
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon/H-ZSM-5 composites as supports for bi-functional Fischer–Tropsch synthesis catalysts†
M. J.
Valero-Romero
a,
S.
Sartipi
b,
X.
Sun
b,
J.
Rodríguez-Mirasol
a,
T.
Cordero
a,
F.
Kapteijn
b and
J.
Gascon
*b
aUniversidad de Málaga, Andalucía Tech, Escuela de Ingenierías, Campus de Teatinos s/n, 29071 Málaga, Spain
bCatalysis Engineering, Dept. Chemical Engineering, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: J. Gascon@tudelft.nl
First published on 7th January 2016
Abstract
Mesoporous H-ZSM-5–carbon composites, prepared via tetrapropylammonium hydroxide (TPAOH) post treatment of H-ZSM-5 followed by deposition of pyrolytic carbon, have been used as the support for the preparation of Co-based Fischer–Tropsch catalysts. The resulting catalysts display an improved performance during Fischer–Tropsch synthesis (FTS), with higher activity, higher selectivity towards C5–C9 (gasoline range) hydrocarbons and lower selectivity towards C1 (and C2) than Co/mesoH-ZSM5 (without pyrolytic carbon). This is due to the weaker metal–support interaction caused by the deposited carbon (as revealed by XPS) leading to a higher reducibility of the Co species. Further, the partial deactivation of the Brønsted acid sites by pyrolytic carbon deposition, as was observed by NH3-TPD, allows the modification of the zeolite acidity. Both the olefin to paraffin (O/P) and the isoparaffin to normal paraffin (I/N) ratios decrease with the increase in the carbon content, opening the door to further tune the catalytic performance in multifunctional FTS operations.
Introduction
In the past decades, the limited worldwide oil reserves together with the new requirements for quality specifications of fuels have boosted research into technological solutions for energy supply. In this context, the Fischer–Tropsch synthesis (FTS) plays an important role due to its versatility in the transformation of different non-petroleum carbon resources such as natural gas, coal and biomass, to high quality clean hydrocarbon fuels and valuable chemicals from synthesis gas (CO + H2).1In spite of recent advances in the development of Fe-based systems,2 when high purity syngas is used Co-based catalysts are preferred because Co exhibits superior chain propagation and a higher intrinsic activity than Fe under similar conditions. Moreover, cobalt catalysts are more selective towards the formation of waxes, are more stable against deactivation by water and have a low activity for the competing water-gas shift reaction.3,4 In this respect, much effort has been devoted to tune the low-temperature Fischer–Tropsch (LT-FTS) product selectivity of Co-based catalysts, which generally follows the Anderson–Schulz–Flory (ASF) distribution.
Nanostructured zeolites with hierarchical pore arrangements have been recently considered as one of the most suitable catalyst supports for the direct synthesis of gasoline from syngas via FTS.5–10 It is suggested that FTS waxes are hydrocracked to shorter chain hydrocarbons on the acid sites of these bifunctional catalysts, increasing the product selectivity towards the gasoline-range hydrocarbons (C4–C11).11,12 On the one hand, the micropore system of the hierarchical zeolite provides shape selective features and active acid sites. On the other hand, the mesopore network of these materials provides shorter diffusion pathways along the zeolite particle,13 enhancing the diffusivity of the reactants and products to/from the active sites, making the inner zeolite surface more accessible than that in microporous zeolites. Additionally, the larger mesopore surface area allows the dispersion of larger amounts of cobalt per mass of catalyst7,14 and improves metal dispersion.9,15 Many approaches to improve transport in microporous zeolites have been developed, including desilication,13,16 dealumination,17 synthesis of wide-pore zeolites and nanosized zeolites,18–20 and carbon-templated synthesis.21 One of the most effective approaches to create the so-called ‘hierarchical pore structure’ is the desilication or basic leaching method, which implies the selective extraction of framework silicon by treatment in alkaline solutions. Desilication has been known and applied for decades in industry and more recently rationalized by several groups.16,22
Sartipi et al.9 studied the catalytic performance of different Co/zeolite FTS catalysts with respect to their mesoporous hierarchy and the amount of extra framework aluminium (EFAl). They found that the zeolite topology and, most importantly, the amount and strength of acid sites are the key parameters in the performance of bifunctional catalysts to maximize the FTS product selectivity towards liquid9 hydrocarbons. The most promising results were obtained with H-ZSM-5 since it has fair isomerization and oligomerization activities at low temperature and a relatively stable catalytic performance, especially under LT-FTS process conditions. However, the selectivity to CH4 over these bifunctional catalysts is usually high.7–10 This is believed to arise partly from the low-coordination Co surface sites stabilized over the zeolite surface, which seem to be derived from the strong interactions between the FTS phase and the zeolite.8,23 These low-coordination Co sites are reported to be very active in CO hydrogenation and the development of synthetic protocols able to avoid their formation is one of the biggest challenges in multifunctional FTS.23
Herein we report a detailed characterization of carbon/zeolite composites prepared by deposition of pyrolytic carbon from pyrolysis of propylene on H-ZSM-5 zeolite, and the utilization of these composites as the supports for Co-based FTS catalysts. The objectives of this work are to reduce the strong Co–zeolite interactions by depositing pyrolytic carbon and to study their effects on the acidity of the carbon/zeolite composites, the cobalt particle size and the catalyst reducibility, with special emphasis on their catalytic performance in FT synthesis.
Experimental
Materials
ZSM-5 zeolite in ammonium form with a nominal Si/Al ratio of 40 was purchased from Zeolyst (CBV 8014). A 1 M tetrapropylammonium hydroxide (TPAOH) solution, a 70% HNO3 solution and Co(NO3)2·6H2O were purchased from Sigma-Aldrich. All chemicals were used without any further purification.Support preparation and catalyst synthesis
After heat treatment, a fraction of the mesoporous H-ZSM-5 zeolite was treated with a 1 M HNO3 aqueous solution (volumeacid solution/weightzeolite = 28.6 cm3 g−1) at 343 K for 2 h under stirring in order to remove the extraframework alumina and the amorphous phase formed during the leaching procedure. After quenching, the samples were washed thoroughly with deionized water, dried and calcined similarly to after the desilication procedure. Mesoporous H-ZSM-5 before the acid treatment was denoted as ‘mesoH-ZSM-5’, and the acid washed zeolite was denoted as ‘mesoH-ZSM-5(w)’.
The zeolite samples and the carbon/zeolite composites were loaded with 17–24 wt% Co via incipient wetness impregnation with an aqueous solution of Co(NO3)2·6H2O. All supports were dried overnight at 393 K before impregnation. After impregnation, the samples were kept overnight in a desiccator at room temperature and dried under air flow at 393 K for 12 h followed by calcination at 523 K for 2 h. All the above-mentioned drying and calcination steps were performed at a heating rate of 2 K min−1 and under static air conditions. The bifunctional catalysts are denoted as ‘Co/mesoH-ZSM-5(wnnc)’.
Characterization
Surface chemistry characterization and elemental composition
Elemental analysis was performed using a PerkinElmer Optima instrument. The samples were first digested in ca. 50 ml of a 1.25% H2SO4 and 1.00% HF mixture. After dilution the liquid analysis was done by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES).The surface chemistry of the samples was analyzed by X-ray photoelectron spectroscopy (XPS) using a 5700C model Physical Electronics apparatus with Mg-Kα radiation (1253.6 eV). For the analysis of the XPS data, the C 1s peak position was set at 284.5 eV and used as reference. The deconvolution of the peaks was done using Gaussian–Lorentzian curves and a Shirley type background line. The difference between the experimental and calculated curves was minimized by using the least-squares method.
The X-ray diffraction (XRD) patterns were recorded in the Bragg–Brentano geometry in a Bruker D8 Advance diffractometer equipped with a Vantec position sensitive detector and a graphite monochromator. Measurements were performed at room temperature, using monochromatic Co-Kα radiation (λ = 0.179026 nm) in the 2θ region between 10° and 90°. The samples were placed on a Si substrate and rotated during measurements. All patterns were background-subtracted to eliminate the contribution of air scatter and possible fluorescence radiation. The average particle size of Co3O4 in the catalysts was estimated from the Scherrer equation applied to the most intense (311) diffraction (2θ = 36.9°) using the shape factor K = 0.9. The mean Co0 particle size in the reduced catalysts, d(Co0), was then obtained from the corresponding Co3O4 particle size by applying the molar volume correction:31d(Co0) = 0.75d(Co3O4).
NH3-Temperature Programmed Desorption (NH3-TPD) was performed using an AutoChem II Chemisorption Analyzer (Micromeritics). Ca. 0.200 g of the zeolite containing samples was first degassed under a He flow at 673 K for 1 h and then saturated with NH3 at 473 K for 1 h, using a flow of 1.65% NH3 in He. The gas mixture was then switched back to He and the sample was purged at 473 K for ca. 1 h to remove the weakly adsorbed NH3 molecules until no ammonia was detected. TPD was subsequently recorded in a He flow, from 473 to 873 K. All the flow rates were adjusted to 25 cmSTP3 min−1 and the heating rates were 10 K min−1 during different stages of the experiment.
Temperature Programmed Reduction by H2 (TPR(H2)) was performed on homemade equipment. Ca. 0.100 g of the Co containing samples was mounted in a temperature controlled reactor where a 27 cmSTP3 min−1 flow of 7.4% H2 in Ar was fed over the samples. The reactor temperature was then ramped from room temperature to 1223 K with a heating rate of 5 K min−1 and the H2 consumption was monitored by a TCD. Calibration was performed with CuO (Alfa Aesar), and the total H2 consumption values were obtained from the TPR(H2) patterns. The ratio between the H2 consumption up to 673 K and the corresponding theoretical value, calculated for the full reduction of each catalyst (assuming all Co atoms to be initially in the form of Co3O4), was reported as the degree of reduction. TEM images were obtained using a PHILIPS CM-200 transmission electron microscope at an accelerating voltage of 200 kV.
Fischer–Tropsch synthesis
The FTS experiments were performed in a six-flow fixed-bed microreactor setup described elsewhere.32 For all the experiments, 250 mg of the fresh catalyst (100–212 μm particle size) was diluted with silicon carbide (100–212 μm) to attain a constant bed volume of approximately 1.3 cm3. The catalysts were first activated in situ by 80 cmSTP3 min−1 of H2 at 623 for 10 h at atmospheric pressure followed by cooling down to 453 K under H2 flow. After increasing the pressure to the process value (15 bar), CO was gradually introduced to the feed stream at 453 K in order to reach its final concentration (H2/CO = 2) in 1 h. Subsequently, the reactor was heated to the process temperature (523 K). A rate of 2 K min−1 was applied for all the heating/cooling steps.During the experiment, heavy hydrocarbons (waxes) were collected by the gas/liquid separators at 448 K and reaction pressure. Once expanded to atmospheric pressure the lighter hydrocarbons and water were collected in cold traps at ca. 278 K. After separation from water, these liquid hydrocarbons as well as the waxes were weighed, dissolved in CS2 and analyzed offline by a simulated distillation (SimDis) GC (Hewlett Packard 5890, Series II) equipped with an FID and a HP-1 column (7.5 m × 0.53 mm, film thickness 2.65 μm), using He as the carrier gas. During the analysis, the oven temperature was ramped from 308 to 673 K (14 K min−1) and kept at the final temperature for 5 min.
Permanent gases as well as light hydrocarbons in the gas phase were analyzed online by a Compact GC (Interscience) equipped with three columns and detectors in parallel using He as the carrier gas. In the first column (Carboxen 1010, 10 m × 0.32 mm) N2, CO, CH4 and CO2 were separated at 333 K and detected by a TCD. In the second column (Al2O3/KCl, 10 m × 0.32 mm) with FID detection, the separation between all the C1–C4 components was achieved at 434 K. In the third column (RTx-1 0.5μm, 15 m × 0.32 mm) the C5–C10 hydrocarbons were separated at 353 K and analyzed by an FID.
