
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mesocrystals: structural and morphogenetic aspects†
Elena V.
Sturm (née Rosseeva)
* and
Helmut
Cölfen
*
Physical Chemistry, Department of Chemistry, University of Konstanz, Universitätsstr. 10, D-78457 Konstanz, Germany. E-mail: elena.sturm@uni-konstanz.de; helmut.coelfen@uni-konstanz.de
First published on 9th August 2016
Abstract
Mesocrystals are a fascinating class of crystalline nanostructured materials since they combine the properties of nanoparticles with order on the microscopic or even macroscopic length scale. This tutorial review deals with the structural aspects of mesocrystals as well as their formation mechanisms known so far. The goal is to reach an understanding about what is special about the structuration principles of mesocrystals and their complex structures and what are the possibilities to control their formation processes.
 Elena V. Sturm | Elena V. Sturm (née Rosseeva) is a junior group leader at the University of Konstanz in the Department of Chemistry and a research fellow of the Zukunftskolleg. She received her PhD from the Department of Crystallography at Saint-Petersburg State University in 2010 (supervisor Prof. O. V. Frank-Kamenetskaya) based on work carried out in cooperation with the group of Prof. R. Kniep at the Max Planck Institute for Chemical Physics of Solids in Dresden. From 2010 to 2012 she was a PostDoc at MPI CPfS. Current research interests include the investigation of the structure and morphogenesis of nanocomposite materials from biological and biomimetic systems as well as nanoparticle self-assemblies. |
 Helmut Cölfen | Helmut Cölfen is a full professor of physical chemistry at the University of Konstanz. His research interests are in the area of nucleation, classical and non-classical crystallization, biomineralization, synthesis of functional polymers, directed self-assembly of nanoparticles and fractionating methods of polymer and nanoparticle analysis – especially analytical ultracentrifugation. His group has made contributions in high-resolution particle size analysis with Angström resolution in solution, mesocrystals, nonclassical Crystallization, CaCO3 crystallization, synthesis of double hydrophilic block copolymers and additive controlled crystallization. He is the editor-in-chief of “Crystals” and a co-editor of “Current Nanoscience” and serves on the editorial/advisory boards of 9 further scientific journals. He has received several awards including the Academy Award of the Berlin-Brandenburg Academy of Science and Humanities. He has also been listed in the Thomson Reuters and Times Higher Education Index lists of the top 100 chemists worldwide for the years 2000–2010 – a list including several Nobel laureates. In his private time, he enjoys loud music. |
Key learning points• Mesocrystals are kinetically stabilized nanostructured crystalline materials combining the properties of crystallographically aligned individual nanocrystals allowing enhanced collective properties by near-field coupling within ordered arrays and the micro/macroscopic size of the aggregates.• Mesocrystals are formed by non-classical particle mediated crystallization (including self-assembly, templated simultaneous nucleation scenarios, etc.), therefore, their morphology can significantly deviate from that determined by the symmetry of the crystal structure and external influences (i.e. Curie's principle, growth regime, etc.). • The physical properties of mesocrystals originate from their special structures, which are in some cases impossible for a crystal of the same size. • Various strategies can be employed for nanocrystal alignment and assembly into a mesocrystal. |
1. Introduction
Mesocrystals are fascinating class of crystalline materials, which are composed of aligned nanocrystals in crystallographic register and order over a microscopic size regime. They are formed by non-classical crystallization which is crystallization mediated by nanoparticles rather than single atoms, ions or molecules like in classical crystallization pathways.1–3 Such an assembly scenario is highly modular allowing for variations of the particle size, shape and chemical nature as well as for that of the organic stabilizing molecules. Mesocrystals were first systematically described about 10 years ago4 and since then, the interest in these materials has been rapidly increasing.5 The reasons for this are the special properties of mesocrystals, some of which cannot be realized by any other crystalline structure or crystals with the size of the mesocrystal. If anisotropic nanocrystals with directional physical properties are aligned in a mesocrystal, coupling and amplification of these properties can be expected. Also collective and emergent properties can arise.6 This can, for example, be advantageous for electrical or magnetic dipoles. Another example is very small superparamagnetic magnetite nanoparticles, which often have too low magnetization to be controlled or separated by a magnetic field. Increasing their size would increase the saturation magnetization but due to the superparamagnetic to ferromagnetic transition at a size of ca. 30 nm, the superparamagnetism would not be retained. However, the controlled assembly of 6–10 nm magnetite nanoparticles led to superparamagnetic mesocrystals with a much higher magnetization than that of the individual nanoparticles.7 This makes these mesocrystals ideal candidates for bioseparation, drug delivery and magnetic resonance imaging and shows in an impressive way that a mesocrystal arrangement can yield physical properties, which would be impossible for the given particle size of the mesocrystal. Similar possibilities exist for mesocrystals assembled from metal nanoparticles, which would show the surface plasmon resonance of nanoparticles, or quantum dots, while the mesocrystals would show the band gap and thus the optical and electrical properties of the individual nanoparticles. Besides the physical properties, the chemical properties of a mesocrystal can also be different from those of the constituting nanoparticles like the catalytic properties of metal mesocrystals, which can show improved specific activities as summarized in ref. 8.In addition, the physical properties of a mesocrystal can also benefit from its structure itself rather than from the properties of the aligned nanoparticles. One such property would be porosity, which originates from the space in-between nanocrystals. For calcite mesocrystals, a rather significant inner surface of more than 500 m2 g−1 was reported.9 However, not all mesocrystals show porosity because the assembled nanocrystals are stabilized by organic molecules on their surface, which can form a nonporous interlayer between the nanocrystals (Fig. 1). This makes them organic–inorganic hybrid structures, which increase the possibilities of mesocrystals due to the combination of two phases at the nanoscale. Therefore, for example, the hardness of inorganic nanocrystals can be combined with the ductility of polymeric interlayers. An obvious advantage of such hybrid mesocrystal structures is the improvement of the mechanical properties since crack propagation is largely hindered by the ductile and elastic organic layers. This principle is exploited in many biominerals like bone, for example, which are highly evolutionary optimized materials with optimized material properties.
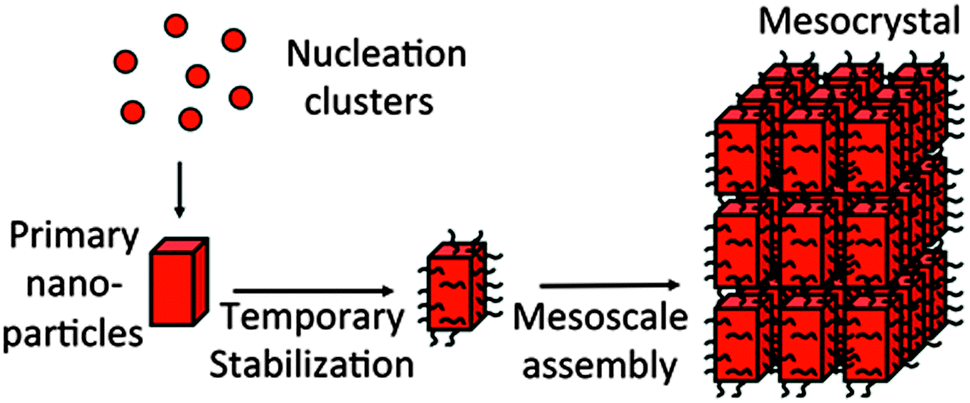 | ||
| Fig. 1 Formation of a mesocrystal by nanoparticle self-assembly. | ||
These few examples cited above already show the huge potential of mesocrystals in materials science, and mesocrystals have already been reported for a number of functional nanocrystals.1,10–13 The first reported applications are in catalysis and electrocatalysis,8,11,14 photocatalysis,13,15,16 surface enhanced Raman scattering,11 optoelectronic applications,14–16 terahertz radiation generation,8 as sensors,8,14,15 electrodes,11,14–16 and lithium ion batteries,13–15 in biomedical applications,15,16 and mechanical as well as structural applications.15,16 The listed references are review papers which give an overview of (not only) the named applications, and the references therein are recommended to access the primary literature.
Besides their huge potential, mesocrystals are not yet commonly applied in technological applications because their formation mechanisms and ways to control them are largely unexplored and not well understood yet. The reason is the huge analytical challenge to observe a self-assembly or other ordering process from the nm to the μm or even mm size within the associated time scales.1 Possibly new analytical developments like liquid cell transmission electron microscopy can improve this situation in the future but up to now, the main knowledge about mesocrystal formation relies on snapshots taken during their formation process. Therefore, it is beneficial to summarize the existing knowledge about mesocrystals and their formation mechanisms to open the door for future applications of these exiting materials. This is the goal of this tutorial review.
2. Mesocrystal definition and structural aspects
2.1. Mesocrystal definition
The word “mesocrystal” is an abbreviation for “mesoscopically structured crystal” which implies its aligned building units from nanocrystals as well as the crystalline mesostructure arising from this arrangement. The definition of a mesocrystal has developed with time starting with the very first definition given in 2005:4“A mesocrystal is defined as a superstructure of crystalline nanoparticles with external crystal faces on the scale of some hundred nanometers to micrometers” and the relation was given to a structure and formation mechanism as shown in Fig. 1 and a vectorial alignment of the building units was mentioned.4
This definition was restricted since it reduced mesocrystals to morphologies with external faces, which is not a requirement (see Section 3.2.) and 1D and 2D mesocrystals are also known. Moreover, due to the fact that nanostructured materials are widely spread in nature (including biological hard tissues), the characterization of these complex structures is very demanding and was sometimes performed by “non-experts” in the field of structural chemistry, crystallography and crystal growth. This induced even more confusion and contradictions in using the term “Mesocrystals” within the last few years.
To describe “Mesocrystals” crystallographically, a number of specific variables have to be specified. The most essential questions in this case are: can we classify a “mesocrystal” as a crystal? Which criterion allows us to identify them as a unique class of solid state material?
