
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and applications of porous non-silica metal oxide submicrospheres
Yash
Boyjoo
a,
Meiwen
Wang
a,
Vishnu K.
Pareek
a,
Jian
Liu
 *a and
Mietek
Jaroniec
*b
*a and
Mietek
Jaroniec
*b
aDepartment of Chemical Engineering, Curtin University, Perth, WA 6845, Australia. E-mail: jian.liu@curtin.edu.au
bDepartment of Chemistry & Biochemistry, Kent State University, Kent, Ohio 44242, USA. E-mail: jaroniec@kent.edu
First published on 12th September 2016
Abstract
Nowadays the development of submicroscale products of specific size and morphology that feature a high surface area to volume ratio, well-developed and accessible porosity for adsorbates and reactants, and are non-toxic, biocompatible, thermally stable and suitable as synergetic supports for precious metal catalysts is of great importance for many advanced applications. Complex porous non-silica metal oxide submicrospheres constitute an important class of materials that fulfill all these qualities and in addition, they are relatively easy to synthesize. This review presents a comprehensive appraisal of the methods used for the synthesis of a wide range of porous non-silica metal oxide particles of spherical morphology such as porous solid spheres, core–shell and yolk–shell particles as well as single-shell and multi-shell particles. In particular, hydrothermal and low temperature solution precipitation methods, which both include various structure developing strategies such as hard templating, soft templating, hydrolysis, or those taking advantage of Ostwald ripening and the Kirkendall effect, are reviewed. In addition, a critical assessment of the effects of different experimental parameters such as reaction time, reaction temperature, calcination, pH and the type of reactants and solvents on the structure of the final products is presented. Finally, the practical usefulness of complex porous non-silica metal oxide submicrospheres in sensing, catalysis, biomedical, environmental and energy-related applications is presented.
 Yash Boyjoo | Yash Boyjoo received his PhD in 2013 from Curtin University, Australia. After two years as a research associate at Curtin University working with Dr Jian Liu and Prof. Vishnu Pareek, he currently works as a postdoctoral research fellow at Université Lille 1 in France, where his studies deal with the synthesis of materials for VOC elimination from polluted air. His other research interests include photocatalytic wastewater treatment, synthesis of activated carbons for application as CO2 adsorbents and supercapacitors, and the design and modelling of photocatalytic reactors. |
 Meiwen Wang | Meiwen (Sharon) Wang received her bachelor's degree with first class honours in Chemical Engineering at Curtin University, where she is now pursuing PhD research under the supervision of Dr Jian Liu. She is interested in the design and fabrication of yolk–shell particles and their use as nanoreactors. |
 Vishnu K. Pareek | Vishnu Pareek is a Professor in the Department of Chemical Engineering at Curtin University, Australia. He received his PhD degree from the University of New South Wales, an MTech from IIT Delhi and a BE from the University of Rajasthan, Jaipur, all in Chemical Engineering and with high distinction. Prof. Pareek's expertise lies in the simulation and design of chemical processes with particular emphasis on the use of these tools in industrial-scale processes. He is an ardent cricket fan and an enthusiastic reader especially on subjects related to politics, environment and economy. In his spare time, he is a bird watcher with special interest in understanding the behaviour of birds of prey. |
 Jian Liu | Jian Liu obtained his PhD degree in Physical Chemistry from the Dalian Institute of Chemical Physics, Chinese Academy of Science, in 2008. Subsequently, he moved to Australia and worked as a Postdoctoral Research Fellow at AIBN, the University of Queensland. He started as a Lecturer at Curtin University in 2013 and was promoted to Senior Lecturer in 2014. Dr Liu has published more than 120 peer reviewed journal articles with over 6300 citations (H-index of 43). He has been honoured with a prestigious UQ Foundation Research Excellence Award and Australian Postdoctoral Fellowship. His current research interests are nanoreactor design, green chemical processes, and utilization of CO2. |