Results and discussion
Characterization of the catalytic supports
Treatment with 4% C3H6 in N2 at 973 K at different reaction times results in different amounts of pyrolytic carbon deposited over the mesoporous zeolite mesoH-ZSM-5(w) as evidenced by the TGA patterns of these carbon/zeolite composites (Fig. S1†). As a result, ca. 5, 10, 15 and 22 wt% (gcarbon/100gzeolite) are deposited over mesoH-ZSM5-(w) after 3, 6, 12 and 20 h, respectively.The N2 adsorption–desorption isotherms for the zeolites and the carbon/zeolite composites are presented in Fig. 1a and b, respectively, whereas the chemical properties and the values of the structural parameters that characterize the porous structure of the samples, as derived from the N2 and CO2 adsorption–desorption isotherms, are reported in Table 1. This table summarizes the Si/Al ratio, the values of the apparent surface area obtained by the BET method, the effective micropore volumes and the external surface areas calculated by the t-method and the mesopore volumes derived from the N2 adsorption data. In addition, the micropore volumes obtained by the application of the Dubinin–Radushkevich (DR) equation to both the N2 and CO2 adsorption isotherms are also included in Table 1.
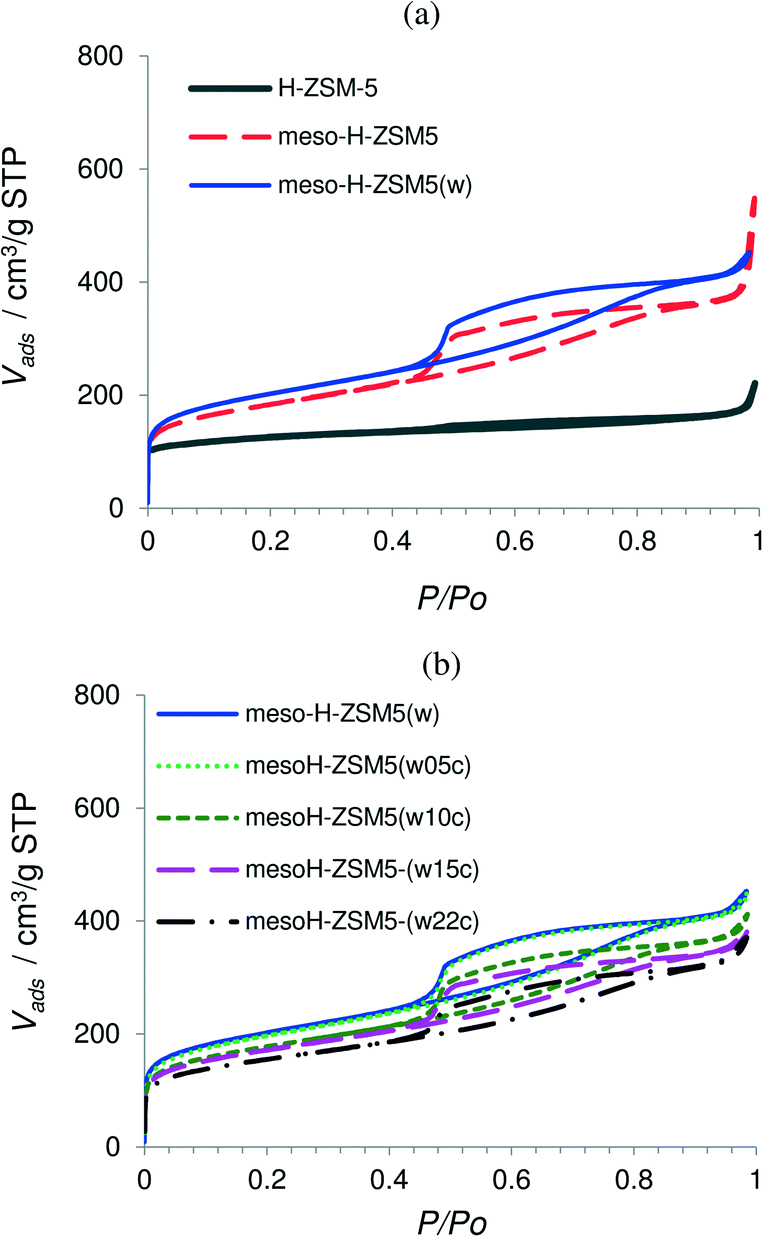 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms at 77 K of the micro- and mesoporous zeolites (a) and the acid washed zeolite and carbon/zeolite composites (b). | ||
| Support | DR methodf | |||||||
|---|---|---|---|---|---|---|---|---|
| Si/Ala | S Total (m2 g−1) | S meso (m2 g−1) | V t (cm3 g−1) | V meso (cm3 g−1) | V DR (N2) (cm3 g−1) | V DR (CO2) (cm3 g−1) | S DR (CO2) (m2 g−1) | |
| a Obtained from ICP-OES. b BET surface area. c Mesopore surface area obtained from the t-plot applied to the N2 isotherm. d Micropore volume obtained from the t-plot. e Mesopore volume calculated as Vmeso = Vtotal (P/P0 = 0.95) − Vt. f Micropore volume and surface area obtained by the Dubinin–Radushkevich (DR) method applied to the N2 and CO2 isotherm. g Not determined. | ||||||||
| H-ZSM-5 | 41 | 425 | 60 | 0.170 | 0.070 | 0.170 | 0.168 | 421 |
| mesoH-ZSM-5 | 22 | 700 | 440 | 0.113 | 0.487 | 0.267 | 0.179 | 459 |
| mesoH-ZSM-5(w) | 41 | 723 | 473 | 0.105 | 0.546 | 0.271 | 0.190 | 475 |
| mesoH-ZSM-5(w05c) | n.d.g | 707 | 478 | 0.093 | 0.547 | 0.265 | 0.176 | 440 |
| mesoH-ZSM-5(w10c) | n.d. | 640 | 430 | 0.090 | 0.498 | 0.239 | 0.165 | 412 |
| mesoH-ZSM-5(w15c) | n.d. | 620 | 418 | 0.086 | 0.458 | 0.231 | 0.161 | 390 |
| mesoH-ZSM-5(w22c) | n.d. | 562 | 370 | 0.081 | 0.437 | 0.215 | 0.150 | 371 |
The isotherm of the parent zeolite H-ZSM-5 is of type I and exhibits an almost horizontal plateau starting at a very low relative pressure, indicating that the porous structure consists predominantly of very narrow micropores (Fig. 1a). After the TPAOH treatment and Si extraction from the zeolite framework the shape of the zeolite isotherm changes from type I to a clear type E hysteresis, suggesting a high degree of hierarchy with large cavities connected by smaller mesopores.33 The mesopore surface area of H-ZSM-5 increases from 60 to 440 m2 g−1 for mesoH-ZSM-5 and the mesopore volume increases from 0.07 to 0.49 cm3 g−1. Alongside, the micropore volume decreased from 0.17 to 0.11 m2 g−1, which indicates a minor collapse of the zeolite structure during the base treatment.34 However, the hierarchical zeolite obtained preserves the long-range crystallinity, as was previously shown by X-ray diffraction.35
The yield of this treatment, defined as the weight of the TPAOH treated sample relative to the raw material, on a dry basis, was ca. 50%, indicating a moderate silicon dissolution when compared with strong inorganic bases, such as NaOH, KOH and LiOH.8,16,36 For instance, the yield of the treatment using NaOH as a desilication agent and under the same base leaching conditions was only 20% for H-ZSM-5 zeolite.8 The Si/Al ratio of H-ZSM-5 decreases from 41 to 22 for mesoH-ZSM-5 due to the Si extraction from the zeolite framework (Table 1). This ratio is readjusted by acid treatment to the original value (41), resulting in mesoH-ZSM-5(w). It was worthy to note that the acid wash treatment did not significantly alter the textural properties of mesoH-ZSM-5(w) which suggests that the employed acid wash does not leach out the zeolite framework Al, in line with previous observations.8,10
The N2 adsorption–desorption isotherms for the carbon/zeolite composites show lower N2 uptake at low relative pressures (Fig. 1b) and a lower micropore volume and surface area, most probably due to partial filling of the pores by pyrolytic carbon (Table 1). There are some differences between the micropore volumes obtained from the N2 and CO2 isotherms. N2 adsorption is carried out within the entire range of relative pressures and cooperative filling of wider micropores takes place, while CO2 adsorption at relative pressures smaller than 0.03 allows only the primary filling of narrow micropores, where a high potential exists.37 The micropore volumes of these carbon/zeolite composites derived from the N2 isotherms are larger than those obtained from the CO2 isotherms. This observation indicates that these composites still possess a predominantly wider microporosity, even after a pyrolytic carbon deposition of 0.22 g of carbon per g of zeolite.