In order to answer these questions Fig. 2 schematically illustrates and brings together different types of solid state materials (i.e. single crystals and different types of colloidal solids) to a common focus which helps to highlight general criteria to put mesocrystals into a materials concept.12 For illustrational purposes the crystalline material (core of a nanoparticle) presented in Fig. 2 has a rock salt type crystal structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) while in colloidal crystals the crystalline nanoparticles which are stabilized by organic molecules (shown as blue shell) are arranged in an fcc superlattice (space group Fmm). Further specific examples of mesocrystalline materials are provided in the following sections of this tutorial review with a detailed description of their structure and formation principles.
m) while in colloidal crystals the crystalline nanoparticles which are stabilized by organic molecules (shown as blue shell) are arranged in an fcc superlattice (space group Fmm). Further specific examples of mesocrystalline materials are provided in the following sections of this tutorial review with a detailed description of their structure and formation principles.
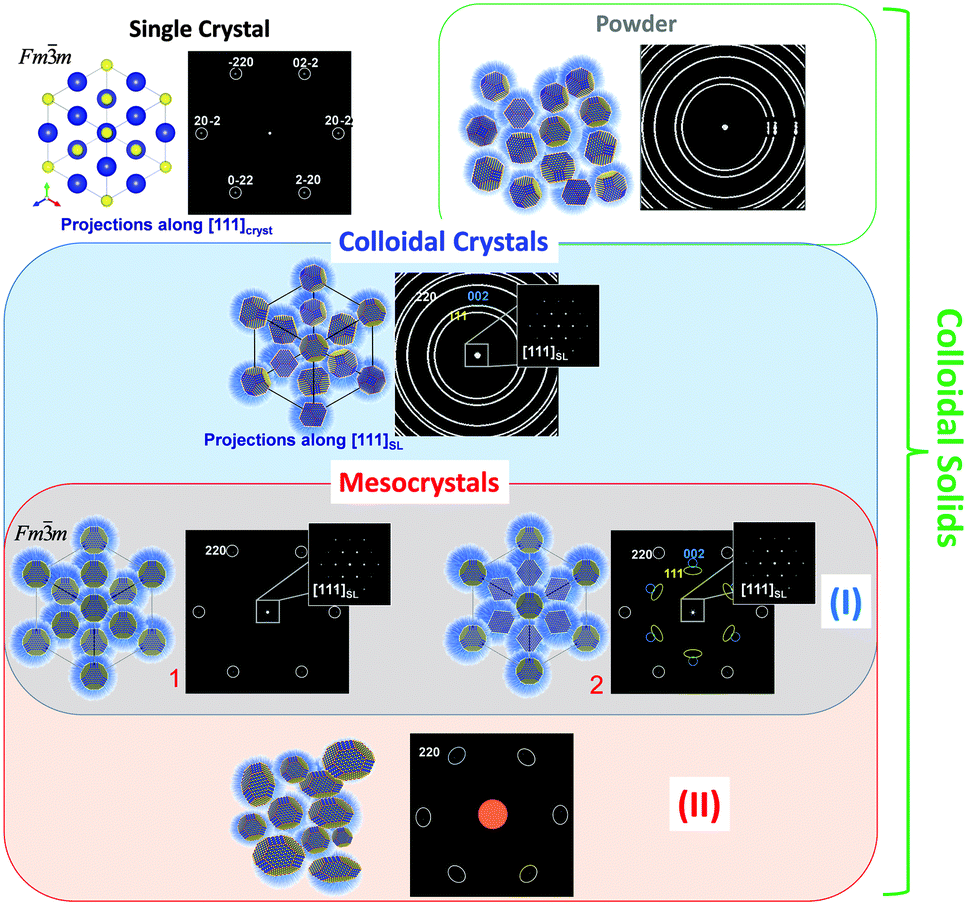 | ||
| Fig. 2 Schematic illustration of different types of crystalline materials with the corresponding diffraction patterns. (Top rows) Single crystal, disordered colloidal aggregates e.g. “powder” (green frame), colloidal crystal (blue frame) with the corresponding diffraction patterns. (Bottom rows) Mesocrystals (red frame): type I – colloidal crystals with mutually oriented monodisperse nanocrystals (characterized by a single crystal-like diffraction pattern in the small angle region and a single crystalline (1) or textured (2) pattern in the wide angle region); type II – colloidal aggregates with mutually oriented polydispersed nanocrystals with a possible certain degree of orientational mismatch. | ||
In general, mesocrystals can consist of both monodisperse and polydisperse crystalline nanoparticles if the material still demonstrates specific mutual crystallographic orientation resulting in a single crystal- or texture-like diffraction pattern with sharp Bragg peaks in the wide-angle region. In the case of type I mesocrystals (Fig. 2), which are composed of monodisperse nanoparticles with a well-defined shape, it is possible that the packing arrangements of the nanosized building blocks have a long range order (essential criterion for a “colloidal crystal”) and at the same time these crystalline building blocks can be arranged in a way that they have a specific mutual crystallographic orientation (such examples are given in: Section 3.2.2, Fig. 8; Section 3.2.3; Section 3.2.5). Thus, this type of solid state material fulfills the definition of “crystals” at least on two hierarchical levels and has a diffraction pattern with sharp maxima both in small and wide-angle regions. For other mesocrystals (type II), the small-angle diffraction pattern can represent the whole range of arrangements from well-ordered to disordered (single crystalline to amorphous-like) colloidal solids (examples are given in: Section 3.2.1; Section 3.2.2, Fig. 7; Section 3.2.4; Section 3.2.6). Nevertheless, mesocrystalline materials can also be defined as mosaic-dominated superstructures.17
Building on the definition of a “crystal” given by IUCr, recently,18 we propose that mesocrystals are defined as “a nanostructured material with a defined long-range order on the atomic scale (in at least one direction), which can be inferred from the existence of an essentially sharp wide angle diffraction pattern (with sharp Bragg peaks) together with clear evidence that the material consists of individual nanoparticle building units”.
In order to classify a material as a mesocrystal it is important to combine several techniques to clearly verify the long-range order at the atomic scale (in at least one crystallographic direction) as well as the existence of nanoparticles in the entire solid state material (e.g. by combining electron microscopy imaging and diffraction techniques).
2.2. Structuration principles of mesocrystals as a special type of colloidal crystal
Monodisperse nanoparticles with a well-defined size and shape provide a chance to fabricate self-assembled materials with various packing arrangements and specific orientational ordering within superlattices. These materials (fabricated using a so-called “bottom-up” approach) are especially interesting in the field of materials science, since their properties can be easily tuned by modifying the size, shape and composition of their nanosized building blocks.19–26 Therefore, in this Section our aim is to clarify, which of them can be classified as a mesocrystal and which cannot. In relatively common cases of crystalline nanoparticles stabilized by organic molecules, the structure of the formed self-assemblies strongly depend on the shape of the crystalline cores (i.e. inorganic) and the degree and thickness of their coverage by organic molecules (stabilizers).27 Previous experimental observations suggest that nanoparticles within a colloidal crystal tend to arrange in such a way that the optimal packing efficiency is achieved (principle of maximum space filling). The space filling can be realized as packing, covering and tiling.28 Therefore, in the case of anisotropic nanoparticles, the formation of a particular type of superlattice array (depending on the degree of coverage of nanocrystals by organic molecules) can be rationalized by considering four phenomenological models, which are schematically illustrated in Fig. 3: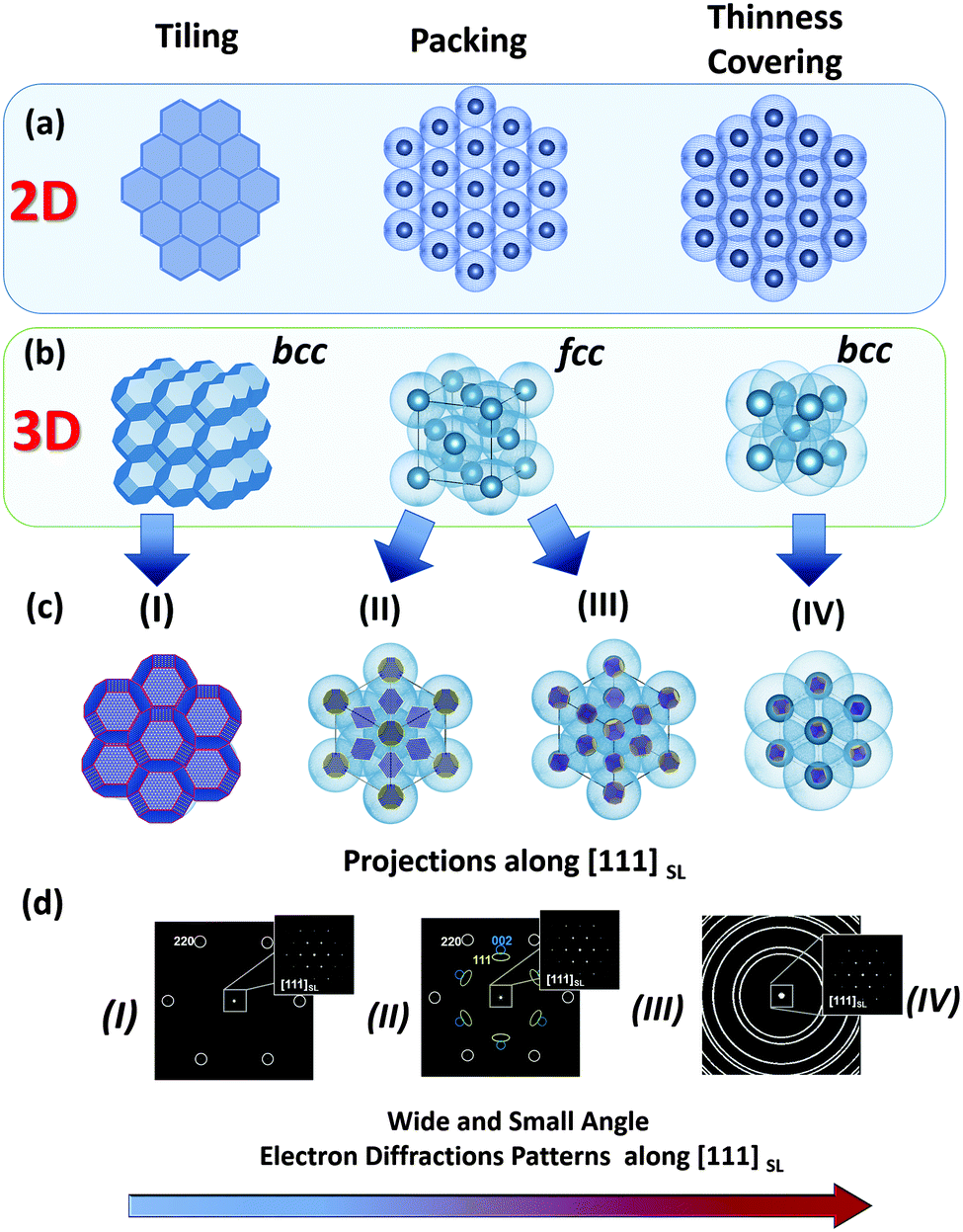 | ||
| Fig. 3 Schematic illustration of different types of “space filling” principles realized in 2D and 3D colloidal crystals depending on the shape of the inorganic core and the thickness (including the degree of coverage) of an organic stabilizer (shown as light blue shell). For illustrational reasons the shape of the inorganic core (in (b) (left) and (c)) is chosen as an ideal truncated octahedron material of rock salt crystal structure. (a and b) Illustrate the topology and symmetry of 2D and 3D superlattices realized for tiling, packing and thinness covering principles of space filling. (c and d) Illustrate the arrangements of building blocks within superlattices in accordance with the proposed phenomenological models (I–IV) and their respective small and wide angle diffraction patterns. The bottom arrow shows the decrease of the orientational order of the crystalline cores of the modelled superlattices (I → IV). | ||
(i) Rigid, anisotropically shaped nanoparticles (i.e. inorganic part) without or with a tiny shell of organic molecules;
(ii) Hard spheres/cylinders/ellipsoids with a comparatively large anisotropic core (inorganic part) covered by a relatively thin shell of organic molecules;
(iii) Hard spheres/cylinders/ellipsoids with a comparatively small anisotropic core (inorganic part) and a thick shell of organic molecules;
(iv) Soft and easily deformable spheres/cylinders/ellipsoids with a small anisotropic core (inorganic part) and an even thicker shell of organic molecules.
