
Highly selective oxidation of cyclohexene to 2-cyclohexene-1-one in water using molecular oxygen over Fe–Co–g-C3N4†
Dexin
Yang
,
Tao
Jiang
,
Tianbin
Wu
*,
Peng
Zhang
,
Hongling
Han
and
Buxing
Han
*
Beijing National Laboratory for Molecular Sciences (BNLMS), CAS Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wtb@iccas.ac.cn; hanbx@iccas.ac.cn; Fax: +86 10 62562821; Tel: +86 10 62562821
First published on 11th September 2015
Abstract
Efficient and greener oxidation of cyclohexene to 2-cyclohexene-1-one is an interesting topic. In this work, we prepared a series of Fe–Co doped graphitic carbon nitride (Fe–Co–g-C3N4) catalysts through simple impregnation and calcination methods. The catalysts were characterized by different techniques, such as transmission electron microscopy (TEM), Fourier-transform infrared spectroscopy (FTIR), nitrogen adsorption–desorption measurement, powder X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). The selective oxidation of cyclohexene to 2-cyclohexene-1-one was carried out in different solvents over the catalysts using molecular oxygen as an oxidant. The influence of supports, solvents, Fe/Co molar ratio in the catalysts, pressure of oxygen, reaction temperature and time of the reaction was investigated. It was revealed that the bimetallic Fe–Co–g-C3N4 catalysts were very efficient for the reaction. More interestingly, the selectivity of the reaction in water was much higher than that in other solvents. Under optimized conditions, the selectivity to 2-cyclohexene-1-one could reach 95% at a cyclohexene conversion of 36%. The Fe–Co–g-C3N4 catalyst could be reused at least four times without obvious loss of efficiency.
Introduction
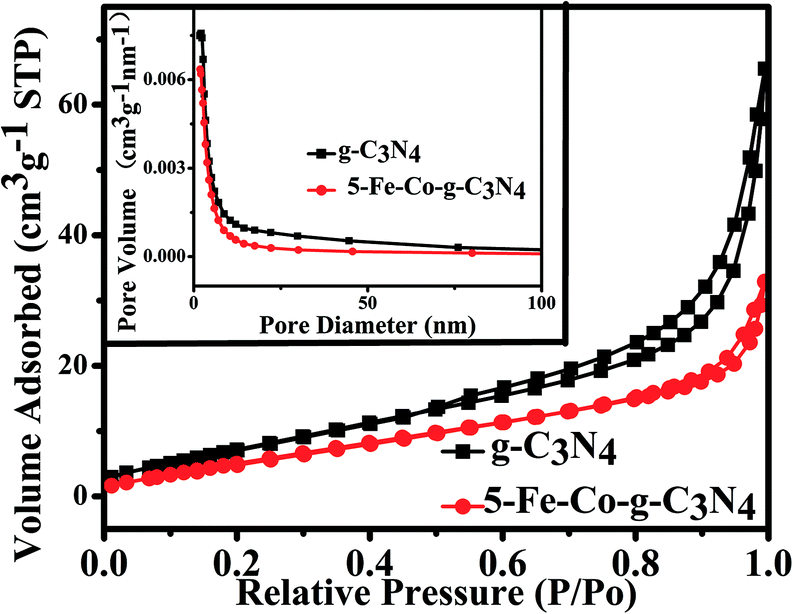
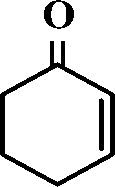
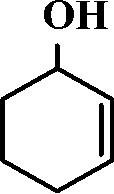
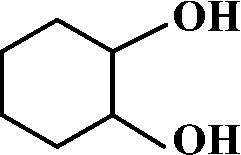
The selective allylic oxidation of alkenes into high value-added chemical products occupies an important position in the chemical industry.1 Among the oxidation products, 2-cyclohexene-1-one, which is obtained by the allylic oxidation of cyclohexene, is a very important intermediate because it is extensively used in spices, medication, pesticides and insect pheromone.2 However, it is very difficult to control the selectivity to oxygenated products in the allylic oxidation of cyclohexene due to the existence of two active sites on the cyclohexene molecule, where there are a C–H bond of the allylic site and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Some compounds, such as cyclohexanone, cyclohexanediol and dialdehyde, will be produced if the CC bond is oxygenated. On the contrary, 2-cyclohexene-1-ol, 2-cyclohexene-1-one or cyclohexene hydroperoxide will be generated when the C–H bond of the allylic site is oxygenated.3 Therefore, it is still challenging and attractive to obtain high selectivity to the target product in the allylic oxidation of cyclohexene.
C bond. Some compounds, such as cyclohexanone, cyclohexanediol and dialdehyde, will be produced if the CC bond is oxygenated. On the contrary, 2-cyclohexene-1-ol, 2-cyclohexene-1-one or cyclohexene hydroperoxide will be generated when the C–H bond of the allylic site is oxygenated.3 Therefore, it is still challenging and attractive to obtain high selectivity to the target product in the allylic oxidation of cyclohexene.
The traditional oxidants for the oxidation of cyclohexene are usually iodosylbenzene, sodium hypochlorite, chromium trioxide, etc.4 Although these strong oxidants can enhance the yield of the product, their applications are limited due to high cost, inconvenient transportation, explosibility, environmental pollution and lower atomic economy.5,6 Thus, the development of green processes for the catalytic oxidation of cyclohexene is becoming increasingly important. Recently, some greener oxidants have been applied for the oxidation of cyclohexene, including t-butyl hydroperoxide (TBHP),7 H2O2 (ref. 8) and molecular oxygen.9 Among these oxidants, molecular oxygen, which is cheap, abundant, and environmentally benign, has received much attention in academic research and industrial application.10 For example, the allylic oxidation of cyclohexene over cobalt resinate using oxygen as an oxidant was carried out under solvent-free conditions. It was shown that cobalt resinate exhibited high catalytic activity, but the selectivity to 2-cyclohexen-1-one was only 44.4%.2 More recently, magnetic core–shell Fe3O4@chitosan-Schiff base Co(II), Cu(II) and Mn(II) complexes have been proven to be active catalysts for cyclohexene oxidation with oxygen in the absence of solvents.11 A conversion of 50.2% and a selectivity of 80.3% to 2-cyclohexene-1-one were obtained over this catalyst. Organic solvents were often used when oxygen was used to oxygenate cyclohexene. For instance, the allylic oxidation of cyclohexene over the CoO catalyst in cyclohexane was studied, and high conversion and selectivity to 2-cyclohexene-1-one could be achieved.12 Generally, improving the selectivity to 2-cyclohexene-1-one in the allylic oxidation of cyclohexene using oxygen as an oxidant is still a very interesting topic.