Density functional theory (DFT) was applied to obtain the pore size distribution curves assuming a slit pore model and using the adsorption branch of the CO2 and N2 isotherms at 273 and 77 K, respectively, plotted on logarithmic scales. It is interesting to remark that at the pore sizes below 0.7 nm, N2 adsorption is kinetically restricted, thus the use of Ar and/or CO2 molecules is more appropriate for the characterization of microporous materials with micropores of dimensions between 0.5 and 1 nm.30,38Fig. 2a and b show the CO2- and the N2-DFT pore size distributions, respectively, of mesoH-ZSM-5(w) and that of the parent zeolite, for comparison. H-ZSM-5 shows a broad peak at 0.6 nm, which is approximately the pore size for the MFI zeolite topology,39 and a very sharp peak at about 0.9 nm indicating the presence of wider micropores. The pore size distributions for mesoH-ZSM-5 present less intense peaks at 0.6 (Fig. 2a) and at about 1.1 nm (Fig. 2b). Furthermore, a new and broader peak is observed in the micropore range of 1.1–2 nm. In addition, two broad peaks can be distinguished in the N2-DFT mesopore region, with the first maximum at a pore size of around 3 nm, and the second maximum at around 6 nm. This higher contribution of mesoporosity, observed for mesoH-ZSM-5(w), is caused by the silicon extraction from the zeolite framework during the TPAOH treatment. A higher solid yield and a more precise control of the mesopore formation are the major advantages of using organic hydroxides as desilicating agents, due to their slower desilication kinetics when compared with inorganic hydroxides. Against the application of this method at larger scales is the higher cost of organic hydroxides compared to inorganic hydroxides, although the extra ion-exchange step with NH4NO3, necessary in the case of treatment with inorganic hydroxides, would be avoided.36,40
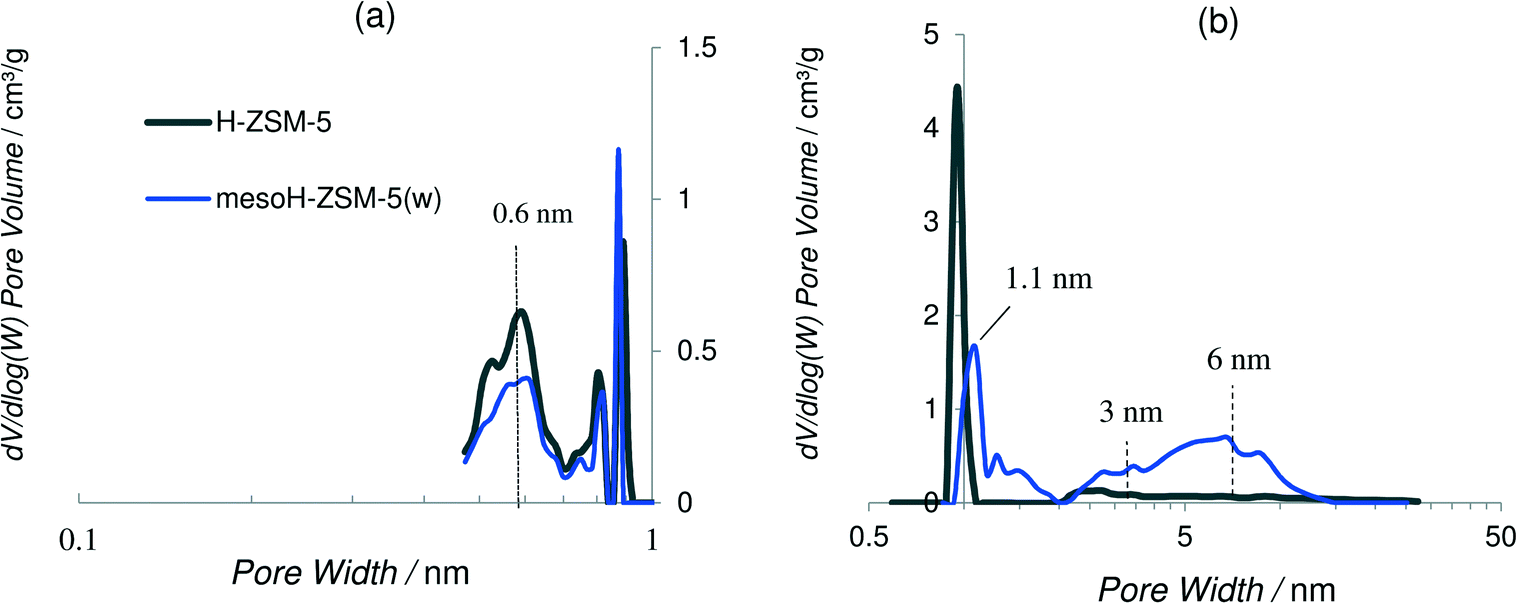 | ||
| Fig. 2 CO2-DFT (a) and N2-DFT (b) pore size distributions of the zeolites. | ||
The DFT pore size distributions of the carbon/zeolite composites were also studied and compared to that of mesoH-ZSM-5(w) in Fig. 3a and b for the CO2- and N2-DFT models, respectively. The DFT model for CO2 remains unchanged after carbon deposition on mesoH-ZSM-5, suggesting that a propylene molecule does not access the narrower micropores (below 0.7 nm). On the other hand, the DFT model for N2 shows how the peak at 1.1 nm is gradually narrowing after the CVD process, and the small shoulder at around 1.5 nm becomes a well-defined peak at 1.3 nm. Besides, the dV/dlog(W) intensity of the carbon/zeolite composites in the pore size region between 2 and 10 nm clearly decreases with an increasing carbon content, resulting in a decrease of the mesopore volume (Table 2). As a reference, the mesopore volume of mesoH-ZSM-5(w) is reduced from 0.55 to 0.44 cm3 g−1 for mesoH-ZSM-5(w22c) after the treatment with propylene at 700 °C for 20 h. Thus, these results suggest that propylene reacts on the surface of the zeolite depositing pyrolytic carbon in the wider micropores and on the walls of the mesopores, but also covering the external crystallite surface reducing both the micro- and mesopore volumes (Table 2). It must be emphasized that in any case the pyrolytic carbon deposits seem to block the zeolite micropore entrance. Related to this, Rodriguez-Mirasol et al.24 pointed out that once a first layer of pyrolytic carbon has covered the zeolite surface, the deposition continues on the surface of the pyrolytic carbon already deposited, which provides new active sites for carbon deposition. As the reaction proceeds, the pyrolytic carbon reduces the pore accessibility and leads to a slower diffusion of propylene into the pores. Therefore, the deposition continues only on the ‘external’ (mesoporous) surface of the zeolite, increasing the thickness of the deposited layer and thus reducing the mesopore volume, as we have observed (Table 1).
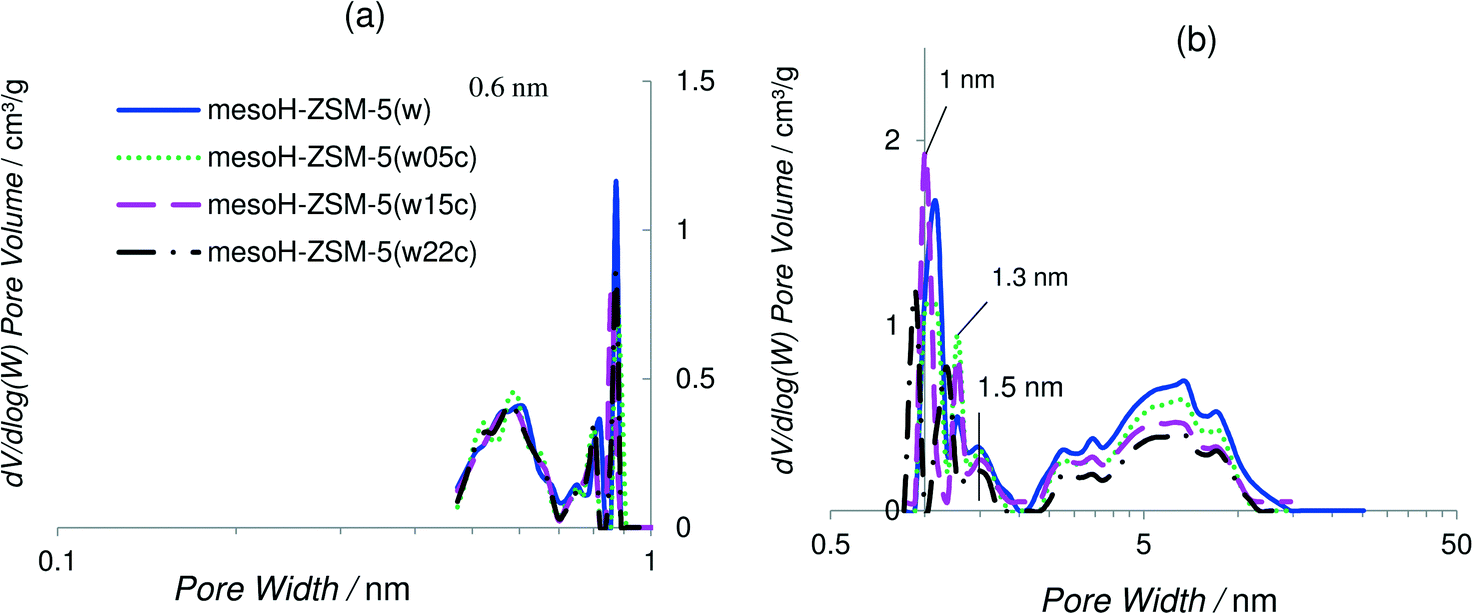 | ||
| Fig. 3 CO2-DFT (a) and N2-DFT (b) pore size distributions of the acid washed zeolite and the carbon/zeolite composites. | ||
| Catalyst | N2 isotherm | Co wt%e | XRD | ||||
|---|---|---|---|---|---|---|---|
| S Total (m2 g−1) | S meso (m2 g−1) | V t (cm3 g−1) | V meso (cm3 g−1) | d Co3O4 (nm) | d Co (nm) | ||
| a BET surface area. b Mesopore surface area obtained from the t-plot applied to the N2 isotherm. c Micropore volume obtained from the t-plot. d Mesopore volume calculated as Vmeso = Vtotal − Vmicro. e Obtained from ICP-OES. f d(Co0) = 0.75d(Co3O4). g From XRD after H2-reduction | |||||||
| Co/mesoH-ZSM-5 | 549 | 355 | 0.079 | 0.405 | 20.7 | 16 | 11 |
| Co/mesoH-ZSM-5(w) | 645 | 412 | 0.095 | 0.477 | 23.8 | 13 | 10 (10)g |
| Co/mesoH-ZSM-5(w05c) | 564 | 364 | 0.084 | 0.420 | 17.9 | 11 | 8 |
| Co/mesoH-ZSM-5(w10c) | 595 | 368 | 0.087 | 0.428 | 17.5 | 12 | 9 |
| Co/mesoH-ZSM-5(w15c) | 585 | 346 | 0.074 | 0.410 | 17.0 | 16 | 12 (14)g |
| Co/mesoH-ZSM-5(w22c) | 470 | 310 | 0.054 | 0.398 | 16.8 | 17 | 13 (17)g |
The acidities of the micro and mesoporous zeolites and the carbon/zeolite composites are investigated by NH3-TPD. The desorption profiles are presented in Fig. 4a and b, respectively. Fig. 4a shows that the parent H-ZSM-5 exhibits a broad NH3 desorption band at around 680 K. This zeolite usually exhibits two NH3 desorption peaks;5,8 a lower-temperature peak at 560 K that may arise from the weakly adsorbed NH3 molecules and a higher temperature peak, observed at 680 K, which is assigned to the desorption of NH3 molecules chemisorbed on the strong Brønsted acid sites. In this work, desorption temperatures higher than 474 K are studied. After desilication and thus, Si extraction from the zeolite framework, the Brønsted acidity disappears and only the weak Lewis acid sites, associated with extra framework aluminum (EFAl), are detected over meso-HZSM-5. Acid washing of the mesoH-ZSM-5 sample (Fig. 5b) removes the EFAl and the Brønsted acids sites are almost fully restored.
