
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect dehydration of 1,3-butanediol into butadiene over aluminosilicate catalysts†
F.
Jing
ab,
B.
Katryniok
ab,
M.
Araque
ab,
R.
Wojcieszak
a,
M.
Capron
a,
S.
Paul
ab,
M.
Daturi
c,
J.-M.
Clacens
d,
F.
De Campo
d,
A.
Liebens
d,
F.
Dumeignil
ae and
M.
Pera-Titus
*d
aUnité de Catalyse et Chimie du Solide (UCCS, CNRS UMR 8181), Université Lille 1 - Sciences et Technologies, F-59655, Villeneuve d'Ascq Cedex, France
bEcole Centrale de Lille, ECLille, F-59655, Villeneuve d'Ascq, France
cLaboratoire Catalyse et Spectrochimie (LCS), UMR 6506 CNRS – ENSICAEN, F-14000, Caen, France
dEco-Efficient Products and Processes Laboratory (E2P2L), UMI 3464 CNRS – Solvay, 3066 Jin Du Road, Xin Zhuang Ind. Zone, 201108 Shanghai, China
eInstitut Universitaire de France, Maison des Universités, 103 Bd St-Michel, Paris, F-75005, France. E-mail: marc.pera-titus-ext@solvay.com
First published on 5th February 2016
Abstract
The catalytic dehydration of 1,3-butanediol into butadiene was investigated over various aluminosilicates with different SiO2/Al2O3 ratios and pore architectures. A correlation between the catalytic performance and the total number of acid sites and acid strength was established, with a better performance for lower acid site densities as inferred from combined NH3-TPD, pyridine adsorption and 27Al-NMR MAS spectroscopy. The presence of native Brønsted acid sites of medium strength was correlated to the formation of butadiene. A maximum butadiene yield of 60% was achieved at 300 °C over H-ZSM-5 with a SiO2/Al2O3 ratio of 260 with the simultaneous formation of propylene at a BD/propylene selectivity ratio of 2.5. This catalyst further exhibited a slight deactivation during a 102 h run with a decrease in the conversion from 100% to 80% due to coke deposition as evidenced by XPS and TGA-MS, resulting in a 36% loss of the specific surface area.
1. Introduction
1,3-Butadiene (BD) is a relevant industrial intermediate used for the production of synthetic rubbers and resins.1 The BD consumption has significantly increased over the past years, closely related to the rapid growth of the world population. Approximately 95% of the BD world production is obtained as a by-product of steam cracking of naphtha and gas oil for ethylene and propylene production (C4 cut), whereas the remaining 5% is produced via the catalytic oxidative dehydrogenation of n-butane and n-butene.2 The strong dependence on ethylene and propylene production might become problematic in the near future due to the extensive use of shale gas, especially in North America, as less naphtha will be cracked in refineries and thus lower BD volumes will be produced. In a dynamic progress to greener and more sustainable processes,3,4 the development of new bio-derived organic building blocks affording on-purpose BD in a reduced number of catalytic steps might offer an attractive alternative to naphtha cracking.The production of biosourced BD has been reported so far using (bio)ethanol as a raw material.5,6 Bioethanol is one of the most abundant sustainable platform molecules that can be produced by fermentation of sugars.7,8 The synthesis route of BD from ethanol, also known as the Lebedev process, was first reported in 1928, but was not further developed because of the high availability and low cost of naphtha.9 It is commonly accepted that the reaction mechanism in the Lebedev process proceeds first via the generation of acetaldehyde from ethanol dehydrogenation and further aldol condensation into hydroxybutanal (i.e., acetaldol). The latter can undergo reduction with ethanol to generate 1,3-butanediol, the dehydration of which can lead either to crotyl alcohol or 3-butene-1-ol. Both compounds can be further dehydrated into BD. The reaction is usually catalyzed at high temperatures (>400 °C) over MgO/SiO2 catalysts in the presence of promoters (e.g., Zn, Zr, Ag, Cu) including a small amount of strong basic sites (O2−) combined with an intermediate amount of moderate acid (Si–OH) and weak basic sites (Mg–OH).10–16 The maximum BD yield reported so far on MgO/SiO2 formulations is about 50%, but at low BD productivities up to 0.2 gBD gcat−1 h−1 in the presence of Ag and Cu dopants.13 Alternative formulations with balanced acid–base properties such as Ta2O5/SiO2,17–19 Ag/ZrO2/SiO2,20 ZnO/ZrO2/SiO2,21 ZnO/γ-Al2O3,22,23 ZnO/talc,24 Na-doped ZnxZryOz25 and Ag and Zr-doped molecular sieves (e.g., Ag/ZrBEA26) have also been reported, but with modest BD productivities.
Recently, it has been demonstrated that butanediol (BDO) isomers (1,2-, 1,3-, 1,4- and 2,3-) can be obtained in high yields by fermentation of glucose, sucrose, glycerol and mixtures of glucose and xylose,27–32 paving the way to alternative routes to the Lebedev process for bio-BD synthesis. Indeed, the catalytic dehydration and/or dehydrogenation of BDO into BD have been reported so far, but usually offering poor yields and selectivities. On the contrary, a rich yet undesired panel of products can be generated depending on the starting BDO isomer. For instance, 1,2-BDO dehydrogenation over Cu-supported catalysts yields 1-hydroxy-2-butanone with high selectivities.33 In the case of 1,3-BDO, dehydration over CeO2 leads to unsaturated alcohols at moderate to high temperatures,34–36 whereas 1-hydroxy-2-butanone and 3-hydroxybutanal, as well as unsaturated alcohols, are obtained as main products by combined dehydration/dehydrogenation over bifunctional CuO/SiO2 catalysts (Fig. 1).37,38 The synthesis of BD by dehydration of 1,3-BDO has also been attempted over acid catalysts (e.g., SiO2–Al2O3), but affording very low yields (up to 11% at 225 °C39). A few catalysts have been reported for 1,4-BDO and 2,3-BDO dehydration reactions. On the one hand, dehydrogenative cyclization of 1,4-BDO over CuO/SiO2 catalysts yields γ-butyrolactone,40,41 whereas dehydration over CeO2 and ZrO2 or aluminosilicates yields in each case either unsaturated alcohols,42 or a mixture of THF and BD43 (Fig. 1). On the other hand, the catalytic dehydration of 2,3-BDO into methyl ethyl ketone (MEK) and 3-buten-2-ol has been reported over H(B)ZSM-5 zeolites and ZrO2–CaO mixed oxides, respectively, but with negligible BD formation.44–46 In contrast, encouraging BD selectivities up to 42% and >90% at full 2,3-BDO conversion have been recently reported over Al2O3 and Sc2O3 operated at 325 °C and 318 °C, respectively.47,48
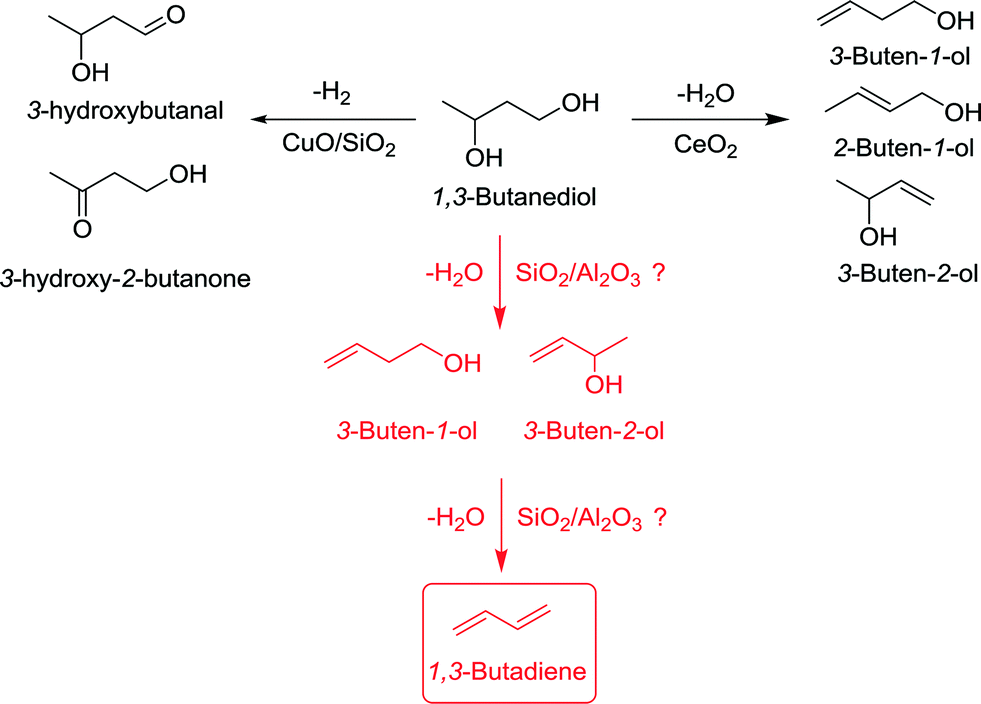 | ||
| Fig. 1 Scheme of the dehydration and dehydrogenation reactions of 1,3-BDO. | ||
Silica–aluminas and acid zeolites have been widely explored for the dehydration of 1- and 2-butanol into butenes, showing the formation of by-products resulting from oligomerization, cracking and aromatization side reactions.49–52 In this study, we investigated the catalytic properties of a series of aluminosilicates with variable textural and surface acid properties for the gas-phase conversion of 1,3-BDO into BD. We show that ZSM-5 zeolites with high SiO2/Al2O3 molar ratios encompassing a low density of Lewis acid sites and the presence of Brønsted acid sites with an intermediate strength can afford the synthesis of BD at moderate-to-high yields and productivities with reasonable catalyst stability against coke formation.