For each case (i–iv) the type of superlattice (translational order) and the orientational ordering of nanoparticles within the superlattice array are significantly different. In case (i) more or less pure inorganic nanoparticles with an anisotropic polyhedral shape assemble into a superlattice with a symmetry determined by the symmetry of the polyhedra. In a relatively simple case of nanoparticles with a shape of space filling polyhedra they have a tendency to assemble into a superlattice with 100% packing efficiency and a strong orientational relationship (crystallographic directions of nanocrystals are coaxial with those of the superlattice, and the symmetry is determined by the shape of the polyhedra) (Fig. 3, left column). In the case of models (ii) and (iii), by increasing the degree of coverage of the anisotropic (inorganic) nanoparticles by organic molecules, their faces are continuously smoothed, thereby introducing a certain orientational mismatch of the nanoparticles within the superlattice array. Interestingly, in the case of hard spheres in both cases (ii) and (iii) nanoparticles have a tendency to self-assemble into an fcc packing. Although the type of superlattice can be the same (fcc), the extent of the orientational ordering of the nanocrystals decreases from (ii) to (iii), such that the orientational ordering of the nanocrystals almost completely disappears in the latter case (Fig. 3, middle column). Therefore, types (i) and (ii) can be characterized as mesocrystals, while type (iii) cannot (see Section 2.1, Fig. 2). In the case of model (iv), quasi-spherical (-cylindrical, -ellipsoidal) nanoparticles with a thick shell of organic molecules (easily deformable spheres) tend to be assembled with the minimal overlap in order to avoid repulsive interactions as much as possible. As an example, if equal spheres (in this case, monodisperse nanoparticles) have to be put together with the minimal possible overlap, this situation corresponds to the most economical covering of space by spheres (the so-called “thinnest covering” in mathematics) and thus they mainly arrange in a superlattice with bcc symmetry.29 Due to the thick organic shell, the orientational ordering of nanocrystals within this type of superlattice is low; therefore this type of crystal is not a mesocrystal.
In addition to these four ideal phenomenological models, we are also expecting the formation of intermediate, defect-rich and multi-domain structures under real experimental conditions. Based on these phenomenological models, we can conclude that types (i) and (ii) can be characterized as mesocrystals, while types (iii) and (iv) are not. Therefore, in the present review we will focus only on those self-assembled materials, which will fulfill the criteria for these first two types of self-assemblies.
3. Formation mechanisms and morphogenesis
The big problem with mesocrystal growth mechanisms is that mesocrystal growth is very difficult to analyze due to the large range of involved time and length scales.1 Therefore, our current knowledge is based on snapshots taken and analyzed during mesocrystal growth or conclusions reached about the growth mechanism taken from the final mesocrystal structure. Often, it is even impossible to take samples during the mesocrystal formation process (e.g. like in hydrothermal syntheses). Therefore, it is likely that not all formation mechanisms of mesocrystals have been explored so far and further mechanisms will be revealed in the future. Nevertheless, even the so far known formation mechanisms are considerably different from each other offering high potential for mesocrystal morphosynthesis on the basis of the properties of the given systems.3.1. Growth scenarios and morphogenesis
Up to now, six different growth scenarios of mesocrystals could be identified, which are schematically shown in Fig. 4.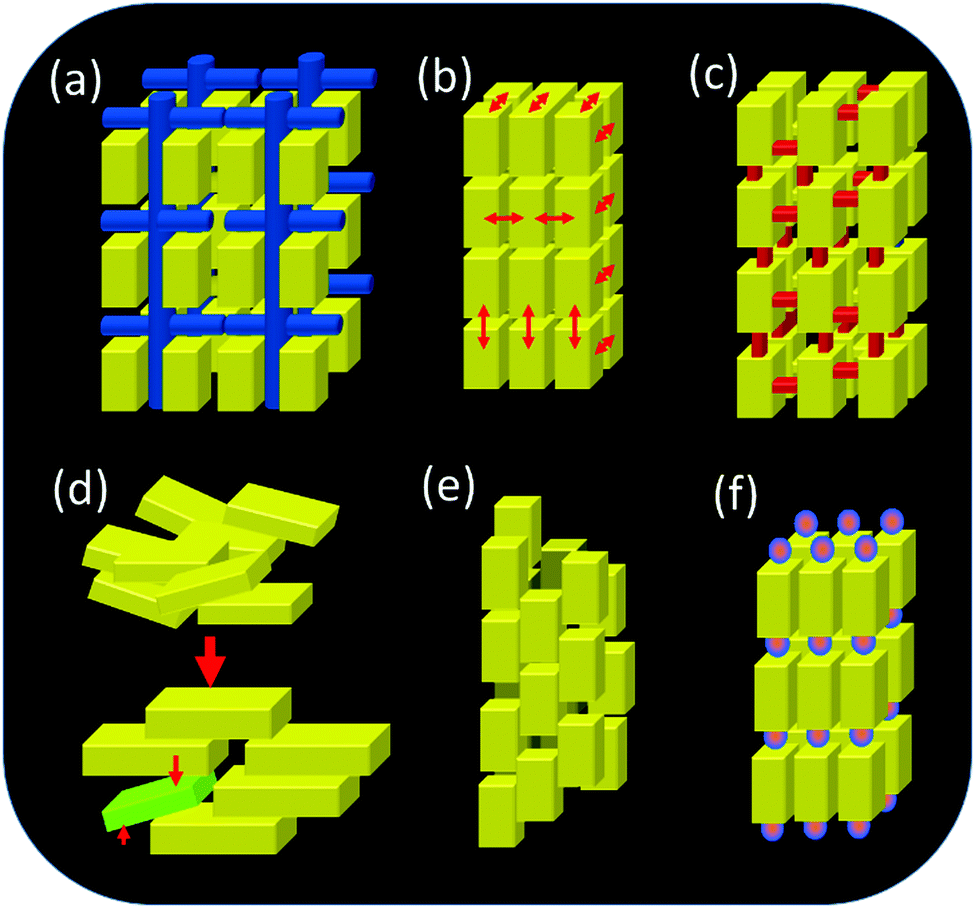 | ||
| Fig. 4 Formation mechanisms of mesocrystals. (a) Alignment by the organic matrix, (b) alignment by physical forces, (c) crystalline bridges, epitaxial growth and secondary nucleation, (d) alignment by spatial constraints, (e) alignment by oriented attachment and (f) alignment by face selective molecules. | ||
Each of these formation mechanisms has its unique pathway involving different physical and chemical forces and properties. The above formation pathways will be described below for an illustrative example highlighting what is special about each mechanism and illuminating the possibilities for the transfer of the mechanism to other materials.