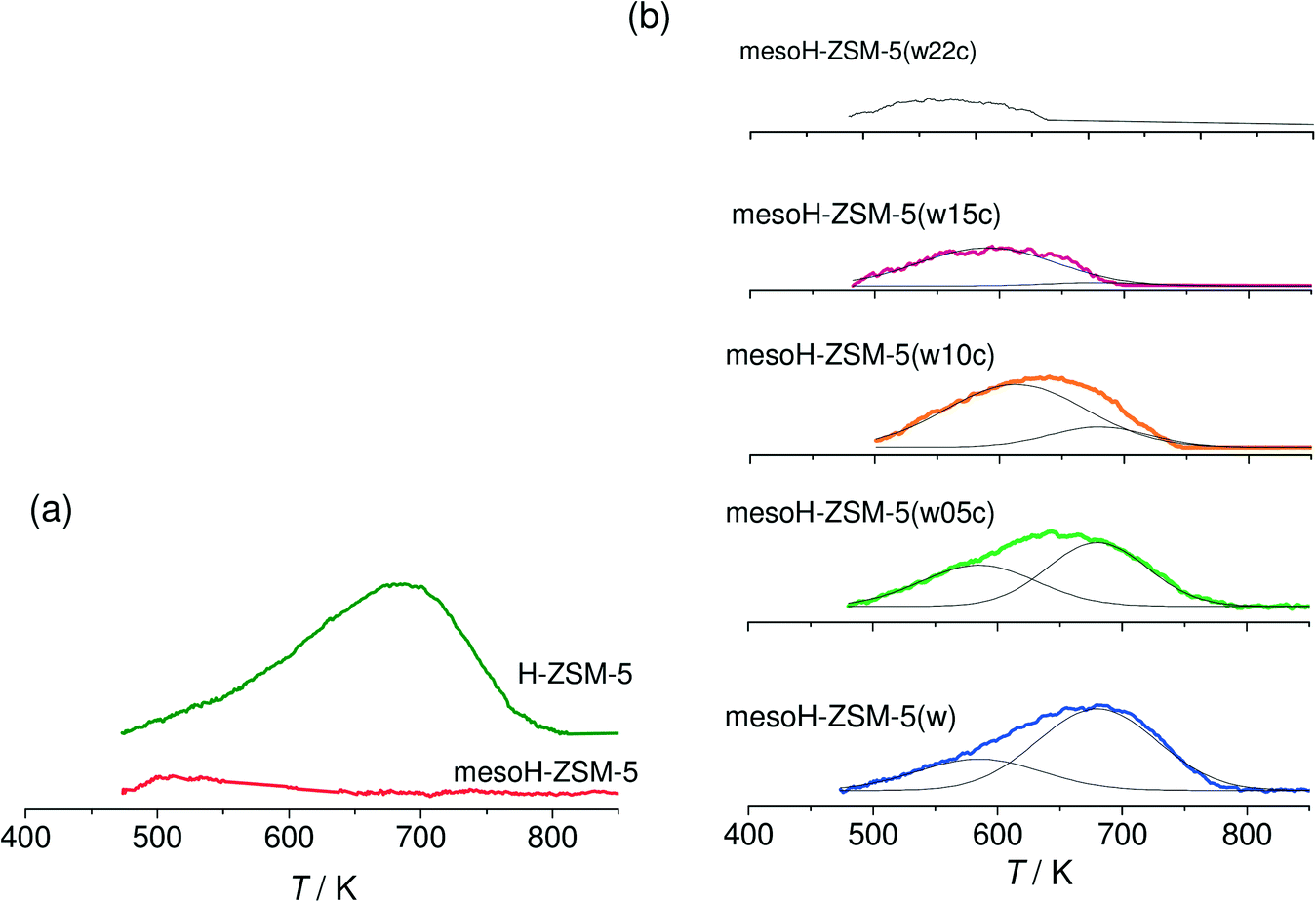 | ||
| Fig. 4 NH3-TPD profiles (10 K min−1) of the micro- and mesoporous zeolites (a) and the acid washed and carbon/zeolite composites (b). NH3 was adsorbed at 473 K. | ||
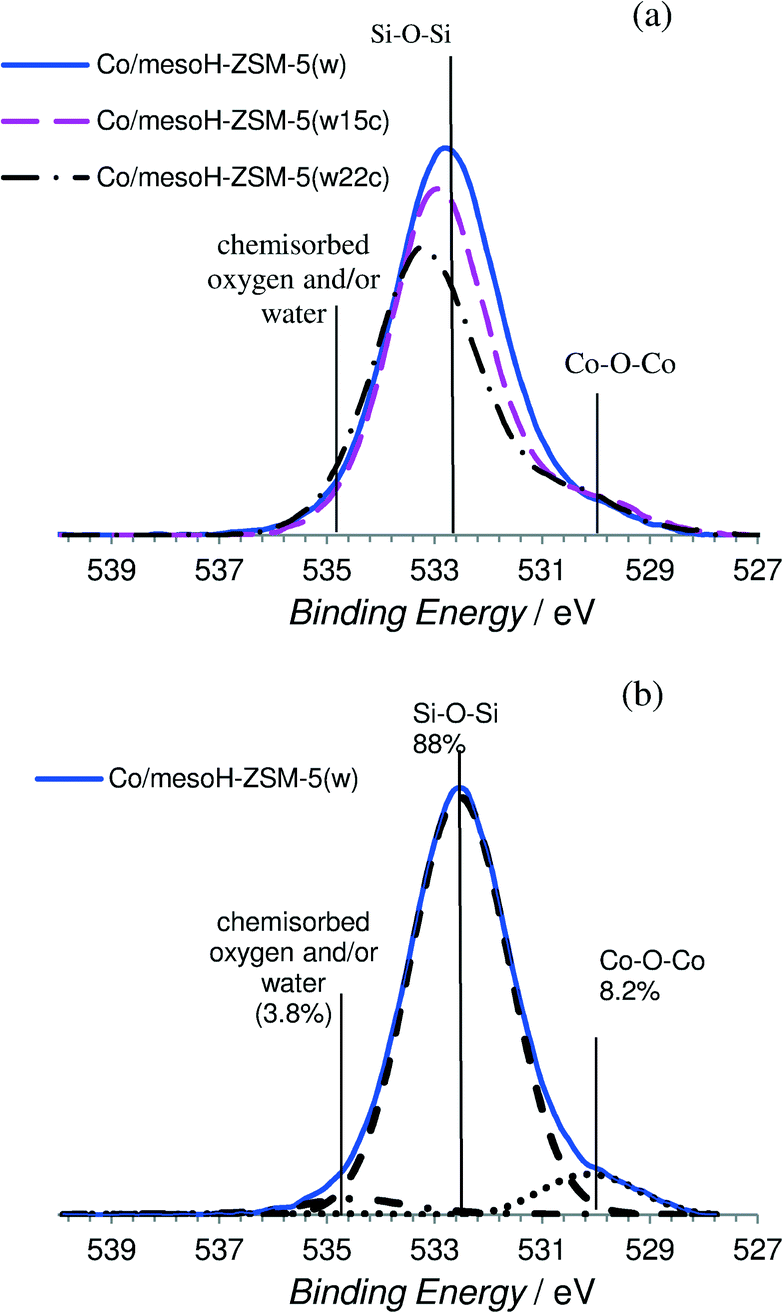 | ||
| Fig. 5 (a) XPS spectra (O 1s region) for the different Co-supported catalysts and (b) the representative XPS spectra of Co/mesoH-ZSM-5(w) showing the deconvolution. | ||
The deposition of pyrolytic carbon decreases considerably the concentration of Brønsted acid sites (Fig. 4b) and this effect reflects acid sites of different strengths on the mesoporous zeolite. In fact, this broad band can be deconvoluted into two components; the component at the relatively higher temperature (680 K), ascribed to strong Brønsted acid sites, is the first to be deactivated by pyrolytic carbon and the component at the lower temperature (585 K) is assigned to weak Brønsted acid sites. Increasing the carbon content gradually shifts the peak to lower temperatures in the NH3-TPD profile indicating a complete deactivation of the strong Brønsted acid sites and an increasing predominance of the weaker acidity, as observed for mesoH-ZSM-5(w15c). These results suggest that by controlling the CVD reaction time and, therefore, the amount of carbon deposited on the mesoporous zeolite, it is possible to fine-tune the acidity of the zeolite.
Co catalysts
The mesoporous supports given in Table 1 were loaded with 17–24 wt% Co. Table 2 summarizes the Co loadings of Co/mesoH-ZSM-5(w) as well as the modified zeolite-supported catalysts, and the parameter values characteristic of their porous structure as derived from the N2 adsorption–desorption isotherms. As expected, after cobalt incorporation, the apparent (SBET) and the external (St) surface area values decrease due to the partial filling of the porous structure. In spite of this partial filling, the remaining porosity remains high and is suitable for catalytic applications.The XRD patterns of the catalysts (data not shown) show the reflections characteristic of the Co3O4 spinel, as reported before for cobalt supported on mesoporous H-ZSM-5.7,8 The crystallite sizes of Co in the catalysts based on the Scherrer equation are also reported in Table 2. The Co3O4 crystallite size does not change as a function of the cobalt loading and the different zeolite supports (before and after the acid wash), which points out the dominant effect of the hierarchical character of zeolites. However, the average Co3O4 crystallite size in the carbon/zeolite composites shows a clear dependence on the carbon content and thus, on the pore volume of the supports (see Table 1), with larger particles being formed in the samples with lower micro- and mesopore volumes. For instance, at 5 wt% carbon content the mean diameter of Co3O4 was 11 nm, and it increased up to 17 nm for the sample containing ca. 20 wt% carbon, despite the lower metal loading compared to the reference samples without carbon. Khodakov41 proposed that “the templating effect” of mesoporous structures was responsible for the size of the supported cobalt oxide particles rather than the surface area of the support, which was supported later by Fu et al.42 Furthermore, Co sintering might be favored on the carbon/zeolite composites (less interacting supports), particularly with increasing carbon contents, during the activation step. In order to verify this, the average size of the cobalt crystallites of the catalyst after H2 reduction has been calculated using the Scherrer equation applied to the FCC crystallite cobalt reflection (2θ = 51.8°) using the shape factor K = 0.9 (Fig. S2†). The Co crystallite size increases by 16% (from 12 to 14 nm) for Co/mesoH-ZSM-5(w15c) and 30% (from 13 to 17 nm) for Co/mesoH-ZSM-5(w22c), whereas it remains invariable over Co/mesoH-ZSM-5(w) (Table 2).
The particle distribution of Co on the activated catalysts was characterized by using TEM analysis as shown in Fig. S3.† This figure shows the TEM images for Co/mesoH-ZSM-5(w) and Co/mesoH-ZSM-5(w22c) after activation in H2 at 673 K for 4 h. The Co particles in the mesoporous H-ZSM-5 show a homogeneous distribution of the FTS active phase on this support (Fig. S3a and S3b†). However, the TEM images of Co/mesoH-ZSM-5(w22c) (Fig. S3c and S3d†) show higher Co concentrations at the outer surface (edge) of the carbon/zeolite composite crystallites, which indicates that most of the FTS active phase is deposited on the external surface of mesoH-ZSM-5(w22c).
The surface element distributions of the catalysts were studied by XPS before H2 reduction. According to the survey spectrum for Co/mesoH-ZSM-5(W15c) shown in Fig. S4,† the peaks detected at 780.3, 532.9, 284.5, 103.5 and 74.4 eV can be assigned to (Co 2p), (O 1s), (C 1s), (Si 2p) and (Al 2p), respectively. Quantitative analysis of the peaks was performed to obtain the mass surface concentrations; the results are summarized in Table 3. The main elements found on the surfaces of the cobalt-supported catalysts were silicon and oxygen, with lower amounts of cobalt and carbon. Aluminum was also detected, but at very low concentrations. As expected, the amount of surface carbon increases as the pyrolytic carbon deposition time increases, at the cost of lower amounts of surface silicon and oxygen. It is necessary to take into account the limited escape depth of the photoelectrons (1–2 nm) that makes XPS a surface sensitive technique.43 Therefore, since the total amount of carbon measured by the TGA analysis (Fig. S1†) is significantly higher than the carbon at the surface measured by XPS, these results support the fact that carbon is not only deposited on the zeolite crystal external surface, but also in the inner mesoporous walls of the zeolite (Fig. 3). On the other hand, the amount of surface cobalt for Co/mesoH-ZSM-5(w) is significantly lower than the total amount of cobalt supported, as shown by the elemental analysis results (Table 2). This result seems to indicate that most of the Co loaded on mesoporous H-ZSM-5 zeolite is located in the internal porous structure. In contrast, the amount of surface cobalt significantly increases when the content of carbon deposited on mesoH-ZSM-5(w) is above 15 wt%. The reduction of the zeolite mesopore volume and mean mesopore size observed for zeolites with high carbon loadings (Fig. 3b), as well as the higher mean Co3O4 crystallite diameter measured by XRD, all suggests that a higher amount of Co3O4 is located mostly at the external surface of the carbon/zeolite composite supports, as suggested by TEM. In addition to this, the decrease of the concentration of terminal silanol groups associated with the Brønsted acid sites on the support surface after carbon deposition may also have decreased the penetration of the Co nitrate solution into the zeolite during impregnation.