2. Experimental
2.1. Materials
Siral 1 and Siral 40 aluminosilicates with a SiO2/Al2O3 molar ratio of 0.03 and 1.79, respectively, as measured by X-ray fluorescence (XRF) spectroscopy were commercially obtained from SASOL (Table S1†). These catalysts are referred hereinafter as SA@0.03 and SA@1.79, respectively. Three ammonium-substituted ZSM-5 zeolite catalysts (CBV23, CBV55 and CBV280) were supplied by Zeolyst International, whereas a fourth catalyst (MFI90) was purchased from ChemSud. These catalysts are labeled hereinafter as ZSM-5@30 (CBV23), ZSM-5@64 (CBV55), ZSM-5@260 (CBV280) and ZSM-5@106 (MFI90), respectively, where the number in the label refers to the SiO2/Al2O3 molar ratio (Table S1†). The ZSM-5@260 was further subjected to Na-exchange (3 times) at 60 °C for 6 h using an aqueous solution of NaNO3 (1 M), thus obtaining Na-ZSM-5@260. The Al-free silicalite-1 (i.e. ZSM-5@∞) was prepared using the protocol described elsewhere.53 Briefly, 4 mL of TEOS was mixed with 4 mL of an aqueous solution of tetrapropylamine hydroxide (TPAOH, 20 wt%) for 2 h under mild stirring. The mixture was then transferred into a Teflon-lined stainless steel autoclave for hydrothermal crystallization at 80 °C overnight followed by 120 °C for 4 h. The as-synthesized nanocrystals were collected after centrifugation (8000 r.p.m, 10 min), dried at 100 °C for 12 h, and calcined at 550 °C for 5 h under air. Before use, all the ZSM-5 catalysts were activated at 550 °C for 5 h under static air to remove ammonia and/or water.2.2. Synthesis of Al-SBA-15 catalysts
Two Al-doped SBA-15 catalysts with a SiO2/Al2O3 molar ratio of 100 and 200 were synthesized by the evaporation-induced self-assembly (EISA) method.54,55 In a typical preparation, 2.0 g of Pluronic P-123 triblock co-polymer was dissolved in 38 mL of anhydrous ethanol. Then, 4.16 g (0.02 mol) of tetraethylorthosilicate (TEOS), an appropriate amount of aluminium isopropoxide [(CH3)2CHO]3Al and deionized water were added under vigorous stirring. After stirring for 24 h at room temperature, the mixture was poured into a Petri dish to initiate the EISA process. The as-obtained gels were calcined at 550 °C for 6 h under static air with a heating rate of 1 °C min−1. The SiO2/Al2O3 molar ratios of the final catalysts were 102 and 190, respectively, as determined by XRF, which were close to the theoretical values (Table S1†). Both catalysts are denoted hereinafter as Al-SBA-15@102 and Al-SBA-15@190, respectively.2.3. Catalyst characterization
The SiO2/Al2O3 molar ratio of the different aluminosilicates was measured by X-ray Fluorescence Spectroscopy (XRF) on a TORNADO instrument from Bruker.The textural properties of the calcined and spent catalysts were measured from N2 adsorption/desorption isotherms at 77 K using a Micromeritics ASAP 2010 surface area analyzer. The specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) method in the relative pressure range 0.05 < P/P0 < 0.25, while the pore volumes were measured at P/P0 = 0.995. The Barret–Joyner–Halenda (BJH) method was used for measuring inter- and intracrystalline mean pore sizes, whereas the t-plot method was used to measure the micropore surface area. Prior to the measurements, the samples were outgassed at 150 °C for 3 h under vacuum (50 Pa).
Thermogravimetric analysis (TGA) was employed to study the thermal behavior of the spent catalysts. The experiments were performed on a TA 2960 SDT 2960 V3.0F instrument. The samples were heated from room temperature to 700 °C at a rate of 3 °C min−1 under a diluted O2 stream (5 v/v% in He). A mass spectrometer was used to identify the effluent gas. The signal at m/z = 44 was used to track the generation of CO2 during the measurements.
NH3-TPD was used to measure the amount and strength of acid sites in the catalysts. The measurements were performed on a Micromeritics AutoChem II 2920 system equipped with a quartz U-type tubular reactor and a thermal conductivity detector (TCD). A typical test was carried out as follows: 50 mg of catalyst was pre-treated at 250 °C for 2 h under a He flow [30 mL (STP) min−1] to remove adsorbed moisture and vapors, and then cooled down to room temperature. Subsequently, NH3 was chemisorbed at 130 °C using pulsed injections until saturation. The temperature was then increased up to 600 °C at a heating rate of 10 °C min−1 and the TPD profiles were recorded from 130 to 600 °C. The quantification method used for measuring the number of acid sites can be found elsewhere.56,57
In situ pyridine adsorption experiments were carried out using a FT-IR spectrometer from Thermo Nicolet (NEXUS 470) equipped with a KBr beam splitter and a DTGS detector. In a typical test, the sample (∼20 mg) was pressed into a wafer of 16 mm diameter. The wafer was then loaded into the cell and outgassed at 450 °C for 2 h under vacuum. After activation, pyridine (133 Pa) was introduced into the cell at room temperature and the wafer was heated to 150 °C for 15 min to ensure optimal pyridine diffusion. The wafer was then evacuated at 200 °C and the IR spectra were collected every 50 °C until 450 °C using 64 scans with a resolution of 4 cm−1. For comparison, the IR spectra were collected on the parent catalysts before pyridine adsorption. All the spectra were normalized to a constant mass of wafer.
27Al-NMR solid-state experiments were carried out on a DSX Bruker spectrometer operated with a magnetic field strength of 18.8 T and a 27Al Larmor frequency of 208.6 MHz using a 3.2 mm MAS probe head. The 27Al-NMR MAS spectra were recorded at a spinning rate of 20 kHz using a single pulse excitation sequence with a small pulse angle (π = 12°) to ensure a quantitative excitation of central transitions (see ref. 58) and a recycle delay of 4 s. The 27Al chemical shifts were referenced relative to a 1 M Al(NO3)3 aqueous solution.
Powder X-ray diffraction (XRD) patterns were collected on a Bruker D8 Advance diffractometer using a Cu Kα radiation (λ = 1.5418 Å) as the X-ray source. The angle range (10° < 2θ < 80°) was scanned with steps of 0.02° s−1 and a 2 s acquisition time.
X-ray photoelectron spectroscopy (XPS) surface analyses were carried out on a Kratos Axis Ultra DLD spectrometer (Kratos Analytical) equipped with a hemispherical analyzer and a delay line detector. The analyses were performed using Al-Kα radiation (10 mA–15 kV) with a pass energy of 40 eV (0.1 eV per step) for high resolution spectra and a pass energy of 160 eV (1 eV per step) for the survey spectrum in hybrid mode and slot lens mode, respectively. The C-(C,H) component of the C1s peak of adventitious carbon was fixed to 284.5 eV as a reference.
2.4. Catalytic tests
The catalytic dehydration of 1,3-BDO into BD was carried out in a fixed-bed reactor (stainless steel, i.d.: 10 mm, length: 200 mm) under atmospheric pressure. In a typical test, 200 mg of the given catalyst was diluted with 400 mg of SiC (ϕ = 0.21 mm) and loaded into the reactor center to ensure thermal stabilization. The feed reactant (1,3-BDO, 10 wt% in water) was pumped into a vaporization chamber kept at 190 °C with a flow rate of 2.8 mL h−1 (WHSV = 14 h−1) ensuring the absence of external diffusion limitations. The vaporized mixture was then introduced into the reactor under a He flow (60 mL (STP) min−1). The lighter products such as 1,3-BDO and propylene were analyzed online using a GC equipped with two semi-capillary columns (WAX plus: 0.53 mm i.d., 30 m length; and Porapak Q: 0.53 mm i.d., 30 m length) and a flame ionization detector (FID). Heavier products such as methyl ethyl ketone (MEK), methyl vinyl ketone (MVK), 3-buten-1-ol (3B1ol), 3-buten-2-ol (3B2ol), 1-butanol and 2-butanol were condensed in a cold trap and further analyzed using an offline GC-FID equipped with a semi-capillary column (Alltech EC-1000: 0.53 mm i.d., 30 m length). The reaction data were collected after 8 h of stabilization to ensure that the reaction reached the steady state.3. Results
3.1. Screening of catalytic conditions
In the first series of experiments, the dehydration of 1,3-BDO was investigated as a function of the temperature in the range 250–350 °C at a standard space velocity (WHSV = 14 h−1) over 8 different aluminosilicates showing variable textural and surface acid properties (i.e. SA@0.03, SA@1.79, ZSM-5@30, ZSM-5@64, ZSM-5@106, ZSM-5@260, Al-SBA-15@102 and Al-SBA-15@190). In all cases, the main products detected were BD, propylene and 3B1ol, suggesting a reaction mechanism proceeding first by a dehydration step encompassing the formation of 3B1ol and 3B2ol as intermediates followed by two parallel reactions (Fig. 1): (1) dehydration of 3B1ol into BD, and (2) C–C cleavage of either 3B1ol or 3B2ol into propylene. The formation of propylene might further promote the generation of heavy products via oligomerization, which together with BD oligomerization might promote in turn the generation of coke. Minor by-products were also detected including MEK, MVK, 3B2ol, 1-butanol and 2-butanol. Unlike previous studies on 1,3-BDO dehydration over silica–aluminas,39 no ether formation was detected.Fig. S1† plots the main trends obtained for the 1,3-BDO conversion and the BD, propylene and 3B1ol selectivities over the different aluminosilicates, whereas Tables S2–S4† list the BD yields, BD/propylene selectivity ratios and sum of selectivities, respectively. The last parameter provides an indication of the level of coke formation for the different catalysts. Overall, the best catalytic performance was observed in the temperature range 250–300 °C, affording higher BD yields and BD/propylene selectivity ratios at high 1,3-BDO conversions and moderate 3B1ol yields. Also noteworthy, a modest effect of temperature on the BD/propylene selectivity ratios was observed for the different zeolites, especially at high Si/Al ratios, suggesting similar activation energies for BD and propylene generation from 3B1ol. In contrast, for the SA and Al-SBA-15 catalysts, an increase in the temperature favored the generation of propylene at the expense of BD. This observation suggests a higher activation energy for propylene generation from 3B1ol for these series of catalysts.