 Mietek Jaroniec | Mietek Jaroniec received his MS and PhD from M. Curie-Sklodowska University, Poland, in 1972 and 1976, respectively. Since 1991 he has been a Professor of Chemistry at Kent State University, Kent, Ohio (USA). Before joining Kent State he was a Professor of Chemistry at M. Curie-Sklodowska University, Poland. His research interests revolve primarily around inter-disciplinary topics of interfacial chemistry, and chemistry of materials, including physical adsorption at the gas/solid and liquid/solid interfaces, adsorbents, and catalysts. At Kent State he has established a vigorous research program in the area of ordered nanoporous materials such as ordered mesoporous silicas, organosilicas, inorganic oxides and carbons, focusing on their synthesis and environmental and energy-related applications. |
1. Introduction
The design of functional nano- and microsized particles is a hot topic due to their application in a wide range of fields such as energy conversion and storage, catalysis, biomedicine, and environmental remediation. These tiny particles feature a high surface area to volume ratio, which is beneficial for diffusion and transport of reactants and products. Furthermore, depending on the material used, the particle size, crystal size, surface area, pore size distribution, and morphology can be tailored for specific applications by adjusting the chemical composition and synthesis conditions.Metal oxides (MeO) exhibit several attractive features such as high mechanical strength, thermal stability, chemical inertness, non-toxicity, biocompatibility, oxygen vacancies, semiconductor properties and high isoelectric point, and can act as supports for noble metals such as Au, Pt and Pd, or rare earth metals to achieve synergistic catalytic activity toward specific chemical reactions. Furthermore, for nanosized crystals, quantum effects become important as reflected by significantly different optical and electronic properties from those observed for the bulk phases,1 which can have favorable outcomes for their application in catalysis and photocatalysis; for example, new physicochemical phenomena such as ferromagnetism and paramagnetism can be achieved for otherwise antiferromagnetic systems, as in the case of NiO spheres.2 As a result, countless studies have been performed to synthesize submicrosized porous non-silica metal oxide particles with different morphologies, phases, sizes, crystal sizes and pore size distributions. These particles are useful for a variety of industrial, biomedical, environmental and energy-related applications as illustrated in Fig. 1.
 | ||
| Fig. 1 Spherical metal oxide particles and their potential applications. | ||
The porous non-silica metal oxide submicrospheres can be designed to have large surface areas and well-developed porosity to enhance interfacial interactions with reactants and facilitate the transport and diffusion of reactants and products. Also, the crystal size and growth directions need to be considered for specific catalytic and semiconductor applications. Furthermore, a proper balance between micropores, mesopores and macropores that respectively act as reaction sites, distribution/evacuation pathways and reservoirs is desired. The spherical morphology is the most stable shape that is achieved in nature. In the case of metal oxides, the advantages offered by spherical structures are high mechanical strength, short pathways for diffusion of species, dispersion enhancement due to the stabilization of electrostatic charges, high surface area to volume ratios, easy coating with other species or metal oxides, minimization of viscous effects and predictable hydrodynamics. Typical spherical morphologies of metal oxides discussed in this review can be classified into three groups: porous solid spheres (Fig. 2Aa), core–shell spheres (Fig. 2Ab) and hollow spheres (Fig. 2Ac). The core–shell spheres shown in Fig. 2Ab can be further extended into single-core particles with a multi-particle (raspberry-like) shell (Fig. 2Ab1), multi-core particles with a single shell (Fig. 2Ab2), single-core particles with a multi-shell (Fig. 2Ab3), or a combination of particles shown in Fig. 2Ab and b1 (see Fig. 2Ab4). Also, hollow spheres can be further classified into yolk–shell spheres (Fig. 2Ac1) and multi-shell hollow spheres (Fig. 2Ac2). In principle, each compartment of these spheres can be nonporous or porous with different pore sizes (micropores, mesopores, and macropores). More complex metal oxide spheres can be proposed and synthesized by combining and modifying the aforementioned nine patterns as shown in Fig. 2A.
