
Molybdenum-doped α-MnO2 as an efficient reusable heterogeneous catalyst for aerobic sulfide oxygenation†
Tsubasa
Uematsu
,
Yumi
Miyamoto
,
Yoshiyuki
Ogasawara
,
Kosuke
Suzuki
,
Kazuya
Yamaguchi
and
Noritaka
Mizuno
*
Department of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: tmizuno@mail.ecc.u-tokyo.ac.jp
First published on 14th October 2015
Abstract
Oxygenation of sulfides to sulfoxides and/or sulfones is an important transformation, and the development of efficient heterogeneous catalysts for oxygenation, which can utilize O2 as the terminal oxidant, is highly desired. In this study, we have successfully developed manganese oxide-based efficient heterogeneous catalysts for aerobic oxygenation of sulfides. Firstly, we prepared four kinds of manganese oxides possessing different crystal structures, such as α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2, and their structure–activity relationships were examined for the aerobic oxygenation of thioanisole. Amongst them, α-MnO2 showed the best catalytic performance for the oxygenation. Moreover, α-MnO2 was highly stable during the catalytic oxygenation possibly due to the tunnel K+ ions. In order to further improve the catalytic performance of α-MnO2, substitutional doping of transition metal cations, such as Mo6+, V5+, Cr3+, and Cu2+, into the framework was carried out. Undoped α-MnO2 possessed a fibrous morphology. When high-valent transition metal cations were doped, especially Mo6+, the lengths of the fibers drastically shortened to form grain-like aggregates of ultrafine nanocrystals, resulting in an increase in specific surface areas and the numbers of catalytically active surface sites. In the presence of Mo6+-doped α-MnO2 (Mo–MnO2), various kinds of sulfides could efficiently be oxidized to the corresponding sulfoxides as the major products. The observed catalysis was truly heterogeneous, and Mo–MnO2 could repeatedly be reused while keeping its high catalytic performance. Besides sulfide oxygenation, Mo–MnO2 could efficiently catalyze several aerobic oxidative functional group transformations through single-electron transfer oxidation processes, namely, oxygenation of alkylarenes, oxidative α-cyanation of trialkylamines, and oxidative S-cyanation of benzenethiols.
Introduction
The development of highly efficient catalytic systems for oxygenation of sulfides has attracted much attention because the corresponding oxygenation products, both sulfoxides and sulfones, have widely been utilized as oxygen-transfer reagents, ligands in asymmetric catalysis, and important synthetic intermediates for natural products, biologically important molecules, and functional materials.1 In addition, oxidative desulfurization has emerged as a promising method for removal of sulfur compounds from fuels and industrial effluents.1g,h Generally, oxygenation of sulfides has been performed using metal oxide-, peracid-, or oxoacid-based stoichiometric oxidants. Instead of using such stoichiometric oxidants, the choice of greener oxidants, such as H2O2 and O2, in combination with suitable catalysts has recently become the mainstream for oxygenation of sulfides.Up to the present, a number of efficient transition metal catalysts have been developed for H2O2-based oxygenation of sulfides.2 Importantly, the use of ubiquitous and the greenest O2 is more desirable from the standpoint of green chemistry. Although several catalytic systems for aerobic oxygenation of sulfides have been reported to date,3 they have several disadvantages in some cases; for example, (i) sacrificial reductants, e.g., aldehydes, are required, (ii) photo-irradiation is required, and/or (iii) most of them are homogeneous systems, and thus recovery and reuse of catalysts are very difficult.3 As far as we know, there are only a few reports on heterogeneously catalyzed oxygenation of sulfides using O2 as the terminal oxidant without any additives and photoirradiation; polyoxometalates,4a MoS2/Ta3N5,4b and Au/MnO2−x (ref. 4c) are examples of the reported heterogeneous catalysts. However, there is still quite a bit of room for improvement of their catalytic activities, selectivities, substrate scopes, and in particular reusabilities. Therefore, the development of reliable heterogeneous catalysts is still an important and challenging research subject.
Manganese oxides, including mixed oxides, have attracted tremendous research interests because of their potential use as electrode materials, magnetic materials, adsorbents, oxidants, catalysts, electrocatalysts, and catalyst supports.5 In laboratory scale organic synthesis, manganese oxides have traditionally been utilized as stoichiometric oxidants. In recent years, the use of manganese oxides as heterogeneous catalysts for aerobic oxidation reactions has received a lot of attention, and several efficient systems have been developed to date.6 For example, α-MnO2, including a manganese oxide octahedral molecular sieve (OMS-2), has been typically utilized for oxidative dehydrogenation and single-electron transfer oxidation processes, such as oxidative dehydrogenation of alcohols and its related transformations,6c–e oxygenation of alkylarenes,6f oxidative α-cyanation of tertiary amines,6g and oxidative homocoupling of thiols and its related transformations.6h,i We have recently developed an aerobic oxidative amidation reaction of primary alcohols using aqueous NH3 as the nitrogen source.6j–l For the oxidative amidation, α-MnO2 (OMS-2) showed high catalytic performance, while β-MnO2, γ-MnO2, and δ-MnO2 were not effective. For the oxidation of formaldehyde, the structure–activity relationships were carefully examined using α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2, and amongst them, δ-MnO2 was found to be the most active catalyst.6m In these manners, dramatic differences in catalytic performances among manganese oxides with different crystal structures have been observed with considerable frequency. Thus, the choice of kinds of manganese oxides for the target reaction is very crucial. In addition, the redox stability is also a very important factor for the catalytic use and repeated reuse of manganese oxides.
In this study, we prepared four kinds of manganese oxides with α-, β-, γ-, and δ-phase structures, and their structure–activity relationships were examined for the aerobic oxygenation of thioanisole. The difference in catalytic performance was clearly observed among them; α-MnO2 showed the best catalytic performance and was highly stable during oxygenation under aerobic and even anaerobic conditions. Furthermore, the performance of α-MnO2 could be much improved by substitutional doping of Mo6+ into the octahedral framework. In the presence of Mo6+-doped α-MnO2 (Mo–MnO2), various kinds of structurally diverse sulfides including aromatic and aliphatic ones could efficiently be oxidized to the corresponding sulfoxides as the major products. The observed catalysis was truly heterogeneous, and Mo–MnO2 could be reused for the oxygenation of thioanisole at least four times without an appreciable loss of its high catalytic performance. The structure of Mo–MnO2 was intrinsically preserved after repeated reuse for oxygenation. Besides sulfide oxygenation, Mo–MnO2 could act as an efficient heterogeneous catalyst for several aerobic oxidative functional group transformations through single-electron transfer oxidation processes, such as oxygenation of alkylarenes, oxidative α-cyanation of trialkylamines, and oxidative S-cyanation of benzenethiols.