The body of results obtained during the screening tests confirms that the WHSV chosen was adapted to ensure the formation of BD while mitigating the formation of propylene and intermediate 3B1ol, as well as coking (see section 3.4). Accordingly, no further optimization of the WHSV was carried out.
3.2. Catalytic performance (250 and 300 °C)
Table 1 lists the conversion and selectivities to the main products obtained for the different catalysts at 300 °C. Except ZSM-5@30 with a 1,3-BDO conversion of 90%, almost full conversion was achieved over all the other catalysts. A very low BD selectivity of 3.7% and high propylene selectivity (58%) were obtained for SA@0.03 indicating a superior cracking capacity. Upon increase of the SiO2/Al2O3 molar ratio (i.e. for SA@1.79), the cracking reaction appears to be less favored with a shift of the BD selectivity (33%). Unlike the SA catalysts, ZSM-5 zeolites showed much higher BD selectivities, especially in the case of ZSM-5@260 (SiO2/Al2O3 = 260) with a BD selectivity (and yield) as high as 60%. Significant amounts of the 3B1ol intermediate were also detected for all ZSM-5 catalysts, which together with propylene constituted the two main by-products. Interestingly, control experiments on Na-exchanged ZSM-5@260 (i.e. Na-ZSM-5@260) and pure silicalite-1 (i.e. ZSM-5@∞ or SIL-1) revealed in both cases much lower 1,3-BDO conversions (<38%), lower BD selectivities (46% and 36% for Na-ZSM-5@260 and ZSM-5@∞, respectively) and a higher selectivity to propylene (ca. 40% for both catalysts). Finally, the Al-SBA-15 family also showed a high BD selectivity (>50%), but exhibited a higher propylene selectivity (>39%) and a lower 3B1ol selectivity (<1%) compared to ZSM-5@260.| Sample | 1,3-BDO conversion (%) | Selectivitya (%) | BD productivity (gBD gcat−1 h−1) | Carbon balance (—) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MEK | MVK | 1Bol | 2Bol | 3B1ol | 3B2ol | Propylene | BD | ||||
| Reaction conditions: T, 300 °C; ambient pressure, time on stream, 8 h; catalyst loading, 200 mg; liquid flowrate, 2.8 mL h−1; carrier gas flowrate, 60 mL (STP) min−1.a Nomenclature: MEK, methyl ethyl ketone; MVK, methyl vinyl ketone; 1Bol, 1-butanol; 2Bol, 2-butanol; 3B1ol, 3-buten-1-ol; 3B2ol, 3-buten-2-ol. | |||||||||||
| SA@0.03 | >95 | 1.1 | 8.3 | — | 1.7 | 7.3 | 0.7 | 58 | 3.7 | 0.45 | 0.87 |
| SA@1.79 | >95 | 2.5 | 1.9 | — | — | — | — | 35 | 33 | 2.8 | 0.86 |
| ZSM-5@30 | 90 | 2.1 | 0.7 | 0.7 | 0.3 | 12 | 2.5 | 25 | 40 | 3.0 | 0.85 |
| ZSM-5@64 | >95 | 2.0 | 1.2 | 1.2 | 0.4 | 10 | 2.6 | 30 | 43 | 3.6 | 0.88 |
| ZSM-5@106 | >95 | 8.2 | 1.2 | 0.8 | 0.4 | 9.4 | 2.6 | 23 | 45 | 3.8 | 0.89 |
| ZSM-5@260 | >95 | 1.8 | 1.3 | 1.0 | 0.3 | 8.7 | 2.3 | 24 | 60 | 5.1 | 0.97 |
| Na-ZSM-5@260 | 36 | Trace | Trace | Trace | Trace | Trace | Trace | 43 | 46 | 1.4 | 0.96 |
| ZSM-5@∞ (SIL-1) | 38 | Trace | Trace | Trace | Trace | Trace | Trace | 41 | 36 | 1.2 | 0.91 |
| Al-SBA-15@102 | >95 | 2.1 | 0.1 | 1.1 | 2.2 | 0.6 | — | 39 | 53 | 4.5 | 0.97 |
| Al-SBA-15@190 | >95 | 2.3 | 0.3 | 1.2 | 0.2 | 0.3 | — | 39 | 51 | 4.3 | 0.97 |
The catalytic tests carried out at 250 °C reflected in all cases lower 1,3-BDO conversions (Table S5†). SA@0.03 and SA@1.79 showed 1,3-BDO conversions of 77% and 88%, respectively, with a variable product distribution. SA@1.79 displayed a much higher production of BD and the 3B1ol intermediate than SA@0.03 (33% vs. 14% and 14% vs. 6.8%), whereas the latter catalyst exhibited a higher selectivity to propylene (34% vs. 17%). In the case of the ZSM-5 catalysts, the 1,3-BDO conversion and BD selectivity were enhanced from 75% to 92% and from 39% to 47%, respectively, when increasing the SiO2/Al2O3 molar ratio from 30 to 260. In contrast, the 3B1ol and propylene selectivities showed increasing and decreasing trends with the SiO2/Al2O3 ratio, respectively, until a ratio of 106, whereas the opposite trend was observed at higher ratios. Finally, Al-SBA-15@102 and Al-SBA-15@190 displayed very similar catalytic properties, achieving full 1,3-BDO conversion and BD, 3B1ol and propylene selectivities of 57%, 4.9–6.0% and 24%, respectively.
3.3. Catalytic stability over ZSM-5@260
From the series above, ZSM-5@260 revealed the most promising catalytic performance for 1,3-BDO dehydration into BD at 300 °C. Accordingly, a long-term stability test was conducted on this catalyst under the very same reaction conditions (Fig. 2). The catalyst exhibited almost full conversion during the initial 24 h and a stable BD selectivity of 60%. After this period, the catalyst displayed a continuous but slow deactivation trend with a drop of the 1,3-BDO conversion to 80% after 102 h of reaction, but keeping the BD selectivity almost unchanged. This observation suggests that the deactivation process did not alter the reaction mechanism, this being most likely ascribed to a lower accessibility to the acid sites.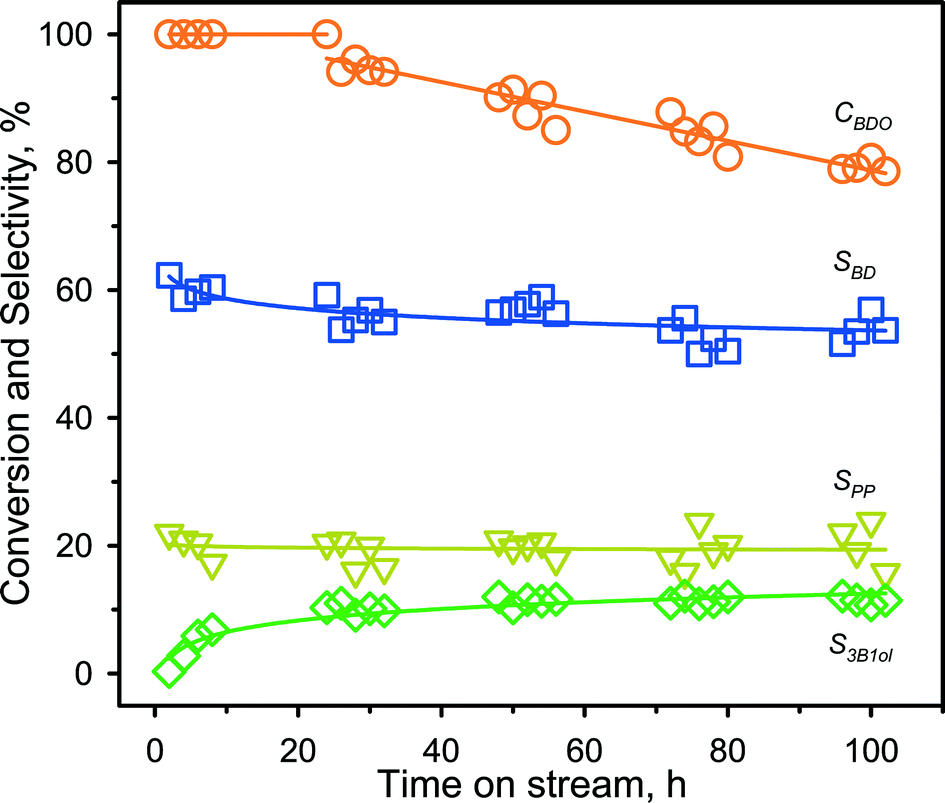 | ||
| Fig. 2 Catalytic stability over ZSM-5@260 at 300 °C. Reaction conditions: T, 300 °C; ambient pressure, catalyst loading, 200 mg; liquid flowrate, 2.8 mL h−1; carrier gas flowrate, 60 mL (STP) min−1. Nomenclature: CBDO, 1,3-BDO conversion; SBD, selectivity to BD; SPP, selectivity to PP; S3B1ol, selectivity to 3-buten-1-ol. | ||
3.4. Structural evolution during reaction
To gain insight into the deactivation mechanism, TGA-MS analyses were performed on the spent ZSM-5@260 catalyst after 102 h of reaction at 300 °C (denoted hereinafter as ZSM-5@260_LR). The TGA profiles of this catalyst showed a continuous weight loss from 330 °C to 700 °C (Fig. 3). Combining this technique with MS, carbon combustion appears to be responsible for the weight loss, leading to the formation of CO2 (Fig. S2†). The temperature range for carbon combustion shifted to lower temperatures (ca. 40 °C) for the spent ZSM-5@260 catalyst after only 8 h on stream (Table 2). In line with reported studies on dehydration reactions,59–61 this observation supports the formation of heavier carbonaceous species over the catalyst surface after long-term reaction (102 h). Furthermore, the amount of carbon deposited on the catalyst increased significantly after the long-term reaction with a value of 65 mgC g−1, which was ca. 1.3 times higher than the value measured after 8 h of reaction (Table 2 and Fig. 3). Interestingly, while a similar thermal behavior was observed for ZSM-5@64, the latter catalyst showed the formation of a much higher amount of carbon deposits just after 8 h of reaction (103 mgC g−1, Table 2 and Fig. S2†).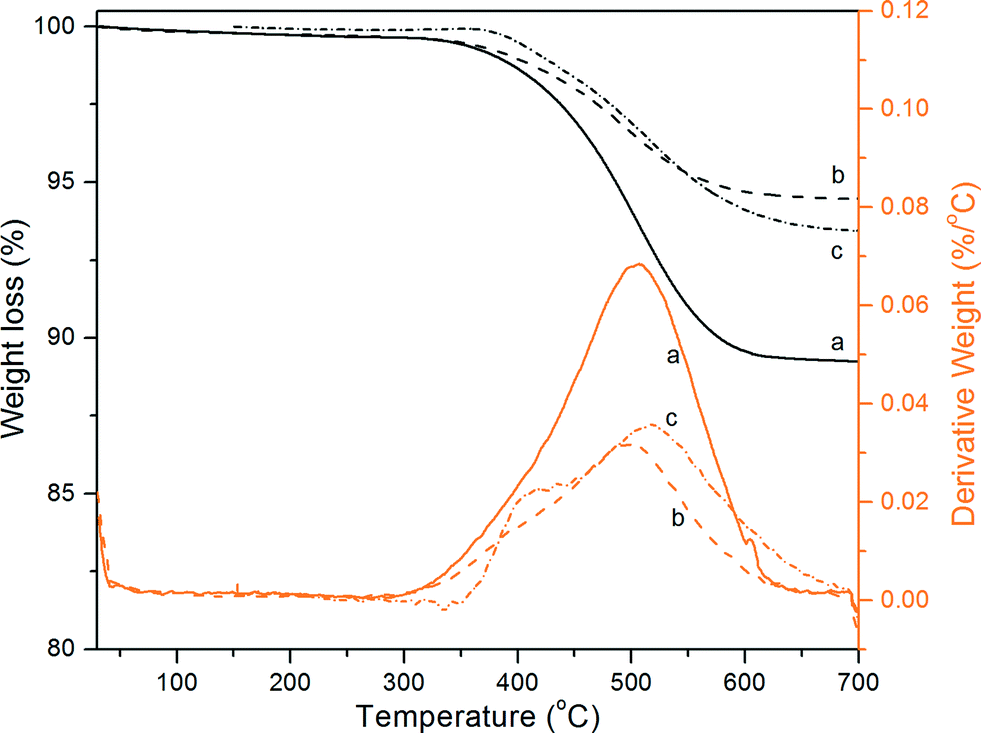 | ||