Traditionally, most of the catalysts used for the allylic oxidation of cyclohexene are complexes based on Co, Fe, Mn, Ir and Cu.13–16 However, it is very difficult to recover these catalysts from homogeneous systems because these oxidation reactions are usually carried out in the liquid phase. Hence, heterogeneous catalysts have been developed by immobilizing transition metals on solid supports, such as SiO2, zeolites and Fe3O4 for the oxidation of cyclohexene.11,17,18 It has been reported that noble metal Au nanoparticles immobilized on different supports can improve the catalytic performance for the oxidation of cyclohexene. Donoeva et al. found that the size of gold nanoparticles deposited on a SiO2 support played a crucial role for catalytic activity in aerobic oxidation of cyclohexene without solvent.19 Recently, gold nanoparticles supported on modified silica and bentonite have also shown high conversion and selectivity to 2-cyclohexene-1-one for cyclohexene oxidation with oxygen in the absence of solvents.20 Graphitic C3N4 (g-C3N4) as a special support has recently attracted much attention due to its extreme chemical and thermal stability and easy accessibility. g-C3N4 exhibited outstanding potential in materials science, catalysis, and electronic and optical fields due to its excellent optical and photoelectric properties.21,22 Interestingly, g-C3N4 itself has been reported to be an excellent catalyst for the oxidation of alcohols, cyclohexene and amines by oxygen under visible light irradiation in the absence of any metal derivatives.23–25 Moreover, some organic compounds or metal derivatives could bind or intercalate into the matrix of g-C3N4 through the numerous anchoring sites on the surface, which undoubtedly greatly improved the catalytic performance and expanded the applications of g-C3N4 in catalysis.21,26 For example, Fe or Pd doped g-C3N4 catalysts exhibited excellent catalytic performance in the oxidation of benzene and selective hydrogenation of phenol and its derivatives.27,28
Water has been widely applied as a greener reaction medium in catalysis.29–32 From an environmental point of view, it would be much better to replace organic solvents with water because it is abundant, environmentally friendly, and low-cost. It is well known that transition metals are of great importance and have played an irreplaceable role in chemical reactions.33 Fe and Co are abundant and cheap transition metals. In this work, we designed Fe–Co doped g-C3N4 (Fe–Co–g-C3N4) catalysts for the selective allylic oxidation of cyclohexene using oxygen as an oxidant (Scheme 1). The results showed that the bimetallic Fe–Co–g-C3N4 catalysts were highly effective for the oxidation of cyclohexene to 2-cyclohexene-1-one. Especially, the selectivity of the reaction in water could be very high and was much higher than that in other solvents at a similar conversion. As far as we know, this is the first work to use the bimetallic Fe–Co–g-C3N4 as a catalyst for the selective oxidation of cyclohexene.
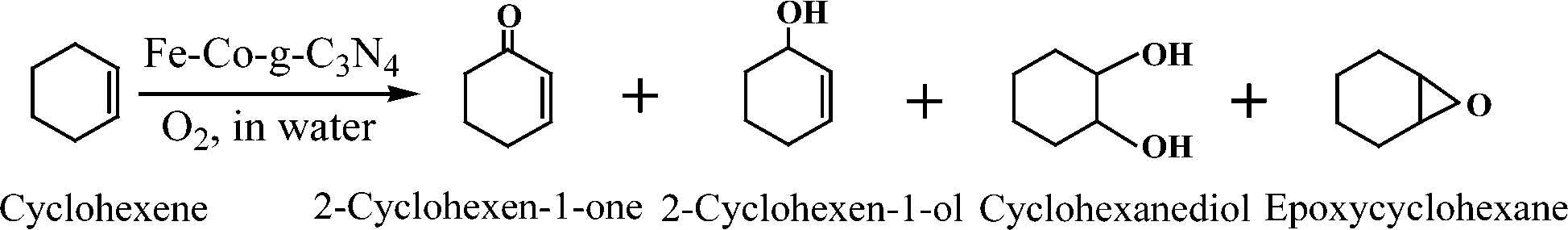 | ||
| Scheme 1 The oxidation of cyclohexene catalyzed by the Fe–Co–g-C3N4 catalyst with molecular oxygen in water. | ||
Results and discussion
Characterization of the catalysts
In order to understand well the existent state of bimetallic Fe–Co on the surface of a g-C3N4 support, the morphologies of g-C3N4 and 5-Fe–Co–g-C3N4 were characterized by TEM techniques, as shown in Fig. 1(a) and (b). It can be seen that there are no significant differences between the morphologies of g-C3N4 and 5-Fe–Co–g-C3N4. Both of them showed a similar sheet-like structure of graphene, indicating that Fe–Co doped g-C3N4 did not significantly change the texture of the carbon nitride polymer. Although Fe and Co species could not be observed on the surface of g-C3N4 by TEM, the element mapping images of 5-Fe–Co–g-C3N4 in Fig. 1(c) proved that they existed, probably due to the interaction between Fe or Co and g-C3N4 by the formation of Fe–N or Co–N bonds.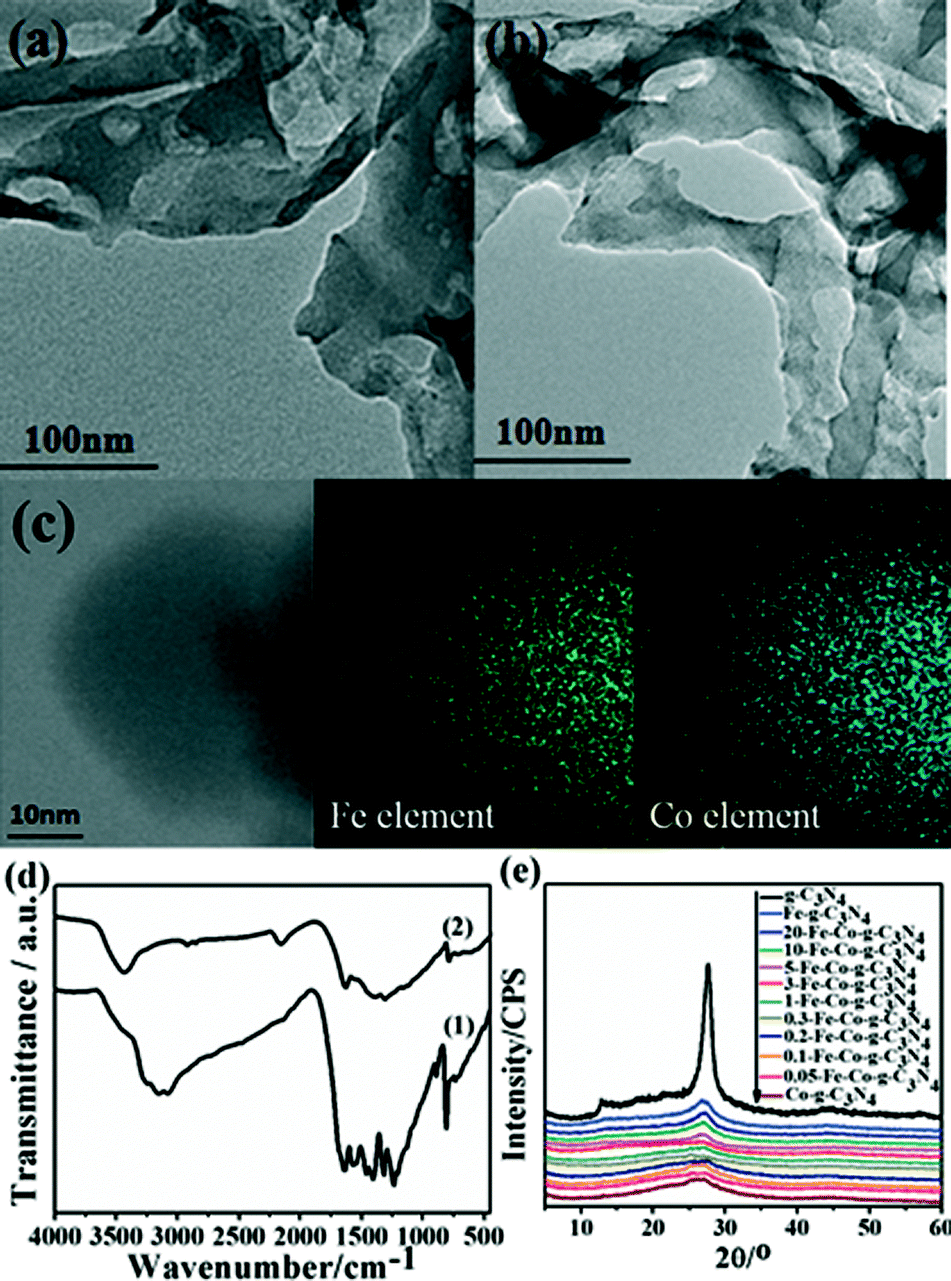 | ||
| Fig. 1 TEM images of g-C3N4 (a) and the 5-Fe–Co–g-C3N4 catalyst (b); element mapping images of 5-Fe–Co–g-C3N4 (c); FT-IR spectra of g-C3N4 (1) and the 5-Fe–Co–g-C3N4 (2) catalyst (d); XRD patterns of g-C3N4 and the Fe–Co–g-C3N4 catalyst with different molar ratios of Fe to Co (e). | ||