Results and discussion
Structure–activity relationships for thioanisole oxygenation
To begin with, we prepared four kinds of manganese oxides with α-, β-, γ-, and δ-phase structures according to previously reported procedures with minor modifications (see the Experimental section).6k,m,7 Their X-ray powder diffraction (XRD) patterns are shown in Fig. 1. The XRD pattern of α-MnO2 could be well indexed to a cryptomelane-type manganese oxide with a 2 × 2 tunnel (4.6 Å × 4.6 Å) (JCPDS 29-1020). The XRD patterns of β-MnO2 and γ-MnO2 were in good agreement with those of pyrolusite-type (JCPDS 24-0735) and nsutite-type (JCPDS 14-0644) manganese oxides, respectively. The XRD pattern of δ-MnO2 was characteristic of a birnessite-type manganese oxide with a layered structure (JCPDS 43-1456). Therefore, four types of manganese oxides with different crystal structures were successfully prepared. The peak widths and intensities of these XRD patterns imply that α-MnO2 and β-MnO2 possess relatively high crystallinities in comparison with γ-MnO2 and δ-MnO2. The specific surface areas of α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2 were 80 m2 g−1, 18 m2 g−1, 73 m2 g−1, and 124 m2 g−1, respectively (Table 1). The manganese contents in α-MnO2 (52.5 wt%) and δ-MnO2 (47.4 wt%) were somewhat lower than those in β-MnO2 (64.3 wt%) and γ-MnO2 (62.0 wt%) (Table 1), which is mainly due to the accommodation of K+ ions into the 2 × 2 tunnel of α-MnO2 or into the interlayer of δ-MnO2.8 The average oxidation state (AOS, determined by redox titration) of α-MnO2 was 3.77, slightly lower than those of the others (3.90–3.92) (Table 1), suggesting the compensation of the mixed valency of manganese by the tunnel K+ ions (or H+).8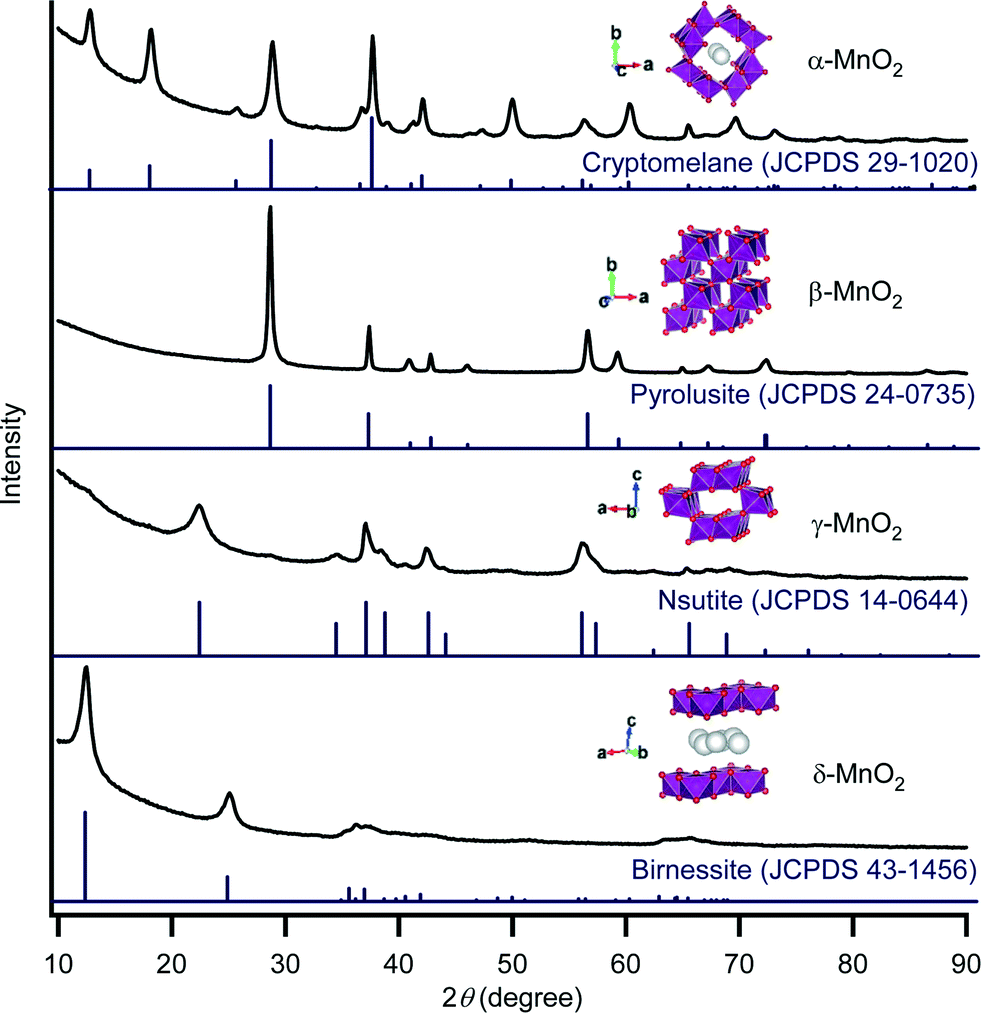 | ||
| Fig. 1 XRD patterns and polyhedral representations of α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2. Purple octahedra represent {Mn3+O6} or {Mn4+O6}. Red spheres represent oxygen atoms. White spheres represent accommodated K+ ions or water molecules. | ||
| Entry | Catalyst | BET surface area (m2 g−1) | Content (wt%) | AOS | |
|---|---|---|---|---|---|
| Mn | K | ||||
| 1 | α-MnO2 | 80 | 52.5 | 4.07 | 3.77 |
| 2 | β-MnO2 | 18 | 64.3 | — | 3.90 |
| 3 | γ-MnO2 | 73 | 62.0 | — | 3.90 |
| 4 | δ-MnO2 | 124 | 47.4 | 7.78 | 3.92 |
By using these manganese oxide catalysts, their structure–activity relationships were examined for the oxygenation of thioanisole (1a). The reactions were performed in o-dichlorobenzene at 150 °C (bath temperature) under aerobic (O2: 5 atm) or anaerobic (Ar: 1 atm) conditions. The results are summarized in Table 2. Under aerobic conditions, the oxygenation selectively proceeded to afford methyl phenyl sulfoxide (2a) with trace amounts of methyl phenyl sulfone (3a) in all cases, and the order of the catalytic performances from the viewpoint of the product yields was as follows: α-MnO2 (41%) ≈ γ-MnO2 (42%) > δ-MnO2 (29%) > β-MnO2 (12%) (the values in the parentheses are the total yields of 2a and 3a; Table 2, entries 1, 3, 5, and 7). The yields did not simply increase with increasing specific surface areas. The low catalytic performance of β-MnO2 is probably due to its low specific surface area. Under anaerobic conditions, the oxygenation hardly proceeded when α-MnO2 and δ-MnO2 (Table 2, entries 2 and 8) were used, indicating that O2 is effectively utilized as the terminal oxidant in these cases. In other words, the electron-transfer from the reduced manganese species to O2 can smoothly proceed in the case of α-MnO2 and δ-MnO2. In the case of γ-MnO2, 2a was produced in a significant amount (11%) even under anaerobic conditions (Table 2, entry 6), thus suggesting that γ-MnO2 can effectively utilize its lattice oxygen species for the oxygenation of 1a in addition to the use of O2.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalysta (mol%) | Atmosphereb | Time (h) | Conv. of 1a (%) | Yield (%) | |
| 2a | 3a | |||||
| Reaction conditions: catalyst (25 mg), 1a (0.5 mmol), o-dichlorobenzene (1 mL), 150 °C (bath temp.).a The values in the parentheses are based on the surface exposed manganese species estimated from the specific surface areas and the crystal structures.b O2 (5 atm) or Ar (1 atm).c Reuse experiments. These experiments used the retrieved catalyst. Conversions and yields were determined by GC analysis using naphthalene as an internal standard. | ||||||
| 1 | α-MnO2 (9.8) | O2 | 24 | 45 | 40 | 1 |
| 2 | α-MnO2 (9.8) | Ar | 96 | 4 | 4 | <1 |
| 3 | β-MnO2 (2.3) | O2 | 24 | 15 | 12 | <1 |
| 4 | β-MnO2 (2.3) | Ar | 96 | 6 | 1 | <1 |
| 5 | γ-MnO2 (9.0) | O2 | 24 | 43 | 41 | 1 |
| 6 | γ-MnO2 (9.0) | Ar | 96 | 18 | 11 | <1 |
| 7 | δ-MnO2 (14.1) | O2 | 24 | 34 | 24 | 5 |
| 8 | δ-MnO2 (14.1) | Ar | 96 | 9 | <1 | <1 |
| 9c | α-MnO2 | O2 | 24 | 41 | 36 | 1 |
| 10c | β-MnO2 | O2 | 24 | 17 | 13 | <1 |
| 11c | γ-MnO2 | O2 | 24 | 47 | 25 | <1 |
| 12c | δ-MnO2 | O2 | 24 | 36 | 30 | 2 |
The XRD patterns of these manganese oxides retrieved after the oxygenation of 1a under the conditions described in Table 2 were measured in order to confirm their redox stabilities. With regard to α-MnO2, the peak positions, widths, and intensities were almost unchanged after its use for the oxygenation under both aerobic and anaerobic conditions, as shown in Fig. 2. Thus, the crystal structure and crystallinity of α-MnO2 were specifically preserved after the oxygenation. Moreover, α-MnO2 could be reused for the oxygenation of 1a without an appreciable loss of its catalytic performance (Table 2, entry 9). Although the peak positions were almost unchanged after the use of δ-MnO2 for the oxygenation, the peak intensities significantly decreased, suggesting that the crystallinity of δ-MnO2 was lowered after the oxygenation (Fig. S1†). In contrast, different phases and significant peak shifts were observed after the oxygenation in the case of β-MnO2 and γ-MnO2 (Fig. S1†). In particular, γ-MnO2 was partly converted into a redox inactive manganite phase (MnO(OH), JCPDS 41-1379) after its use under anaerobic conditions (Fig. S1†). Even when γ-MnO2 was utilized for the aerobic oxygenation, the XRD peaks significantly shifted toward lower angles (Fig. S1†), suggesting the occurrence of lattice enlargement due to the reduction of manganese species. In addition, we confirmed that the AOS of γ-MnO2 significantly decreased from 3.90 to 3.42 even after its use for the aerobic oxygenation (Table S1†). These analyses imply that the redox stabilities for the oxygenation of 1a decrease in the order of α-MnO2 > δ-MnO2 > β-MnO2 > γ-MnO2. Manganese oxides possessing the accommodated K+ ions, such as α-MnO2 and δ-MnO2, showed relatively high redox stabilities. Temperature-programmed reduction (TPR) measurements using H2 were performed for these manganese oxide samples. The peaks in the TPR profiles correspond to the sequential reduction of MnO2, that is, MnO2 → Mn2O3 → Mn3O4 → MnO. The TPR measurements revealed that the reduction temperature increased in the order of γ-MnO2 < β-MnO2 < δ-MnO2 < α-MnO2 (Fig. S2†). This result showed that the reducibility sequence is γ-MnO2 > β-MnO2 > δ-MnO2 > α-MnO2, which is closely correlated with the redox stabilities.