| Fig. 3 Thermogravimetric analysis (TG: black, DTG, orange) of the spent ZSM-5 catalysts after reaction at 300 °C: (a) ZSM-5@64, (b) ZSM-5@260, and (c) ZSM-5@260_LR. | ||
Further XPS analyses were conducted on the fresh ZSM-5@260 and ZSM-5@64 catalysts after 8 h of reaction. The C 1s peaks clearly supported the formation of coke for both catalysts after the reaction (Fig. S3†). As a matter of fact, the surface carbon concentration (Table 3) showed a 6-fold increase for ZSM-5@64 (18.7 atom% vs. 3.7 atom%; 12.3 wt% vs. 2.3 wt%), whereas a 2-fold increase was only observed for ZSM-5@260 (9.5 atom% vs. 3.2 atom%; 6.0 wt% vs. 2.0 wt%). In all cases, the zeolite structure did not reveal any visible modification by comparison of the XRD patterns (Fig. S4†), the 27Al-NMR MAS spectra (Fig. S5†), and the Si/Al surface ratios (Table 3) measured on the corresponding fresh and spent catalysts.
| Sample | Molar composition (Si/Al/O/C) | Weight composition (Si/Al/O/C) | Si/Al ratio (molar) | Variation C (%) |
|---|---|---|---|---|
| ZSM-5@64 | 28.4/1.4/66.5/3.7 | 41.0/1.9/54.7/2.3 | 41 | — |
| ZSM-5@64_LR | 24.2/1.1/56.0/18.7 | 37.1/1.6/49.0/12.3 | 44 | +430% |
| ZSM-5@260 | 30.1/0.3/66.3/3.2 | 43.3/0.4/54.3/2.0 | 201 | — |
| ZSM-5@260_LR | 28.6/0.3/61.5/9.5 | 42.1/0.4/51.5/6.0 | 191 | +200% |
Also noteworthy, the 27Al-NMR MAS spectrum of the fresh ZSM5@64 catalyst consisted of 2 main peaks centered around 55 ppm which could be fitted to two components located at 56.4 and 54.3 ppm, indicative of fourfold coordinated framework Al atoms in the zeolite framework. The difference between these two sites could be due to a small change in terms of the O–Al–O angles and/or Al–O distances in the Al(O)4 tetrahedra. A second peak was visualized at ca. 0 ppm that is a characteristic of ex-framework Al (sixfold coordinated). A similar structural organization was observed on the spent catalyst after 102 h of reaction at 300 °C (i.e. ZSM5@64 _LR). However, the former peak corresponding to the sixfold coordinated Al was split into two components reflecting the presence of both sixfold and fivefold coordinated Al species.
The spectrum of the fresh ZSM5@260 catalyst also consisted of 2 main peaks centered at around 55 ppm which could be fitted to the same two components similar to the former catalyst (53.7 and 51.8 ppm), also indicative of fourfold coordinated framework Al atoms. A shift of the two initial peaks was observed on the spent catalyst, which can be interpreted by a reorganization of the structure during the reaction. A very low fraction of sixfold coordinated Al was observed on the fresh catalyst (ca. 7%), whereas within the limits of the experimental error, no ex-framework Al was detected on the spent catalyst after 102 h of reaction at 300 °C (i.e. ZSM5@260_LR).
3.5. Textural properties of the fresh and spent catalysts
Table 4 compiles the textural properties of the fresh and spent aluminosilicates after 8 h of reaction at 300 °C measured from N2 adsorption/desorption at 77 K, whereas Fig. S6 and S7† plot the corresponding isotherms and pore size distributions (BJH). The specific surface area of the parent SA catalysts exhibited a marked increase from 207 to 405 m2 g−1 when tuning the SiO2/Al2O3 molar ratio from 0.03 to 1.79, but keeping in both cases a low contribution of micropores (5% and 1% for SA@0.03 and SA@1.79, respectively). The broad hysteresis loop between the adsorption and desorption curves observed for both catalysts (Fig. S6a,† left) suggests the presence of large and non-uniform interparticle mesopores with a broad pore size distribution between 2 and 40 nm centered at about 8 nm (Fig. S6a,† right).| Sample | Before reaction (0) | After reaction (A) | Difference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| S BET (m2 g−1) | V p (cm3 g−1) | D p (nm) | S BET (m2 g−1) | V p (cm3 g−1) | D p (nm) | ΔSBETa (%) | ΔVpb (%) | ΔDpc (%) | |
| a In parentheses, micropore area. ΔSBET = (SBET,0 − SBET,A) × 100/SBET,0. b Total pore volume measured at P/P0 = 0.995. ΔVp = (Vp,0 − Vp,A) × 100/Vp,0. c Measured from the N2 desorption curve by the BJH method. ΔDp = (Dp,0 − Dp,A) × 100/Dp,0. | |||||||||
| SA@0.03 | 207 (12) | 0.57 | 7.9 | 184 (<5) | 0.40 | 6.9 | 12 (58) | 30 | 13 |
| SA@1.79 | 405 (<5) | 0.82 | 6.7 | 216 (20) | 0.51 | 8.3 | 47 (−300) | 38 | −24 |
| ZSM-5@30 | 199 (145) | 0.06 | 5.1 | 20 (<5) | 0.04 | 7.6 | 90 (97) | 33 | −49 |
| ZSM-5@64 | 334 (216) | 0.15 | 5.9 | 24 (<5) | 0.08 | 12.5 | 93 (98) | 47 | −112 |
| ZSM-5@106 | 383 (203) | 0.34 | 11.3 | 109 (22) | 0.43 | 20.1 | 72 (94) | −26 | −78 |
| ZSM-5@260 | 366 (231) | 0.06 | 4.0 | 233 (97) | 0.04 | 3.5 | 36 (58) | 33 | 12.5 |
| Al-SBA-15@102 | 611 (122) | 0.52 | 3.6 | 339 (18) | 0.36 | 3.5 | 45 (85) | 31 | 3.3 |
| Al-SBA-15@190 | 630 (123) | 0.61 | 3.6 | 337 (27) | 0.34 | 3.3 | 47 (78) | 44 | 7.5 |
The ZSM-5 catalysts displayed a volcano trend when representing the specific surface area against the SiO2/Al2O3 molar ratio with a maximum for ZSM-5@106. The micropore surface area showed the opposite trend with a contribution to the overall specific surface area of 73% for ZSM-5@30, 65% for ZSM-5@64 and 63% for ZSM-5@260. Nonetheless, for ZSM-5@106, the contribution of the micropore surface area was only of 53%, which was probably caused by the rather complex porosity of this catalyst as suggested by the isotherm shape (Fig. S6b,† left) and pore size distribution (Fig. S6b,† right). The adsorption branch of the ZSM-5@106 isotherm could be assigned to Type II, reflecting the presence of macropores (Dp > 50 nm), while the type H4 hysteresis loop also indicated the presence of mesopores. Two families of mesopores could be discerned: (1) mesopores between 3 and 7 nm, and (2) mesopores combined with macropores with a wide pore size distribution between 7 and 100 nm. Unlike the other ZSM-5 catalysts, the complex pore system in ZSM-5@106 also led to higher values for both the pore volume (0.34 cm3 g−1) and the mean pore size (11.3 nm).