| %Si (2p) | %O (1s) | %C (1s) | %Co (2p) | %Al (2p) | BE O 1s (eV) | BE Co 2p3/2 (eV) | Reduction degree (%) | |
|---|---|---|---|---|---|---|---|---|
| Co/mesoH-ZSM-5(w) | 40.2 | 47.8 | 4.6 | 6.5 | 0.9 | 532.5 | 780.0 | 27 |
| Co/mesoH-ZSM-5(w05c) | 40.3 | 45.6 | 6.8 | 6.7 | 0.6 | 532.7 | 780.2 | 35 |
| Co/mesoH-ZSM-5(w10c) | 39.8 | 46.1 | 7.0 | 6.4 | 0.6 | 532.8 | 780.3 | 54 |
| Co/mesoH-ZSM-5(w15c) | 36.4 | 43.1 | 10.6 | 9.3 | 0.7 | 532.9 | 780.3 | 53 |
| Co/mesoH-ZSM-5(w22c) | 32.5 | 39 | 16 | 11.7 | 0.8 | 533.2 | 780.7 | 52 |
Fig. 5 and 6 show some representative XPS spectra for the O 1s and Co 2p regions, respectively, and the values of the binding energies are summarized in Table 3. Fig. 5a shows the XPS O 1s spectra of the different catalysts and Fig. 5b shows the deconvolution of the O 1s spectrum for Co/mesoH-ZSM-5(w). The O 1s spectrum of the Co/mesoH-ZSM-5(w) sample shows the high contribution of oxygen at a binding energy of 532.4 eV (88%) which is a characteristic of single bonded oxygen in Si–O–Si.44 The deposition of carbon on the mesoporous zeolite produces a slight increase in the binding energy of the maxima of the peaks, suggesting distortion of the Si–O–Si bonds (Table 3). On the other hand, compared to Co/mesoH-ZSM-5(w), the O 1s spectrum for Co/mesoH-ZSM-5(w22c) shows a more pronounced shoulder at lower binding energies (BE = 530.1 eV) which is a characteristic of the Co–O–Co bonds of cobalt oxide (Co3O4)45 (Fig. 5a). A binding energy of ca. 535.5 eV is a characteristic of chemisorbed oxygen and/or water.43
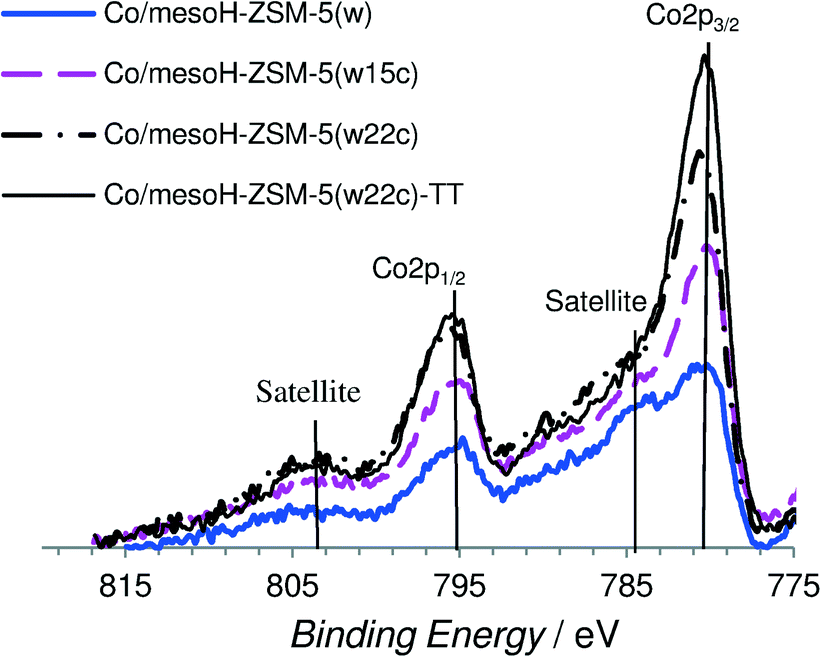 | ||
| Fig. 6 XPS spectra (Co 2p region) of the different Co-supported catalysts. | ||
The Co 2p spectrum of cobalt (Fig. 6) displays the characteristic doublet corresponding to the spin–orbit coupling (2p3/2, 2p1/2) with the most intense peak of the Co 2p3/2 component at a binding energy of about 780.1 eV associated with the Co2+/Co3+ ions in the Co3O4 spinel phase45 and a low intensity of the shake-up satellite peak at ca. 787 eV, which is associated with the presence of Co2+ ions in an octahedral symmetry, typical of CoO.46 As we observed, cobalt supported on mesoH-ZSM-5(w) is characterized by a very high intensity of the shake-up satellite peak. In line with previous observations,44,47 the presence of the Co2+ species in an octahedral symmetry evidenced a strong interaction of the cobalt species with the silica network (Si–O–Co–O) of the mesoporous zeolite support. On the other hand, in the case of cobalt supported on carbon/zeolite, the Co3O4 component at a higher binding energy is more intense compared to the shake-up satellite peak and it is even more intense when the amount of carbon deposited on the zeolite support increases. This result together with the O 1s spectrum shown in Fig. 8a seems to indicate that a larger fraction of the Co2+ ions interacts with the siliceous substrate of mesoHZSM-5(w) whereas a higher amount of the Co3O4 phase is present on the surface of the carbon/zeolite supports. Martínez et al.47 pointed out that the strong cobalt–support interaction leading to the formation of the Co2+ species in an octahedral symmetry is favored at the high temperatures usually required for decomposing organic cobalt salts.
However, the XPS results obtained for the sample Co/mesoH-ZSM-5(w22c) after a thermal treatment in a N2 atmosphere at 673 K for 4 h, sample Co/mesoH-ZSM-5(w22c)-TT (Fig. 6), clearly suggest that the deposition of coke prevents, to some extent, these interactions between the cobalt and the zeolite even after a thermal treatment at higher temperatures.
Fig. 7 shows the TPR(H2) profiles for the calcined Co catalysts before H2 reduction. Clearly, all the catalysts exhibit a reduction peak at temperatures below 600 K, attributed to the reduction of Co3O4 to metallic cobalt.48 This assignment is also supported by the XRD and XPS results where Co3O4 was detected in all the catalysts. This sharp peak slightly moves towards lower temperatures when increasing the carbon content and the d(Co3O4), suggesting that the presence of carbon between the metal and the zeolite support enhances the rate of Co reduction, probably due to the better accessibility of H2 to the Co3O4 crystallites located on the outer composite surface and to their bigger size.42 In the case of the cobalt catalysts supported on mesoH-ZSM-5 and mesoH-ZSM-5(w), two broad peaks at 600–900 K and above 900 K are visible, whereas a significant reduction feature in the higher temperature region (>1000 K) is found. This suggests that, in addition to the crystalline Co3O4 phase, different cobalt species are present on the zeolite surface, and these are more difficult to reduce due to the strong interaction of cobalt with the zeolite.8 Co–Si–O mixed oxides, such as Co2SiO4, in which cobalt is divalent in tetrahedral coordination, have been identified as interacting species on the surface of amorphous SiO2 (ref. 49) and silica delaminated zeolites,50 which are reducible only at temperatures above 900 K. Thus, it appears that the formation of a surface cobalt silicate may be possible for Co supported on mesoH-ZSM-5(w) due to the expected high stability of the silicate and also to the higher Si/Al ratio (40), when compared to mesoH-ZSM-5 (Si/Al = 21). The degrees of reduction, as calculated from the consumption of H2 up to 673 K, are listed in Table 3. The degree of reduction is 27% for Co/mesoH-ZSM-5(w), and increases considerably to above 50% for the samples with 10 or more wt% carbon.
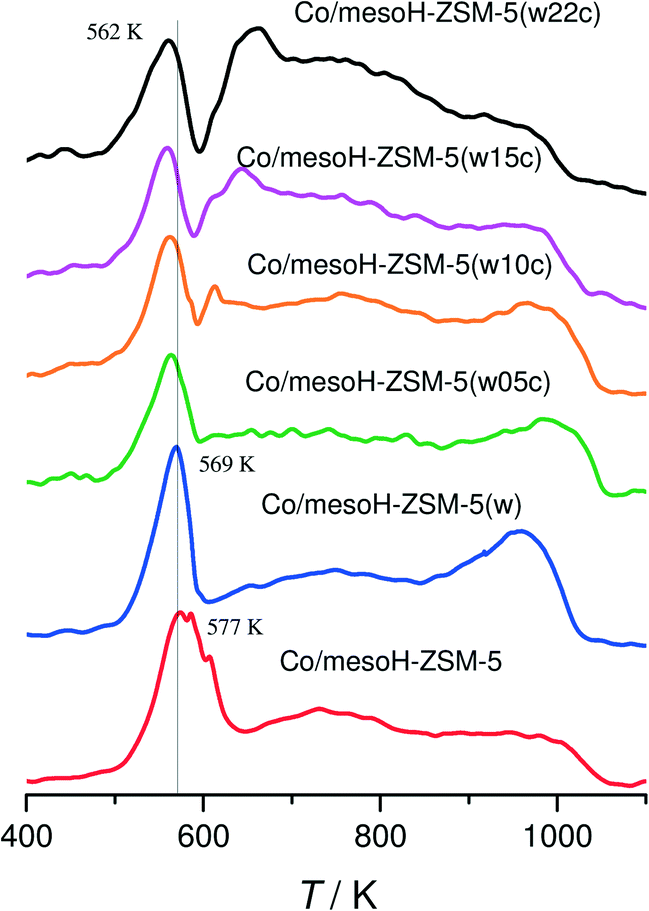 | ||
| Fig. 7 TPR(H2) profiles of the Co-supported catalysts. | ||
On the other hand, a new peak gradually appears at around 650 K with a shoulder at higher temperatures for the Co catalysts when the amount of carbon deposited on the mesoporous zeolite increases. Moreover, it is apparent that the hydrogen consumption above 900 K decreases upon carbon addition. These results reveal that the reducibility of Co is higher over the carbon/zeolite composites than over the mesoporous zeolites due to less metal–support interactions between the cobalt and the composites, as evidenced by the XPS results. Next to the reduction of Co in interaction with the zeolite, the broad band above 600 K in the TPR profiles can be assigned to some gasification of the carbon support,8,42 explaining the high H2 consumption during the TPR for the sample Co/mesoH-ZSM-5(w22c) above 650 K.