 | ||
| Fig. 2 (A) Graphically illustrated classification of metal oxide spheres: (a) porous spheres; (b) core–shell spheres: (b1) raspberry-like core–shell spheres; (b2) multi-core spheres with a single shell; (b3) single-core spheres with multi-shells; (b4) raspberry-like single-core with multi-shells; and (c) hollow spheres: (c1) yolk–shell spheres; (c2) multi-shell hollow spheres. (B) SEM and TEM images of various metal oxide spherical particles: (a) dense spheres; (b) porous spheres; (c) core–shell spheres; (d and e) yolk–shell spheres; (f) hollow spheres. Panel B reproduced with permission from ref. 3. Copyright © 2007, American Chemical Society. | ||
Several excellent reviews have been published previously focusing on the synthesis and applications of metal oxides. However, these reviews consider either a specific metal oxide such as TiO24,5 and iron oxides,6 or a specific application such as gas sensors,7 energy storage and conversion applications8,9 or a specific morphology such as hollow spheres10 and multi-shell structures.11 Indeed, a concise and up-to-date review on the synthesis strategies of different types of porous non-silica metal oxide submicrospheres with various spherical morphologies as well as their emerging applications is timely as this research field continuous to rapidly grow. This review is focused on the synthesis and applications of non-silica-based metal oxide spheres based on the literature for the past five years. One of the main sections of this review is devoted to the major strategies frequently used for the preparation of MeO particles such as hydrothermal/solvothermal synthesis at elevated temperatures (>100 °C), solution precipitation synthesis at low temperatures (<100 °C), and the aerosol-type synthesis. Other methods are only briefly mentioned. The hydrothermal and low temperature precipitation syntheses are general methods that allow for the development of more sophisticated structures by hard templating, soft templating or controlled hydrolysis, or by taking advantage of Ostwald ripening and the Kirkendall effect.11,12 The aforementioned section on the synthesis methods is supplemented by a critical appraisal of the effect of different experimental parameters such as reaction time, reaction temperature, calcination, pH and the type of reactants and solvents on the structure of the resulting metal oxide particles. Finally, the last section presents the major applications of metal oxide spheres in industrially relevant sensing and catalysis, in the development of biomedically relevant photoluminescent devices and drug delivery vehicles, in environmentally relevant photocatalysis and adsorption, and in energy-relevant applications such as lithium-ion batteries, supercapacitors and dye sensitized solar cells.
2. Synthesis methods
Metal oxide particles have been generated by using different synthetic strategies, for example, colloidal synthesis, sol–gel process, aerosol process, precipitation, hydrothermal/solvothermal synthesis, hot injection, and non-aqueous and non-hydrolytic chemistry method (e.g., solid-state reactions, solid–gas reactions). In this section we summarize the main methods for the preparation of MeO particles with spherical structures such as the hydrothermal synthesis at elevated temperatures (>100 °C), low temperature solution precipitation synthesis (<100 °C) and the spray method. The first two methods include structure defining approaches such as hard templating, soft templating, hydrolysis, Ostwald ripening and the Kirkendall effect. This section ends with a brief overview of other methods involving electrodeposition, laser irradiation or ultrasonic irradiation.2.1 Hydrothermal and solution precipitation methods
The hydrothermal/solvothermal synthesis of metal oxide particles involves the use of a batch reactor at elevated temperatures (above 100 °C) and pressures. It offers a controlled environment for interactions of the salts used as metal oxide with other reactants in solution to form nanosized crystallites. The elevated pressure and temperature conditions facilitate dissolution of precursors and recrystallization of materials that can be insoluble under ambient conditions. As a result, high purity, homogeneous, metastable and often crystalline products are formed with unique properties and narrow particle size distribution.13 This synthesis method is flexible and well suited for the control of morphology and crystallinity of the porous metal oxide spheres by varying experimental parameters such as reaction temperature and time, type of reactants and solvents, and the chemical composition of the synthesis mixture. During the hydrothermal process, the nanosized crystallites self-assemble (with or without additives such as polymers and surfactants) into more complex architectures with optimal stability and lowest surface energy; hence, spherical shapes are favored. Also, metal oxide spherical structures can be synthesized via solution precipitation using milder conditions (i.e., temperature below 100 °C and atmospheric pressure) as reported elsewhere.13–20Tables 1 and 2 present the pertinent experimental conditions and characteristic features of solid spheres and shell-type particles obtained by hydrothermal synthesis together with relevant references.21–119 As can be seen from these tables, the hydrothermal method is very popular for the synthesis of various categories of metal oxides such as alkaline earth metals (MgO), rare-earth metals (CeO2, Y2O3), transition metals (TiO2, V2O5, Cr2O3, MnO2, Fe2O3, Co3O4, NiO, CuO, ZnO, ZrO2, Nb2O5, MoO2, Ta2O5 and WO3) and post-transition metals (Al2O3, Ga2O3, In2O3, Bi2O3 and SnO2). Composites, perovskites and doped metal oxides can also be created by controlling the ratio of the precursors.