The FT-IR spectra of g-C3N4 and the 5-Fe–Co–g-C3N4 catalyst are shown in Fig. 1(d). The bands between 1200 and 1650 cm−1 corresponding to the typical stretching modes of CN heterocycles and the characteristic mode of the triazine units (C6N7) at 803 cm−1 are observed for g-C3N4,34,35 indicating the presence of a typical structure of g-C3N4. It is also notable that the spectrum of 5-Fe–Co–g-C3N4 was similar to that of g-C3N4, with the expectation that the characteristic bands were relatively weak, suggesting that the doping of bimetallic Fe–Co had no obvious influence on the typical structure of the g-C3N4 support. This can be further confirmed by the XRD patterns of g-C3N4 and the Fe–Co–g-C3N4 catalyst with different molar ratios of Fe and Co. As shown in Fig. 1(e), the strong peak at 27.6° and the weak peak at 12.9° were observed for g-C3N4, corresponding to the graphitic materials as the (002) peak of the layered stacking of the conjugated aromatic system and the in-plane structural packing motif of tri-s-triazine units, respectively.36 Interestingly, the XRD patterns of Fe–Co–g-C3N4 catalysts with different molar ratios of Fe and Co were similar to that of pristine g-C3N4, but the overall intensity of the peaks decreased with increasing content of Co in the Fe–Co–g-C3N4. There were no peaks originating from Fe or Co species. This is presumably caused by the formation of Fe–N or Co–N bonds through the chemical coordination of Fe or Co species with g-C3N4 in the preparation of Fe–Co–g-C3N4 catalysts under flowing nitrogen atmosphere.
The 5-Fe–Co–g-C3N4 catalyst was further investigated by XPS analysis, as shown in Fig. 2. The surface catalyst concentrations of Fe and Co species obtained from XPS were 1.91% and 1.1%, respectively. It can be seen from the full XPS spectrum that the main elements were C, N, O, Fe and Co in the 5-Fe–Co–g-C3N4 catalyst. The photoelectron peaks of these elements were clearly observed at binding energies of 288.0 eV (C1s), 399.0 eV (N1s), 530.9 eV (O1s), 710.3 eV (Fe2p) and 781.3 eV (Co2p), respectively. The fitted spectrum of C in Fig. 2(b) shows three main peaks with binding energies of 287.9 eV, 285.8 eV and 284.5 eV, corresponding to sp2-bonded carbon (N–CN), C–O and graphitic carbon (C–C), respectively.37 The high resolution N 1s spectra (Fig. 2(c)) for 5-Fe–Co–g-C3N4 displayed typical heptazine repeating units with several N species at different binding energies, including triazine rings (CN–C, 398.8 eV), tertiary nitrogen (N–(C)3, 400.1 eV), and a handful of amino functions (N–H, 401.2 eV).34,38 These representative signals indicated that the framework of g-C3N4 was not changed with the doping of Fe and Co species. The high resolution Fe 2p XPS spectrum is shown in Fig. 2(e). A typical binding energy peak at 710.3 eV indicates the existence of the Fe(III) valence state in the 5-Fe–Co–g-C3N4 catalyst, which may be due to the stabilization of Fe species in the electron-rich g-C3N4 structure mainly through Fe–N bonds.27,39 Moreover, the binding energy peaks at 781.3 eV and 783.4 eV were observed in the high resolution Co 2p XPS spectrum (Fig. 2(f)), suggesting the existence of the Co(II) valence state in the form of Co–N moieties.40 These results reasonably explained the fact that Fe and Co species in the 5-Fe–Co–g-C3N4 catalyst could not be observed through XRD and TEM methods.
 | ||
| Fig. 2 XPS spectra of the 5-Fe–Co–g-C3N4 catalyst; survey spectra (a), high resolution spectra of C1s (b), N1s (c), O1s (d), Fe2p (e), and Co2p (f). | ||
The N2 adsorption–desorption isotherms and pore size distribution plots for g-C3N4 and the 5-Fe–Co–g-C3N4 catalyst are shown in Fig. 3. Both samples exhibited type H4 sorption hysteresis loops, indicating the existence of narrow slit-like pores. The pore size distribution of pure g-C3N4 and the 5-Fe–Co–g-C3N4 catalyst calculated by the original DFT method showed a main peak centered at approximately 2–5 nm. The 5-Fe–Co–g-C3N4 catalyst had a Brunauer–Emmett–Teller (BET) surface area of 22.9 m2 g−1 and a pore volume of 0.050 cm3 g−1, while the BET surface area and pore volume of pure g-C3N4 were 29.9 m2 g−1 and 0.101 cm3 g−1, respectively.