Catalytic activity
Table 4 summarizes the CO conversion, carbon selectivity to different product ranges and the olefin to paraffin and the isoparaffin to n-paraffin ratios of the FTS products after 20 h on-stream and at different GHSV values. The time-on-stream (TOS) evolution of the cobalt-time-yield (CTY; number of CO moles converted per kilogram of Co per hour) of Co/mesoH-ZSM-5 and Co/mesoH-ZSM-5(w) is presented in Fig. 8a. The CTY of Co/mesoH-ZSM-5(w) is 13% higher than that of Co/mesoH-ZSM-5, which may be associated with the removal of EFAl formed in mesoH-ZSM-5 after acid washing, as evidenced by the NH3-TPD results of the zeolite supports (Fig. 4). The EFAl species may react with Co to form cobalt aluminates which are inactive in Fischer–Tropsch synthesis.48 The carbon selectivity to the different product ranges over these catalysts is compared in Fig. 8b under iso-conversion conditions (as depicted in the figure inset). Under the applied process conditions Co/mesoH-ZSM-5 presents a relatively high selectivity to the gasoline-range hydrocarbons (C5–C11, 40%), and the selectivity to C12–20 and waxes (C21+) is 20% and 5%, respectively. On the other hand, the production of waxes is reduced completely over Co/mesoH-ZSM-5(w), while the selectivity to the C5–C11 fraction increases to 50%. Moreover, this catalyst is less selective to lower hydrocarbons (C2–C4) and the diesel range hydrocarbons (C12–C20). However, the selectivity to C1 is high, about 20% for both catalysts, which is undesirable for FTS.| Process conditions | Catalyst | X CO/% | S/% | O/P (C2–8)a | I/N (C4)b | ||
|---|---|---|---|---|---|---|---|
| C1 | C5–C11 | CO2 | |||||
| a Olefin to paraffin ratio of C2–C8. b Iso-to-normal C4 ratio. | |||||||
| 513 K, 15 bar, H2/CO = 2 | |||||||
| 12.0 mSTP3 kgcat−1 h−1 | |||||||
| Co/mesoH-ZSM-5 | 57 | 19 | 41 | 2 | 0.31 | 0.01 | |
| Co/mesoH-ZSM-5(w) | 83 | 17 | 51 | 2 | 0.65 | 0.09 | |
| 513 K, 15 bar, H2/CO = 2 | |||||||
| 24.0 mSTP3 kgcat−1 h−1 | |||||||
| Co/mesoH-ZSM-5(w) | 55 | 21 | 48 | 1 | 0.80 | 0.13 | |
| Co/mesoH-ZSM-5(w05c) | 53 | 19 | 48 | 1 | 0.74 | 0.09 | |
| Co/mesoH-ZSM-5(w10c) | 63 | 17 | 57 | 1 | 0.44 | 0.07 | |
| Co/mesoH-ZSM-5(w15c) | 58 | 17 | 56 | 0.6 | 0.42 | 0.06 | |
| Co/mesoH-ZSM-5(w22c) | 48 | 17 | 52 | 0.8 | 0.39 | 0.05 | |
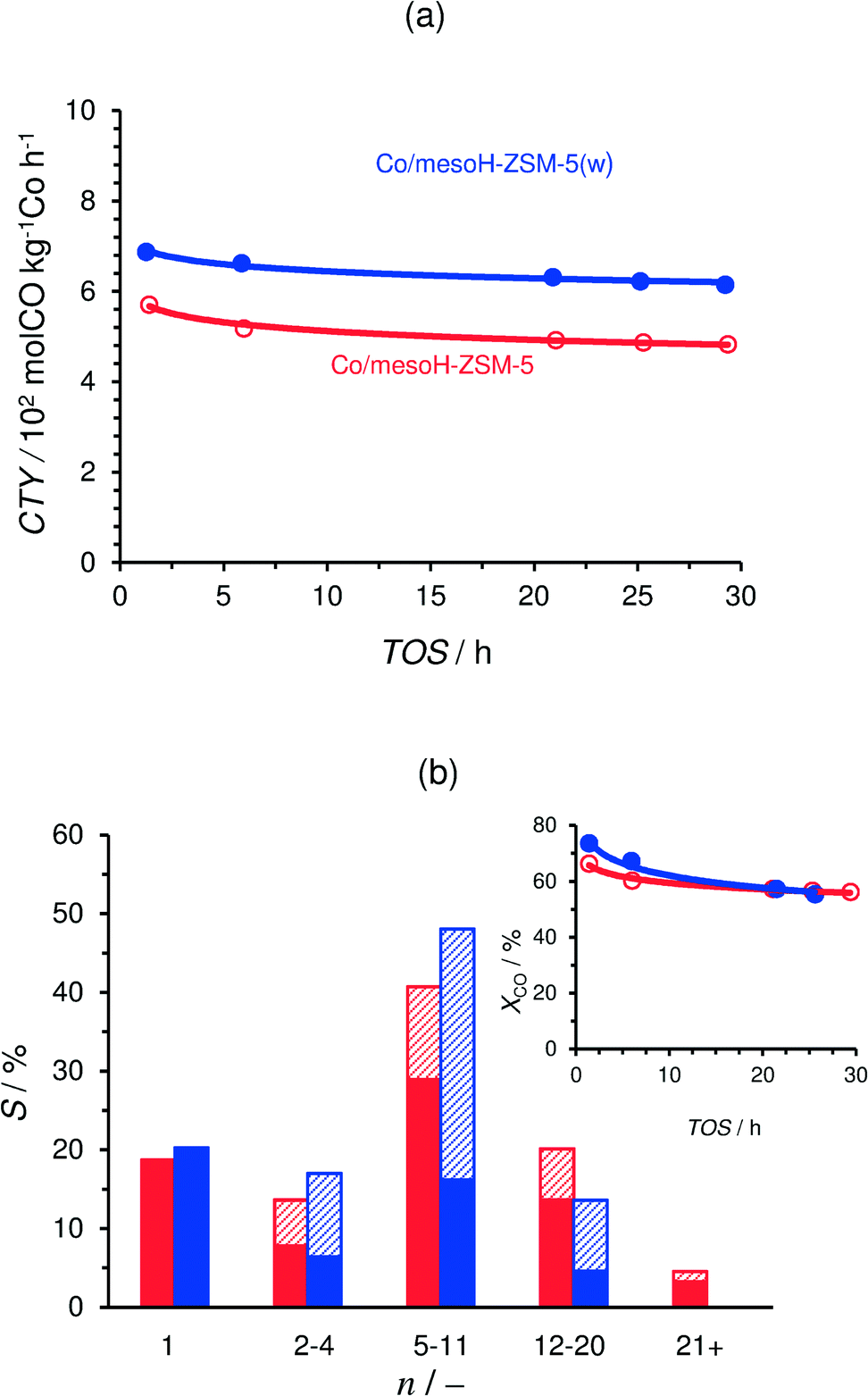 | ||
Fig. 8 Time-on-stream (TOS) evolution of the cobalt-time-yield (CTY) during FTS where GHSV = 12.0 mSTP3 kgcat−1 h−1 (a). Carbon selectivity of the FTS products after 20 h on-stream under iso-conversion conditions (as depicted in the inset) where GHSV = 12.0 mSTP3 kgcat−1 h−1 for Co/mesoH-ZSM-5 and 24 for Co/mesoH-ZSM-5(w) (b); in each carbon number group from left to right: Co/mesoH-ZSM-5 and Co/mesoH-ZSM-5-(w);  : n-paraffins, ///: sum of isoparaffins and olefins. Experiments were performed at 513 K, 15 bar, H2/CO = 2. : n-paraffins, ///: sum of isoparaffins and olefins. Experiments were performed at 513 K, 15 bar, H2/CO = 2. | ||
The olefin to paraffin ratio (O/P) of the C2–C8 hydrocarbons is highest over Co/mesoH-ZSM-5(w) and decreases by 24% for Co/mesoH-ZSM-5, under iso-conversion conditions (XCO = 57 and 55% for Co/mesoH-ZSM-5 and Co/mesoH-ZSM-5(w), respectively). On the other hand, the iso-to-normal ratio (I/N) of C4 increases by a factor of ten over the catalyst supported on acid-washed mesoporous zeolite compared to Co/mesoH-ZSM-5 (Table 4).
The molar product distribution over Co/mesoH-ZSM-5 and Co/mesoH-ZSM-5(w) under iso-conversion conditions is presented in Fig. 9. The molar fraction of the FTS hydrocarbons versus their carbon number follows a linear trend for the Co/mesoH-ZSM-5 representative for an ASF distribution with a chain growth probability (α) of 0.77. In contrast, the hydrocarbon distribution over Co/mesoH-ZSM-5(w) does not follow this linear trend. The ASF distribution breaks at around C13 and the molar fractions of higher hydrocarbons drop considerably. In line with the literature,5–8,11 these results with mesoH-ZSM-5(w) show that a close proximity of the acid sites and the Co sites clearly promotes acid-catalyzed reactions, i.e. cracking and isomerization, of the formed olefinic hydrocarbons.8
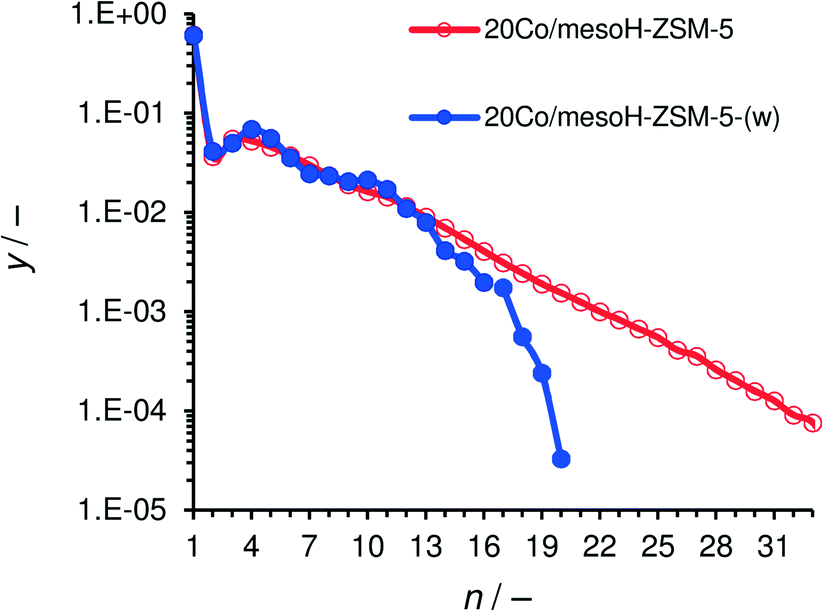 | ||
| Fig. 9 Molar distribution of the FTS products after 20 h on-stream at 513 K, 15 bar, H2/CO = 2 and iso-conversion conditions where GHSV = 24.0 mSTP3 kgcat−1 h−1 for Co/mesoH-ZSM-5-(w) and 12 for Co/mesoH-ZSM-5. | ||
Fig. 10 and 11 display the catalytic performance of the Co catalysts based on the carbon/zeolite composites. The time-on-stream (TOS) evolution of the CTY of these catalysts is depicted in Fig. 10. The CTY for Co/mesoH-ZSM-5(w) is included for comparison. The CTY increases with an increase in the carbon content (up to 15 wt%) of the employed support. These results further confirm that the weaker metal–support (zeolite) interactions and thus an enhanced reducibility of the Co species (Fig. 7) are the main origins of the enhanced catalytic activity of the catalysts supported over the carbon/zeolite composites compared to that of Co/mesoH-ZSM-5(w). Furthermore, the possible generation of cobalt carbide during catalyst preparation cannot be disregarded. Although cobalt carbide by itself is not active in FTS, the reduction of supported cobalt carbide promotes the formation of the hexagonal cobalt phase (hcp), which is more active than the face-centered cubic cobalt phase (fcc) in FTS.51,52 Sartipi et al.8 evidenced the presence of the hcp in Co supported over a carbon/zeolite composite by IR-assisted CO adsorption, which further supports this interpretation.
