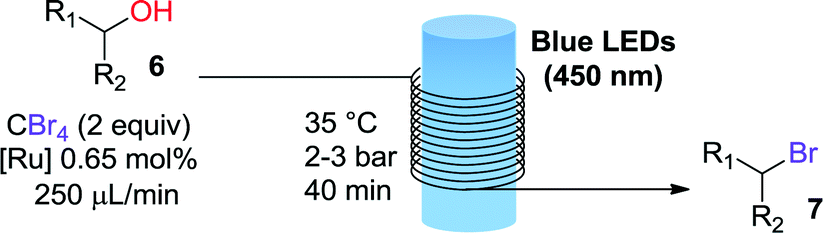
We chose the bromination of 1-nonanol as model to assess the photoredox catalyst recyclability. The pressure required to perform an efficient solvent nanofiltration in the presence of DMF with the membrane employed (EXP-133-LP, Solsep) was in the range from 12 to 20 bar. This pressure is above the working limit of the Vapourtec photoreactor (max. 10 bar). Thus, we decided to perform an “off-line”, continuous catalyst retrieval from the crude reaction mixture obtained from the photoreactor output using a separate syringe pump. Recycling of the G2-PAMAM(Ru)16(PF6)32 catalyst was difficult due to precipitation of the catalyst and clogging of the system during filtration. This problem was not observed for G2-PAMAM(Ru)16Cl32 and the filtration could be successfully carried out. Gratifyingly, ICPMS analysis of the filtrate containing the reaction product revealed that only <1% of the total ruthenium amount had been lost through the filter. Fresh reactants were added to the obtained catalyst solution and processed in the photochemical reactor. Full conversion was again obtained. Unfortunately, when the catalyst was further recycled a drop in catalytic activity of ca. 20% was observed, although significant amounts of ruthenium were not detected in the filtrate (ICPMS). Visual inspection of the inner part of the liquid–liquid separator and the size-exclusion membrane (see Fig. S6†) revealed that the dendrimer catalyst had been partially retained on the membrane surface, possible due to the low solubility of the macromolecular catalyst in the solvent mixtures employed. The decrease in catalytic activity was therefore mainly ascribed to this factor as no ruthenium was found in the filtrate.
The second set of photochemical reactions tested comprised the reductive opening of chalcone-α,β-epoxide 8 and the reduction of 4-chlorophenylazide 9 (Table 2). In both cases the HCOOH/iPr2NEt reagents system was used in the absence of the Hantzsch ester as described by Seeberger.15 Our G2-PAMAM-Ru dendrimer with chloride or hexafluoro-phosphate anions delivered similar catalytic results for both reactions. Excellent conversions to the desired reduced products 10 and 11 were obtained in all cases. The efficiency was similar to that observed for the corresponding catalyst monomers (Table 2, entries 1 and 3) even employing a lower catalyst loading. Hence, anchoring on the dendrimer surface does not diminish the catalytic efficiency.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Conc. [M] | Catalyst | Conv.b (%) |