| Ref. | Particle type | Reactants | Solvents | Hydrothermal conditions | Calcination conditions | Particle size | BET SA (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|---|---|---|---|---|---|
| a Microwave heating. b By varying the concentration of NbCl5 between 0.3 g and 0.5 g in 25 ml solution. c Prepared by an antisolvent method. d For nanowire-, flower- and urchin-like spheres respectively by increasing urea concentration. e For nanowire-, flower- and urchin-like spheres respectively. f For W(CO)6 concentration between 4.26 mM and 28.4 mM. g For WO3/TiO2 between 2% and 10%. h Supercritical temperature. NR: not reported. | |||||||||
| 22 | Bi2O3 spheres | Bi(NO3)3·5H2O, PVP | EG | 180 °C, 0.17 ha | 400 °C, 3 h in air | 10 μm | NR | NR | NR |
| 23 | Bi2O3 spheres | Bi(NO3)3·5H2O, HNO3, urea | Water, EG | 150 °C, 3 h | None | 350 nm | 8 | 0.018 | NR |
| Bi(NO3)3·5H2O, NaOH, HNO3, PVP | 100 nm | 23 | 0.15 | ||||||
| 24 | Bi2WO6 spheres | Na2WO4·2H2O, Bi(NO3)3·5H2O, PVP K30 | Water | 180 °C, 12 h | None | 4 μm | NR | NR | NR |
| 25 | Bi2WO6 perovskite spheres | Bi(NO3)3·5H2O, Na2WO4·2H2O, NaHCO3, citric acid | Water | 200 °C, 18 h | None | 2 μm | 24 | NR | NR |
| 26 | CeO2 spheres | Ce(NO3)3·6H2O | Water, C2H5COOH, EG | 180 °C, 3.3 h | None | 130 nm | 216 | NR | 3.8 |
| 27 | Co3O4 spheres | Co(CH3COO)2·4H2O, NH3 | Water, EG | 180 °C, 12 h | 500 °C, 4 h in air | 2–5 μm | 13 | NR | NR |
| 28 | Co3O4 spheres | Co(NO3)2·6H2O, urea | Water | 160 °C, 6 h | 300 °C, 2 h in air | 8–20 μm | 30 | 0.245 | 17 |
| 29 | CoFe2O4 spheres | CoCl2·6H2O, FeCl3·6H2O, urea | Water, ethanol | 170 °C, 0.42 ha | 500 °C in air | 1 μm | 25 | 0.18 | 25 |
| 30 | Cr2O3 spheres | C15H21CrO6, NH4HCO3 | Ethanol | 250 °C, 2 h | 500 °C, 4 h | 1–1.2 μm | 15 | NR | 20–80 |
| 31 | Cr2O3 spheres | Cr(NO3)3·9H2O, H2C2O4, urea | Ethanol, PEG | 180 °C, 5 h | 500 °C, 2 h in air | 2–3 μm | NR | NR | NR |
| 32 | CuO spheres | Cu(CH3COO)2 | Water, EG | 160 °C, 1 h | None | 412 nm | 168 | NR | 5 |
| 33 | CuO spheres | Cu(CH3COO)2, NH3, sodium alginate | Water | 160 °C, 2 h | None | 500 nm | 21 | NR | NR |
| 34 | α-Fe2O3 spheres | FeCl3·6H2O, ascorbic acid, urea | Water | 160 °C, 4 h | 500 °C, 4 h in air | 0.5–5 μm | 20 | 0.11 | 2–50 |
| 35 | α-Fe2O3 spheres | Fe(NO3)3·9H2O | Water, 2-butanone | 140 °C, 12 h | None | 100 nm | NR | NR | NR |
| 36 | α-Ga2O3 spheres | Ga(NO3)3, oxalic acid | Water | 200 °C, 10 h | 450 °C, 3 h | 0.5–4 μm | 62 | 0.193 | 12.3 |
| 37 | Cubic-In2O3 spheres | InCl3·4H2O, citric acid | Water, ethylenediamine | 180 °C, 7 h | 400 °C, 0.17 h in air | 150–200 nm | 88 | NR | NR |