The parent Al-SBA-15 catalysts showed a slight increase of the specific surface area from 611 to 630 m2 g−1 when raising the SiO2/Al2O3 ratio from 102 to 190 with a micropore contribution of ca. 20% to the overall specific surface area. Both catalysts showed a hysteresis loop in the P/P0 range between 0.4 and 0.65 (Fig. S6c,† left), reflecting a narrower pore size distribution between 3 and 6 nm centered at about 4 nm (Fig. S6c,† right).
Although the textural properties of the catalysts were considerably altered after the reaction due to the formation of carbon deposits (Table 4), the main porous features of the catalysts were preserved without any qualitative change on the shape of the isotherms (Fig. S7†). The specific surface area showed a marked decrease by 90% for ZSM-5@30 and ZSM-5@64 and by 45% and 47% for Al-SBA-15@102 and Al-SBA-15@190, respectively, whereas the specific surface area for SA@0.03 and SA@1.79 only showed a moderate decline of 12% and 47%, respectively. Noticeably, ZSM-5@30, ZSM-5@64 and ZSM-5@106 exhibited a pronounced decline of the micropore surface area (>94%), whereas this decline was more moderate for ZSM-5@260 (58%). The lower reduction of the specific surface area for the latter catalyst is consistent with a smaller formation of carbon deposits during the reaction. All the catalysts showed a decline of the pore volume after the reaction except for ZSM-5@106, for which the pore volume increased from 0.34 to 0.43 cm3 g−1, suggesting the generation of new porosity. In the meantime, the different catalysts displayed an increase of the mean mesopore size except for SA@0.03, showing only a slight pore contraction (7.9 nm vs. 6.9 nm), and for ZSM-5@260, Al-SBA-15@102 and Al-SBA-15@190, the mean pore size keeping essentially unchanged.
3.6. Surface acidity of the catalysts
Fig. 4 plots the NH3-TPD desorption profiles in the temperature range 130–600 °C for the different aluminosilicates. It is generally accepted that NH3 is a good probe molecule for titrating acid hydroxyl groups accessible through pores, channels or windows with a size <4 Å in aluminosilicates and zeolites because of its small molecular dimensions (3.70 × 3.99 × 3.11 Å3).62,63 In this study, the acid site strength was broadly classified in three groups after peak deconvolution according to the NH3 desorption temperature (Fig. S8†): (1) weak sites between 130 °C and 280 °C, (2) medium sites between 280 °C and 450 °C, and (3) strong sites between 450 °C and 600 °C. All the catalysts contained three types of sites except ZSM-5@106, which did not contain strong acid sites. In the case of ZSM-5 catalysts, the maximum peak position shifted to lower temperatures at higher SiO2/Al2O3 ratios in both the low- and high-temperature regions, reflecting a decrease of the acid site strength.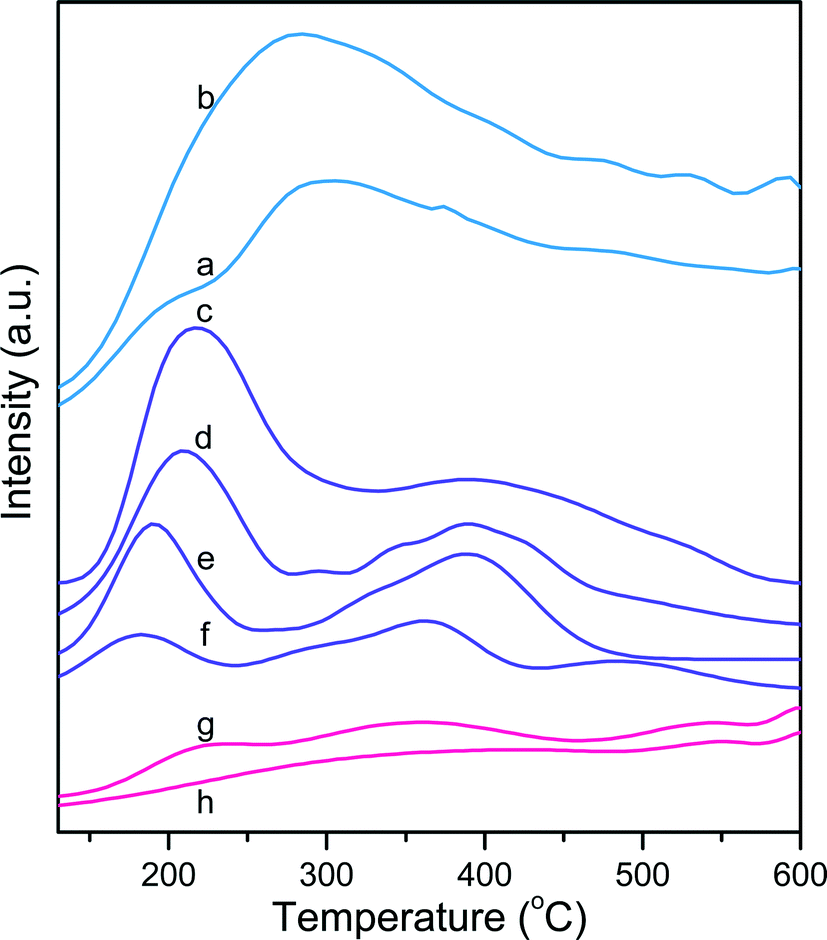 | ||
| Fig. 4 NH3-TPD curves of the fresh aluminosilicate catalysts: (a) SA@0.03, (b) SA@1.79, (c) ZSM-5@30, (d) ZSM-5@64, (e) ZSM-5@106, (f) ZSM-5@260, (g) Al-SBA-15@102 and (h) Al-SBA-15@190. | ||
Table 5 summarizes the surface acidities measured for the different sites in the different aluminosilicates. The highest acidity was observed for SA@0.03 and SA@1.79 with a noticeably larger amount of medium and strong acid sites compared to the other catalysts. The ZSM-5 family showed predominantly weak to medium acidity with the total acidity decreasing from 0.88 to 0.26 mmol g−1 with an increase of the SiO2/Al2O3 ratio from 30 to 260. Similarly, for the Al-SBA-15 catalysts, the total acidity decreased with the SiO2/Al2O3 ratio. However, in these catalysts, medium and strong sites were predominant. Noteworthy, irrespective of the catalyst family, the NH3 uptake increased inversely with the SiO2/Al2O3 ratio (Fig. 5). The NH3 uptake decreased sharply until the threshold SiO2/Al2O3 ratio of 64, whereas this decline was much less pronounced at higher ratios.