| a Conditions: 2 mL reaction mixture, 10 equiv. HCOOH, 10 equiv. of DIPEA in MeCN. b Determined by HPLC (215 nm). c Reactions performed with only 0.6 mol% of Ru instead of 1.0 mol%. | ||||
| 1 | 8 | 0.2 | Ru(bpy)2(Me2bpy)(PF6)2 | 98 |
| 2c | 8 | 0.2 | G2-PAMAM(Ru)16(PF6)32 | 99 |
| 3 | 9 | 0.1 | Ru(bpy)2(Me2bpy)(PF6)2 | 99 |
| 4c | 9 | 0.1 | G2-PAMAM(Ru)16(PF6)32 | 99 |
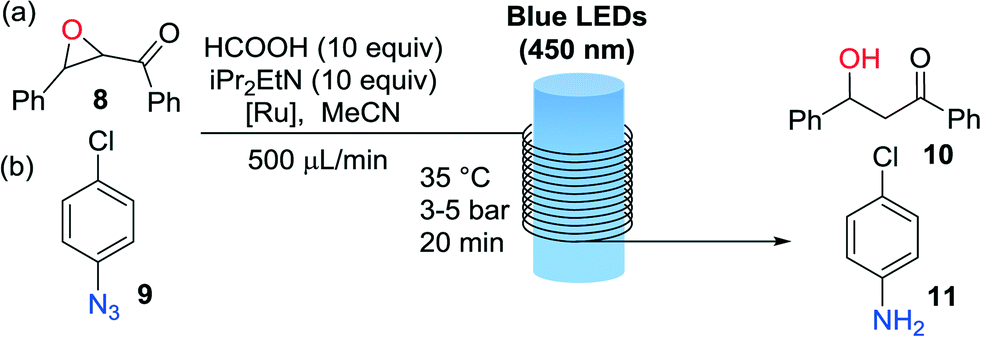
Importantly, after the epoxide and azide reductions the catalyst could be successfully retrieved from the crude reaction mixture but not reused for further catalytic cycles (a significant drop in the reaction conversion of 70% was observed after the second cycle). The decrease in catalytic activity was probably due to degradation of the dendrimeric structure in the presence of the large excess of formic acid. A specific degradation of the G2-PAMAM-Ru structure under irradiation with light cannot be discarded, as processing through the flow reactor of a solution containing only the catalyst showed a decrease in the Ru concentration after each irradiation/nanofiltration cycle. To solve this issue hydrazine hydrate was tested as reducing agent for the transformation of 4-chlorophenyl azide to the aniline (Fig. 3), using methanol as solvent. Light-mediated reduction of nitroaromatics with N2H4·H2O catalyzed by Ru(bpy)3Cl2 had been previously reported,18 but application to the reduction of azides constitutes a new transformation. The reaction was not as efficient as with the HCOOH/iPr2EtN system and around 50% conversion was obtained after a residence time of 20 min in the photoreactor. However, in this case the G2-PAMAM(Ru)16(Cl)32 dendrimer could be recycled 5 times without apparent decrease on the catalytic efficiency.
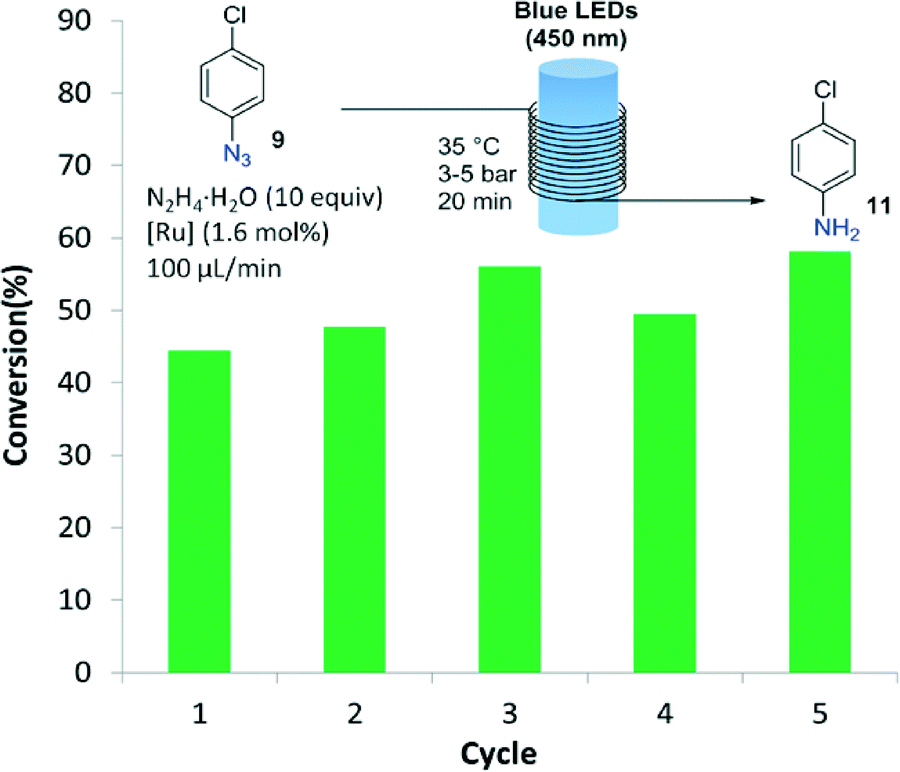 | ||
| Fig. 3 Recyclability study of the G2-PAMAM-Ru catalyst for the reduction of 9. | ||
In summary, we have described a novel macromolecular Ru(bpy)32+ based dedrimeric catalyst having a high efficiency as photoredox catalyst. The catalytic activity has been tested under continuous flow conditions for a set of three model reactions displaying excellent results comparable to the common monomeric Ru(bpy)32+ catalysts. The macromolecular nature of the catalysts permits its facile retrieval and recycling using size-exclusion membrane technology. Recyclability of the catalyst has been successfully proven although decrease in catalytic activity has been observed in some cases. This proof-of-concept study has shown that judicious choice and optimization of solvents and reagents in terms of membrane19 and dendrimer compatibility is important and is the key for the development of fully integrated continuous flow photochemical reactors with inline catalyst recovery.