 | ||
| Fig. 3 N2 adsorption–desorption isotherms and the corresponding pore size distribution plots (inset) for pure g-C3N4 and the 5-Fe–Co–g-C3N4 catalyst. | ||
The contents of C, N, and H in the 5-Fe–Co–g-C3N4 catalyst determined by elemental analysis were 32.55%, 56.34%, and 1.15%, respectively. An approximate value of 0.77 was calculated for the C/N ratio, which is close to the theoretical value of 0.75 for g-C3N4, indicating that the 5-Fe–Co–g-C3N4 catalyst was highly enriched in nitrogen. The results further suggest that the doping of Fe–Co did not change the chemical structure of the g-C3N4 support. The minor amount of hydrogen (1.15%) and oxygen (5.75%) may be attributed to adsorbed water and imperfections in the condensation process, as confirmed by XPS analyses. The loading of Fe and Co in the catalyst determined by ICP was 3.28 wt% and 0.93 wt%, respectively. The loading of Fe and Co determined by XPS was less than that measured by ICP, perhaps due to some Fe and Co existing inside the catalysts in the form of Fe–N and Co–N which were not detected through XPS.
Catalytic performance of catalysts with different supports
The oxidation of cyclohexene was firstly conducted without a catalyst in water using oxygen as an oxidant, as shown in Table 1 (entry 1). It is found that the catalytic activity and selectivity to 2-cyclohexene-1-one were considerably dissatisfactory. It is well known that the support often plays an important role for the performance of a catalyst. So we loaded Co on activated C, TiO2, SiO2, and g-C3N4 and studied the selective oxidation of cyclohexene to 2-cyclohexene-1-one in water using oxygen as an oxidant, and the results are shown in Table 1. The catalytic activity and selectivity to 2-cyclohexene-1-one over the Co–g-C3N4 catalyst were much higher than those over Co–C, Co–TiO2 and Co–SiO2 catalysts under similar reaction conditions. This indicates that g-C3N4 was an excellent support of Co for the selective oxidation of cyclohexene to 2-cyclohexene-1-one, probably due to the fact that the interaction between Co and N in g-C3N4 could inhibit the generation of by-products (such as 2-cyclohexene-1-ol and cyclohexanediol) effectively. As a result, g-C3N4 can be regarded as a promising support for the oxidation of cyclohexene.| Entry | Catalysts | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|
|
|
|
|
Others | |||
| a Reaction conditions: cyclohexene, 0.5 mL; oxygen pressure, 4 MPa; deionized water, 2.0 mL; catalyst, 0.023 g; reaction time, 5 h; reaction temperature, 90 °C. | ||||||
| 1 | — | 5.5 | 51.4 | 18.1 | 21.9 | 8.6 |
| 2 | Co–C | 16.7 | 63.8 | 25.7 | 7.6 | 2.9 |
| 3 | Co–TiO2 | 24.6 | 67.3 | 18.6 | 5.2 | 8.9 |
| 4 | Co–SiO2 | 19.9 | 65.0 | 25.6 | 5.2 | 4.2 |
| 5 | Co–g-C3N4 | 55.9 | 71.7 | 23.0 | 1.9 | 3.4 |
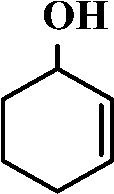
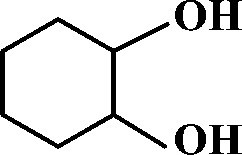
Catalytic performance of g-C3N4 doped with different metals
We investigated the effect of different metals doped in g-C3N4 on the oxidation of cyclohexene using oxygen as an oxidant and water as solvent, and the results are summarized in Table 2. Without a doping metal, g-C3N4 showed a relatively high activity for the oxidation of cyclohexene, but the selectivity to 2-cyclohexene-1-one was very low (Table 2, entry 1). The V or W doped g-C3N4 also showed low activity and selectivity (Table 2, entries 2 and 3). Although the Cu and Co doped g-C3N4 catalysts exhibited higher activity than the V and W doped g-C3N4, the selectivity to 2-cyclohexene-1-one was still low (Table 2, entries 4 and 5). However, when g-C3N4 was doped with Fe metal, the selectivity to 2-cyclohexene-1-one increased to 90.4%, but the conversion of cyclohexene was relatively low (Table 2, entry 6). Based on the results of higher activity of Cu–g-C3N4 and Co–g-C3N4 for the oxidation of cyclohexene and higher selectivity of the Fe–g-C3N4 catalyst to 2-cyclohexene-1-one, we attempted to combine Cu and Fe or Co and Fe with g-C3N4 for the reaction. The results showed that Fe–Co–g-C3N4 had higher activity and selectivity than Fe–Cu–g-C3N4 (Table 2, entries 7 and 8), i.e., the Fe–Co–g-C3N4 catalyst was a very effective catalyst for the oxidation of cyclohexene to 2-cyclohexene-1-one with oxygen as an oxidant and water as solvent.| Entry | Catalysts | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|
|
|
|
|
Others | |||
| a Reaction conditions: cyclohexene, 0.5 mL; oxygen pressure, 4 MPa; deionized water, 2.0 mL; catalyst, 0.023 g; reaction time, 5 h; reaction temperature, 90 °C. | ||||||
| 1 | g-C3N4 | 37.6 | 60.3 | 26.2 | 2.9 | 10.6 |
| 2 | V–g-C3N4 | 22.2 | 62.6 | 25.7 | 3.3 | 8.4 |
| 3 | W–g-C3N4 | 13.8 | 53.4 | 29.1 | 14.0 | 3.5 |
| 4 | Cu–g-C3N4 | 45.9 | 65.2 | 26.6 | 5.9 | 2.8 |
| 5 | Co–g-C3N4 | 55.9 | 71.7 | 23.0 | 1.9 | 3.4 |