| Hexagonal-In2O3 spheres | InCl3·4H2O, tartaric acid | Water, ethylenediamine | 180 °C, 7 h | 400 °C, 0.17 h in air | 150–200 nm | 85 | NR | NR | |
| 38 | In2O3 spheres | InCl3·4H2O, urea, sodium citrate | Water, EG | 200 °C, 16 h | 400 °C, 2 h in air | 600–700 nm | 19 | NR | NR |
| 39 | Nb2O5 spheres | NbCl5 | Ethanol | 200 °C, 24 h | 550 °C, 2 h in air | 200–900 nmb | 23–68b | NR | NR |
| 40 | Nb2O5 spheres | Glycolated Nb2O5 spheresc | Water | 180 °C, 12 h | None | 400–500 nm | 312 | 0.567 | 2 |
| 41 | NiO spheres | Ni(NO3)2·6H2O, Na2SO4, NaOH, glycine | Water | 180 °C, 0.5 ha | 300 °C, 3 h in air | 2 μm | 202 | NR | 25 |
| 42 | NiO spheres | Ni(NO3)2·6H2O, NaCl, sodium acetate | EG | 190 °C, 8 h | 300 °C, 2 h in air | 600 nm | 222 | NR | 4–10 |
| 2 | NiO spheres | NiCl2, sodium acetate, polyethyleneimine | Water, triethanolamine | 200 °C, 8 h | 270 °C, 0.5 h in air | 500 nm | 60 | NR | 10–30 |
| 43 | NiO spheres | NiCl2·H2O, urea | Water | 100 °C, 20 h | 300 °C, 2 h in air | 3–4 μm | 200–240d | NR | 3.2, 8.9, 4e |
| 44 | La doped NiO spheres | Ni(NO3)2·6H2O, La(NO3)2·6H2O, NH3, glucose | Water | 140 °C, 12 h | 550 °C, 4 h in air | 1–2 μm | 278 | 0.79 | 2–50, >50 |
| 45 | SnO2 spheres | Na2SnO3·3H2O, sodium alginate | EG, water | 180 °C, 24 h | None | 200–400 nm | 29 | NR | 15 |
| 46 | SnO2 spheres | SnCl2·2H2O, NaClO, HCl | Ethanol | 180 °C, 12 h | None | 150 nm | 62 | NR | 4 |
| 47 | SnO2 spheres | SnCl4·5H2O, PVP | Methanol | 180 °C, 3 h | 500 °C, 2 h in air | 400–700 nm | 78 | NR | 10 |
| 48 | SnO2 spheres | SnCl4·5H2O, PVP | Methanol | 180 °C, 3 h | 500 °C, 2 h in air | 500–700 nm | 78 | NR | 10 |
| 49 | SnO2@C spheres | K2SnO3·3H2O, glucose | Water | 180 °C, 4 h | 450 °C, 4 h in N2 | 100 nm | NR | NR | NR |
| 50 | C–V2O3 spheres | NH4VO3, citric acid | Ethanediol, water | 180 °C, 24 h | 600 °C, 3 h in N2 | 2 μm | 45 | NR | 20 |
| 51 | V2O5 spheres | VO(OiPr)3 | Acetic acid | 200 °C, 1.5 h | 350 °C, 0.5 h in air | 4–10 μm | 42 | NR | NR |
| 52 | WO2 spheres | W(CO)6 | Ethanol | 200 °C, 24 h | None | 0.7–1.5 μmf | 78–114f | NR | NR |
| 53 | WO3 spheres | WCl6, carbon microspheres | Dimethylformamide | 120 °C, 4 h | 420 °C, NR | 150–220 nm | 22 | 0.0447 | NR |
| 54 | WO3/TiO2 spheres | (NH4)10H2(W2O7)6, TiOSO4, P123 | Water, ethanol | 140 °C, 16 h | 500 °C, 6 h in air | NR | 45–64g | 0.21–0.26g | 12.2–15.2g |
| 55 | Eu3+:Y2O3 spheres | Y(NO3)3·6H2O, Eu(NO3)3, KOH | Water, 1-propanol | 400 °Ch, 0.17 h | 1000 °C, 1 h in air | 2–3 μm | NR | NR | NR |