Notes and references
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152 RSC; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350 CrossRef CAS PubMed.
- M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576 CrossRef CAS PubMed.
- (a) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC; (b) J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- (a) Y. Su, N. J. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562 CrossRef CAS PubMed; (b) Z. J. Garlets, J. D. Nguyen and C. R. J. Stephenson, Isr. J. Chem., 2014, 54, 351 CrossRef CAS PubMed.
- I. V. Gürsel, T. Noël, Q. Wang and V. Hessel, Green Chem., 2015, 17, 2012 RSC.
- A. Kajetanowicz, J. Czaban, G. R. Krishnan, M. Malińska, K. Woźniak, H. Siddique, L. G. Peeva, A. G. Livingston and K. Grela, ChemSusChem, 2013, 6, 182 CrossRef CAS PubMed.
- N. Priyadarshani, Y. Liang, J. Suriboot, H. S. Bazzi and D. E. Bergbreiter, ACS Macro Lett., 2013, 2, 571 CrossRef CAS.
- D. Rackl, P. Kreitmeier and O. Reiser, Green Chem., 2016, 18, 214 RSC.
- J. F. B. Hall, X. Han, M. Poliakoff, R. A. Bourne and M. W. George, Chem. Commun., 2012, 48, 3073 RSC.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Macromolecules, 1986, 19, 2466 CrossRef CAS.
- G. D. Storrier, K. Takada and H. C. D. Abruña, Langmuir, 1999, 15, 872 CrossRef CAS.
- A. Ruggi, C. Beekman, D. Wasserberg, V. Subramaniam, D. N. Reinhoudt, F. W. B. van Leeuwen and A. H. Velders, Chem. – Eur. J., 2011, 17, 464 CrossRef CAS PubMed.
- (a) J. Guerra, A. C. Rodrigo, S. Merino, J. Tejeda, J. C. García-Martínez, P. Sánchez-Verdú, V. Ceña and J. Rodríguez-López, Macromolecules, 2013, 46, 7316 CrossRef CAS; (b) A. C. Rodrigo, I. Rivilla, F. C. Pérez-Martínez, S. Monteagudo, V. Ocaña, J. Guerra, J. C. García-Martínez, S. Merino, P. Sánchez-Verdú, V. Ceña and J. Rodríguez-López, Biomacromolecules, 2011, 12, 1205 CrossRef CAS PubMed.
- F. R. Bou-Hamdan and P. H. Seeberger, Chem. Sci., 2012, 3, 1612 RSC.
- R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801 CrossRef.
- C. Dai, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2011, 3, 140 CrossRef CAS PubMed.
- T. Hirao, J. Shiori and N. Okahata, Bull. Chem. Soc. Jpn., 2004, 77, 1763 CrossRef CAS.
- M. Janssen, C. Muller and D. Vogt, Green Chem., 2011, 13, 2247 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and supplementary figures and tables. See DOI: 10.1039/c6cy00070c |
| This journal is © The Royal Society of Chemistry 2016 |