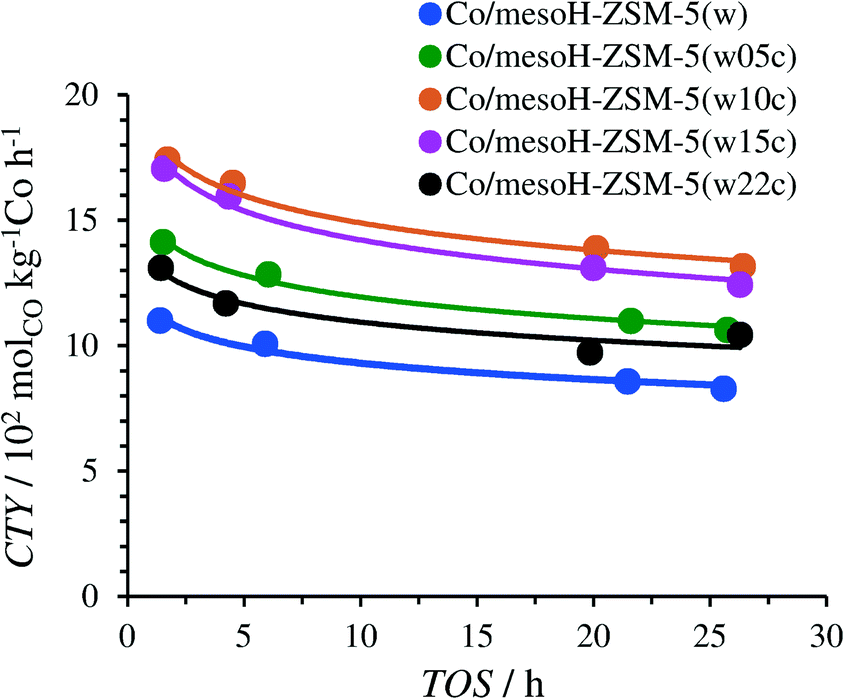 | ||
| Fig. 10 Time-on-stream (TOS) evolution of the cobalt-time-yield (CTY) during FTS at 513 K, 15 bar, H2/CO = 2 and GHSV = 24.0 mSTP3 kgcat−1 h−1. | ||
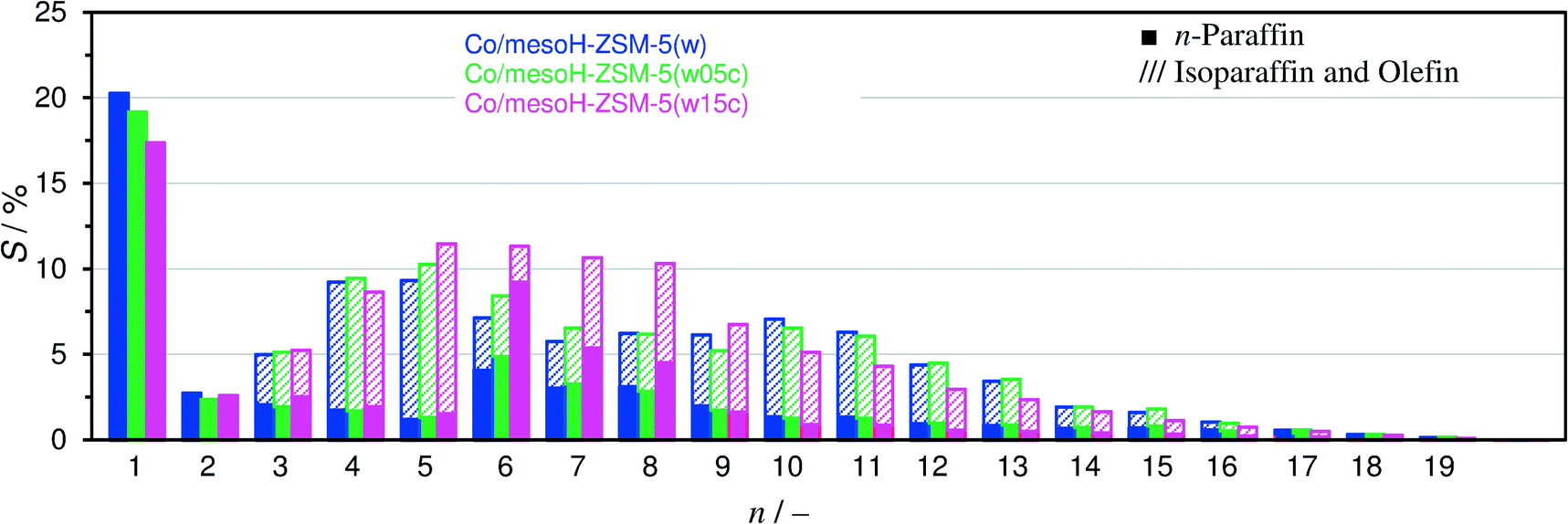 | ||
Fig. 11 Carbon selectivity of the FTS products after 20 h on-stream under iso-conversion conditions (XCO = 55 ± 3%). In each carbon number group from left to right: Co/mesoH-ZSM-5(w), Co/mesoH-ZSM-5(w05c) and Co/mesoH-ZSM-5(w15c);  : n-paraffins, ///: sum of isoparaffins and olefins. Experiments were performed at 513 K, 15 bar, H2/CO = 2 and GHSV = 24.0 mSTP3 kgcat−1 h−1. : n-paraffins, ///: sum of isoparaffins and olefins. Experiments were performed at 513 K, 15 bar, H2/CO = 2 and GHSV = 24.0 mSTP3 kgcat−1 h−1. | ||
The CTY of Co/mesoH-ZSM-5(w22c), however, decreases significantly compared to all the carbon/zeolite supported catalysts, which may be attributed to both the lower cobalt reduction degree (Table 3) and the lower dispersion of the active phase. The average size of the cobalt crystallites of the Co/mesoH-ZSM-5(w22c) sample was larger than that of the crystallites of Co/mesoHZSM5(w) (Table 2 and Fig. S2†).
Fig. 11 depicts the carbon selectivity of the FTS products after 20 h on-stream under iso-conversion conditions (see Fig. 10) for Co/mesoH-ZSM-5(w05c), Co/mesoH-ZSM-5(w15c) and Co/mesoH-ZSM-5(w), for comparison. For the Co/mesoH-ZSM-5(w15c) catalyst, the C5–C8 selectivity is higher and that of C10+ lower. Moreover, the selectivity to C1 decreases to 17%. On the other hand, both the O/P and I/N ratios (Table 4) decrease with an increase in the carbon content. For instance, the I/N ratio decreases by a factor of 4 over Co/mesoH-ZSM-5(w22c) compared to Co/mesoH-ZSM-5(w) (Table 4). Both the decrease in the concentration and strength of the acid sites of the carbon/zeolite support and the larger distance between the surface Co and these acid sites may decrease the chance of the primary olefinic FT products to adsorb on the acid isomerization and cracking sites. Hence, smaller amounts of branched hydrocarbons (isoparaffins) are produced over the carbon/zeolite composite-based catalysts. At the same time, if the acid site domains are far from Co, the formed hydrocarbons are more likely to be hydrogenated on a neighbouring metal site and higher amounts of paraffins may be also produced. The FTS selectivity using bifunctional catalysts is undoubtedly directed by proximity of the (de)hydrogenation and acid functions.53 For all catalysts the CO2 carbon selectivity is lower than 2% (Table 4), evidencing a very low water-gas shift activity under the applied reaction conditions.
The carbon containing catalysts display a similar break in the ASF distribution as Co/mesoHZSM-5(w) up to C19, but the molar fractions of the heavier hydrocarbons relatively increase with increasing carbon content (Fig. S5†). This is also attributed to the partial deactivation or blocking of the zeolite acid cracking sites by pyrolytic carbon deposition.
In summary, the high C5–C11 (gasoline fraction) selectivity of Co supported on mesoporous zeolites is attributed to both the close proximity of the Co crystallites and acid sites, which promote acid-catalyzed reactions (cracking and isomerization), and to an enhancement in the diffusion of hydrocarbons out of the catalyst particle when compared to the purely microporous catalyst particles.5–10,23 However, the selectivity to methane is higher than expected for the calculated chain growth probabilities, implying that direct CO hydrogenation and hydrogenolysis take place in addition to FTS. Recently, Sartipi et al.8,23 attributed the origin and nature of the above-mentioned hydrogenation and hydrogenolysis sites to the low-coordination Co sites, which seem to be stronger when Co is loaded on the H-ZSM-5 supports. This is also associated with the Co crystallite size, since the density of the lower index surface crystallographic planes or steps and corners increases as the Co crystallite size decreases.50 Apparently, in mesoporous H-ZSM-5 the formation of highly dispersed cobalt with such coordinatively unsaturated sites is promoted, in contrast to native HZSM-5 where larger Co crystallites are only formed at the outer surface of the crystals.8 This accounts for the high C1 selectivity observed in Fig. 8b and 11.
In the present work, the selectivity to C1–C2 is reduced after increasing the carbon content in the mesoporous zeolite support (Fig. 11), in spite of a reduction in the micropore and mesopore volumes.
This deposition of carbon on the zeolite support prior to impregnation seems to reduce the Co–support interactions as was evidenced by XPS and H2-TPR, resulting in the formation of larger Co crystallites (which are reducible at lower temperatures) and a lower selectivity towards C1 (and C2) is achieved. The partial deactivation of the Brønsted acid sites by pyrolytic carbon deposition, as was observed by NH3-TPD (Fig. 4), allows to tune the zeolite acidity, and thus the product distribution shifts to the heavier hydrocarbons as observed for Co/mesoH-ZSM-5(w15c) and Co/mesoH-ZSM-5(w22c) with respect to the Co/mesoH-ZSM-5(w) catalyst (Fig. 11 and S4†). This delicate relationship among the Co particle size, their reducibility, their interactions with the zeolite, their proximity to the acid sites and the strength of the acid sites demands a precise balance in order to be able to control the productivity and selectivity of this bifunctional FTS process.
Conclusions
A detailed characterization of carbon/zeolite composites prepared by deposition of pyrolytic carbon over mesoporous H-ZSM-5 and the utilization of these composites as the supports of Co for Fischer–Tropsch synthesis is reported. Propylene reacts on the surface of the zeolite depositing pyrolytic carbon in the micropores, on the walls of the mesopores and covering the external crystallite surface. The resulting composites display a promising catalytic performance in the direct synthesis of the gasoline range hydrocarbons from syngas, with higher selectivity to the C5–C9 products. The deposition of carbon on the zeolite support prior to Co impregnation improves considerably the reducibility of supported Co oxide, resulting in an enhanced catalytic activity and a lower selectivity towards C1 (and C2) during FTS. This is due to the lower Co–support interactions as revealed by TPR and XPS analyses. Both the O/P and I/N ratios in the FTS products are lower at an increasing carbon content due to the decrease in the concentration and strength of the acid sites of the carbon/zeolite support.Acknowledgements
The authors gratefully acknowledge the support of the Spanish Ministry of Economy and Competitiveness (MINECO) and FEDER (Project CTQ2012-36408) and the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. M. J. V. R. gratefully thanks the Spanish Ministry of Economy and Competitiveness (MINECO) for a research stay fellowship (EEBB-I-14-08633).References
- I. Wender, Reactions of synthesis gas, Fuel Process. Technol., 1996, 48, 189–297 CrossRef CAS.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaen, A. I. Dugulan, M. A. Nasalevich, H. U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Metal–organic framework-mediated synthesis of highly active and stable Fischer–Tropsch catalysts, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- A. d. Klerk and E. Furimsky, Catalysis in the Refining of Fischer–Tropsch Syncrude, Royal Society of Chemistry Publishing, Cambridge, UK, 2010 Search PubMed.
- R. Luque, A. R. de la Osa, J. M. Campelo, A. A. Romero, J. L. Valverde and P. Sanchez, Design and development of catalysts for Biomass-To-Liquid-Fischer–Tropsch (BTL-FT) processes for biofuels production, Energy Environ. Sci., 2012, 5, 5186–5202 CAS.
- J. C. Kang, K. Cheng, L. Zhang, Q. H. Zhang, J. S. Ding, W. Q. Hua, Y. C. Lou, Q. G. Zhai and Y. Wang, Mesoporous Zeolite-Supported Ruthenium Nanoparticles as Highly Selective Fischer–Tropsch Catalysts for the Production of C-5–C-11 Isoparaffins, Angew. Chem., Int. Ed., 2011, 50, 5200–5203 CrossRef CAS PubMed.
- K. Cheng, J. C. Kang, S. W. Huang, Z. Y. You, Q. H. Zhang, J. S. Ding, W. Q. Hua, Y. C. Lou, W. P. Deng and Y. Wang, Mesoporous Beta Zeolite-Supported Ruthenium Nanoparticles for Selective Conversion of Synthesis Gas to C-5–C-11 Isoparaffins, ACS Catal., 2012, 2, 441–449 CrossRef CAS.