| 56 | ZnO spheres | Zn(NO3)2·6H2O, L-asparagine, urea | Water | 100 °C, 3 h | 300 °C, 0.5 h in air | Several μm | 194 | NR | 5 |
| 57 | ZnO spheres | Zn(NO3)2, urea | Water | 120 °C, 2 h | 450 °C, 2 h in air | 10 μm | 38 | NR | 8.67 |
| 58 | ZnO spheres | Zn(NO3)2·6H2O, trisodium citrate, urea | Water | 120 °C, 6 h | 300 °C, 2 h in air | 4–6 μm | 40 | NR | 20–60 |
| 59 | ZnO spheres | Zn(CH3COOH)2·2H2O, MEA, urea | Water | 120 °C, 12 h | 450 °C, 2 h in air | 1–2 μm | 40 | NR | 5–50 |
| 60 | ZnO spheres | Zn(CH3COO)2·2H2O, NaOH, citric acid | Water, ethanol | 120 °C, 24 h | None | 2–3 μm | 42 | NR | 2–30 |
| 61 | ZnO spheres | Zn(CH3COO)2, thiourea | Water | 180 °C, 10 h | 500 °C, 3 h in air | 3–5 μm | 21 | NR | 22.6 |
| 60 | Ag loaded ZnO spheres | Zn(CH3COO)2·2H2O, AgNO3, NaOH, citric acid | Water, ethanol | 120 °C, 24 h | None | 2–3 μm | 37 | NR | NR |
| 62 | ZrO2 spheres | ZrOCl2·8H2O, HCl, urea | Ethanol, water | 160 °C, 2 h | None | 1–2 μm | 102 | 0.09 | 2–102 |
| Ref. | Particle type | Reactants | Hollowing mechanism | Solvents | Hydrothermal conditions | Calcination conditions | Particle and shell dimensions | BET SA (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
a Microwave heating.
b For F/Fe fractions between 0 and 1.
c By varying the fraction of glycerol in water between 0.05 and 0.125.
d (acac) = CH3COCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(O−)CH3.
e Ratio SiO2 C(O−)CH3.
e Ratio SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ta2O5 = 1:0.85.
f Ratio SiO2:Ta2O5 = 1:1.7.
g Nanosheet shell.
h Porous shell. NR: not reported. :Ta2O5 = 1:0.85.
f Ratio SiO2:Ta2O5 = 1:1.7.
g Nanosheet shell.
h Porous shell. NR: not reported. |
||||||||||
| 63 | γ-Al2O3 hollow spheres | KAl(SO4)2·12H2O, urea | Ostwald ripening | Water | 170 °C, 3 h | 600 °C, 2 h in air | 4–6 μm, shell thickness 700–900 nm | 149 | 0.45 | 12.3 |
| 64 | Perovskite BaZrO3 hollow spheres | Ba(NO3)2, ZrOCl2·8H2O, KOH | Ostwald ripening | Water | 200 °C, 24 h | None | 160 nm, shell thickness 15 nm | NR | NR | NR |
| 65 | Bi2O3/Co3O4 hollow spheres | Bi(NO3)3·5H2O, Co(NO3)3·6H2O, PEG, NaAc | Ostwald ripening | EG | 180 °C, 12 h | 500 °C, 2 h in air | 2–6 μm | 46 | 0.16 | NR |
| 66 | BiFeO3 hollow spheres | Bi(NO3)3·5H2O, Fe(NO3)3·9H2O, citric acid | Ostwald ripening | Glycerol, ethanol | 160 °C, 24 h | 500 °C, 24 h in air | 1.5 μm, shell thickness 0.2 μm | 15 | NR | NR |
| 67 | CeO2 hollow spheres | Ce(NO3)3·6H2O, HCl, citric acid | Ostwald ripening | Water | 160 °C, 24 h | 365 °C, 1.5 h in air | 2–4 μm | 56 | NR | NR |
| 68 | CeO2 hollow spheres | Ce(NO3)3·6H2O, PVP, H2O2, urea | Ostwald ripening | Water | 180 °C, 24 h | None | 126 nm | 21 | NR | 4 |