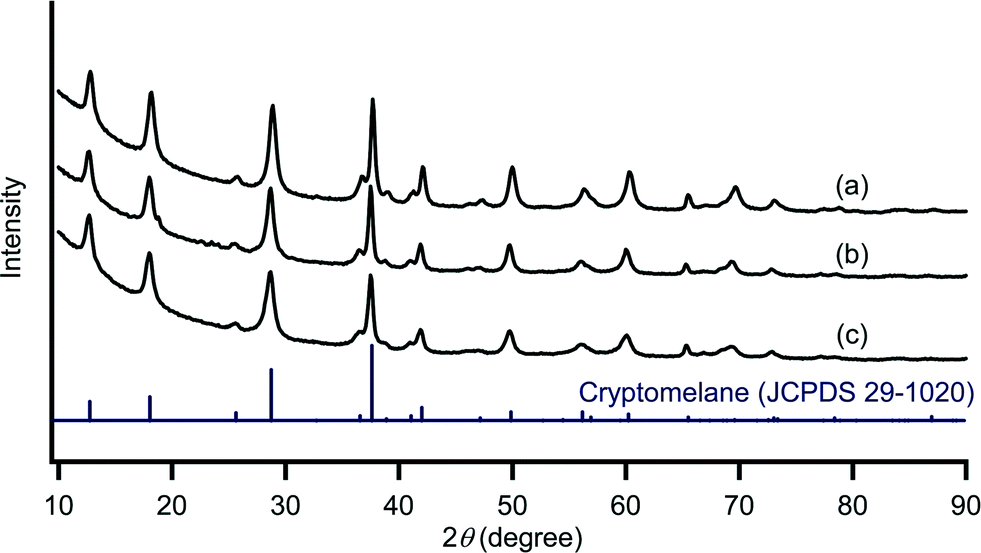 | ||
| Fig. 2 XRD patterns of (a) as-prepared α-MnO2, (b) α-MnO2 retrieved after the oxygenation of 1a under aerobic conditions, and (c) α-MnO2 retrieved after the oxygenation of 1a under anaerobic conditions. | ||
As mentioned above, γ-MnO2 can act as an effective “oxidant” likely because its lattice oxygen species can effectively be utilized for the oxygenation of 1a (Table 2, entry 6). The result of TPR analysis of γ-MnO2 also supports this idea (Fig. S2†). From the data of the oxygenations under aerobic and anaerobic conditions (Table 2, entries 5 and 6), the AOSs of γ-MnO2 before and after the oxygenation (Table S1†), and the elemental analysis (Table 1), we estimated that the production of 2avia the “stoichiometric” oxygenation using the lattice oxygen species was 12% and the production of 2avia the “catalytic” oxygenation using O2 was 29% in the case of γ-MnO2. However, once the active lattice oxygen species were consumed for the oxygenation, their regeneration (reoxidation) using O2 was quite difficult under the present conditions, resulting in the formation of reduced redox inactive phases, e.g., manganite phase. In addition, when γ-MnO2 was reused, a significant decrease in performance was observed; the yield of 2a dropped to 25% (Table 2, entry 11). Therefore, the catalytic use and repeated recycling of γ-MnO2 for the oxygenation are difficult. We consider that the redox stability of manganese oxides is one of the most important factors for their catalytic use for this type of oxygenation and that the accommodated K+ ions would play an important role in the stabilization of the manganese oxide framework structures. Indeed, in the case of stable α-MnO2, the yield of 2a for the reuse experiment was almost the same as that of the first run with as-prepared fresh α-MnO2 (Table 2, entries 1 and 9). As mentioned above, α-MnO2 gave 2a mostly through the catalytic oxygenation using O2. Consequently, α-MnO2 was the best catalyst for the oxygenation of 1a among the manganese oxides examined from the viewpoints of both the product yield (catalytic activity) and the redox stability. In order to further improve the performance of α-MnO2, substitutional doping of additional transition metal cations into the framework was next carried out.
Substitutional doping into the framework of α-MnO2
Substitutional doping of additional metal cations into the metal oxide framework is one of the most commonly utilized strategies to improve the catalytic properties. To date, substitutional doping of various transition metal cations into the framework of α-MnO2 (including OMS-2) has been studied.9 Doping of metal cations into α-MnO2 possibly occurs in the octahedral framework and/or in the tunnel, which is largely dependent on the crystal radii (CR) and the coordination geometries of the dopant cations. Six-coordinated cations with similar sizes to those of Mn4+ (the Shannon–Prewitt CR:10 0.67 Å) and Mn3+ (0.72 Å for low spin species, 0.785 Å for high spin species) can readily be introduced into the octahedral framework, while relatively larger cations tend to be introduced into the tunnel.11a,b,e,g In this study, we attempted to introduce several transition metal cations with different CR and valences, that is, Mo6+, V5+, Cr3+, and Cu2+. According to previous reports, the following substitution patterns can be expected.9j,11 With regard to six-coordinated high-valent dopant cations, such as Mo6+ (0.73 Å) and V5+ (0.68 Å), framework substitution may occur, which likely causes the formation of manganese vacancies.9j,l,11c,d On the other hand, when doping of six-coordinated low-valent cations, such as Cr3+ (0.755 Å) and Cu2+ (0.87 Å), is performed, the net negative charge of the α-MnO2 octahedral framework increases, and thereby protonation of oxygen atoms to form surface OH species and/or increase in amounts of tunnel cations would occur for charge compensation.9j,11a There is some possibility of introducing relatively larger Cu2+ (0.87 Å) into the tunnel of α-MnO2.11f,gFour kinds of metal-doped α-MnO2 catalysts (given in the format: M–MnO2) were prepared essentially by the same procedure for α-MnO2; 5 mol% (with respect to total metal content) precursor solutions of K2MoO4, NaVO3, Cr(CH3COO)3·H2O, and CuSO4·5H2O were utilized for the preparation of Mo–MnO2, V–MnO2, Cr–MnO2, and Cu–MnO2, respectively (see the Experimental section). As shown in Fig. 3, the XRD patterns of M–MnO2 were specifically the same as that of α-MnO2, and no additional peaks attributed to segregated phases of MoO3, V2O5, Cr2O3, and CuO were observed. The XRD peak intensities of Mo–MnO2 were slightly weaker than those of the others (Fig. 3), suggesting the low crystallinity and/or the formation of smaller crystals of Mo–MnO2. Fig. 4 shows the Raman scattering spectra of α-MnO2 and M–MnO2. All the observed Raman bands could be attributed to the Mn–O lattice vibrations within the MnO6 octahedral double chains in α-MnO2, and the strong bands typically observed for the segregated phases of MoO3 (around 820 cm−1), V2O5 (around 990 cm−1), Cr2O3 (around 550 cm−1), and CuO (around 290 cm−1) were not detected.12 The Raman band around 390 cm−1 was assignable to the Mn–O bending vibrations.8a The two intense bands around 575 cm−1 and 635 cm−1 were attributed to the symmetric Mn–O vibrations, thus indicating the formation of a well-developed tetragonal structure with an interstitial space consisting of 2 × 2 tunnels in these M–MnO2 samples.8a It has been reported that the Raman band around 635 cm−1 is related to the Mn–O vibrations perpendicular to the direction of the MnO6 octahedral double chains and that this band is significantly damped by the presence of heavy tunnel cations.8a As shown in Fig. 4, the relative intensities of the bands around 575 cm−1 and 635 cm−1 for M–MnO2 were almost the same as those for α-MnO2, indicating that these dopant cations are introduced not into the tunnels but mostly into the octahedral frameworks. From these XRD and Raman analyses, we consider that all four M–MnO2 samples possess pure cryptomelane-type phases with dopant cations in their octahedral frameworks. The contents of the dopant cations in Mo–MnO2 and V–MnO2 were 5.0 mol% and 5.2 mol% (Table 3), respectively, the same as those of the precursor solutions (5 mol%). In contrast, the contents of the dopant cations in Cr–MnO2 and Cu–MnO2 were 2.0 mol% and 2.5 mol%, respectively (Table 3), smaller than those of the precursor solutions (5 mol%). This is likely because of the difference in the substitution patterns of the dopant cations, as mentioned above. The contents of potassium in these M–MnO2 were 3.53–4.15 wt% (Table 3).