| Sample | Acidity, NH3 uptakea (mmol g−1) | Total acidity (NH3 uptake)b (mmol g−1) | B/L (pyr) | AlF (atoms/uc) | AlEFc (atoms/uc) | %Bd (weak) (NH3 uptake) | ||
|---|---|---|---|---|---|---|---|---|
| 130–280 °C (weak) | 280–450 °C (medium) | 450–600 °C (strong) | ||||||
| a In parentheses, percentage over the total acidity. b In parentheses, μmol m−2. c Measured from the B/L ratios (in parentheses, values measured from 27Al-NMR MAS). d Number of weak Brønsted acid centers measured from the B/L ratios and the density of ex-framework Al (AlEF). | ||||||||
| SA@0.03 | 0.26 (17.3%) | 0.58 (38.7%) | 0.66 (44.0%) | 1.50 (7.14) | 0.0 | — | — | |
| SA@1.79 | 0.71 (30.1%) | 0.73 (30.9%) | 0.92 (39.0%) | 2.36 (5.83) | 0.06 | — | — | |
| ZSM-5@30 | 0.41 (46.6%) | 0.34 (38.6%) | 0.13 (14.8%) | 0.88 (4.42) | 1.25 | 2.95 (—) | 2.35 (—) | 4.6% |
| ZSM-5@64 | 0.24 (43.6%) | 0.20 (36.4%) | 0.11 (20.0%) | 0.55 (1.65) | 3.28 | 2.40 (2.10) | 0.51 (0.81) | 46% |
| ZSM-5@106 | 0.25 (59.5%) | 0.17 (40.5%) | — (0%) | 0.42 (1.10) | 1.94 | 1.00 (—) | 0.78 (—) | 48% |
| ZSM-5@260 | 0.06 (23.1%) | 0.16 (61.5%) | 0.04 (15.4%) | 0.26 (0.71) | 6.16 | 0.68 (0.61) | 0.05 (0.12) | 40% |
| Al-SBA-15@102 | 0.10 (17.5%) | 0.16 (28.1%) | 0.31 (54.4%) | 0.57 (0.93) | 0.27 | — | — | — |
| Al-SBA-15@190 | 0.02 (4.4%) | 0.13 (28.9%) | 0.30 (66.7%) | 0.45 (0.71) | 0.38 | — | — | — |
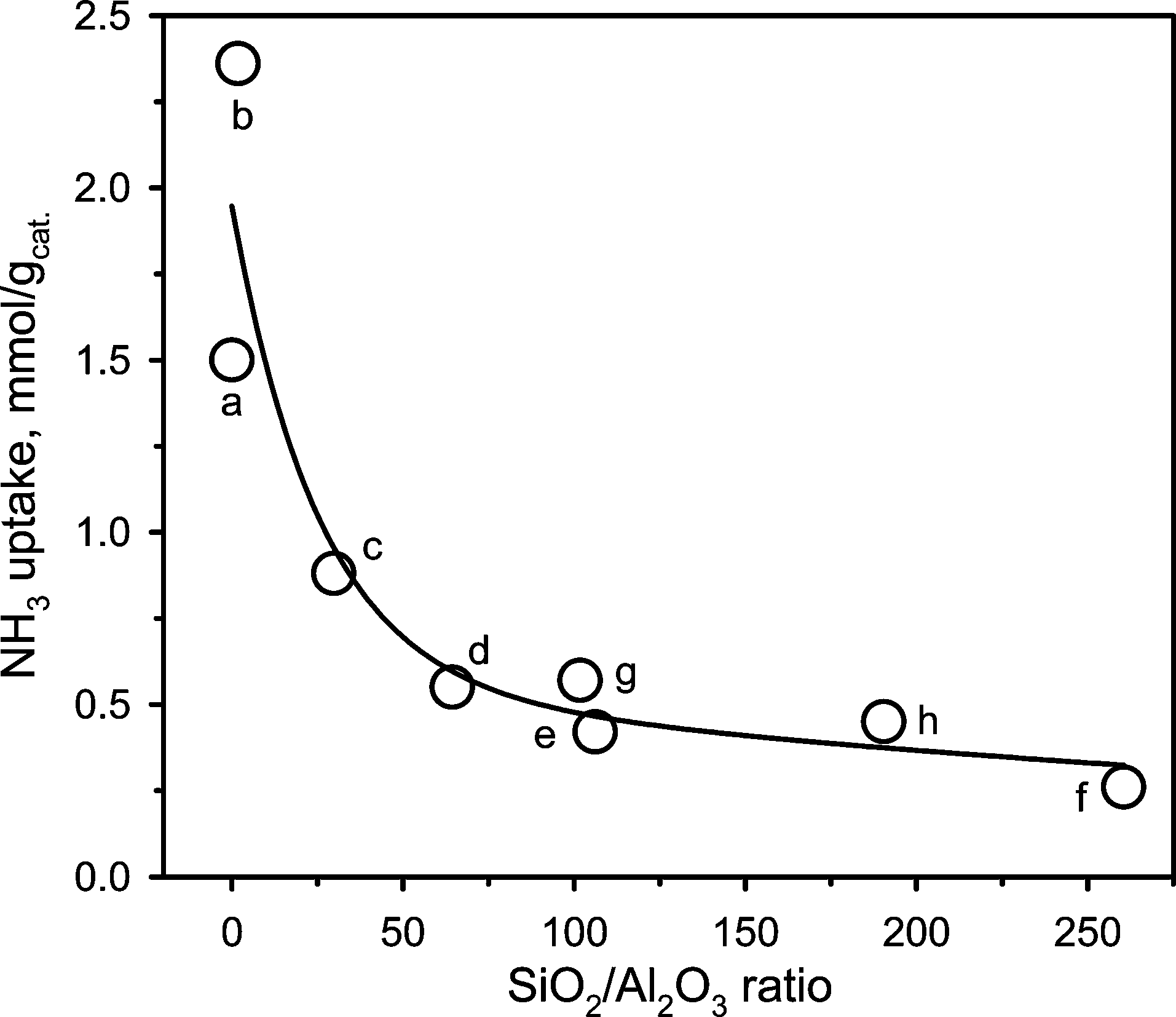 | ||
| Fig. 5 Evolution of the total acidity on the fresh aluminosilicate catalysts measured by NH3-TPD as a function of the SiO2/Al2O3 ratio: (a) SA@0.03, (b) SA@1.79, (c) ZSM-5@30, (d) ZSM-5@64, (e) ZSM-5@106, (f) ZSM-5@260, (g) Al-SBA-15@102 and (h) Al-SBA-15@190. | ||
Although NH3-TPD is a relevant tool for measuring the amount and strength of acid sites in aluminosilicates, this technique cannot provide direct information on their Brønsted (B) or Lewis (L) nature. FT-IR spectroscopy in combination with a specific probe molecule such as pyridine offers a powerful tool for discriminating the nature of acid sites. The characteristic band at around 1545 cm−1 can be attributed to the formation of pyridinium ions by interaction of pyridine with the Brønsted acid sites (e.g., bridged Si–OH–Al centers), while the band at around 1455 cm−1 originates from the interaction between pyridine and the Lewis acid sites.64,65 Furthermore, the comparison of the IR spectra in the stretching region (i.e. >3000 cm−1) before and after pyridine adsorption can provide valuable information about the Brønsted acidity of hydroxyl groups and their accessibility to pyridine. As a matter of fact, hydroxyl groups remaining unaffected after pyridine adsorption can be regarded as weak acid sites belonging most often to silanol groups reminiscent of the tetrapropylamine (TPA) template after calcination/extraction.
Fig. 6 collects the IR spectra measured on the different catalysts before and after pyridine adsorption, whereas Table 5 lists the measured B/L ratios after peak integration. The SA catalysts displayed an intense band at 1456 cm−1 irrespective of the SiO2/Al2O3 ratio, reflecting a large proportion of Lewis acid sites (B/L < 0.1). In contrast, the ZSM-5 and Al-SBA-15 families showed the simultaneous presence of Lewis and Brønsted acid sites, but with variable proportion. In the case of ZSM-5 catalysts, the intensity of the Lewis acid band at 1456 cm−1 weakened and became broader with an increase of the SiO2/Al2O3 ratio, whereas the Brønsted acid band at 1547 cm−1 showed the opposite trend. As a result, the B/L ratio was enhanced from 1.25 to 6.16 with an increase of the SiO2/Al2O3 ratio from 30 to 260. Similarly, the intensity of the Lewis acid band decreased remarkably when the SiO2/Al2O3 ratio doubled up to 200 for the Al-SBA-15 catalysts, whereas the Brønsted acid band kept essentially unchanged. As a result, the B/L ratios on these latter samples were significantly lower with values of about 0.27 and 0.38 for Al-SBA-15@102 and Al-SBA-15@190, respectively.
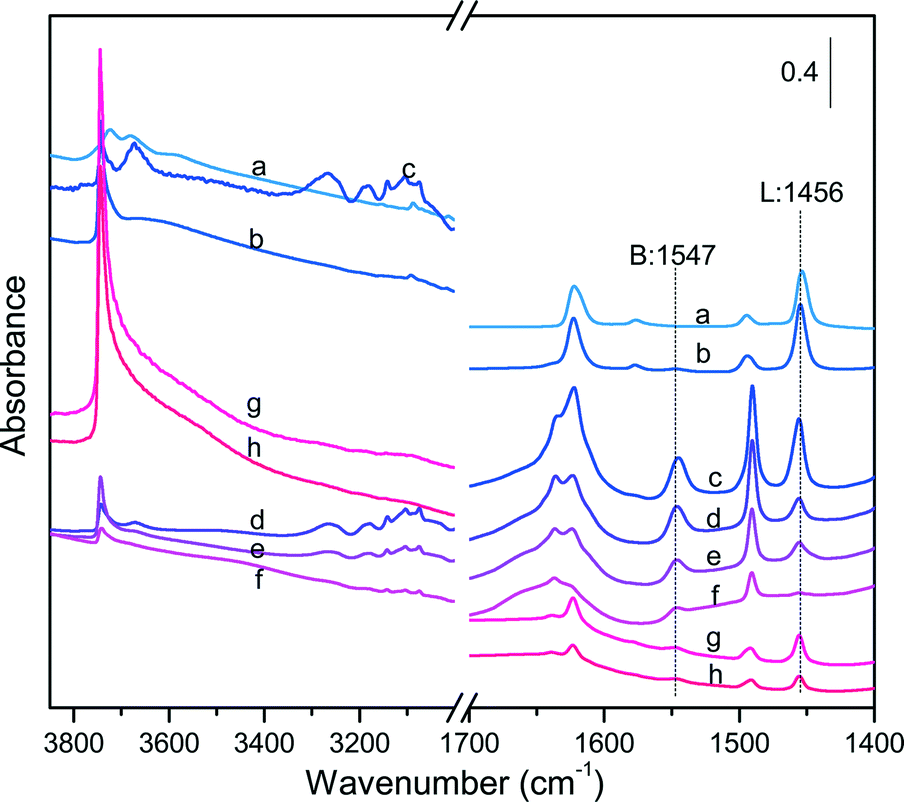 | ||
| Fig. 6 FT-IR spectra of pyridine evacuated at 200 °C on the fresh aluminosilicate catalysts: (a) SA@0.03, (b) SA@1.79, (c) ZSM-5@30, (d) ZSM-5@64, (e) ZSM-5@106, (f) ZSM-5@260, (g) Al-SBA-15@102 and (h) Al-SBA-15@190. The labels ‘B’ and ‘L’ refer to Brønsted and Lewis acid sites, respectively. | ||
All the catalysts exhibited resilient bands in the stretching region after pyridine adsorption that can be attributed to weakly acidic hydroxyl groups (Fig. S9†). In particular, the catalysts with a larger density of Lewis acid centers exhibited more pronounced bands in the stretching region, especially SA@1.79, Al-SBA-15, SBA-15@102 and Al-SBA-15@190, showing a unique and large band centered at 3745 cm−1. This band was also observed for SA@0.03 and the ZSM-5 catalysts, but with a much lower intensity. An additional band centered at 3670 cm−1 was visualized for SA@0.03 and ZSM-5@30 after pyridine adsorption, which can be attributed to weakly acidic hydroxyl groups on ex-framework alumina.