| 69 | CeO2 hollow spheres | Ce(NO3)3·6H2O, adipic acid | Ostwald ripening | Water, EG | 180 °C, 5 h | None | 135 nm | 145 | NR | 4 |
| 70 | CeO2 hollow spheres | CeCl3·7H2O, H2O2, urea | Ostwald ripening | Water | 180 °C, 10 h | None | 300 nm, shell thickness 50 nm | 85 | 0.23 | 3–10 |
| 71 | CeO2 hollow spheres | Ce(NO3)3·6H2O, PVP | Ostwald ripening | EG, ethanol, water | 180 °C, 24 h | None | 160 nm | 66 | 0.181 | 3–30 |
| 72 | CeO2 hollow spheres | CeCl3·7H2O, urea | Ostwald ripening | Water | 180 °C, 4 h | None | 300 nm, shell thickness 30 nm | 37 | NR | 36 |
| 73 | Yolk–shell CeO2 | Ce(NO3)3·6H2O, PVP, NH4Ac·2H2O | Carbon spheres | Ethanol | 180 °C, 12 h | 600 °C, 3 h in air | 180 nm | NR | NR | NR |
| 74 | Multi-yolk–shell Pd@CeO2 spheres | Pd@SiO2, Ce(NO3)3·9H2O | SiO2 etching | EG, CH3COOH, water | 130 °C, 12 h | 350 °C, 2 h in H2 | 150–200 nm | 104 | 0.078 | 2–25 |
| 75 | Co3O4 hollow spheres | Co(NO3)2·6H2O, sodium citrate, HMT, sucrose | In situ carbon from sucrose | Water | 140 °C, 24 h | 500 °C, 5 h in air | Shell thickness 130 nm | 60 | NR | 7.8 |
| 76 | Co3O4 hollow spheres | Co(NO3)2 | Ostwald ripening | Glycerol, isopropanol | 180 °C, 6 h | 200 °C, 2 h in air | 1 μm | 180 | NR | 2–150 |
| 77 | CoFe2O4 double shell spheres | CoSO4·7H2O, (NH4)2Fe(SO4)2·6H2O, sucrose | In situ carbon from sucrose | Water | 180 °C, 24 h | 600 °C, 2 h in air | 0.5–1.5 μm, 200–500 nm hollow core | NR | NR | NR |
| Same as above with half sucrose concentration | NR | 38 | NR | 30 | ||||||
| 33 | CuO hollow spheres | Cu(CH3COO)2, NH3, sodium alginate | Ostwald ripening | Water | 160 °C, 6 h | None | 500 nm | 72 | NR | NR |
| 78 | CuO hollow spheres | Cu(NO3)2, urea | Ostwald ripening | Water | 180 °C, 18 h | 400 °C, 2 h in air | 4.5–6.5 μm | NR | NR | NR |
| 79 | CuO hollow spheres | Cu(CH3COO)2·H2O | Ostwald ripening | Water | 120 °C, 24 h | None | 3.5 μm, shell thickness 1.25 μm | NR | NR | NR |
| 32 | Cu2O hollow spheres | Cu(CH3COO)2, glucose | In situ carbon from glucose | Water, EG | 160 °C, 1 h | None | 1.5 μm, shell thickness 400 nm | 37 | NR | 50 |
| 15 | Cu/Cu2O hollow spheres | Cu(Oac)2·H2O, PVP | Ostwald ripening | Ascorbic acid | 100 °C, 0.5 ha | None | 150–500 nm | 19 | 0.118 | 2–100 |
| 80 | CuO/Cu2O composite hollow spheres | Cu(NO3)2·3H2O, ethanolamine | Ostwald ripening | Water | 180 °C, 12 h | None | 1.5–3 μm | 16 | NR | NR |
| 31 | Cr2O3@C core shell spheres | Cr(NO3)3·9H2O, H2C2O4, urea | Controlled annealing | Ethanol, PEG | 180 °C, 5 h | 750 °C, 6 h in 5% Ar and 95% H2 | 2–3 μm | NR | NR | NR |