3.2. Examples of growth scenarios
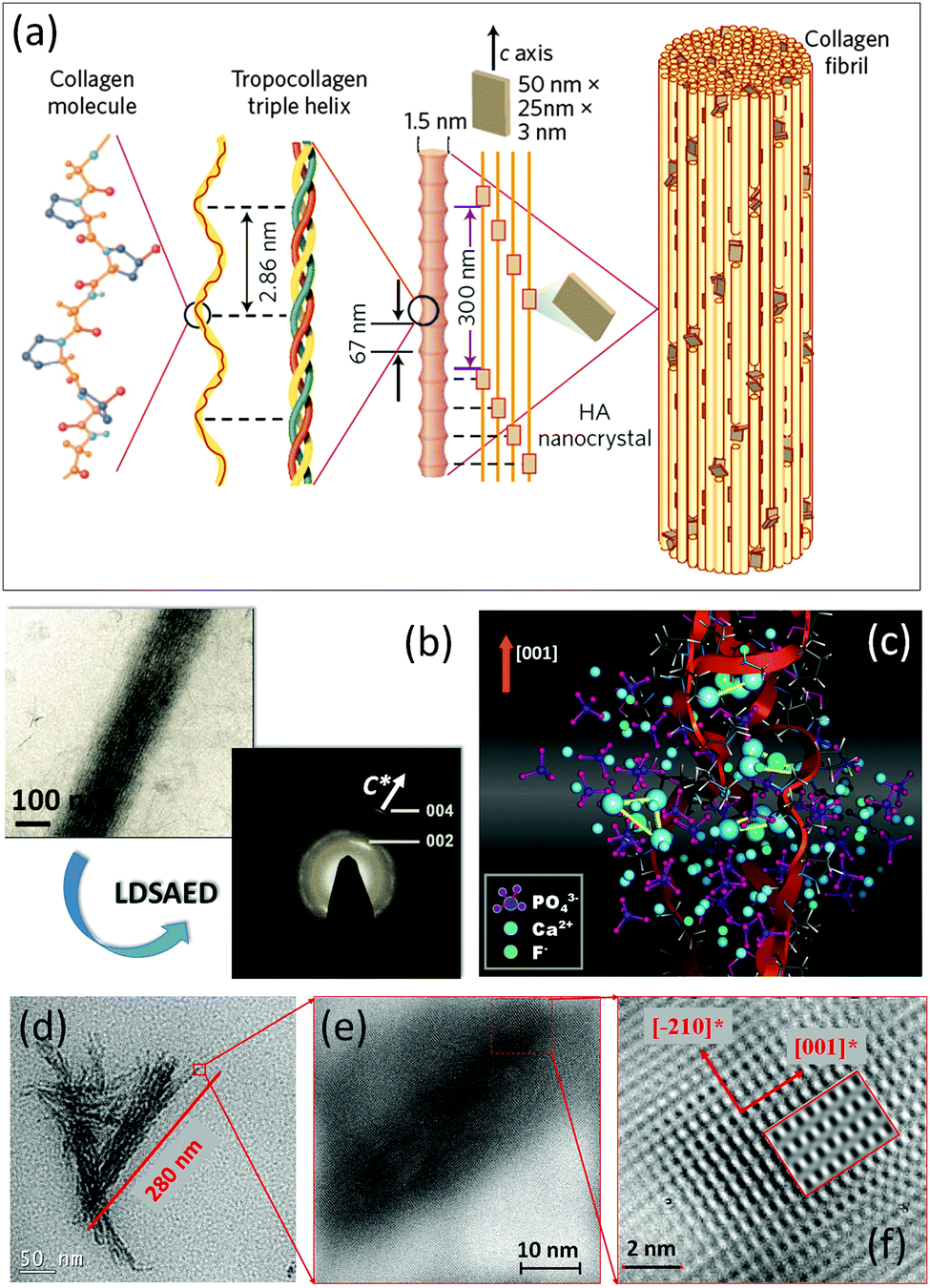 | ||
| Fig. 5 (a) Schematic illustration of mineralized collagen fibril by apatite nanoparticles within bone hard tissues. The figure is reproduced from ref. 31 with permission of the Macmillan Publishers Limited. (b) Cryo-TEM image and the corresponding LDSAED pattern of the collagen fibril mineralized by apatite nanoplates in the presence of pAsp. The figure is reproduced from ref. 32 with permission of the Macmillan Publishers Limited. (c) The results of atomistic simulations: snapshot taken from the late stages of ion aggregation (Ca87[PO4]49F26+) on/in a collagen triple helix (red ribbons). (d) TEM image of mineralized bundle of triple helical protein molecules. (e and f) Sequence of zoomed (HR)TEM images of mineralized triple helical protein molecules, showing the mosaic arrangement and the preferred crystallographic orientation of apatite nanoplates. The figure is adapted from ref. 17 with permission of the WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
To date, many strategies have been developed for the synthesis of biomimetic analogues of natural biominerals.30 Recently, in vitro mineralization of collagen fibrils was achieved by performing crystallization of apatite nanoparticles from aqueous solution in the presence of polyaspartic acid.32 The resulting nanocomposite structure bears a strong relation with that found in bone hard tissues. The most exciting feature is that plate-like apatite nanocrystals (2–5 nm thick, 15–55 nm long and 5–25 nm wide) intergrown with collagen fibrils show a preferable crystallographic orientation (apatite c-axis is parallel to fibril elongation) (Fig. 5b), reflecting a mesocrystalline structure.
Another example of successful biomimetic growth of analogues of apatite-based biomaterials is the fluorapatite–gelatin nanocomposite system grown by double-diffusion in gelatin gels17 (Fig. 5c–f and 6). The morphogenesis starts with the mineralization of triple-helical protein fibers, however, it is quite difficult to experimentally visualize the nucleation of fluorapatite on protein macromolecules. Therefore, atomistic simulations give a first hint at how the motif of the fluorapatite crystal structure emerges by attaching the corresponding ions to the triple-helical model collagen molecule.33 The collagen triple-helices induce an orientation control of Ca3F-motifs in the apatite crystal structure. These Ca3F-triangles are preferably oriented with their planes normal to the long axis of the protein triple-helix (Fig. 5c). The first experimental evidence for the morphogenesis of the fluorapatite–gelatin nanocomposite is characterized by triple-helical protein bundles (about 300 nm in length), which are partially mineralised by fluorapatite. The fluorapatite nanoplatelets cover the protein fibrils in a mosaic arrangement. The crystallographic c-axis of fluorapatite runs preferably parallel to the long-axis of the protein molecules (Fig. 5d–f) similar to the biological bone structure (Fig. 5a). Further morphogenesis of flourapatite–gelatin nanocomposites is schematically illustrated in Fig. 6.34
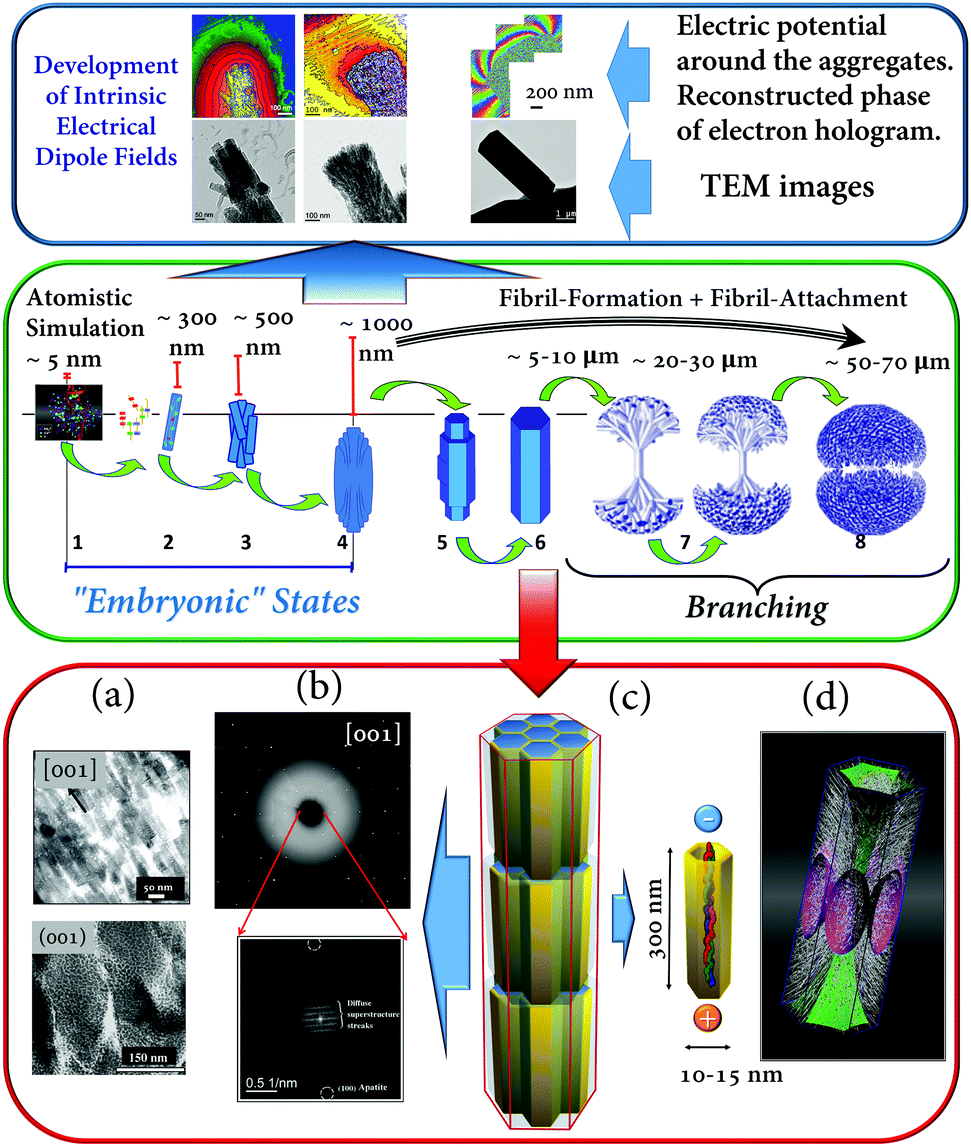 | ||
| Fig. 6 (Blue box): (a–c) Fluorapatite–gelatin composites in different states of morphogenesis. Bottom row: TEM images; top row: reconstructed phase images of electron holograms showing the electric potential distribution around the corresponding composite aggregates. (Green box): Schematic illustration of the morphogenesis process of fluorapatite–gelatin composites. (Red box): Structural features of the hexagonal-prismatic entities: (a) TEM images of ultra-thin slices oriented along and perpendicular to [001]; (b) X-ray diffraction pattern (wide angle) and FFT of the HRTEM image (superstructure – small angle) obtained perpendicular to [001]; (c) schematic illustration of the nanocomposite superstructure; (d) simulated 3D fibril pattern revealing the areas separating fibrils with different orientations. Figure adapted from ref. 17 with permission of the WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
After complete mineralization of the protein fibrils the bundles form elongated composite nanoboards. In the next step, these nanoboards form bundles of boards with a more or less parallel alignment with respect to each other. Once a critical size is reached an electric dipole field develops (detected by means of electron holography (Fig. 6 (blue box))). This electric field can direct a further growth process and induces a more perfect parallel alignment of the elongated polar nanoboards and additionally controls the formation and inclusion of a hierarchical pattern of protein fibrils within the composite aggregate (Fig. 6 (red box, (d))). Further growth leads to a considerable increase of the thickness and volume (3D growth) until the state of a perfect hexagonal prism is reached. Finally, a branching process leads to the development of a notched sphere via several growing dumbbell states (Fig. 6 (green box)). The detailed structural characterisation of hexagonal prismatic crystal-like entities of fluorapatite–gelatin nanocomposites (containing 2.3(3) wt% gelatin) reveals that the material can be described as a mesocrystal (mosaic dominated nanocomposite superstructure17) (Fig. 6 (red box)). In this case, the wide angle diffraction pattern of the specimen is consistent with a fluorapatite single crystal, however, the small angle region of the digital diffractogram (obtained by Fast Fourier Transform (FFT) of the HRTEM image recorded along the [001] zone axis of fluorapatite) reveals diffuse streaks with a periodicity of about 10 nm (and multiples of this value) (Fig. 6 (red box (b))). It was shown that this periodicity originated from the incorporation of the organic component (gelatin macromolecules inside the nanoboards) on which the fluorapatite is nucleated. The real inner architecture of the hexagonal-prismatic entities is consistent with parallel rod-stacking of elongated nanosubunits with their long axes running parallel to [001] shown in TEM images (Fig. 6 (red box (a))) and as schematically presented in Fig. 6 (red box (c)). In addition this superstructure matrix is overlaid with a meso/macroscopic pattern of gelatin microfibrils, which did not cause significant distortion of the mesocrystal matrix. Finally, it has been proposed that the intrinsic electric dipole fields, generated by the dipolar character of the aligned gelatin protein helices, not only cause the integration of the gelatin microfibril pattern, but also control the morphogenesis of fluorapatite–gelatin nanocomposite aggregates. More examples of field-induced morphogenesis of mesocrystals will be described in Section 3.2.2.