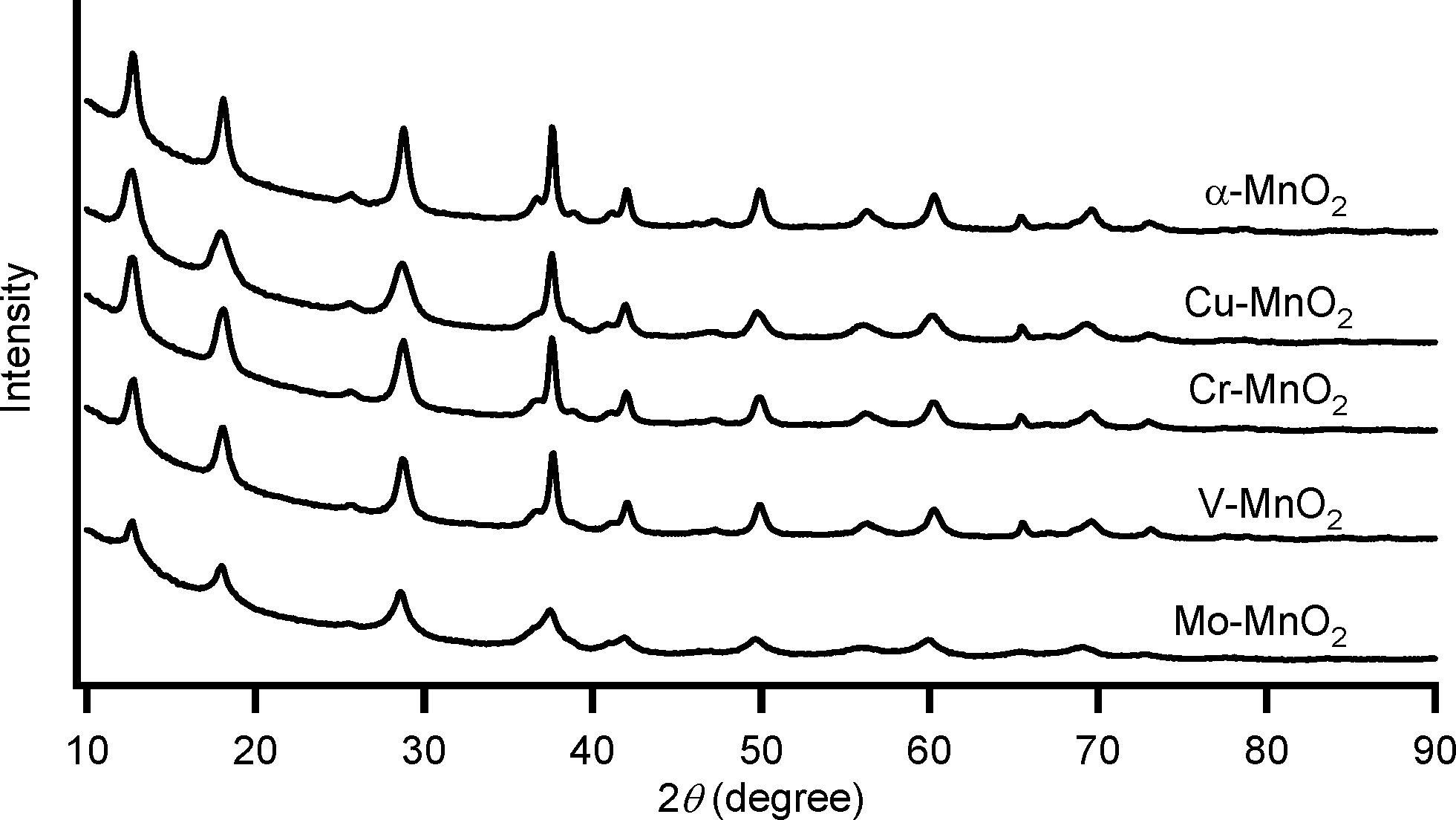 | ||
| Fig. 3 XRD patterns of α-MnO2 and M–MnO2. | ||
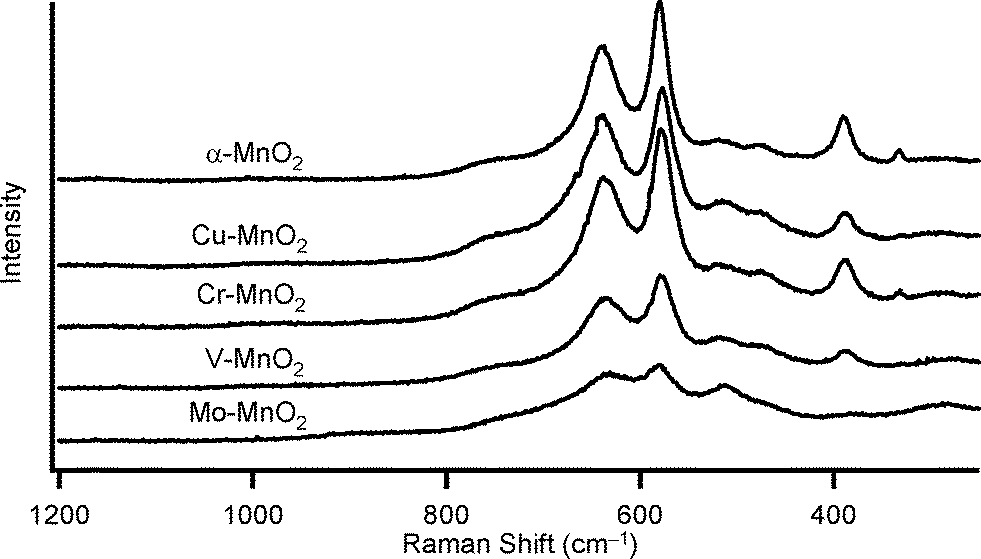 | ||
| Fig. 4 Raman spectra of α-MnO2 and M–MnO2. | ||
| Entry | Catalyst | BET surface area (m2 g−1) | Content (wt%) | M/(Mn+M) (mol%) | |
|---|---|---|---|---|---|
| Mn | K | ||||
| 1 | α-MnO2 | 80 | 52.5 | 4.07 | — |
| 2 | Mo–MnO2 | 212 | 51.1 | 3.60 | 5.0 |
| 3 | V–MnO2 | 120 | 55.3 | 4.15 | 5.2 |
| 4 | Cr–MnO2 | 91 | 57.6 | 4.04 | 2.0 |
| 5 | Cu–MnO2 | 109 | 56.9 | 3.53 | 2.5 |
The specific surface areas of Mo–MnO2, V–MnO2, Cr–MnO2, and Cu–MnO2 were 212 m2 g−1, 120 m2 g−1, 91 m2 g−1, and 109 m2 g−1, respectively (Table 3), larger than that of undoped α-MnO2 (80 m2 g−1). The significantly large surface area of Mo–MnO2 (212 m2 g−1) is likely attributed to its small crystalline size (Fig. 5b). The XPS spectra in the Mn 2p region showed no significant difference among α-MnO2 and M–MnO2 (Fig. S3†). This indicated that the AOSs on these surfaces were almost the same. The XPS spectra in the O 1s region are shown in Fig. S4.† Each O 1s spectrum can be deconvoluted into three peaks corresponding to three types of surface oxygen species; the low (around 530 eV), medium (around 531 eV), and high binding energy peaks (around 532 eV) are ascribed to the coordinatively saturated lattice oxygen species (described as Osat), the coordinatively unsaturated oxygen species (e.g., OH and adsorbed oxygen species on the surface, described as Ounsat), and adsorbed molecular H2O on the surface, respectively.6d The curve-fitting analyses of these XPS spectra showed that the Ounsat/(Osat + Ounsat) values for α-MnO2, Mo–MnO2, V–MnO2, Cr–MnO2, and Cu–MnO2 were 0.24, 0.18, 0.25, 0.26, and 0.31, respectively (Table S2†). In addition, the content of potassium in Cu–MnO2 was significantly lower than those in the others (Table 3). Thus, the surface concentration of the coordinatively unsaturated oxygen species (possibly OH species) in Cu–MnO2 was somewhat larger than those in the others.
 | ||
| Fig. 5 TEM images of (a) α-MnO2, (b) Mo–MnO2, (c) V–MnO2, (d) Cr–MnO2, and (e) Cu–MnO2. | ||
The transmission electron microscopy (TEM) images indicated that undoped α-MnO2 possessed a fibrous morphology (Fig. 5a). The average length and width of the fibers were 500 ± 100 nm and 20 ± 5 nm, respectively, and a lattice fringe spacing of 4.9 Å attributed to the (200) plane was observed throughout the α-MnO2 sample. Similar fibrous morphologies were also observed when low-valent metal cations were doped, such as Cr3+ and Cu2+, and in these cases a lattice fringe spacing of 6.9 Å due to the (110) planes was clearly observed (Fig. 5d and e). The average length of the fibers in Cr–MnO2 and Cu–MnO2 was almost the same as that in α-MnO2, while the widths (15 ± 5 nm) were somewhat thinner than that in α-MnO2 (20 ± 5 nm). As shown in Fig. 5b and c, when high-valent metal cations were doped, such as Mo6+ and V5+, the lengths of the fibers drastically shortened, especially in the case of Mo–MnO2, resulting in the formation of grain-like aggregates of ultrafine nanocrystals. Their larger specific surface areas (Table 3) are possibly caused by the formation of the aggregates of nanocrystals. Such morphologies were also observed in Mo–MnO2 and V–MnO2 samples possessing lower dopant contents (2.5 mol%) (Fig. S5†). The TEM image of Mo–MnO2 displayed clear lattice fringes throughout the sample. Fortunately, we could successfully observe the c-axis view (2 × 2 tunnel view) of Mo–MnO2, which was well consistent with the simulated structure (Fig. 5b).