- S. Sartipi, K. Parashar, M. Makkee, J. Gascon and F. Kapteijn, Breaking the Fischer–Tropsch synthesis selectivity: direct conversion of syngas to gasoline over hierarchical Co/H-ZSM-5 catalysts, Catal. Sci. Technol., 2013, 3, 572–575 CAS.
- S. Sartipi, K. Parashar, M. J. Valero-Romero, V. P. Santos, B. van der Linden, M. Makkee, F. Kapteijn and J. Gascon, Hierarchical H-ZSM-5-supported cobalt for the direct synthesis of gasoline-range hydrocarbons from syngas: Advantages, limitations, and mechanistic insight, J. Catal., 2013, 305, 179–190 CrossRef CAS.
- S. Sartipi, M. Alberts, M. J. Meijerink, T. C. Keller, J. Perez-Ramirez, J. Gascon and F. Kapteijn, Towards Liquid Fuels from Biosyngas: Effect of Zeolite Structure in Hierarchical-Zeolite-Supported Cobalt Catalysts, ChemSusChem, 2013, 6, 1646–1650 CrossRef CAS PubMed.
- S. Sartipi, M. Alberts, V. P. Santos, M. Nasalevich, J. Gascon and F. Kapteijn, Insights into the Catalytic Performance of Mesoporous H-ZSM-5-Supported Cobalt in Fischer–Tropsch Synthesis, ChemCatChem, 2014, 6, 142–151 CrossRef CAS.
- A. Martinez and G. Prieto, The Application of Zeolites and Periodic Mesoporous Silicas in the Catalytic Conversion of Synthesis Gas, Top. Catal., 2009, 52, 75–90 CrossRef CAS.
- B. Sun, M. H. Qiao, K. N. A. Fan, J. Ulrich and F. Tao, Fischer–Tropsch Synthesis over Molecular Sieve Supported Catalysts, ChemCatChem, 2011, 3, 542–550 CrossRef CAS.
- J. C. Groen, W. D. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Perez-Ramirez, Direct demonstration of enhanced diffusion in mesoporous ZSM-5 zeolite obtained via controlled desilication, J. Am. Chem. Soc., 2007, 129, 355–360 CrossRef CAS PubMed.
- S. Sartipi, J. E. van Dijk, J. Gascon and F. Kapteijn, Toward bifunctional catalysts for the direct conversion of syngas to gasoline range hydrocarbons: H-ZSM-5 coated Co versus H-ZSM-5 supported Co, Appl. Catal., A, 2013, 456, 11–22 CrossRef CAS.
- J. A. Martens, D. Verboekend, K. Thomas, G. Vanbutsele, J. P. Gilson and J. Perez-Ramirez, Hydroisomerization of Emerging Renewable Hydrocarbons using Hierarchical Pt/H-ZSM-22 Catalyst, ChemSusChem, 2013, 6, 421–425 CrossRef CAS PubMed.
- J. C. Groen, J. A. Moulijn and J. Perez-Ramirez, Desilication: on the controlled generation of mesoporosity in MFI zeolites, J. Mater. Chem., 2006, 16, 2121–2131 RSC.
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Generation, characterization, and impact of mesopores in zeolite catalysts, Catal. Rev.: Sci. Eng., 2003, 45, 297–319 CAS.
- A. Corma, M. J. Diaz-Cabanas, J. L. Jorda, C. Martinez and M. Moliner, High-throughput synthesis and catalytic properties of a molecular sieve with 18-and 10-member rings, Nature, 2006, 443, 842–845 CrossRef CAS PubMed.
- G. T. Vuong and T. O. Do, A new route for the synthesis of uniform nanozeolites with hydrophobic external surface in organic solvent medium, J. Am. Chem. Soc., 2007, 129, 3810–3811 CrossRef CAS PubMed.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Stable single-unit-cell nanosheets of zeolite MFI as active and long-lived catalysts, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- K. Egeblad, C. H. Christensen, M. Kustova and C. H. Christensen, Templating mesoporous zeolites, Chem. Mater., 2008, 20, 946–960 CrossRef CAS.
- R. Chal, C. Gerardin, M. Bulut and S. van Donk, Overview and Industrial Assessment of Synthesis Strategies towards Zeolites with Mesopores, ChemCatChem, 2011, 3, 67–81 CrossRef CAS.
- S. Sartipi, M. Makkee, F. Kapteijn and J. Gascon, Catalysis engineering of bifunctional solids for the one-step synthesis of liquid fuels from syngas: a review, Catal. Sci. Technol., 2014, 4, 893–907 CAS.
- J. Rodriguez-Mirasol, T. Cordero, L. R. Radovic and J. J. Rodriguez, Structural and textural properties of pyrolytic carbon formed within a microporous zeolite template, Chem. Mater., 1998, 10, 550–558 CrossRef CAS.
- T. Kyotani, Z. X. Ma and A. Tomita, Template synthesis of novel porous carbons using various types of zeolites, Carbon, 2003, 41, 1451–1459 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of gases in multimolecular layers, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- M. M. Dubinin, E. D. Zaverina and L. V. Radushkevich, Sorption and structure of active carbons. Adsorption of organic vapors, Zh. Fiz. Khim., 1947, 21, 1351–1362 CAS.
- K. Kaneko and C. Ishii, Superhigh Surface-Area Determination of Microporous Solids, Colloids Surf., 1992, 67, 203–212 CrossRef CAS.
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and V. L. Zholobenko, Pore size effects in Fischer–Tropsch synthesis over cobalt-supported mesoporous silicas, J. Catal., 2002, 206, 230–241 CrossRef CAS.
- J. Landers, G. Y. Gor and A. V. Neimark, Density functional theory methods for characterization of porous materials, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS.
- D. Schanke, S. Vada, E. A. Blekkan, A. M. Hilmen, A. Hoff and A. Holmen, Study of Pt-Promoted Cobalt Co Hydrogenation Catalysts, J. Catal., 1995, 156, 85–95 CrossRef CAS.
- S. Sartipi, H. Jansma, D. Bosma, B. Boshuizen, M. Makkee, J. Gascon and F. Kapteijn, Six-flow operations for catalyst development in Fischer–Tropsch synthesis: Bridging the gap between high-throughput experimentation and extensive product evaluation, Rev Sci Instrum, 84, 2013 Search PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Reporting Physisorption Data for Gas Solid Systems with Special Reference to the Determination of Surface-Area and Porosity (Recommendations 1984), Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- D. Verboekend, G. Vile and J. Perez-Ramirez, Mesopore Formation in USY and Beta Zeolites by Base Leaching: Selection Criteria and Optimization of Pore-Directing Agents, Cryst. Growth Des., 2012, 12, 3123–3132 CAS.
- S. Mitchell, N. L. Michels, K. Kunze and J. Perez-Ramirez, Visualization of hierarchically structured zeolite bodies from macro to nano length scales, Nat. Chem., 2012, 4, 825–831 CrossRef CAS PubMed.
- S. Abello, A. Bonilla and J. Perez-Ramirez, Mesoporous ZSM-5 zeolite catalysts prepared by desilication with organic hydroxides and comparison with NaOH leaching, Appl. Catal., A, 2009, 364, 191–198 CrossRef CAS.
- J. Garrido, A. Linaressolano, J. M. Martin Martinez, M. Molinasabio, F. Rodriguezreinoso and R. Torregrosa, Use of N2 vs CO2 in the Characterization of Activated Carbons, Langmuir, 1987, 3, 76–81 CrossRef CAS.
- D. Cazorla Amoros, J. Alcaniz Monge and A. Linares Solano, Characterization of activated carbon fibers by CO2 adsorption, Langmuir, 1996, 12, 2820–2824 CrossRef CAS.
- H. van Koningsveld, J. C. Jansen and H. van Bekkum, The Monoclinic Framework Structure of Zeolite H-ZSM-5 - Comparison with the Orthorhombic Framework of as-Synthesized ZSM-5, Zeolites, 1990, 10, 235–242 CrossRef CAS.
- M. S. Holm, M. K. Hansen and C. H. Christensen, “One-Pot” Ion-Exchange and Mesopore Formation During Desilication, Eur. J. Inorg. Chem., 2009, 1194–1198 CrossRef CAS.
- A. Y. Khodakov, R. Bechara and A. Griboval-Constant, Fischer–Tropsch synthesis over silica supported cobalt catalysts: mesoporous structure versus cobalt surface density, Appl. Catal., A, 2003, 254, 273–288 CrossRef CAS.
- T. J. Fu, Y. H. Jiang, J. Lv and Z. H. Li, Effect of carbon support on Fischer–Tropsch synthesis activity and product distribution over Co-based catalysts, Fuel Process. Technol., 2013, 110, 141–149 CrossRef CAS.
- Practical surface analysis, 2nd edn., vol I, auger and X-ray photoelectron spectroscopy, ed. D. Briggs and M. P. Seah, Wiley, Chichester, UK, 1990 Search PubMed.
- A. L. C. Pereira, J. M. Gonzalez-Carballo, F. J. Perez-Alonso, S. Rojas, J. L. G. Fierro and M. D. Rangel, Effect of the Mesostructuration of the Beta Zeolite Support on the Properties of Cobalt Catalysts for Fischer–Tropsch Synthesis, Top. Catal., 2011, 54, 179–189 CrossRef CAS.
- Y. Brik, M. Kacimi, M. Ziyad and F. Bozon-Verduraz, Titania-supported cobalt and cobalt-phosphorus catalysts: Characterization and performances in ethane oxidative dehydrogenation, J. Catal., 2001, 202, 118–128 CrossRef CAS.
- V. I. Nefedov, M. N. Firsov and I. S. Shaplygin, Electronic-Structures of MRhO2, MRh2O4, RhMO4 and Rh2MO6 on the Basis of X-Ray Spectroscopy and Esca Data, J. Electron Spectrosc. Relat. Phenom., 1982, 26, 65–78 CrossRef CAS.
- A. Martinez, C. Lopez, F. Marquez and I. Diaz, Fischer-Tropsch synthesis of hydrocarbons over mesoporous Co/SBA-15 catalysts: the influence of metal loading, cobalt precursor, and promoters, J. Catal., 2003, 220, 486–499 CrossRef CAS.
- P. Arnoldy and J. A. Moulijn, Temperature-Programmed Reduction of CoO/Al2O3 Catalysts, J. Catal., 1985, 93, 38–54 CrossRef CAS.
- Y. Okamoto, K. Nagata, T. Adachi, T. Imanaka, K. Inamura and T. Takyu, Preparation and Characterization of Highly Dispersed Cobalt Oxide and Sulfide Catalysts Supported on SiO2, J. Phys. Chem., 1991, 95, 310–319 CrossRef CAS.
- G. Prieto, A. Martinez, P. Concepcion and R. Moreno-Tost, Cobalt particle size effects in Fischer–Tropsch synthesis: structural and in situ spectroscopic characterisation on reverse micelle-synthesised Co/ITQ-2 model catalysts, J. Catal., 2009, 266, 129–144 CrossRef CAS.
- O. Ducreux, B. Rebours, J. Lynch, M. Roy-Auberger and D. Bazin, Microstructure of Supported Cobalt Fischer–Tropsch Catalysts, Oil Gas Sci. Technol., 2009, 64, 49–62 CrossRef CAS.
- D. I. Enache, B. Rebours, M. Roy-Auberger and R. Revel, In situ XRD study of the influence of thermal treatment on the characteristics and the catalytic properties of cobalt-based Fischer–Tropsch catalysts, J. Catal., 2002, 205, 346–353 CrossRef CAS.
- G. V. Jovana Zečević, K. P. de Jong and J. A. Martens, Nanoscale intimacy in bifunctional catalysts for selective conversion of hydrocarbons, Nat. Chem., 2015, 528, 245 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01942g |
| This journal is © The Royal Society of Chemistry 2016 |