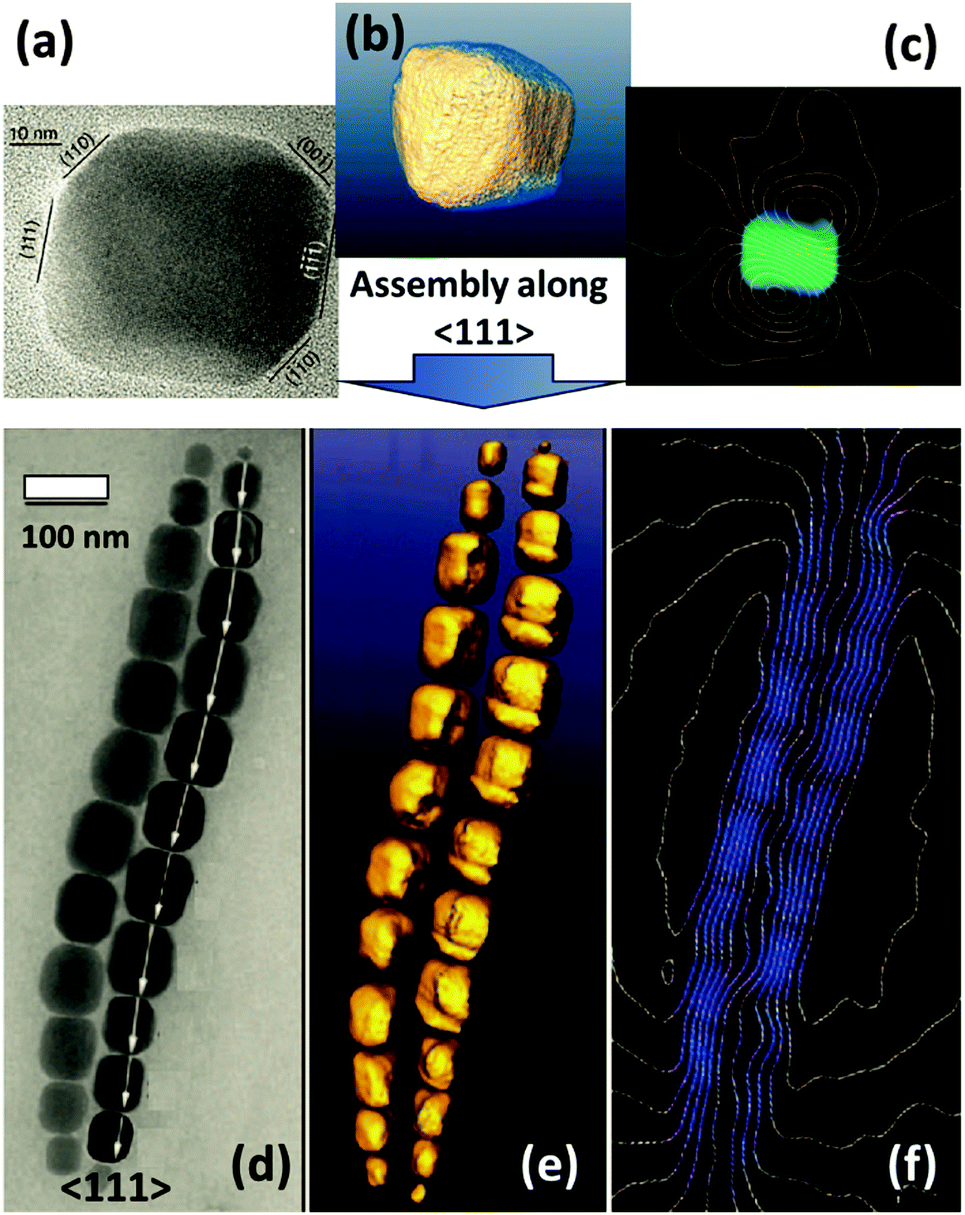 | ||
| Fig. 7 (HR)TEM image, reconstructed high angle annular dark-field (HAAD) tomogram (isosurface visualization) and reconstructed magnetic induction maps (using off-axis electron holography) of a single magnetosome (a, b and c – respectively) and double chains of magnetite nanocrystals (d, e and f – respectively) isolated from a magnetotactic bacterium. Figures are adapted from ref. 36 with permission of the American Chemical Society. | ||
Furthermore, a self-assembly process can be controlled not only by the intrinsic magnetic properties of nanoparticles but also by an external directional magnetic field. In the following example, the induction of a dipolar magnetic attraction during the initial stage of a drying mediated particle assembly process of magnetite octahedra is applied for the synthesis of mesocrystals (Fig. 8).37 Another example involving cubes was also reported.38
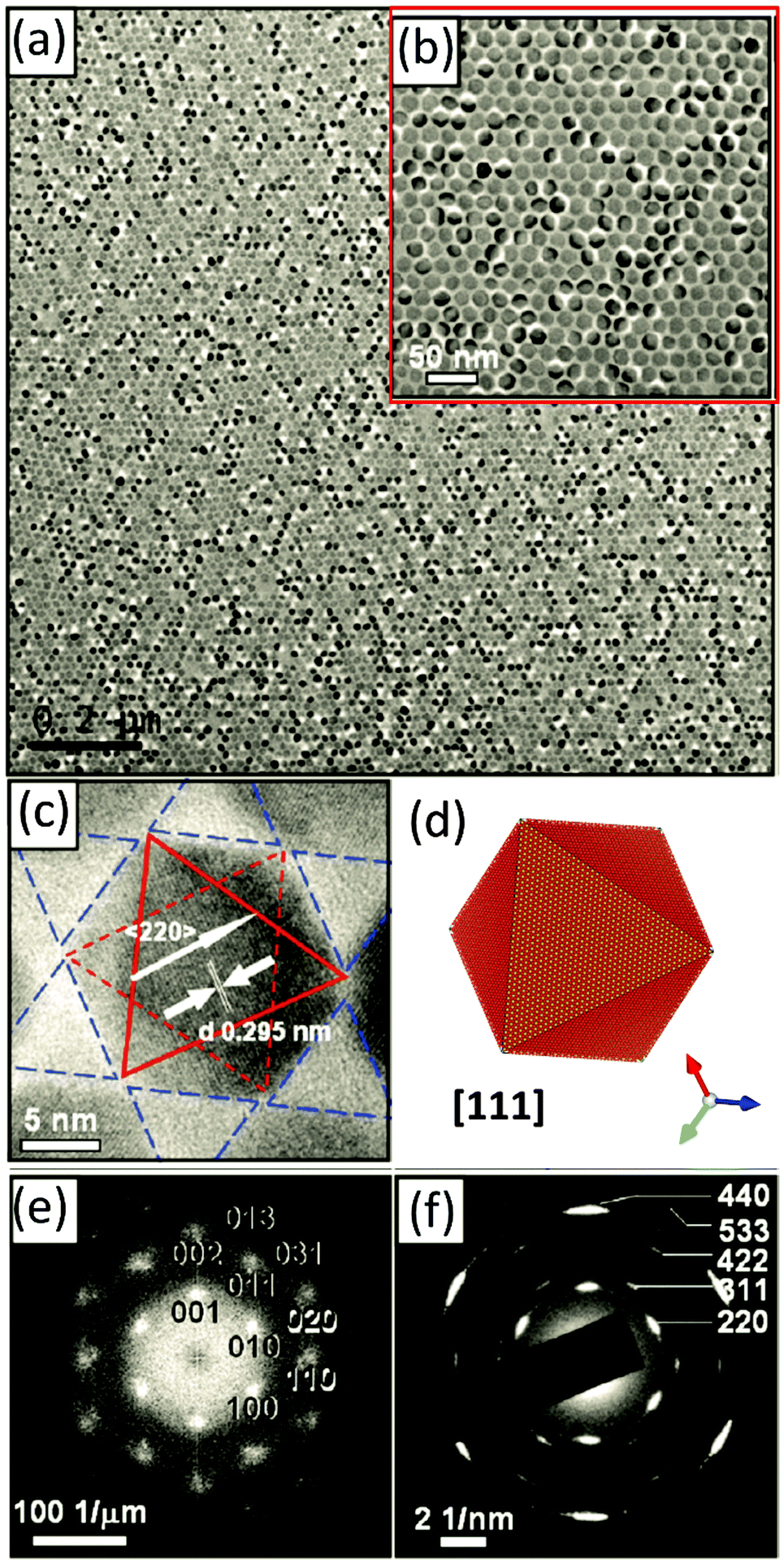 | ||
| Fig. 8 (a and b) TEM images of the 2D monolayer assembly of 21 nm magnetite octahedra in a magnetic field. (c) HRTEM image of one single nanoparticle in the assembly (solid red triangle: (111) plane at the top; dashed red triangle: (111) plane at the bottom); solid blue triangles: interparticle spaces. (d) Model of the magnetite octahedra viewed along the [111] zone axis. (e) Fast Fourier transformation (FFT) pattern and (f) selected area electron diffraction (SAED) pattern of the 2D mesocrystals shown in (b). The figure is reproduced from ref. 37 with permission of the American Chemical Society. | ||
If a droplet of an oleic acid stabilized magnetite octahedral particle dispersion in toluene is evaporated slowly in a weak magnetic field (0.06 T) on a water phase and is later transferred to a substrate like a TEM grid, a mosaic-like pattern of the monodisperse superparamagnetic nanoparticles is obtained (Fig. 8a and b). The HRTEM image in Fig. 8c shows the exposed {111} faces of the octahedra as visualized in a model of a magnetite particle with an octahedral shape in Fig. 8d. The Fourier transformation (Fig. 8e) and the electron diffraction (Fig. 8f) show the high, but not perfect order (arcs in Fig. 8f) of the 2D mesocrystal. This example shows that there is a delicate balance between the attractive van der Waals forces, which assemble the magnetite nanoparticles into a structure as shown in Fig. 8a and b and the ordering magnetic forces, which lead to the crystallographic alignment of the nano-octahedra37 or nanocubes38 into a mesocrystal even if only applied in the nucleation phase38 leading to a mesocrystal nucleus, which propagates and grows by further attachment of nanoparticles to a larger mesocrystal.37,38 A similar scenario can be expected for all nanoparticles which interact with a physical field like a magnetic, electrical or dipole field.