Catalytic performance and scope of metal-doped α-MnO2
The catalytic activities of Mo–MnO2, V–MnO2, Cr–MnO2, and Cu–MnO2 were compared for the aerobic oxygenation of 1a under the conditions described in Table 4. Among the M–MnO2 catalysts examined, Mo–MnO2 showed the highest catalytic performance, which was much superior to that of undoped α-MnO2; the oxygenation of 1a using Mo–MnO2 gave the corresponding sulfoxide 2a and sulfone 3a in 75% and 3% yields, respectively (Table 4, entry 2), while undoped α-MnO2 gave 2a and 3a in 40% and 1% yields, respectively (Table 4, entry 1). The total yield of 2a and 3a obtained with V–MnO2 (46%) was slightly higher than that with α-MnO2 (41%) (Table 4, entry 3). The substantial improvement of the catalytic activity of α-MnO2 by doping of low-valent metal cations, such as Cr3+ and Cu2+, was hardly observed (Table 4, entries 4 and 5), thus suggesting that increasing the surface concentration of OH species is not crucial for the oxygenation. Therefore, the substitutional doping of high-valent metal cations, especially Mo6+, was an effective strategy to improve the catalytic performance of α-MnO2 for this type of oxygenation reaction.|
|
||||
|---|---|---|---|---|
| Entry | Catalysta (mol%) | Conv. of 1a (%) | Yield (%) | |
| 2a | 3a | |||
| Reaction conditions: catalyst (25 mg), 1a (0.5 mmol), o-dichlorobenzene (1 mL), 150 °C (bath temp.), O2 (5 atm), 24 h. Conversions and yields were determined by GC analysis using naphthalene as an internal standard.a The values in the parentheses are based on the surface exposed manganese species estimated from the specific surface areas and the crystal structures. | ||||
| 1 | α-MnO2 (9.8) | 45 | 40 | 1 |
| 2 | Mo–MnO2 (25.9) | 82 | 75 | 3 |
| 3 | V–MnO2 (14.6) | 46 | 45 | 1 |
| 4 | Cr–MnO2 (11.1) | 47 | 40 | 1 |
| 1 | Cu–MnO2 (13.3) | 40 | 38 | 1 |

K2MoO4 (precursor for Mo–MnO2) showed no activity for the oxygenation of 1a (Table 5, entry 4). The catalytic activities of simple physical mixtures of K2MoO4 and α-MnO2 (Table 5, entry 5) as well as MoO3 and α-MnO2 (Table 5, entry 6) were intrinsically the same as that of α-MnO2 (Table 5, entry 1). Moreover, a supported catalyst, Mo6+/α-MnO2, prepared by impregnation of Mo6+ species onto α-MnO2, was not effective for the oxygenation of 1a (Table 5, entry 3). Thus, molybdenum compounds themselves are essentially inactive for the oxygenation. Again, we emphasize that the substitutional doping of Mo6+ into the framework is crucial for the improvement of the catalytic performance of α-MnO2. As described above, the α-MnO2 structure showed the best performance among the examined manganese oxides with various crystal structures. This is principally due to the structure highly stabilized by the tunnel K+ ions. We consider that the manganese vacancies formed by the doping of high-valent cations,9j,l,11c,d especially Mo6+, may prevent the growth of α-MnO2 crystals along the c-axis direction. Although the c-axis growth was significantly suppressed, the local crystallinity was intrinsically preserved, as evidenced by the above-mentioned XRD, Raman, and TEM analyses. As a result, grain-like aggregates of ultrafine nanocrystals of metal-doped α-MnO2 were formed, and the specific surface areas increased in these cases. This kind of morphology possibly provides a large number of catalytically active surface sites in α-MnO2, e.g., vacancy sites, effective for the oxygenation, that is, electron-transfer from the substrate to the catalyst and from the reduced catalyst to O2.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Mo/(Mn+Mo) (mol%) | Conv. of 1a (%) | Yield (%) | |
| 2a | 3a | ||||
| Reaction conditions: catalyst (25 mg), 1a (0.5 mmol), o-dichlorobenzene (1 mL), O2 (5 atm), 150 °C (bath temp.), 3 h. Conversions and yields were determined by GC analysis using naphthalene as an internal standard.a α-MnO2 (25 mg) + K2MoO4 (2.8 mg).b α-MnO2 (25 mg) + MoO3 (1.8 mg). | |||||
| 1 | α-MnO2 | — | 27 | 24 | 1 |
| 2 | Mo–MnO2 | 5.0 | 40 | 37 | 1 |
| 3 | Mo/α-MnO2 | 4.7 | 9 | 3 | <1 |
| 4 | K2MoO4 | — | 6 | <1 | <1 |
| 5a | α-MnO2 + K2MoO4 | 5.0 | 28 | 25 | 1 |
| 6b | α-MnO2 + MoO3 | 5.0 | 25 | 25 | <1 |
By using the most effective Mo–MnO2, the scope of aerobic oxygenation with respect to various kinds of structurally diverse sulfides was next investigated. The results are summarized in Table 6. Thioanisole (1a) and its derivatives, which possess electron-donating as well as electron-withdrawing substituents at each position of the benzene rings (1b–1e), could efficiently be converted into the corresponding sulfoxides in moderate to high yields as the major products with concomitant formation of the corresponding sulfones (Table 6, entries 1–5). Diphenyl sulfide (1f) afforded the corresponding sulfoxide in a high yield (Table 6, entry 6). It should be noted that not only aryl sulfides but also a less reactive alkyl one (1g) could efficiently be oxygenated (Table 6, entry 7). In order to verify whether the observed catalysis was derived from solid Mo–MnO2 or leached metal species (manganese and/or molybdenum), Mo–MnO2 was removed by filtration during the reaction at 3 h, and then the reaction was again carried out with the filtrate under the same reaction conditions. As shown in Fig. S6,† the production of 2a was completely stopped by the removal of Mo–MnO2. In addition, we confirmed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis that manganese and molybdenum species were not present in the filtrate (below 0.1%). This experimental evidence can rule out any contribution to the observed catalysis from metal species that leached into the reaction solution, and the observed catalysis for the present oxygenation is truly heterogeneous.13 After the reaction of 1a was completed, Mo–MnO2 could readily be retrieved from the reaction mixture by simple filtration. The XRD analysis revealed that the crystal structure and crystallinity of Mo–MnO2 were intrinsically preserved after its repeated reuse for the oxygenation of 1a (Fig. 6). Furthermore, Mo–MnO2 could be reused for the same reaction at least four times while keeping its high catalytic performance (Fig. 7).
| Entry | Substrate | Product | Time (h) | Yielda (%) | ||
|---|---|---|---|---|---|---|
| Reaction conditions: Mo–MnO2 (50 mg), 1 (0.5 mmol), o-dichlorobenzene (1 mL), 150 °C (bath temp.), O2 (5 atm). Yields were determined by GC analysis using naphthalene as an internal standard.a The values in the parentheses are the yields of the corresponding sulfones. | ||||||
| 1 |
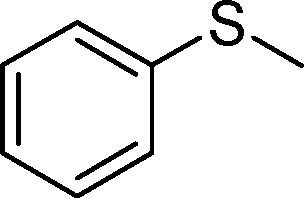
|
1a |
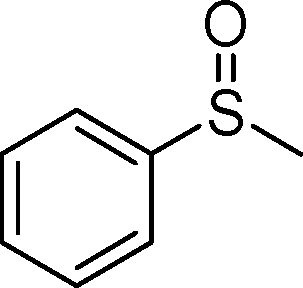
|
2a | 6 | 73 (4) |
| 2 |
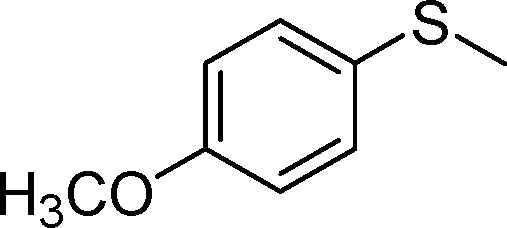
|
1b |
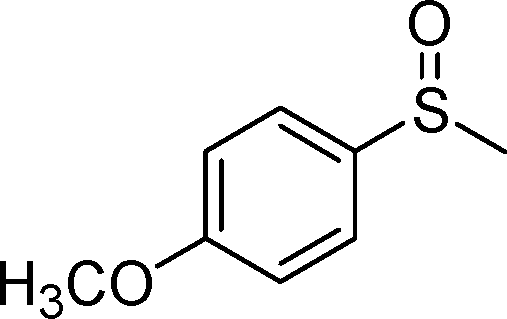
|
2b | 6 | 91 (6) |
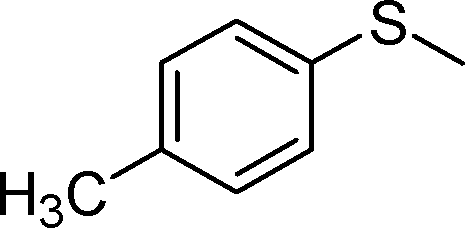
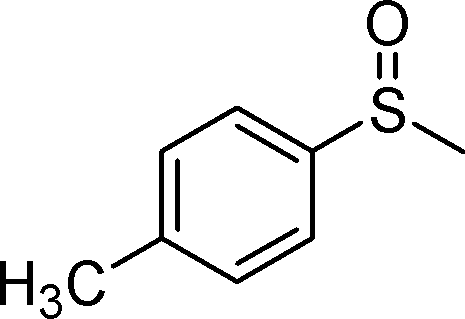
| 3 |
|
1c |
|
2c | 24 | 76 (4) |
| 4 |
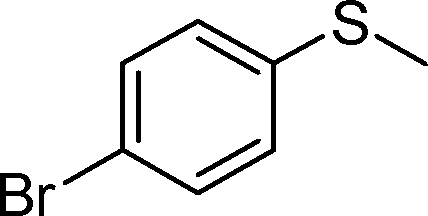
|
1d |
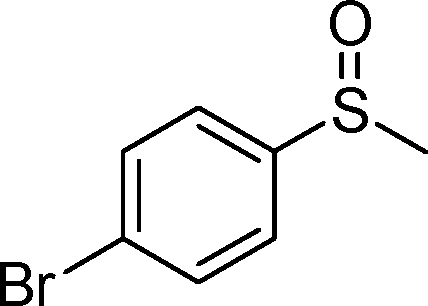
|
2d | 24 | 77 (11) |
| 5 |
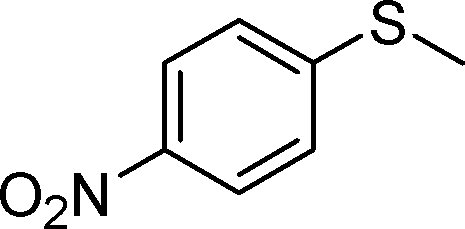
|
1e |
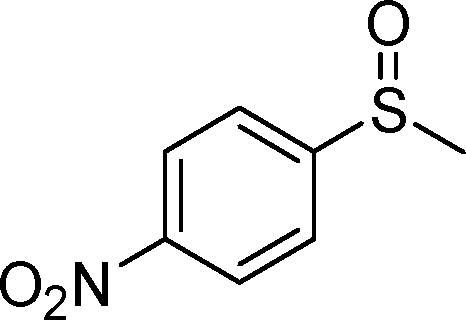
|
2e | 24 | 40 (11) |
| 6 |
|
1f |

|
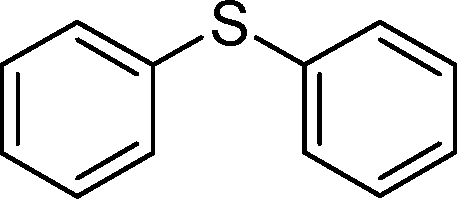
2f | 24 | 90 (10) |
| 7 |
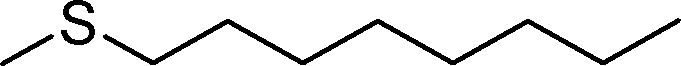
|
1g |
|
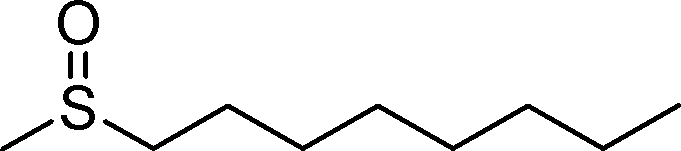
2g | 24 | 53 (1) |
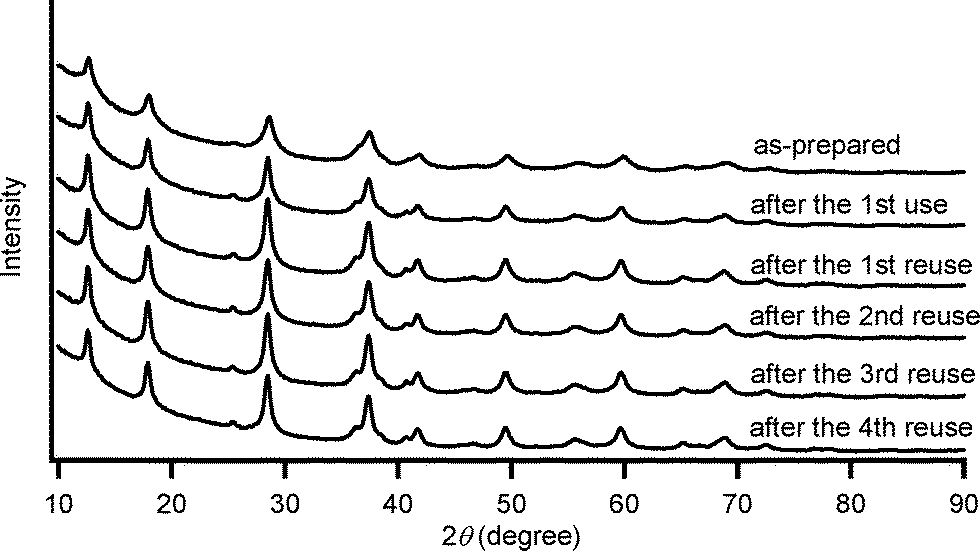 | ||
| Fig. 6 XRD patterns of as-prepared Mo–MnO2 and the retrieved Mo–MnO2 after their use for the oxygenation of 1a under the conditions described in Table 6 for 6 h. | ||
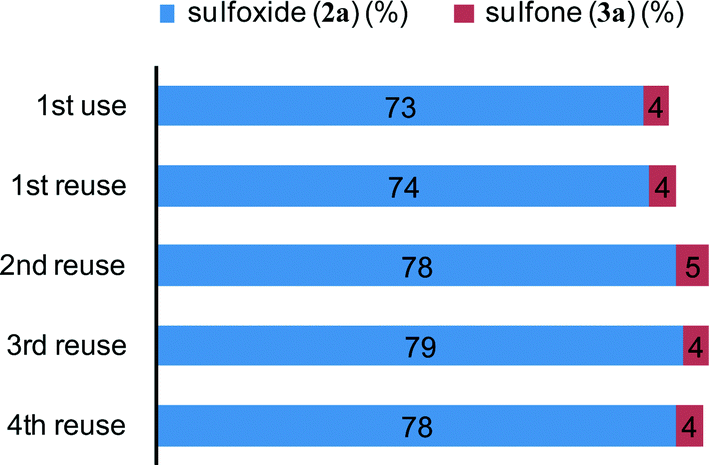 | ||