| 6 | Fe–g-C3N4 | 13.6 | 90.4 | 6.3 | 1.4 | 1.9 |
| 7 | 1-Fe–Cu–g-C3N4 | 16.2 | 79.9 | 15.2 | 1.9 | 3.0 |
| 8 | 1-Fe–Co–g-C3N4 | 27.6 | 89.9 | 7.8 | 0.8 | 1.5 |
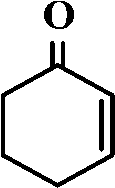

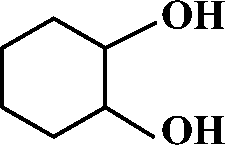
Effect of different solvents on the oxidation of cyclohexene
It is known that solvents can often affect reactions significantly. Herein, we studied the effect of different solvents on the catalytic oxidation of cyclohexene over the 1-Fe–Co–g-C3N4 catalyst with oxygen as an oxidant. The results are shown in Fig. 4. It is found that the selectivity to 2-cyclohexene-1-one was kept about 90% in the cyclohexene conversion range from 4 to 36% using water as reaction solvent. This indicates that water can suppress the formation of by-products (such as 2-cyclohexene-1-ol and 1,2-cyclohexanediol). However, when acetone or acetonitrile was used as solvent, the selectivity to 2-cyclohexene-1-one was only 65–70% at the conversion range, which is much lower than that in water. Similarly, the resulted selectivity to 2-cyclohexene-1-one was also poor under solvent-free conditions. The results suggest that Fe–Co–g-C3N4 and water had a cooperative effect for the effective catalytic oxidation of cyclohexene to 2-cyclohexene-1-one. In addition, water is environmentally benign, abundant, and cheap compared with organic solvents.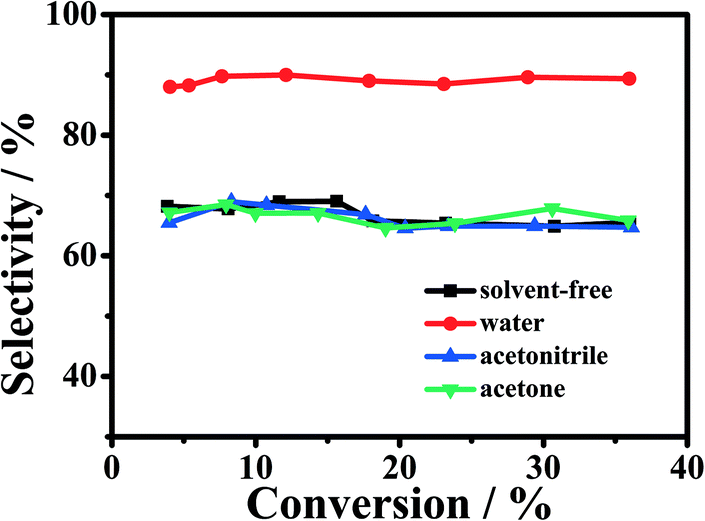 | ||
| Fig. 4 Effect of the different solvents on the oxidation of cyclohexene. Reaction conditions: cyclohexene, 0.5 mL; oxygen pressure, 4 MPa; different solvent, 2.0 mL; 1-Fe–Co–g-C3N4 catalyst, 0.023 g; reaction temperature, 90 °C. | ||
Effect of molar ratio of Fe to Co in Fe–Co–g-C3N4
We also studied the effect of the molar ratio of Fe and Co in the Fe–Co–g-C3N4 catalyst for the oxidation of cyclohexene with oxygen and water as solvent (Table 3). The molar ratio of Fe and Co influenced the selectivity to 2-cyclohexene-1-one dramatically. Although the Co–g-C3N4 catalyst showed the highest activity, the selectivity to 2-cyclohexene-1-one was poor (Table 3, entry 1). With the increase of Fe content in the catalyst, the catalytic activity continuously decreased, while the selectivity to 2-cyclohexene-1-one obviously increased (Table 3, entries 2–7). When the molar ratio of Fe and Co was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the Fe–Co–g-C3N4 catalyst exhibited the highest selectivity to 2-cyclohexene-1-one, which was about 95.5% (Table 3, entry 8). With a further increase in the molar ratio of Fe and Co in the Fe–Co–g-C3N4 catalyst, the activity and selectivity slightly decreased (Table 3, entries 9 and 10). In the absence of Co content in the catalyst, the conversion of cyclohexene decreased to 13.6%, while the selectivity to 2-cyclohexene-1-one decreased to 90.4% (Table 3, entry 11). The results indicate that suitable molar ratios of Fe and Co are required for satisfactory performance of the catalyst. For comparison, Fe, Co and Fe–Co supported g-C3N4 catalysts, which were synthesized by a conventional impregnation method, were used to catalyze the oxidation of cyclohexene under the same reaction conditions, as shown in the ESI (Table S1, Fig. S1–S3†). It can be seen from the results that the catalytic activity and selectivity to 2-cyclohexene-1-one were obviously not as good as those of the catalysts discussed above. This may be attributed to the fact that Fe and Co existed in the form of Fe3O4 and Co3O4 on the pristine g-C3N4 and the active sites on the support were partly occupied by the metal oxide.
:1, the Fe–Co–g-C3N4 catalyst exhibited the highest selectivity to 2-cyclohexene-1-one, which was about 95.5% (Table 3, entry 8). With a further increase in the molar ratio of Fe and Co in the Fe–Co–g-C3N4 catalyst, the activity and selectivity slightly decreased (Table 3, entries 9 and 10). In the absence of Co content in the catalyst, the conversion of cyclohexene decreased to 13.6%, while the selectivity to 2-cyclohexene-1-one decreased to 90.4% (Table 3, entry 11). The results indicate that suitable molar ratios of Fe and Co are required for satisfactory performance of the catalyst. For comparison, Fe, Co and Fe–Co supported g-C3N4 catalysts, which were synthesized by a conventional impregnation method, were used to catalyze the oxidation of cyclohexene under the same reaction conditions, as shown in the ESI (Table S1, Fig. S1–S3†). It can be seen from the results that the catalytic activity and selectivity to 2-cyclohexene-1-one were obviously not as good as those of the catalysts discussed above. This may be attributed to the fact that Fe and Co existed in the form of Fe3O4 and Co3O4 on the pristine g-C3N4 and the active sites on the support were partly occupied by the metal oxide.