The structure of these kinds of mesocrystals is determined by the symmetry and properties of the assembled nanoparticles (see Section 2.2, Fig. 3). Several examples of self-assemblies of anisotropically shaped “rigid” nanoparticles without or with a tiny shell of organic molecules (representing case (i), described in Section 2.2) are nicely summarized in ref. 22, 24 and 26. Therefore, here we would like to describe in more detail the mesocrystals formed by the self-assembly of monodisperse nanoparticles with a faceted inorganic core but a denser (or thicker) shell of organic molecules (representing case (ii) described in Section 2.2).
One of our recent examples of this type of mesocrystal is the facetted 3D PbS-mesocrystals and the 2D superlattice formed by the self-assembly of truncated octahedrally shaped PbS nanocrystals stabilized by oleic acid (OA) molecules.27,39 Several steps of structural characterization of the PbS-organic colloidal superlattice were performed on different length scales using XRD, SEM, HRTEM, electron diffraction and electron holography. The results of these investigations are summarized in Fig. 9. The 3D mesocrystalline specimen (Fig. 9 (green box)) is characterized by long-range translational order of the nanoparticles within an fcc superlattice combined with a preferred crystallographic orientation of the truncated octahedrally shaped PbS-cores of the nanoparticles leading to short range ordering.27 Detailed analyses of the orientational relationships between the PbS nanocrystals within the fcc supperlattice array indicate that the nanoparticles have a tendency towards face-to-face interactions within each close-packed layer (resulting in a [111]SL||[110]PbS crystallographic orientational relation), while allowing for a certain degree of orientational mismatch. A hierarchical scenario of atomistic models was developed describing the multi-domain structure of 3D mesocrystals. It is interesting to point out that these structural features are already prearranged in the 2D state (hexagonal close packed layer) of nanoparticle self-assemblies as was shown by means of HRTEM, ED, and atomistic simulations (Fig. 9 (blue box)).39
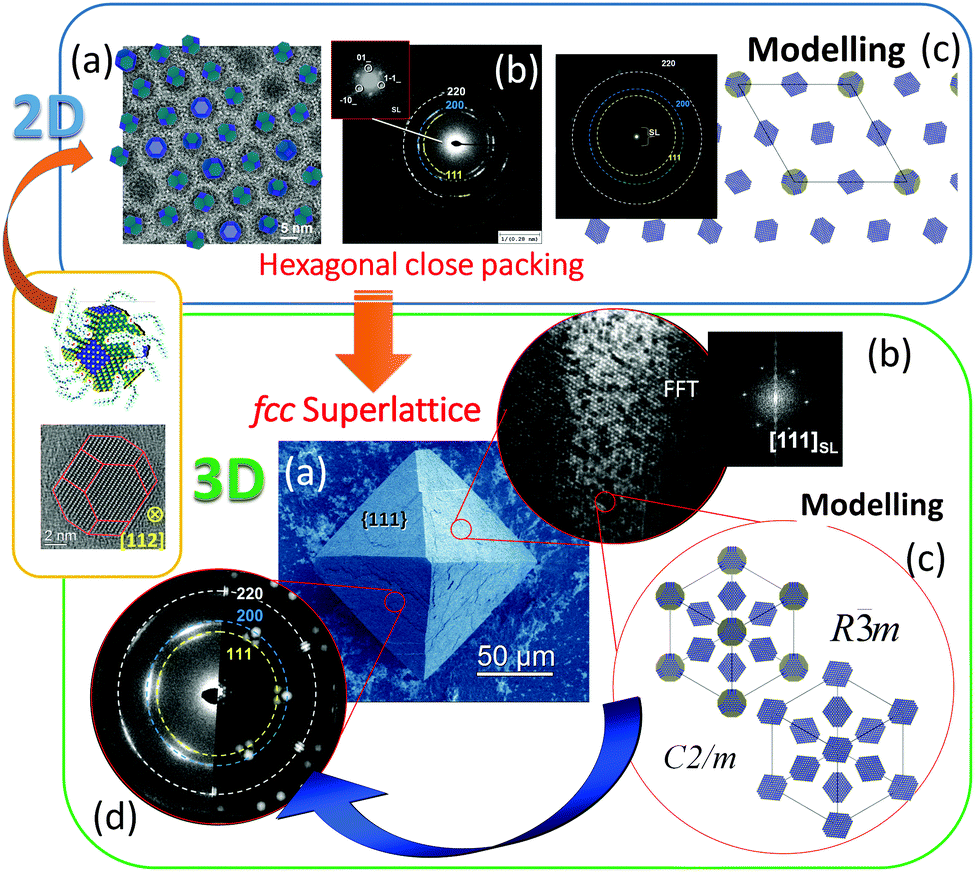 | ||
| Fig. 9 (Orange box): HRTEM image and model of a truncated octahedrally shaped PbS nanocrystal stabilized by oleic acid (OA) molecules. A short summary of experimental observations and the results of simulations on 2D and 3D PbS mesocrystals: (Blue box). (a) HRTEM image of 2D self-assembly overlaid with an orientation map of PbS nanocrystals; (b) ED pattern of a 2D monolayer; (c) structural model of 2D self-assembly and the corresponding simulated ED pattern. (Green box) (a) SEM image of a faceted 3D mesocrystal, (b) TEM image of a FIB slice recorded along [111]SL and the corresponding FFT, (b) simulated structural models of 3D PbS mesocrystals and (d) experimental and simulated ED pattern along [111]SL. Figures are adapted from ref. 27 and 39 with permission of the Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Furthermore, by employing electron microscopy techniques (Cs-corrected HRTEM, electron holography and EFTEM) it was shown that within a 2D array some of the PbS nanocrystals are interconnected by nanobridges, which are partially mineralized by PbS (Fig. 10). This type of ordered array built up by interconnected nanoparticles was previously reported for several systems.19,40 The formation of nanobridges between self-assembled nanoparticles is normally induced and enhanced using careful annealing, plasma treatment, treatment by oxidation agents, etc. Ideally such a “confined-but-connected structure”19 preserves the effects of quantum confinement present within the individual nanoparticles, whilst at the same time having the potential to provide high electron mobility throughout the extended structure, which can be important for several applications.
 | ||
| Fig. 10 (a) Cs-corrected HRTEM image of two nanoparticles (within 2D self-assembly) interconnected by a PbS bridge. The crystallographic orientation is deduced from the FFT (top and bottom insets) corresponding to the [110] zone axis. (b) Digitally zoomed area marked with a yellow frame in (a) overlaid with truncated octahedrally shaped PbS nanoparticle models in the [110] zone axis. The periodicity along the bridge corresponds to 0.34 nm, which is equivalent to the d-spacing of the (111) lattice plane of PbS (galena). (c) Phase image retrieved from the electron hologram. The PbS nanoparticles and the interconnecting sub-nanometer nodes appear bright in the phase image. (d) Elemental map (derived from EFTEM image) showing the distribution of sulfur in an array of PbS nanoparticles. Figures are adapted from ref. 39 with permission of the Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Therefore, the oriented attachment of nanoparticles forming a single crystalline but defect-rich structure via mesocrystalline intermediates might be more common than assumed so far. However, the nanoparticles in a mesocrystal can also connect via crystalline (so-called “mineral”) bridges as shown in Fig. 4c and 10 or crystallographically fuse forming a mesocrystalline structure as shown in Fig. 4e. Crystallographic fusion of the nanoparticles in a mesocrystal was also observed for goethite by Penn and coworkers (Fig. 11).42
 | ||
| Fig. 11 (a) Cryo-TEM image of rod-like goethite mesocrystals after 24 days of aging at 80 °C. (b) The high resolution cryo-TEM image shows the (021) planes of goethite after 14 days of aging. (c) HRTEM image of single-crystalline goethite nanorod with FFT inset (4 days of aging, dry-TEM sample). The figure is reproduced from ref. 42 with permission of the American Chemical Society. | ||
Mesocrystals are formed from ferrihydrite precursor nanoparticles upon aging at elevated temperatures. They were composed of goethite nanoparticles with an inter-particle distance of 1 nm, which was attributed to solvent molecules (Fig. 11a and b). The exact reason for the nanoparticle alignment is not clear but it can be speculated that equal crystal faces recognize each other despite the thin water layer in-between them. This example shows that oriented aggregation does not necessarily directly lead to crystallographic fusion of the nanoparticles despite the thermodynamic driving force resulting from the minimization of high-energy surfaces by crystallographic fusion. However, this fusion finally took place resulting in finally single crystalline goethite nanorods (Fig. 11c).