| Fig. 7 Reuse experiments of Mo–MnO2 for the oxidation of 1a. The reactions were carried out under the conditions described in Table 6 for 6 h. Yields were determined by GC analysis using naphthalene as an internal standard. The retrieved catalyst was washed with acetone and water, and then dried at room temperature prior to being used for the next reuse experiment. | ||
The Mo–MnO2-catalyzed oxygenation of 1a was strongly suppressed by the presence of the radical scavenger 2,6-di-tert-butyl-4-methylphenol (40 mol%) (9% yield of 2a under the conditions described in Table 5), indicating that radical intermediates are possibly involved in the present sulfide oxygenation. It is known that manganese oxides can generate radical cation species from heteroatom-containing compounds, such as thiols and amines, through single-electron transfer (SET) oxidation. Similarly, in the present case, the radical cation species are possibly generated by SET oxidation of sulfides. Then, the reduced Mo–MnO2 is reoxidized by SET to O2, and a superoxide radical anion species is likely formed.6f,g By the reaction of the sulfide radical cation and the superoxide radical anion species, the corresponding sulfoxides are successively obtained as the final products.14
Besides sulfide oxygenation, Mo–MnO2 could efficiently catalyze several aerobic oxidative functional group transformations through SET oxidation processes. Benzylic oxygenation of alkylarenes, such as xanthene, fluorene, and diphenylmethane, efficiently proceeded to give the corresponding ketones in moderate to high yields (Fig. 8a). The oxygenation proceeds through SET oxidation/deprotonation, followed by oxygen insertion.6f Mo–MnO2 could act as an efficient heterogeneous catalyst for oxidative α-cyanation of trialkylamines using trimethylsilyl cyanide (TMSCN) as the cyano source and O2 as the terminal oxidant. The cyanation regioselectively took place at the α-methyl positions, giving the corresponding α-amino nitriles in high yields (Fig. 8b). The cyanation possibly proceeds through the following mechanism.6g Firstly, an amine radical cation is formed by SET. Then, the radical cation was deprotonated to form an α-aminated carbon radical. This step is stereoelectronically controlled and determines the above-mentioned regioselectivity to the α-methyl position.6g Then, the second SET proceeds to form an iminium cation, followed by nucleophilic trapping by CN− species to afford the corresponding α-amino nitrile. Moreover, in the presence of Mo–MnO2, thiocyanates could be synthesized in almost quantitative yields starting from benzenethiols under very mild conditions (Fig. 8c). The present cyanation proceeds through the Mo–MnO2-catalyzed oxidative homocoupling of benzenethiols to the corresponding disulfides through SET oxidation, followed by nucleophilic bond cleavage to produce the desired thiocyanates and thiolate species.6i Mo–MnO2 can catalyze the oxidative homocoupling of the thiolate species, thus resulting in quantitative production of thiocyanates formally from benzenethiols.6i
 | ||
| Fig. 8 Several oxidative functional group transformations using Mo–MnO2: (a) oxygenation of alkylarenes, (b) oxidative α-cyanation of trialkylamines, and (c) oxidative S-cyanation of benzenethiols. | ||
Conclusion
In this work, we have obtained several important findings for the manganese oxide-based catalyst design and the application of manganese oxide catalysts to several aerobic oxidation reactions. Firstly, we prepared four kinds of manganese oxides possessing different crystal structures, namely α-MnO2, β-MnO2, γ-MnO2, and δ-MnO2, and examined their structure–activity relationships for the aerobic oxygenation of thioanisole. Amongst these manganese oxides, α-MnO2 showed the best catalytic performance and was highly durable during the catalytic oxygenation possibly due to the stabilization of its framework structure by the tunnel K+ ions. The performance of α-MnO2 could be further improved by substitutional doping of Mo6+ into the octahedral framework. By the doping of Mo6+, manganese vacancies were likely formed, which would prevent the growth of α-MnO2 crystals along the c-axis direction. Although the c-axis growth was suppressed, the local crystallinity was essentially preserved. Consequently, grain-like aggregates of ultrafine nanocrystals were formed in the case of Mo6+-doped α-MnO2 (Mo–MnO2), resulting in an increase in specific surface area. This kind of morphology would afford a large number of catalytically active surface sites, for example, vacancy sites, effective for the oxygenation. In the presence of Mo–MnO2, various kinds of structurally diverse sulfides including aromatic and aliphatic ones could be oxidized to the corresponding sulfoxides as the major products using O2 as the terminal oxidant. The observed catalysis of Mo–MnO2 was shown to be truly heterogeneous, and Mo–MnO2 could be reused for the oxygenation of thioanisole at least four times while keeping its high catalytic performance. The structure of Mo–MnO2 was intrinsically preserved after its repeated reuse for the oxygenation. The present sulfide oxygenation was likely initiated by a single-electron transfer (SET) oxidation process. Aside from sulfide oxygenation, Mo–MnO2 could efficiently catalyze several aerobic oxidative functional group transformations through SET oxidation processes, such as oxygenation of alkylarenes, oxidative α-cyanation of trialkylamines, and oxidative S-cyanation of benzenethiols.Experimental section
Materials
KMnO4, NaVO3, concentrated HNO3, and KOH were purchased from Kanto Chemical. MnSO4·H2O was purchased from Aldrich. K2MoO4 and MoO3 were purchased from Wako Pure Chemical Industries. Cr(CH3COO)3·H2O was purchased from Nacalai Tesque. (NH4)2S2O8 was purchased from Tokyo Chemical Industry. Substrates, solvents, and naphthalene (internal standard for GC analysis) were purchased from Tokyo Chemical Industry, Kanto Chemical, or Aldrich. All reagents were used as received without further purification. TBA2MoO4 (TBA = tetra-n-butylammonium) was prepared according to a reported procedure with modification.15 The manganese oxide catalysts were prepared by the procedures described below.Preparation of manganese oxide catalysts
α-MnO2 was prepared according to the following procedure.6k An aqueous solution (100 mL) of KMnO4 (5.89 g) was added to an aqueous solution (30 mL) of MnSO4·H2O (8.8 g) and concentrated HNO3 (3 mL). The resulting mixture was refluxed at 100 °C for 24 h. Then, the dark brown solid formed was filtered off, washed with a large amount of water, and dried under open air at 150 °C, affording 7.5 g of α-MnO2. Metal-doped α-MnO2 catalysts (M–MnO2) were prepared intrinsically using the same procedure for non-doped α-MnO2. For the preparation of M–MnO2, a dopant metal source (K2MoO4, NaVO3, Cr(CH3COO)3·H2O, or CuSO4·5H2O) was initially added to the MnSO4·H2O solution (2.5 or 5 mol% with respect to total metal content), followed by the above procedure, affording 7–8 g of M–MnO2. In the case of α-MnO2, the mixed valency of manganese can be compensated by K+ (or H+).β-MnO2 was prepared according to the following procedure.6m An aqueous solution (80 mL) containing MnSO4·H2O (1.69 g) and (NH4)2S2O8 (2.28 g) was stirred at room temperature for 30 min. Then, the mixture was transferred to a Teflon vessel. The vessel was placed inside an autoclave, and the solution was heated at 140 °C for 12 h. The dark brown solid formed was filtered off, washed with a large amount of water, and dried under open air at 150 °C, affording 0.8 g of β-MnO2.