| Entry | Catalyst | Fe/Co ratio | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|---|
|
|
|
|
Others | ||||
| a Reaction conditions: cyclohexene, 0.5 mL; oxygen pressure, 4 MPa; deionized water, 2.0 mL; catalyst, 0.023 g; reaction time, 5 h; reaction temperature, 90 °C. | |||||||
| 1 | Co–g-C3N4 | — | 55.9 | 71.7 | 23.0 | 1.9 | 3.4 |
| 2 | 0.05-Fe–Co–g-C3N4 | 1:20 |
50.1 | 73.5 | 22.1 | 0.8 | 3.7 |
| 3 | 0.1-Fe–Co–g-C3N4 | 1:10 |
45.6 | 77.4 | 17.2 | 2.1 | 3.3 |
| 4 | 0.2-Fe–Co–g-C3N4 | 1:5 |
41.3 | 78.0 | 18.7 | 2.3 | 1.0 |
| 5 | 0.3-Fe–Co–g-C3N4 | 1:3 |
30.3 | 82.5 | 14.0 | 0.8 | 2.6 |
| 6 | 1-Fe–Co–g-C3N4 | 1:1 |
27.6 | 89.9 | 7.8 | 0.8 | 1.5 |
| 7 | 3-Fe–Co–g-C3N4 | 3:1 |
24.6 | 90.3 | 5.3 | 1.9 | 2.5 |
| 8 | 5-Fe–Co–g-C3N4 | 5:1 |
21.8 | 95.5 | 1.2 | 1.9 | 1.4 |
| 9 | 10-Fe–Co–g-C3N4 | 10:1 |
21.3 | 92.5 | 3.3 | 2.7 | 1.6 |
| 10 | 20-Fe–Co–g-C3N4 | 20:1 |
20.8 | 90.2 | 5.1 | 2.9 | 1.8 |
| 11 | Fe–g-C3N4 | — | 13.6 | 90.4 | 6.3 | 1.4 | 1.9 |
Effect of the pressure of molecular oxygen
The effect of oxygen pressure on the conversion and selectivity was studied over the 5-Fe–Co–g-C3N4 catalyst in water, and the results are shown in Fig. 5. With the increase in oxygen pressure, the conversion of cyclohexene increased. The selectivity to 2-cyclohexene-1-one also increased slowly when the pressure was lower than 4 MPa. When oxygen pressure was larger than 4 MPa, the selectivity to 2-cyclohexene-1-one decreased with further increasing pressure.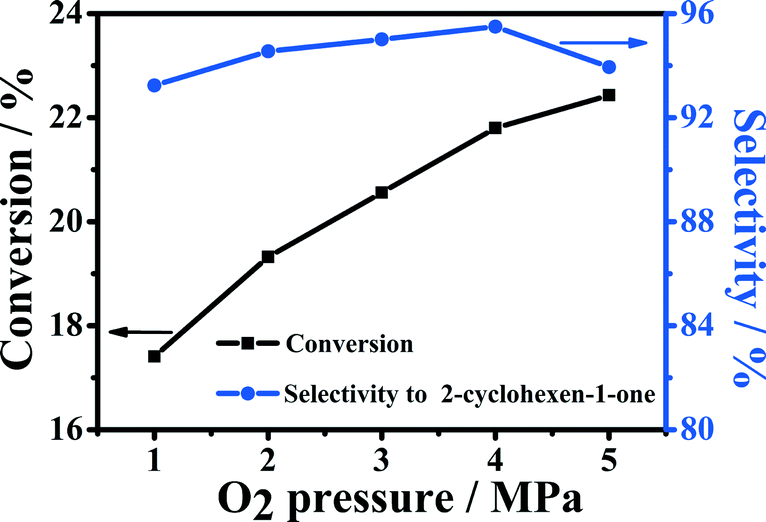 | ||
| Fig. 5 Effect of molecular oxygen pressure on the oxidation of cyclohexene. Reaction conditions: cyclohexene, 0.5 mL; water, 2.0 mL; 5-Fe–Co–g-C3N4 catalyst, 0.023 g; reaction temperature, 90 °C; reaction time, 5 h. | ||
Effect of reaction temperature
The effect of reaction temperature on the oxidation of cyclohexene is shown in Fig. 6. The conversion of cyclohexene was only about 6.7% at 70 °C in 5 h, and the selectivity to 2-cyclohexene-1-one was 95.5%. Increasing the temperature from 70 to 90 °C could improve the conversion significantly, while the selectivity to 2-cyclohexene-1-one was nearly unchanged. When the temperature was increased to 110 °C, the conversion of cyclohexene could reach 36.0% under the experimental conditions, but the selectivity to 2-cyclohexene-1-one declined considerably. This indicates that high temperature led to the formation of more by-products.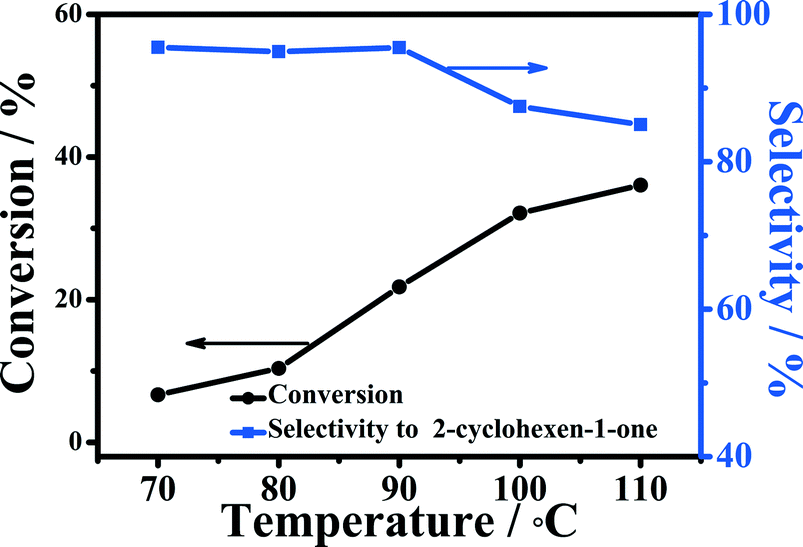 | ||
| Fig. 6 Effect of reaction temperature on the oxidation of cyclohexene. Reaction conditions: cyclohexene, 0.5 mL; oxygen pressure, 4 MPa; water, 2.0 mL; 5-Fe–Co–g-C3N4 catalyst, 0.023 g; reaction time, 5 h. | ||
Effect of reaction time
The influence of reaction time on the oxidation of cyclohexene at 90 °C is shown in Fig. 7. It can be observed that the conversion of cyclohexene increased continuously within 15 h, while the selectivity to 2-cyclohexene-1-one was nearly unchanged. When the reaction time exceeded 15 h, the selectivity to 2-cyclohexene-1-one began to decrease obviously due to the generation of by-products, such as 2-cyclohexene-1,4-dione, adipic acid, and 1,2-cyclohexanediol.9 It can be known from the figure that 5-Fe–Co–g-C3N4 was an excellent catalyst for the reaction in water. The selectivity to 2-cyclohexene-1-one could be as high as 95% at a conversion of 36% at 15 h.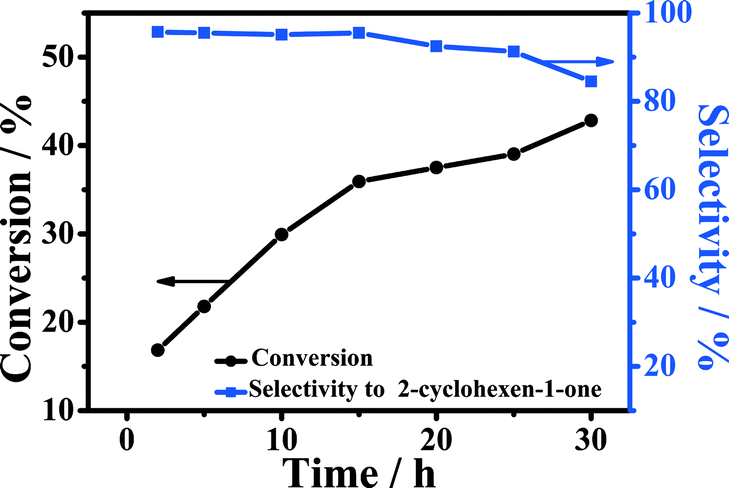 | ||
| Fig. 7 Effect of reaction time on the oxidation of cyclohexene. Reaction conditions: cyclohexene, 0.5 mL; oxygen pressure, 4 MPa; water, 2.0 mL; 5-Fe–Co–g-C3N4 catalyst, 0.023 g; reaction temperature, 90 °C. | ||
Reusability of the catalyst
The reusability of the 5-Fe–Co–g-C3N4 catalyst for the oxidation of cyclohexene was investigated, and the results are given in Fig. 8. The catalyst could be reused at least four times without a considerable change in activity and selectivity to 2-cyclohexene-1-one. The conversion of reaction decreased slightly after recycling. The main reason may be that a small amount of the catalyst was lost when recovering the catalyst by centrifugation and washing with deionized water. Fig. 9(a) gives the TEM images of the fresh and used catalysts after four cycles. The morphology of the catalyst was not changed notably, as can be seen from Fig. 1 and 9(a). Fig. 9(b) compares the XRD pattern of the fresh 5-Fe–Co–g-C3N4 catalyst with that of the catalyst after being used four times. The XRD patterns of the fresh and used catalysts were nearly the same, indicating that the crystal structure of the catalysts was unchanged in the reaction.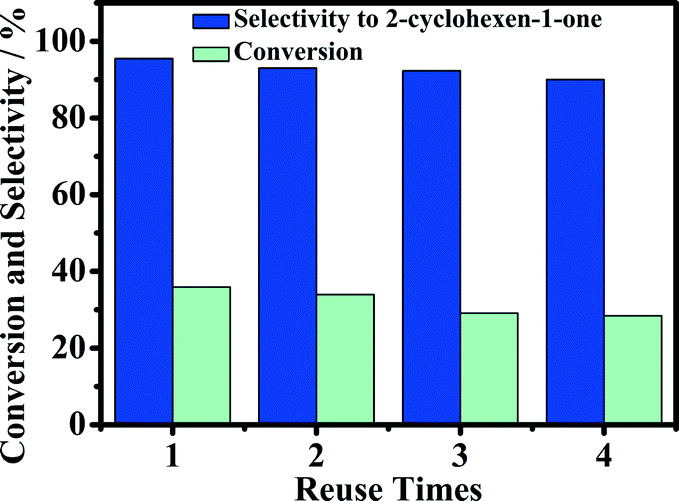 | ||