Finally, this example together with the one given in Section 3.2.3 illustrates that the presence of crystalline inorganic bridges between nanoparticles alone cannot be the indication of a mesocrystalline structure of the material; however, their formation can change the ordering of nanoparticles within a nanostructured material and in some cases leads to the formation of mesocrystalline or even single crystalline materials.
One of the most illustrative examples of this mechanism is the formation of 2D particle self-assembly using the Langmuir–Blodgett technique43,44 (Fig. 12a). This technique allows generation of a quite large area of an ordered 2D nanoparticle monolayer. In addition the interparticle distance and the final superstructure of the assembly can be easily tuned by controlling the compression process. Fig. 12b and c illustrate an example of the formation process (at different stages of compression) of a 2D dense monolayer composed of parallel aligned BaCrO4 nanorods.44
 | ||
| Fig. 12 (a) Schematic illustration of the formation process of 2D assembly using the Langmuir-Blodgett technique. (b and c) The arrangement of BaCrO4 nanorods within a monolayer at different stages of compression. FFT image in c (right bottom) illustrates the long-range positional order of nanorods within the monolayer. Figures are adopted from ref. 44 with permission of the WILEY-VCH-Verlag GmbH. | ||
In particular, in the situation of an evaporating solvent, convection and capillary forces help to minimize the space between nanoparticles leading to their alignment into mesocrystals. This was, for example, demonstrated for monodisperse magnetite nanoparticles with a truncated cubic shape.45 A controlled evaporation of oleic acid stabilized magnetite/maghemite nanocrystals in toluene under a nitrogen flow directly under a light microscope allowed the on-line observation of mesocrystal formation on the micrometer scale.
This process is partly shown in Fig. 13 in the late stage but can be nicely observed on the micron scale in the ESI† video “Iron oxide mesocrystal formation”. In the beginning of the process, a surprisingly ordered nanoparticle array is formed at the rim of the drying droplet. Convection then leads to dendritic mesocrystal growth and despite this condition, which corresponds to the diffusion limited growth regime in classical crystallization characterizing the situations far from equilibrium resulting in dendritic crystals, still a high order of the nanocrystals is observed in the mesocrystal interior but the mesocrystal surface was disordered and defective.45 The final stage of the evaporation experiment led to the formation of individual cuboidal, highly ordered mesocrystals. This stage is shown in Fig. 13 together with the schematic flow pattern leading to this type of mesocrystal formation. A variation of the above reported solvent evaporation method for mesocrystal formation is the application of a second solvent, which is miscible with the nanoparticle dispersion medium but is a non-solvent for the stabilizer molecules on the nanoparticle surface. Slow diffusion of the non-solvent into the stable nanoparticle dispersion via the gas phase leads to gradual destabilization of the nanoparticles and thus controlled aggregation to a mesocrystal.11 By these techniques, the largest amount of the so far reported mesocrystals was formed. This also includes the spectacular examples of 2D mesocrystals formed directly on a TEM grid.6,23
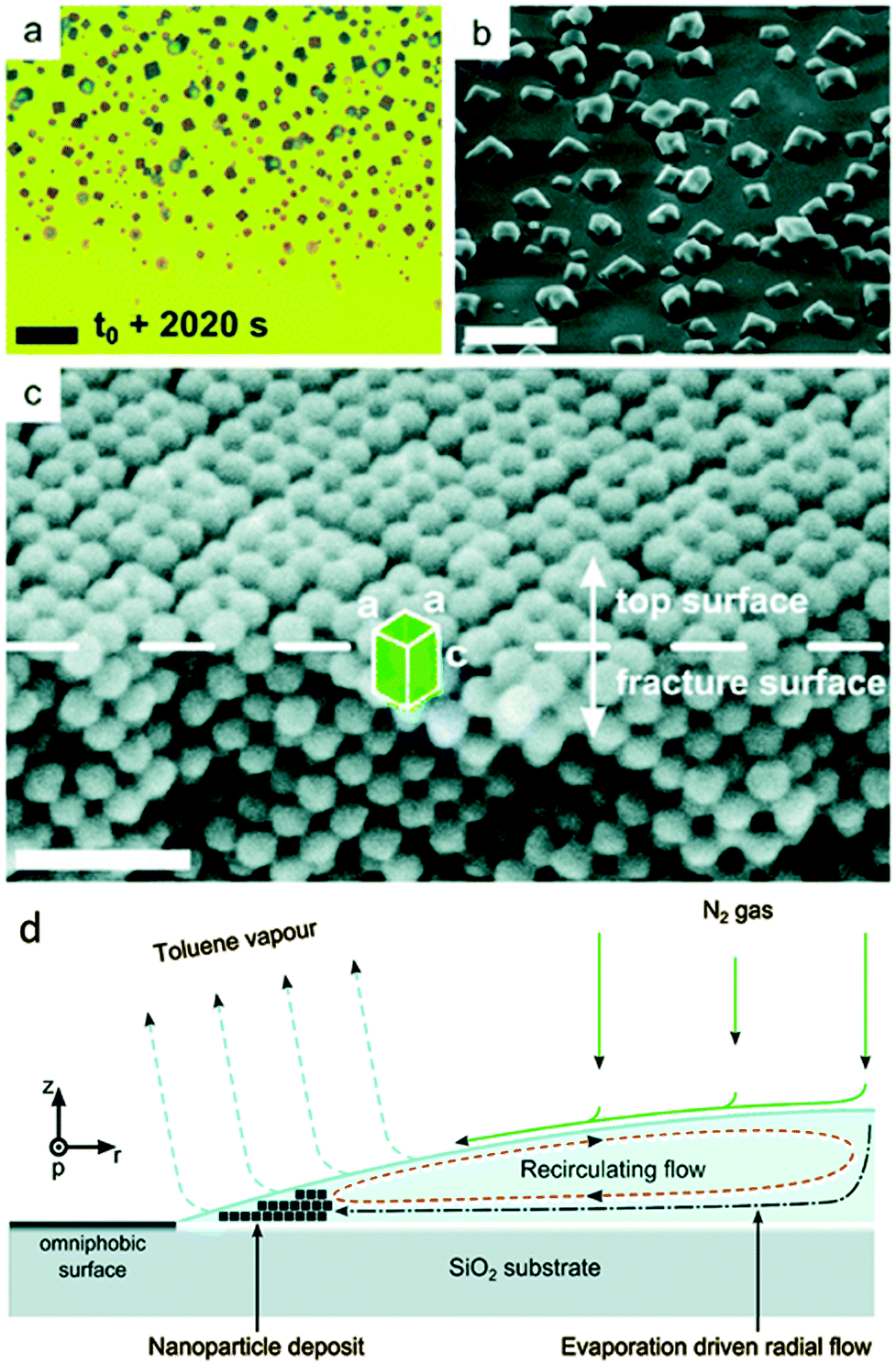 | ||
| Fig. 13 Images illustrating crystallization of mesocrystals during the final stage of drop-casting iron oxide nanocube dispersions. The video microscopy snapshot in (a) was taken about 200 μm inward from the pinned three-phase contact line of the receding droplet. The SEM image in (b) depicts individual cuboidal mesocrystals dispersed on the substrate (sample stage was tilted by 41.8°). In (c), an XHR-SEM image of a fractured mesocrystal, tilted by 30°, clearly shows the bct structure (unit cell with parameters a and c). Scale bar in (a) 20 μm, (b) 10 μm and (c) 50 nm. (d) The side view of one half of a droplet of an evaporating drop positioned on a functionalized silica substrate consisting of oleic acid capped iron oxide nanocubes in a toluene-based fluid carried out in an environmentally controlled chamber. See also ESI† video “Iron oxide mesocrystal formation”. The figure is reproduced from ref. 45 with permission of the Royal Society of Chemistry. | ||
 | ||
| Fig. 14 (a) BaCO3 helical mesocrystals composed of elongated nanoparticles. (b) BaCO3 nanoparticle building unit calculated under vacuum showing the relevant crystal faces (pink {020}, red {011}, blue {111} and green {110}), (c) surface cut (yellow dashed line) of the {110} face showing exposed Ba2+ cations, (d) nanoparticle aggregation possibilities, side view, (e) nanoparticle aggregation possibilities, top view and (f) helical nanoparticle aggregate by an overlay of side and top views. The figure is reproduced from ref. 46 with permission of Nature Publishing Group. | ||
This example shows that mesocrystal morphologies can significantly deviate from the typical crystalline shapes, which are dictated by the symmetry of the crystal structure.
In this example, it is exploited that the BaCO3 orthorhombic building unit (Fig. 14b) has neutral faces except the {110} faces, which can be entirely Ba2+ terminated as shown in the surface cut in Fig. 14c. Therefore, it is possible to selectively address the {110} faces with a polyanion. In this case, a stiff polyphosphonate block copolymer was used46 to selectively adsorb onto the {110} faces shown in green in Fig. 14 by electrostatic attraction sterically stabilizing this face. The four {011} faces as well as the two {020} faces are neutral and can therefore interact with each other via van der Waals attraction leading to aggregation of the particles involving these faces with preference for {011} because of its larger area and occurrence.