γ-MnO2 was prepared according to the following procedure.6m An aqueous solution (80 mL) containing MnSO4·H2O (3.375 g) and (NH4)2S2O8 (4.575 g) was stirred at room temperature for 30 min. Then, the mixture was transferred to a Teflon vessel. The vessel was placed inside an autoclave, and the solution was heated at 90 °C for 24 h. The dark brown solid formed was filtered off, washed with a large amount of water, and dried under open air at 150 °C, affording 1.5 g of γ-MnO2.
δ-MnO2 was prepared according to the following procedure.7 An aqueous solution (200 mL) containing ethanol (92 mL) and KOH (33.6 g) was added dropwise to an aqueous solution (150 mL) of KMnO4 (9.48 g). The mixture was stirred at room temperature for 1 h, followed by heating at 80 °C for 48 h. The dark brown solid formed was filtered off, washed with a large amount of water, and dried under open air at 80 °C, affording 9.0 g of δ-MnO2. It is possible that K+ and OH− can be introduced into the interlayer of δ-MnO2.
A supported catalyst, Mo6+/α-MnO2, was prepared by impregnation of Mo6+ species onto α-MnO2. α-MnO2 (1.0 g) was dispersed in an acetonitrile solution (30 mL) of TBA2MoO4 (0.337 g). After stirring for 30 min, acetonitrile was evaporated to dryness, affording 1.0 g of Mo6+/α-MnO2. The molybdenum content in Mo6+/α-MnO2 was 2.7 wt%.
Characterization of manganese oxide catalysts
The contents of manganese and dopant metals in the manganese oxide samples were determined by ICP-AES analyses using a Shimadzu ICPS-8100 apparatus. The contents of potassium in the manganese oxide samples were determined by atomic absorption spectrometry analyses using a Hitachi Z 2000 apparatus. The AOSs of manganese were determined by redox titration, during which the manganese in the sample was reduced with excess Fe(NH4)2(SO4)2, and unreacted Fe2+ was titrated with an aqueous solution of KMnO4. This redox titration was repeated three times for each sample, and the AOSs were defined as the average values. The BET surface areas were measured by N2 adsorption at −196 °C using a Micromeritics ASAP 2010 instrument. The XRD patterns were recorded using a Rigaku SmartLab instrument under Cu Kα radiation (λ = 1.5418 Å, 45 kV, 200 mA). XPS analyses were performed using a JEOL JPS-9000 apparatus under Mg Kα radiation (hν = 1253.6 eV, 8 kV, 10 mA). The peak positions were calibrated by the Au 4f 7/2 peak (84 eV) of metallic gold deposited on the sample holder. The baselines were subtracted by the Shirley method. The Raman spectra were measured on a Nippon Bunko NRS-5100 Raman apparatus using a 532 nm laser. The TEM images were obtained using a JEM-2000EX II apparatus at an acceleration voltage of 200 kV. The TEM specimens were prepared by dispersing the manganese oxide samples in ethanol and sonicating them for 30 min before deposition on a carbon-coated copper grid. The TPR profiles were measured on a BELCAT apparatus with a quadrupole mass spectrometer. The sample was pretreated at 150 °C in a flow of Ar (30 mL min−1) for 20 min. After the pretreatment, a mixed gas of H2 and Ar (H2: 7 vol%) was allowed to flow into the sample (30 mL min−1) with progress of temperature from 100 to 550 °C at a rate of 10 °C min−1. The amount of consumed H2 was quantified by a mass spectrometer with a fragment of m/z = 2.Catalytic reactions
GC quantitative analyses were performed on a Shimadzu GC-2014 apparatus with a FID detector equipped with a TC-1 or a TC-5 capillary column. GC-MS analyses for product identification were performed on a GCMS-QP2010 apparatus at an ionization voltage of 70 eV equipped with an Inert-Cap 5 capillary column.The oxygenation of sulfides was typically carried out as follows. The manganese oxide catalyst (25–50 mg), sulfide (0.5 mmol), o-dichlorobenzene (1 mL), and naphthalene (internal standard, 0.2 mmol) were placed in a Teflon vessel. The Teflon vessel was placed inside an autoclave, and the reaction was performed at 150 °C (bath temperature) in 5 atm of O2 for 3–96 h. After the reaction was completed, the spent catalyst was separated by filtration and washed with acetone. Then, the filtrate was analyzed. The products were confirmed by comparing their GC retention times and GC-MS spectra with those of authentic data. The spent catalyst was washed with acetone and water, and dried under open air at room temperature before being used for the reuse experiment. As for the oxygenation of sulfides under anaerobic conditions and other oxidation reactions, the reactions were carried out using a Schlenk-type reactor. These procedures were essentially the same as that for aerobic sulfide oxygenation.
Acknowledgements
We thank Mr. S. Torii (The University of Tokyo) for his help with preliminary experiments. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Science, Sports, and Technology of Japan.Notes and references
- (a) H. L. Holland, Chem. Rev., 1988, 88, 357 CrossRef; (b) M. C. Carreño, Chem. Rev., 1995, 95, 1717 CrossRef; (c) I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651 CrossRef PubMed; (d) R. F. D. L. Pradilla, I. Colomer and A. Viso, Org. Lett., 2012, 14, 3068 CrossRef PubMed; (e) K. Hiroi, Y. Suzuki, I. Abe and R. Kawagishi, Tetrahedron, 2000, 56, 4701 CrossRef CAS; (f) S. Caron, W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS PubMed; (g) S. Otsuki, T. Nanoka, N. Takashima, W. Qian, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232 CrossRef CAS; (h) Y. Shiraishi, H. Hara, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 1589 CrossRef CAS.
- (a) D. Edwards and J. B. Stenlake, J. Chem. Soc., 1954, 3272 RSC; (b) F. G. Bordwell and P. J. Boutan, J. Am. Chem. Soc., 1957, 79, 717 CrossRef CAS; (c) M. Madesclaire, Tetrahedron, 1986, 42, 5459 CrossRef CAS; (d) Y. Watanabe, T. Numata and S. Oae, Synthesis, 1981, 204 CrossRef CAS; (e) J. Drabowicz and M. Mikolafczyk, Synthesis, 1978, 758 CrossRef CAS; (f) M. Jafarpour, M. Ghahramoninezhad and A. Rezaeifard, RSC Adv., 2014, 4, 1601 RSC; (g) B. Machura, J. Palion, J. Mroziński, B. Kalińska, M. Amini, M. M. Najafpour and R. Kruszynski, Polyhedron, 2013, 53, 132 CrossRef CAS; (h) A. Rezaeifard, R. Haddad, M. Jafarpour and M. Hakimi, ACS Sustainable Chem. Eng., 2014, 2, 942 CrossRef CAS.
- (a) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (b) I. V. Khavrutskii, G. M. Maksimov and O. A. Kholdeeva, React. Kinet. Catal. Lett., 1999, 66, 325 CrossRef CAS; (c) X. Zhou and H. Ji, Catal. Commun., 2014, 53, 29 CrossRef CAS; (d) H. Kawasaki, S. Kumer, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777 CrossRef CAS; (e) W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417 RSC; (f) W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem. – Eur. J., 2013, 19, 5654 CrossRef CAS PubMed; (g) J. Dad'ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620 CrossRef; (h) H. Zhang and G. Wang, Tetrahedron Lett., 2014, 55, 56 CrossRef CAS; (i) S. E. Martín and L. I. Rossi, Tetrahedron Lett., 2001, 42, 7147 CrossRef; (j) Y. Yuan, X. Shi and W. Liu, Synlett, 2011, 4, 559 CrossRef; (k) H. Zhang, C. Chen, R. Liu, Q. Xu and W. Zhao, Molecules, 2010, 15, 83 CrossRef CAS PubMed; (l) N. Komatsu, M. Uda and H. Suzuki, Chem. Lett., 1997, 1229 CrossRef CAS; (m) H. Zhang, C. Chen and R. Liu, Synth. Commun., 2012, 42, 811 CrossRef CAS; (n) A. M. Khenkin, G. Letius and R. Neurman, J. Am. Chem. Soc., 2010, 132, 11446 CrossRef CAS PubMed; (o) B. Li, A.-H. Liu, L.-N. He, Z.-Z. Yang, J. Gao and K.-H. Chen, Green Chem., 2012, 14, 130 RSC; (p) I. Gamba, S. Palavicini, E. Monzani and L. Casella, Chem. – Eur. J., 2009, 15, 12932 CrossRef CAS PubMed; (q) H. B. Lee and T. Ren, Inorg. Chim. Acta, 2009, 362, 1467 CrossRef CAS; (r) M. Costas, C. W. Cady, S. V. Kryatov, M. J. Ryan, E. V. Rybak-Akimova and L. Que Jr., Inorg. Chem., 2003, 42, 7519 CrossRef CAS PubMed; (s) D. P. Riley and P. E. Correa, J. Chem. Soc., Chem. Commun., 1986, 1097 RSC; (t) K. Suzuki, J. Jeong, K. Yamaguchi and N. Mizuno, New J. Chem. 10.1039/c5nj01045d.