4. Discussion: descriptor of the catalytic activity in ZSM-5 zeolites
As pointed out above, the type of acid sites (Brønsted and Lewis), as well as their concentration and strength, plays a crucial role in the catalytic properties of aluminosilicates for dehydrating 1,3-BDO into BD. Unlike mesoporous SA and Al-SBA-15, where Lewis acid sites are dominant in the fresh catalysts (>70%) and might display a variable strength, Brønsted acidity apparently rules the catalytic properties of ZSM-5 catalysts, especially at higher SiO2/Al2O3 ratios. Direct evidence supporting such a proposition can be found when assessing the catalytic activity of Na-ZSM-5@260 and pure silicalite-1 (i.e. ZSM-5@∞) at 300 °C (Table 1), showing in both cases a drastic decrease of the catalytic activity compared to the parent ZSM-5@260. These results suggest that neither Lewis acidity nor weak Brønsted acidity attributed to silanol groups can catalyze efficiently 1,3-BDO dehydration into BD unless stronger Brønsted acid sites are introduced into the framework.A refined analysis of the acid site nature and strength distribution in the ZSM-5 series was carried out by combining the data obtained by NH3-TPD and pyridine adsorption (Table 5). Using this body of data and taking into account the molecular formula of ZSM-5 (i.e. [Hx(H2O)16][AlxSi96−xO192] for x < 27), the density of framework and ex-framework Al (i.e. AlF and AlEF, respectively) was estimated as a function of the SiO2/Al2O3 ratio. In this analysis, we assumed that all the hydroxyl groups that can be titrated by NH3 in the NH3-TPD tests corresponded to bridged Si–OH–Al sites, but not to the weakly acidic silanol groups (see Table 5 for details). Our calculations reflect a decline of the relative amount of framework Al with the SiO2/Al2O3 ratio with atomic ratios evolving from 56% in ZSM-5@30 to >96% for ZSM-5@260. The validity of this method was assessed by quantitative 27Al-NMR MAS for the ZSM5@64 and ZSM5@260 catalysts by comparing the relative areas between the peaks centered at ca. 55 ppm (fourfold coordinated Al) and at ca. 0 ppm (sixfold coordinated Al) in the spectra (Fig. S5†).
The density of framework and ex-framework Al was further used to estimate the number of Brønsted and Lewis acid sites in the zeolites. It is well established that the low-temperature band (130–280 °C) in the NH3-TPD profiles of ZSM-5 zeolites can be attributed to Lewis acid sites,66 whereas the medium- and high-temperature bands (280–450 °C and 450–600 °C, respectively) can be attributed to Brønsted acid sites.67 With this information in hand, our calculations reflect a relative amount of weak Brønsted acid sites increasing from 4.6% for ZSM-5@30 to 40–48% for ZSM-5@64, ZSM-5@106 and ZSM-5@260. For the latter catalysts, Lewis acid sites might be partially segregated near the external surface of the particles as inferred from the slightly lower Si/Al surface ratio (200) compared to the Si/Al bulk ratio (260) observed on this catalyst (Tables 3 and S1†).
The presence of a higher proportion of Brønsted acid sites in ZSM-5 zeolites with higher SiO2/Al2O3 ratios appears to be at the origin of the higher BD selectivity upon 1,3-BDO dehydration and higher capacity for limiting the cracking reaction. In particular, ZSM-5@260 with the largest amount of Brønsted acid sites with intermediate strength (61%, Table 5) showed the largest BD selectivity at 300 °C. These observations agree well with the conclusions drawn from previous studies on 1-butanol dehydration over acid zeolites, suggesting the major contribution of an E2-type elimination mechanism, but with no ether formation as an intermediate.49,68 In parallel, the lower amount of strong Brønsted acid sites allowed a lower coke formation during the reaction, affording a moderate decrease of the specific surface area (Table 4). Unlike Al-enriched ZSM-5 catalysts, the larger available specific surface area for ZSM-5@260 during operation might favor the accessibility of 1,3-BDO into the internal Brønsted acid sites.
5. Conclusions
A series of aluminosilicates with different porous features and SiO2/Al2O3 ratios were prepared and tested for the dehydration of 1,3-butanediol into butadiene. All the catalysts offered good butadiene yields with the simultaneous formation of lower amounts of unsaturated alcohols such as 3B1ol and propylene, the latter being supposedly issued from the cracking of 3B1ol. Among the different aluminosilicates, acid ZSM-5 zeolites revealed the most promising catalytic properties, showing a higher selectivity to butadiene at higher SiO2/Al2O3 ratios (from 40% for ZSM-5@30 to 60% for ZSM-5@260). Also noteworthy, these catalysts offered a butadiene productivity in the range 2.5–3.6 gBD gcat−1 h−1, which is about one order of magnitude higher than the productivity that can be typically achieved in the Lebedev process (i.e. ethanol → butadiene).The presence of native Brønsted acid sites with medium strength was indispensable to maintain a good selectivity to butadiene as inferred from combined NH3-TPD, pyridine adsorption and 27Al NMR MAS spectroscopy. The formation of carbonaceous species was unavoidable under the reaction conditions as evidenced by XPS and TGA-MS, but could be partially discouraged for ZSM-5@260, showing only 51 mgC g−1 formed at 300 °C after 8 h of reaction and a moderate deactivation at longer reaction times (102 h). As a result, this catalyst suffered from moderate pore blockage during reaction, affording the accessibility of 1,3-BDO into the internal acid sites and in turn the catalytic activity.
Acknowledgements
The authors would like to express their gratitude to G. Cambien, O. Gardoll, M. Trentesaux and V. Ruaux for performing the N2 adsorption/desorption, TGA, XPS, and pyridine adsorption/FT-IR measurements, respectively. The REALCAT platform is also acknowledged for support in the XRF measurements. The REALCAT platform is benefitting from a governmental subvention managed by the French National Research Agency (ANR) within the framework of the ‘Future Investments’ program (PIA) with the contractual reference ‘ANR-11-EQPX-0037’. The Nord-Pas-de-Calais Region and the FEDER are acknowledged for their financial contribution to the acquisition of equipment of the platform. The Central Initiative Foundation and Solvay are also acknowledged for financial support.The Chevreul Institute (FR 2638), Ministère de l'Enseignement Supérieur et de la Recherche, Région Nord – Pas de Calais and FEDER are acknowledged for supporting and funding partially this work.
References
- W. M. C. White, Chem. Biol. Interface, 2007, 166, 10–14 CrossRef CAS PubMed.
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2013, 6, 1595–1614 CrossRef CAS PubMed.
- J. Goldemberg, Science, 2007, 315, 808–810 CrossRef CAS PubMed.
- J. R. Rostrup-Nielsen, Science, 2005, 308, 1421–1422 CrossRef CAS PubMed.
- Z. Wang, Comprehensive Organic Name Reactions and Reagents, 3rd vol, John Wiley & Sons, Inc., 2009, pp. 1728–1730 Search PubMed.
- E. V. Makshina, W. Janssens, B. F. Sels and P. A. Jacobs, Catal. Today, 2012, 198, 338–344 CrossRef CAS.
- M. Lewandowski, G. S. Babu, M. Vezzoli, M. D. Jones, R. E. Owen, D. Mattia, P. Plucinski, E. Mikolajska, A. Ochenduszko and D. C. Apperley, Catal. Commun., 2014, 49, 25–28 CrossRef CAS.
- C. Angelici, M. E. Z. Velthoen, B. M. Weckhuysen and P. C. A. Bruijnincx, Catal. Sci. Technol., 2015, 5, 2869–2879 CAS.
- W. Janssens, E. V. Makshina, P. Vanelderen, F. De Clippel, K. Houthoofd, S. Kerkhofs, J. A. Martens, P. A. Jacobs and B. F. Sels, ChemSusChem, 2015, 8, 994–1008 CrossRef CAS PubMed.
- C. Angelici, M. E. Z. Velthoen, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2014, 7, 2505–2515 CrossRef CAS PubMed.