The key point now is that it is an unfavourable situation to aggregate two particles as shown in Fig. 14d left and right arrangement, since this involves the positioning of an adsorbed polymer at a location, which is occupied by the next aggregating particle. This would result in bending of the stiff polymer at the cost of configurational entropy and/or the stiff polymer would hold the neighbouring nanoparticles apart resulting in a minimized van der Waals attraction. Both contributions would decrease the free enthalpy gain and are thus not favoured. This situation can be completely avoided by the two central particle arrangements shown in Fig. 14d since they allow for enough space for the stiff block copolymer adsorbed onto {011}. This leads to a preferential aggregation direction defined by the aggregation of the first three particles. If one now considers the situation from the top view shown in Fig. 14e, it can be clearly seen that the two {020} faces lead to a linear particle arrangement, while the more probable four {110} interacting faces lead to a clockwise or counter-clockwise aggregation of the next nanoparticle as visualized by the yellow and orange indicated positions for particle adsorption. The orange position would be easily accessible and is thus favoured in kinetic pathways, while the yellow position involves a stronger interaction with two faces and thus a larger gain in free enthalpy for particle aggregation representing the thermodynamic pathway. Regardless of which pathway is taken, a preferential clockwise or counterclockwise particle aggregation is favoured. In Fig. 14e, the kinetic counter-clockwise aggregation is shown. If one overlays the situations shown in Fig. 14d and e, a helical structure results (Fig. 14f) and since the probabilities for defining the two possible axis directions by the aggregation of the first three nanoparticles are equal, there is consequently an equal number of left and right handed mesocrystalline structures.
This example shows in an elegant way how appropriate selection of the reactants and the (slow) nanoparticle assembly conditions can lead to most sophisticated mesocrystal structures. The potential is obvious if such structures can be assembled from functional nanoparticles in the future.
4. Common misunderstandings
There are some common misunderstandings related to mesocrystals, which we would like to clarify here.1. The term mesocrystal defines a structure and does NOT determine a formation mechanism. This misunderstanding arose from figures of the type shown in Fig. 1 and the misunderstanding is that mesocrystals must form via the assembly of nanoparticles. This is not true as Fig. 3 and this entire review shows. Also, mesocrystals found in biominerals like the sea urchin spicule47 are very good examples, which did not form via nanoparticle assembly. Therefore, it is important to state that a mesocrystal is a special type of crystalline structure, which can form via different mechanisms.
2. A mesocrystal is not necessarily porous. Although some mesocrystals show high porosity,9 not every mesocrystal is porous. The interspace between the nanoparticles can be completely filled with amorphous matter,47 or organic stabilizer molecules, which initially stabilized the individual nanoparticles. In such cases, the interspace between the nanoparticles is not accessible.
3. Mesocrystals are always composed of nanocrystal building units. If the building units are larger than nanoparticles, the size range is not mesoscale anymore and therefore the structure cannot be a mesocrystal anymore by definition. If the building units should be smaller than a nanometer, it is in the atomic range and is a cluster at the most but not a crystalline nanoparticle.
4. Nanoparticles in mesocrystals are not always perfectly aligned. Often, the electron diffraction pattern used to prove the crystalline alignment in a mesocrystal is not the ideal spot pattern which is expected for a perfect single crystal-like arrangement (see also Fig. 8f and 9d for example). If the nanocrystal building units are not perfectly aligned, arcs are observed instead in the diffraction pattern. There is no definition as to which degree of misalignment is still tolerable to call a structure a mesocrystal.
5. Conclusions
This overview shows that mesocrystals are a special class of crystalline material with highly ordered nanoparticle superstructures (Fig. 2 and 3). Several pathways exist for their formation and the actual overview (Fig. 4) is certainly not yet complete. However, it is very important to understand that the final mesocrystal structure does not tell us anything about the mesocrystal formation mechanism. Therefore knowledge in this field relies heavily on analytical technical improvements. Liquid cell TEM is such technique, which is promising to reveal mesocrystal formation mechanisms. However, current progress will arise from a combination of different analytical techniques. Only if a solid experimental database on mesocrystal formation is established, will it be possible to reveal their formation mechanisms including a theoretical description of non-classical nucleation and growth.Acknowledgements
The authors thank the DFG (Deutsche Forschungsgemeinschaft) for support of this work within the SFB 1214. They also thank R. Kniep, P. Simon, I. A. Baburin and L. Bergström for useful discussions. ES acknowledges a fellowship of the Zukunftskolleg of the University of Konstanz.Notes and references
- H. Cölfen and M. Antonietti, Mesocrystals and Nonclassical Crystallization, John Wiley & Sons, Chichester, 2008 Search PubMed.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef PubMed.
- L. Zhou and P. O'Brien, Small, 2008, 4, 1566–1574 CrossRef CAS PubMed.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2009, 110, 389–458 CrossRef PubMed.
- J. P. Ge, Y. X. Hu, M. Biasini, W. P. Beyermann and Y. D. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342–4345 CrossRef CAS PubMed.
- J. X. Fang, B. J. Ding and H. Gleiter, Chem. Soc. Rev., 2011, 40, 5347–5360 RSC.
- T. Wang, H. Cölfen and M. Antonietti, J. Am. Chem. Soc., 2005, 127, 3246–3247 CrossRef CAS PubMed.
- R. Q. Song and H. Cölfen, Adv. Mater., 2010, 22, 1301–1330 CrossRef CAS PubMed.
- L. Bahrig, S. G. Hickey and A. Eychmuller, CrystEngComm, 2014, 16, 9408–9424 RSC.
- L. Bergstrom, E. V. Sturm, G. Salazar-Alvarez and H. Cölfen, Acc. Chem. Res., 2015, 48, 1391–1402 CrossRef PubMed.
- T. Tachikawa and T. Majima, NPG Asia Mater., 2014, 6, e100 CrossRef CAS.
- E. Uchaker and G. Z. Cao, Nano Today, 2014, 9, 499–524 CrossRef CAS.
- M. G. Ma and H. Cölfen, Curr. Opin. Colloid Interface Sci., 2014, 19, 56–65 CrossRef CAS.
- L. Zhou and P. O'Brien, J. Phys. Chem. Lett., 2012, 3, 620–628 CrossRef CAS PubMed.
- R. Kniep, P. Simon and E. Rosseeva, Cryst. Res. Technol., 2014, 49, 4–13 CrossRef CAS.
- Report of the Executive Committee for 1991, Acta Crystallogr., Sect. A: Found. Crystallogr., 1992, 48, 922–946 CrossRef.
- T. Hanrath, J. Vac. Sci. Technol., A, 2012, 30, 28 Search PubMed.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef CAS PubMed.
- M. P. Pileni, Phys. Chem. Chem. Phys., 2010, 12, 11821–11835 RSC.
- Z. Quan and J. Fang, Nano Today, 2010, 5, 390–411 CrossRef CAS.
- D. V. Talapin, MRS Bull., 2012, 37, 63–71 CrossRef CAS.
- D. Vanmaekelbergh, Nano Today, 2011, 6, 419–437 CrossRef CAS.
- T. Wang, D. LaMontagne, J. Lynch, J. Zhuang and Y. C. Cao, Chem. Soc. Rev., 2013, 42, 2804–2823 RSC.
- S. Y. Zhang, M. D. Regulacio and M. Y. Han, Chem. Soc. Rev., 2014, 43, 2301–2323 RSC.
- P. Simon, E. Rosseeva, I. A. Baburin, L. Liebscher, S. G. Hickey, R. Cardoso-Gil, A. Eychmüller, R. Kniep and W. Carrillo-Cabrera, Angew. Chem., Int. Ed., 2012, 51, 10776–10781 CrossRef CAS PubMed.
- B. Vainshtein, V. Fridkin and V. Indenbom, Structure of Crystals, Springer, Berlin Heidelberg, 1995, ch. 1, pp. 1–123 DOI:10.1007/978-3-642-97512-7_1.
- V. A. Blatov, Crystallogr. Rev., 2004, 10, 249–318 CrossRef CAS.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
- A. Kawska, O. Hochrein, J. Brickmann, R. Kniep and D. Zahn, Angew. Chem., 2008, 120, 5060–5063 CrossRef.
- P. Simon, E. Rosseeva, J. Buder, W. G. Carrillo-Cabrera and R. Kniep, Adv. Funct. Mater., 2009, 19, 3596–3603 CrossRef CAS.
- R. E. Dunin-Borkowski, M. R. McCartney, R. B. Frankel, D. A. Bazylinski, M. Posfai and P. R. Buseck, Science, 1998, 282, 1868–1870 CrossRef CAS PubMed.
- J. M. Thomas, E. T. Simpson, T. Kasama and R. E. Dunin-Borkowski, Acc. Chem. Res., 2008, 41, 665–674 CrossRef CAS PubMed.
- L. Li, Y. Yang, J. Ding and J. Xue, Chem. Mater., 2010, 22, 3183–3191 CrossRef CAS.
- A. Ahniyaz, Y. Sakamoto and L. Bergstrom, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17570–17574 CrossRef CAS PubMed.
- P. Simon, L. Bahrig, I. A. Baburin, P. Formanek, F. Roeder, J. Sickmann, S. G. Hickey, A. Eychmueller, H. Lichte, R. Kniep and E. Rosseeva, Adv. Mater., 2014, 26, 3042–3049 CrossRef CAS PubMed.
- M. A. Boles and D. V. Talapin, Science, 2014, 344, 1340–1341 CrossRef CAS PubMed.
- D. Schwahn, Y. Ma and H. Coelfen, J. Phys. Chem. C, 2007, 111, 3224–3227 CAS.
- V. M. Yuwono, N. D. Burrows, J. A. Soltis and R. L. Penn, J. Am. Chem. Soc., 2010, 132, 2163–2165 CrossRef CAS PubMed.
- A. R. Tao, J. Huang and P. Yang, Acc. Chem. Res., 2008, 41, 1662–1673 CrossRef CAS PubMed.
- P. Yang and F. Kim, ChemPhysChem, 2002, 3, 503–506 CrossRef CAS PubMed.
- M. Agthe, E. Wetterskog, J. Mouzon, G. Salazar-Alvarez and L. Bergstrom, CrystEngComm, 2014, 16, 1443–1450 RSC.
- S.-H. Yu, H. Cölfen, K. Tauer and M. Antonietti, Nat. Mater., 2005, 4, 51–55 CrossRef CAS PubMed.
- J. Seto, Y. R. Ma, S. A. Davis, F. Meldrum, A. Gourrier, Y. Y. Kim, U. Schilde, M. Sztucki, M. Burghammer, S. Maltsev, C. Jager and H. Cölfen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3699–3704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The video “Iron oxide mesocrystals” showing the formation of mesocrystals on the micrometer scale on-line by light microscopy from ref. 45. See DOI: 10.1039/c6cs00208k |
| This journal is © The Royal Society of Chemistry 2016 |