- (a) N. M. Okun, T. M. Anderson and C. L. Hill, J. Am. Chem. Soc., 2003, 125, 3194 CrossRef CAS PubMed; (b) Q. Gao, C. Giordano and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 11740 CrossRef CAS PubMed; (c) A. Taketoshi, P. Concepción, H. García, A. Corma and M. Haruta, Bull. Chem. Soc. Jpn., 2013, 86, 1412 CrossRef CAS.
- (a) F. Cheng, J. Chen, X. Gou and P. Shen, Adv. Mater., 2005, 17, 2753 CrossRef CAS; (b) D. Zhan, Q. Zhang, X. Hu and T. Peng, RSC Adv., 2013, 3, 5141 RSC; (c) J. Ge, L. Zhuo, F. Yang, B. Tang, L. Wu and C. Tung, J. Phys. Chem. B, 2006, 110, 17854 CrossRef CAS PubMed; (d) L. Espinal, W. Wong-Ng, J. A. Kaduk, A. J. Allen, C. R. Snyder, C. Chiu, D. W. Siderius, L. Li, E. Cockayne, A. E. Espinal and S. L. Suib, J. Am. Chem. Soc., 2012, 134, 7944 CrossRef CAS PubMed; (e) J. Yuan, X. Liu, O. Akbulut, J. Hu, S. L. Suib, J. Kong and F. Stellacci, Nat. Nanotechnol., 2008, 3, 332 CrossRef CAS PubMed; (f) A. J. Fatiadi, Synthesis, 1976, 65 CrossRef CAS; (g) J. R. Kona, C. K. King'ondu, A. R. Howell and S. L. Suib, ChemCatChem, 2014, 6, 749 CrossRef CAS; (h) D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494 CrossRef CAS PubMed; (i) F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2010, 22, 898 CrossRef CAS; (j) Q. Ye, J. Zhao, F. Huo, D. Wang, S. Cheng, T. Kang and H. Dai, Microporous Mesoporous Mater., 2013, 172, 20 CrossRef CAS; (k) T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153 CrossRef CAS PubMed; (l) A. Yamaguchi, R. Inuzuka, T. Takashima, T. Hayashi, K. Hashimoto and R. Nakamura, Nat. Commun., 2014, 5, 4256 CAS.
- (a) L. Jin, C.-H. Chen, V. M. B. Crisostomo, L. Xu, Y.-C. Son and S. L. Suib, Appl. Catal., A, 2009, 355, 169 CrossRef CAS; (b) P. Pal, A. K. Giri, H. Singh, S. C. Ghosh and A. B. Panda, Chem. – Asian J., 2014, 9, 2392 CrossRef CAS PubMed; (c) Y.-C. Son, V. D. Makwana, A. R. Howell and S. L. Suib, Angew. Chem., Int. Ed., 2001, 40, 4280 CrossRef CAS; (d) J. Nie and H. Liu, J. Catal., 2014, 316, 57 CrossRef CAS; (e) Z.-Z. Yang, J. Deng, T. Pan, Q.-X. Guo and Y. Fu, Green Chem., 2012, 14, 2986 RSC; (f) N. N. Opembe, Y.-C. Son, T. Sriskandakumar and S. L. Suib, ChemSusChem, 2008, 1, 182 CrossRef CAS PubMed; (g) K. Yamaguchi, Y. Wang and N. Mizuno, ChemCatChem, 2013, 5, 2835 CrossRef CAS; (h) S. Dharmarathna, C. K. King'ondu, L. Pahalagedara, C.-H. Kuo, Y. Zhang and S. L. Suib, Appl. Catal., B, 2014, 147, 124 CrossRef CAS; (i) K. Yamaguchi, K. Sakagami, Y. Miyamoto, X. Jin and N. Mizuno, Org. Biomol. Chem., 2014, 12, 9200 RSC; (j) K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544 CrossRef CAS PubMed; (k) K. Yamaguchi, H. Kobayashi, Y. Wang, T. Oishi, Y. Ogasawara and N. Mizuno, Catal. Sci. Technol., 2013, 3, 318 RSC; (l) Y. Wang, H. Kobayashi, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 2642 RSC; (m) J. Zhang, Y. Li, L. Wang, C. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305 RSC.
- A. Iyer, J. Del-Pilar, C. K. King'ondu, E. Kissel, H. F. Garces, H. Huang, A. M. El-Sawy, P. K. Dutta and S. L. Suib, J. Phys. Chem. C, 2012, 116, 6474 CAS.
- (a) T. Gao, M. Glerup, F. Krumeich, R. Nesper, H. Fjellvåg and P. Norby, J. Phys. Chem. C, 2008, 112, 13134 CrossRef CAS; (b) J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447 CrossRef CAS PubMed; (c) Y. Ma, J. Luo and S. L. Suib, Chem. Mater., 1999, 11, 1972 CrossRef CAS; (d) A.-C. Gaillot, D. Flot, V. A. Drits, A. Manceau, M. Burghammer and B. Lanson, Chem. Mater., 2003, 15, 4666 CrossRef CAS.
- (a) A. M. A. Hashem, H. A. Mohamed, A. Bahloul, A. Eid and C. Julien, Ionics, 2008, 14, 7 CrossRef CAS; (b) R. Jothiramalingam, B. Viswanathan and T. K. Varadarajan, J. Mol. Catal. A: Chem., 2006, 252, 49 CrossRef CAS; (c) J. Cai, J. Liu, W. S. Willis and S. L. Suib, Chem. Mater., 2001, 13, 2413 CrossRef CAS; (d) C. Calvert, R. Joesten, K. Ngala, J. Villegas, A. Morey, X. Shen and S. L. Suib, Chem. Mater., 2008, 20, 6382 CrossRef CAS; (e) M. Polverejan, J. C. Villegas and S. L. Suib, J. Am. Chem. Soc., 2004, 126, 7774 CrossRef CAS PubMed; (f) S. Ching, P. F. Driscoll, K. S. Kieltyka, M. R. Marvel and S. L. Suib, Chem. Commun., 2001, 2486 RSC; (g) W. Y. Hernández, M. A. Centeno, S. Ivanova, P. Eloy, E. M. Gaigneaux and J. A. Odriozoola, Appl. Catal., B, 2012, 123, 27 CrossRef; (h) X. Tang, Y. Li, J. Chen, Y. Xu and W. Shen, Microporous Mesoporous Mater., 2007, 103, 250 CrossRef CAS; (i) Z. Liu, Y. Xing, C.-H. Chen, L. Zhao and S. L. Suib, Chem. Mater., 2008, 20, 2069 CrossRef CAS; (j) C. K. King'ondu, N. Opembe, C.-H. Chen, K. Ngala, H. Huang, A. Iyer, H. F. Garcés and S. L. Suib, Adv. Funct. Mater., 2011, 21, 312 CrossRef; (k) H. Zhou, Y. F. Shen, J. Y. Wang, X. Chen, C.-L. O'Young and S. L. Suib, J. Catal., 1998, 189, 321 CrossRef; (l) During the peer review of our manuscript, the following very important work on Mo-OMS-2 materials for CO oxidation has been published: C.-H. Chen, E. C. Njagi, S.-Y. Chen, D. T. Horvath, L. Xu, A. Morey, C. Mackin, R. Joesten and S. L. Suib, Inorg. Chem., 2015 DOI:10.1021/acs.inorgchem.5b00906.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- (a) L. R. Pahalagedara, S. Dharmarathna, C. K. King'ondu, M. N. Pahalagedara, Y.-T. Meng, C.-H. Kuo and S. L. Suib, J. Phys. Chem. C, 2014, 118, 20363 CrossRef CAS; (b) Q. Feng, H. Kanoh, Y. Miyai and K. Ooi, Chem. Mater., 1995, 7, 148 CrossRef CAS; (c) M. Polverejan, J. C. Villegas and S. L. Suib, J. Am. Chem. Soc., 2004, 126, 7774 CrossRef CAS PubMed; (d) X. Tang, J. Li and J. Hao, Catal. Commun., 2010, 11, 871 CrossRef CAS; (e) X. Tang, J. Li and J. Hao, Catal. Commun., 2010, 11, 871 CrossRef CAS; (f) Y. Tanaka and M. Tsuji, Solvent Extr. Ion Exch., 1997, 15, 709 CrossRef CAS; (g) X. Chen, Y.-F. Shen, S. L. Suib and C. L. O'Young, Chem. Mater., 2002, 14, 940 CrossRef CAS; (h) Y. Li, Z. Fan, J. Shi, Z. Liu, J. Zhou and W. Shangguan, Catal. Today, 2015, 256, 178 CrossRef CAS.
- (a) L. Seguin, M. Figlarz, R. Cavagnat and J.-C. Lassègues, Spectrochim. Acta, Part A, 1995, 51, 1323 CrossRef; (b) C. V. Ramana, R. J. Smith, O. M. Hussain, M. Massot and C. M. Julien, Surf. Interface Anal., 2005, 37, 406 CrossRef CAS; (c) M. Schraml-Marth, A. Wokaun, H. E. Curry-Hyde and A. Baiker, J. Catal., 1992, 133, 415 CrossRef CAS; (d) D. Chen, G. Shen, K. Tang and Y. Qian, J. Cryst. Growth, 2003, 254, 225 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- (a) E. Baciocchi, T. D. Giac-co, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444 CrossRef CAS PubMed; (b) S. M. Bonsei, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem. – Eur. J., 2006, 12, 4844 CrossRef PubMed.
- H. Sunaba, K. Kamata and N. Mizuno, ChemCatChem, 2014, 6, 2333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01552a |
| This journal is © The Royal Society of Chemistry 2016 |