- A. Chieregato, J. V. Ochoa, C. Bandinelli, G. Fornasari, F. Cavani and M. Mella, ChemSusChem, 2015, 8, 377–388 CrossRef CAS PubMed.
- O. V. Larina, P. I. Kyriienko and S. O. Soloviev, Catal. Lett., 2015, 145, 1162–1168 CrossRef CAS.
- H. E. Jones, E. E. Stahly and B. B. Corson, J. Am. Chem. Soc., 1949, 71, 1822–1828 CrossRef CAS.
- T. W. Kim, J. W. Kim, S. Y. Kim, H. J. Chae, J. R. Kim, S. Y. Jeong and C. U. Kim, Chem. Eng. J., 2014, 278, 217–223 CrossRef.
- H. J. Chae, T. W. Kim, Y. K. Moon, H. K. Kim, K. E. Jeong, C. U. Kim and S. Y. Jeong, Appl. Catal., B, 2014, 150–151, 596–604 CrossRef CAS.
- V. L. Sushkevich, I. I. Ivanova, V. V. Ordomsky and E. Taarning, ChemSusChem, 2014, 7, 2527–2536 CrossRef CAS PubMed.
- M. D. Jones, C. G. Keir, C. D. Iulio, R. A. M. Robertson, C. V. Williams and D. C. Apperley, Catal. Sci. Technol., 2011, 1, 267–272 CAS.
- G. O. Ezinkwo, V. F. Tretjakov, R. M. Talyshinky, A. M. Ilolov and T. A. Mutombo, Catal. Commun., 2014, 43, 207–212 CrossRef CAS.
- S. K. Bhattacharyya and S. K. Sanyal, J. Catal., 1967, 7, 152–158 CrossRef CAS.
- Y. Sekiguchi, S. Akiyama, W. Urakawa, T. R. Koyama, A. Miyaji, K. Motokura and T. Baba, Catal. Commun., 2015, 68, 20–24 CrossRef CAS.
- R. A. L. Baylon, J. Sun and Y. Wang, Catal. Today, 2016, 259, 446–452 CrossRef CAS.
- V. L. Sushkevich, I. I. Ivanova and E. Taarning, Green Chem., 2015, 17, 2552–2559 RSC.
- T. A. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, J. White, A. Manheim, D. Elliot, L. Lasure, S. Jones, M. Gerber, K. Ibsen, L. Lumberg and S. Kelley, Top Value Added Chemicals From Biomass: I. Results of Screening for Potential Candidates from Sugars and Synthesis Gas, PNNL and NREL, 2004, pp. 1–67 Search PubMed.
- X.-J. Ji, H. Huang and P.-K. Ouyang, Biotechnol. Adv., 2011, 29, 351–364 CrossRef CAS PubMed.
- N. Kataoka, A. S. Vangnai, T. Tajima, Y. Nakashimada and J. Kato, J. Biosci. Bioeng., 2013, 115, 475–480 CrossRef CAS PubMed.
- H. Yim, R. Haselbeck, W. Niu, C. Pujol-Baxley, A. Burgard, J. Boldt, J. Khandurina, J. D. Trawick, R. E. Osterhout, R. Stephen, J. Estadilla, S. Teisan, H. Brett Schreyer, S. Andrae, T. H. Yang, S. Y. Lee, M. J. Burk and S. van Dien, Nat. Chem. Biol., 2011, 7, 445–452 CrossRef CAS PubMed.
- N. Itoh, M. Nakamura, K. Inoue and Y. Makino, Appl. Microbiol. Biotechnol., 2007, 75, 1249–1256 CrossRef CAS PubMed.
- J. Baek, T. Y. Kim, W. Kim, W. Kim, H. J. Lee and J. Yi, Green Chem., 2014, 16, 3501–3507 RSC.
- S. Sato, R. Takahashi, T. Sodesawa, H. Fukuda, T. Sekine and E. Tsukuda, Catal. Commun., 2005, 6, 607–610 CrossRef CAS.
- S. Sato, R. Takahashi, T. Sodesawa, N. Honda and H. Shimizu, Catal. Commun., 2003, 4, 77–81 CrossRef CAS.
- H. Gotoh, Y. Yamada and S. Sato, Appl. Catal., A, 2010, 377, 92–98 CrossRef CAS.
- S. Sato, F. Sato, H. Gotoh and Y. Yamada, ACS Catal., 2013, 3, 721–734 CrossRef CAS.
- V. K. Díez, P. A. Torresi, P. J. Luggren, C. A. Ferretti and J. I. Di Cosimo, Catal. Today, 2013, 213, 18–24 CrossRef.
- P. A. Torresi, V. K. Diez, P. J. Luggren and J. I. Di Cosimo, Catal. Sci. Technol., 2014, 4, 3203–3213 CAS.
- N. Ichikawa, S. Sato, R. Takahashi and T. Sodesawa, J. Mol. Catal. A: Chem., 2006, 256, 106–112 CrossRef CAS.
- B. Zhang, Y. Zhu, G. Ding, H. Zheng and Y. Li, Appl. Catal., A, 2012, 443–444, 191–201 CrossRef CAS.
- D. W. Hwang, P. Kashinathan, J. M. Lee, J. H. Lee, U. H. Lee, J.-S. Hwang, Y. K. Hwang and J.-S. Chang, Green Chem., 2011, 13, 1672–1675 RSC.
- R. Takahashi, T. Sodesawa and N. Yamamoto, Catal. Commun., 2004, 5, 397–400 CrossRef.
- N. Yamamoto, S. Sato, R. Takahashi and K. Inui, Catal. Commun., 2005, 6, 480–484 CrossRef CAS.
- W. Zhang, D. Yu, X. Ji and H. Huang, Green Chem., 2012, 14, 3441–3450 RSC.
- A. Multer, N. McGraw, K. Hohn and P. Vadlani, Ind. Eng. Chem. Res., 2013, 52, 56–60 CAS.
- H. Duan, Y. Yamada and S. Sato, Appl. Catal., A, 2014, 487, 226–233 CrossRef CAS.
- H. Duan, D. Sun, Y. Yamada and S. Sato, Catal. Commun., 2014, 48, 1–4 CrossRef CAS.
- H. Duan, Y. Yamada and S. Sato, Appl. Catal., A, 2015, 491, 163–169 CrossRef CAS.
- H. Vinek, J. A. Lercher and H. Noller, J. Mol. Catal., 1985, 30, 353–359 CrossRef CAS.
- M. A. Makarova, C. Williams, J. M. Thomas and K. I. Zamaraev, Catal. Lett., 1990, 4, 261–264 CrossRef CAS.
- M. A. Makarova, C. Williams, K. I. Zamaraev, J. M. Thomas and J. Chem, Soc. Faraday Trans., 1994, 90, 2147–2153 RSC.
- M. A. Makarova, E. A. Paukshtis, J. M. Thomas, C. Williams and K. I. Zamaraev, J. Catal., 1994, 149, 36–51 CrossRef CAS.
- J. Hua and Y. Han, Chem. Mater., 2009, 21, 2344–2348 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- F. Jing, B. Katryniok, E. Bordes-Richard and S. Paul, Catal. Today, 2013, 203, 32–39 CrossRef CAS.
- F. Jing, B. Katryniok, F. Dumeignil, E. Bordes-Richard and S. Paul, J. Catal., 2014, 309, 121–135 CrossRef CAS.
- F. Jing, B. Katryniok, F. Dumeignil, E. Bordes-Richard and S. Paul, Catal. Sci. Technol., 2014, 4, 2938–2945 CAS.
- A. Samoson and E. Lippmaa, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 6567–6570 CrossRef CAS.
- P. A. Russo, M. M. Antunes, P. Neves, P. V. Wiper, E. Fazio, F. Neri, F. Barreca, L. Mafra, M. Pillinger, N. Pinna and A. A. Valente, Green Chem., 2014, 16, 4292–4305 RSC.
- K. K. Ramasamy, M. A. Gerber, M. Flake, H. Zhang and Y. Wang, Green Chem., 2014, 16, 748–760 RSC.
- L. G. Possato, R. N. Diniz, T. Garetto, S. H. Pulcinelli, C. V. Santilli and L. Martins, J. Catal., 2013, 300, 102–112 CrossRef CAS.
- C. V. Hidalgo, H. Itoh, T. Hattori, N. Niwa and Y. Murakami, J. Catal., 1984, 85, 362–369 CrossRef CAS.
- B. Hunger, M. Heuchel, L. A. Clark and R. Q. Snurr, J. Phys. Chem. B, 2002, 106, 3882–3889 CrossRef CAS.
- W. Zhu, H. Yang, J. Chen, C. Chen, L. Guo, H. Gan, X. Zhao and Z. Hou, Green Chem., 2014, 16, 1534–1542 RSC.
- D. P. Serrano, R. A. García, G. Vicente, M. Linares, D. Procházková and J. Čejka, J. Catal., 2011, 279, 366–380 CrossRef CAS.
- Y. Wang, X. Guo, C. Zhang, F. Song and X. Wang, et al. , Catal. Lett., 2006, 107, 209–214 CrossRef CAS.
- G. L. Woolery, G. H. Kuehl, H. C. Timken, A. W. Chester and J. C. Vartuli, Zeolites, 1997, 19, 288–296 CrossRef CAS.
- M. John, K. Alexopoulos, M.-F. Reyniers and G. B. Marin, J. Catal., 2015, 330, 28–45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy02211h |
| This journal is © The Royal Society of Chemistry 2016 |