| Fig. 8 Reuse of the 5-Fe–Co–g-C3N4 catalyst. Reaction conditions: cyclohexene, 0.5 mL; oxygen pressure, 4 MPa; water, 2.0 mL; 5-Fe–Co–g-C3N4 catalyst, 0.023 g; reaction temperature, 90 °C, reaction time, 15 h. | ||
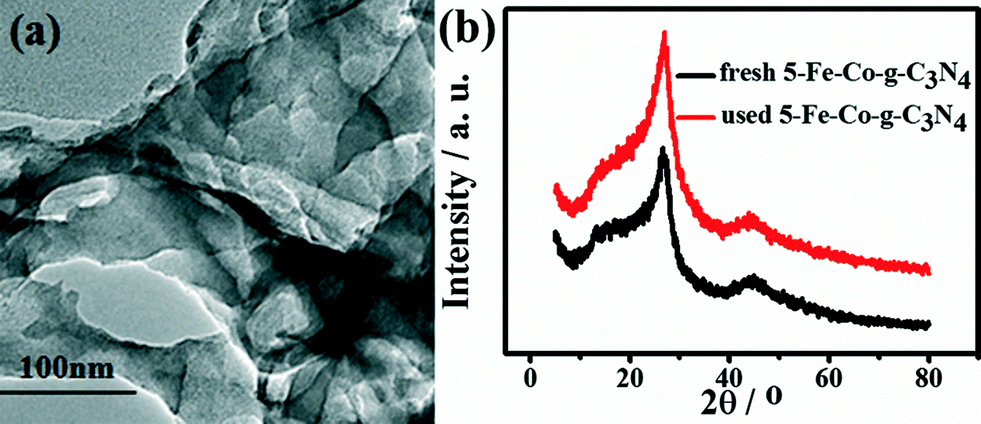 | ||
| Fig. 9 TEM images of the 5-Fe–Co–g-C3N4 catalyst after being used four times (a), and XRD patterns of the fresh and used 5-Fe–Co–g-C3N4 catalysts after being used four times (b). | ||
Conclusions
Fe–Co–g-C3N4 catalysts have been designed and prepared and used to catalyze the selective oxidation of cyclohexene to 2-cyclohexene-1-one with oxygen as an oxidant in water. It is shown that the metals in the Fe–Co–g-C3N4 catalysts are Fe(III) and Co(II) species stabilized by Fe–N bonds and Co–N bonds. The Fe–Co–g-C3N4/water catalytic system is very active and selective for the reaction, and the selectivity to 2-cyclohexene-1-one reaches 95% at a cyclohexene conversion of 36%. The catalyst can be used at least four times without a considerable decrease in activity and selectivity. We believe that the Fe–Co–g-C3N4/water catalytic system, which is highly efficient, environmentally benign, and prepared from abundant feedstocks, has great potential for application in the selective oxidation of cyclohexene to 2-cyclohexene-1-one.Experimental
Materials
Cyclohexene, 2-cyclohexene-1-one, 2-cyclohexene-1-ol, ethyl acetate, n-heptane, acetonitrile, acetone, ethanol, Co(NO3)2·6H2O, NH4VO3, (NH4)10W12O41·xH2O and Cu(NO3)2 were all of analytical grade and purchased from Sinopharm Chemical Reagent Co. Ltd. Anhydrous ferric chloride and dicyandiamide were provided by Aldrich and used as received. Molecular oxygen (99.99%) was obtained from Beijing Analytical Instrument Company.Catalyst preparation
Catalyst characterization
The morphology of the catalyst was characterized by a transmission electron microscope (TEM, JEOL JEM-2100F) equipped with an EDS spectrometer and a HITACHI S-4300 SEM equipped with an EDX spectrometer. The Fourier transform infrared (FT-IR) spectra of the samples through compressing with KBr powder were obtained by using a Bruker Tensor 27 spectrometer with a resolution of 1 cm−1 and 32 scans. The nitrogen adsorption–desorption isotherms were measured on a Micromeritics ASAP 2020 sorptometer at 77 K. Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/max-2500 X-ray diffractometer using Cu-Kα radiation (y = 0.15406 nm). The tube voltage was 40 kV and the current was 200 mA. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific ESCALab 250 Xi using 200 W monochromated Al-Kα radiation. A 500 μm X-ray spot was used for XPS analysis. The base pressure in the analysis chamber was about 3 × 10−10 mbar. Typically, the hydrocarbon C1s line at 284.8 eV from adventitious carbon was used for energy referencing. The content of metal in the catalysts was determined by inductively coupled plasma optical emission spectroscopy (ICP-AES, Vista-MPX). Elemental analysis of the catalyst was performed on a Flash EA 1112 elemental analysis instrument.Reaction
The oxidation of cyclohexene over the Fe–Co–g-C3N4 catalyst was carried out with molecular oxygen as an oxidant and water as solvent, as shown in Scheme 1. In a typical reaction, 0.5 mL of cyclohexene, 2.0 mL of deionized water, and 0.023 g of catalyst were introduced into a 6 mL Teflon-lined stainless steel autoclave equipped with a magnetic stirrer. After being filled with the desired molecular oxygen, the autoclave was heated to the required temperature and stirring was started. After the reaction, the autoclave was placed in ice water and molecular oxygen was slowly released. The reaction product was extracted by ethyl acetate (4 mL × 3) and analyzed by using an Agilent 6820 gas chromatograph equipped with a flame ionization detector and a PEG-20M capillary column (30 m × 0.25 mm × 0.25 mm), using n-heptane as internal standard. In the recycling experiment, the catalyst was separated from the liquid by centrifugation, washed with deionized water, dried at 60 °C under vacuum for 3 h, and then reused for the next run.Acknowledgements
The authors thank the National Natural Science Foundation of China (21373230, 21273253, 21321063) and the Chinese Academy of Sciences (KJCX2.YW.H30).Notes and references
- M. D. Hughes, Y. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS PubMed.
- C. Yin, Z. Yang, B. Li, F. Zhang, J. Wang and E. Ou, Catal. Lett., 2009, 131, 440–443 CrossRef CAS.
- S. E. Dapurkar, H. Kawanami, K. Komura, T. Yokoyama and Y. Ikushima, Appl. Catal., A, 2008, 346, 112–116 CrossRef CAS.
- J. Muzart, Chem. Rev., 1992, 92, 113–140 CrossRef CAS.
- E. M. Serwicka, J. Połtowicz, K. Bahranowski, Z. Olejniczak and W. Jones, Appl. Catal., A, 2004, 275, 9–14 CrossRef CAS.
- R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. Singh, I. Ahmed and R. V. Jasra, J. Mol. Catal. A: Chem., 2004, 218, 141–146 CrossRef CAS.
- K. Leus, I. Muylaert, M. Vandichel, G. B. Marin, M. Waroquier, V. Van Speybroeck and P. Van Der Voort, Chem. Commun., 2010, 46, 5085–5087 RSC.
- K. Li, J. Wang, Y. Zou, X. Song, H. Gao, W. Zhu, W. Zhang, J. Yu and M. Jia, Appl. Catal., A, 2014, 482, 84–91 CrossRef CAS.
- Y. Cao, H. Yu, F. Peng and H. Wang, ACS Catal., 2014, 4, 1617–1625 CrossRef CAS.
- J. Li, X. Li, Y. Shi, D. Mao and G. Lu, Catal. Lett., 2010, 137, 180–189 CrossRef CAS.
- X. Cai, H. Wang, Q. Zhang, J. Tong and Z. Lei, J. Mol. Catal. A: Chem., 2014, 383–384, 217–224 CrossRef CAS.
- F. P. Silva, M. J. Jacinto, R. Landers and L. M. Rossi, Catal. Lett., 2011, 141, 432–437 CrossRef CAS.
- L. X. Alvarez, M. L. Christ and A. B. Sorokin, Appl. Catal., A, 2007, 325, 303–308 CrossRef CAS.
- Z. Yang, Q. Kang, H. Ma, C. Li and Z. Lei, J. Mol. Catal. A: Chem., 2004, 213, 169–176 CrossRef CAS.
- P. J. Baricelli, V. J. Sánchez, A. J. Pardey and S. A. Moya, J. Mol. Catal. A: Chem., 2000, 164, 77–84 CrossRef CAS.
- B. Sun, J. Chen, J. Hu and X. Li, J. Inorg. Biochem., 2006, 100, 1308–1313 CrossRef CAS PubMed.
- N. Malumbazo and S. F. Mapolie, J. Mol. Catal. A: Chem., 2009, 312, 70–77 CrossRef CAS.
- S. Yamaguchi, T. Fukura, K. Takiguchi, C. Fujita, M. Nishibori, Y. Teraoka and H. Yahiro, Catal. Today, 2015, 242, 261–267 CrossRef CAS.
- B. G. Donoeva, D. S. Ovoshchnikov and V. B. Golovko, ACS Catal., 2013, 3, 2986–2991 CrossRef CAS.
- M. Shahabi nejad, G. Ghasemi, M. V. Martínez-Huerta and M. Ghiaci, J. Mol. Catal. A: Chem., 2015, 406, 118–126 CrossRef.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- Y. Gong, M. Li, H. Li and Y. Wang, Green Chem., 2015, 17, 715–736 RSC.
- P. Zhang, Y. Wang, J. Yao, C. Wang, C. Yan, M. Antonietti and H. Li, Adv. Synth. Catal., 2011, 353, 1447–1451 CrossRef CAS.
- F. Z. Su, S. Mathew, G. Lipner, X. Z. Fu, M. Antonietti, S. Blechert and X. C. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- F. Z. Su, S. Mathew, L. Molmann, M. Antonietti, X. C. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657–660 CrossRef CAS PubMed.
- Y. Wang, H. Li, J. Yao, X. Wang and M. Antoniettia, Chem. Sci., 2011, 2, 446–450 RSC.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- Y. Wang, J. Yao, H. R. Li, D. S. Du and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- J. Cao, S. Shen, P. Yang and J. Qu, Org. Lett., 2013, 15, 3856–3859 CrossRef CAS PubMed.
- P. Zhang, T. Wu, T. Jiang, W. Wang, H. Liu, H. Fan, Z. Zhang and B. Han, Green Chem., 2013, 15, 152–159 RSC.
- T. Wu, P. Zhang, T. Jiang, D. Yang and B. Han, Sci. China: Chem., 2015, 58, 93–100 CrossRef CAS.
- H. Liu, T. Jiang, B. Han, S. Liang, W. Wang, T. Wu and G. Yang, Green Chem., 2011, 13, 1106–1109 RSC.
- N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS.
- G. Ding, W. Wang, T. Jiang, B. Han, H. Fan and G. Yang, ChemCatChem, 2013, 5, 192–200 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC.
- J. Mao, T. Y. Peng, X. H. Zhang, K. Li, L. Q. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- Q. Liu and J. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD patterns and EDX spectra of catalysts by a conventional impregnation method. See DOI: 10.1039/c5cy01177a |
| This journal is © The Royal Society of Chemistry 2016 |