| 81 | α-Fe2O3 hollow spheres | FeCl3·6H2O | Ostwald ripening | Water, DMF, TFA | 180 °C, 24 h | None | 2 μm | 4 | NR | 65.8 |
| 82 | α-Fe2O3 hollow spheres | K3[Fe(C2O4)3] | In situ gas bubbles | Water, EG | 150 °C, 48 h | 450 °C, 3 h in air | 190 nm | 41 | NR | 4–12 |
| 83 | α-Fe2O3 hollow spheres | FeSO4·7H2O | Quasi-emulsion droplets | Water, glycerol | 145 °C, NR | None | 1 μm, shell thickness 100–200 nm | 103 | NR | <30 |
| 84 | Double-shelled α-Fe2O3 spheres | K3[Fe(CN)6], NH4H2PO4 | Ostwald ripening | Water | 200 °C, 30 h | None | 350 nm, 200 nm core, 20 nm outer shell, 40 nm inner shell | 98 | NR | 11.2 |
| 85 | Fe3O4 hollow spheres | FeCl3·6H2O, NaOH, SDBS | Precursor templated | EG | 200 °C, 1.5 ha | 300 °C, 1 h in N2 | 2–4 μm | 62 | 0.131 | 10.2 |
| 86 | Fe3O4@TiO2 double shelled yolk–shell spheres | Fe3O4@SiO2@TiO2, NaOH | Ostwald ripening + NaOH etching | Water | 150 °C, 24 h | None | 560 nm | 150 | 0.27 | 7.5 |
| 85 | γ-Fe2O3 hollow spheres | FeCl3·6H2O, SDBS, NaOH | Precursor templated | EG | 200 °C, 1.5 ha | 300 °C, 1 h in air | 2–4 μm | 56 | 0.159 | 16.3 |
| 87 | α-Fe2O3 four shelled hollow spheres | Fe(NO3)3·9H2O, L-histidine | Amino acid templated | Water | 180 °C, 12 h | 600 °C, 2 h in air | 3 μm | 14 | 0.07 | NR |
| 88 | γ-Fe2O3 hollow spheres | FeCl3·6H2O, NH4F, ethylenediamine | Ostwald ripening | EG | 200 °C, 20 h | 250 °C, 5 h in air | 250 nm, shell thickness 20–40 nmb | 9–19b | NR | 13.3–34.5b |
| 89 | Fe3O4 hollow spheres | Fe(NO3)3·6H2O | Kirkendall mechanism | Glycerol, isopropanol, water | 190 °C, 12 h | 350 °C, 3 h in N2 | 900 nm, shell thickness 10 nm | 89 | NR | 4, 5, 7 |
| 90 | α-FeOOH hollow spheres | FeSO4·7H2O | Quasi-emulsion | Water, glycerol | 120 °C, 24 h | None | 1 μm, varied shell thicknessc | 54–97c | 0.28–0.36c | <20 |
| 91 | Perovskite LaFeO3 hollow spheres | La(NO3)3·6H2O, Fe(NO3)3·9H2O, citric acid | Ostwald ripening | Water | 180 °C, 24 h | 800 °C, 2 h in air | 2–5 μm, shell thickness 40–60 nm | 49 | NR | 30–80 and 100–300 |
| 92 | β-Ga2O3 hollow spheres | Metallic Ga, HCl, urea | In situ gas bubbles | Acetone | 200 °C, 4 h | 700–800 °C, 2 h in air | 1–2 μm | 22 | NR | 3 |
| γ-Ga2O3 hollow spheres | 500–600 °C, 2 h in air | 1–2 μm | 31 | NR | 7 | |||||
| 93 | Er doped In2O3 hollow spheres | InCl3·4H2O, Er(NO3)·7H2O | Carbon spheres | Water | 180 °C, 6 h | 500 °C, 3 h in O2 | 300 nm, shell thickness 40 nm | NR | NR | NR |
| 94 | Rh-loaded In2O3 hollow spheres | In(NO3)3·xH2O, RhCl3·xH2O, D(+) glucose monohydrate | In situ carbon from glucose | Water | 180 °C, 24 h | 500 °C, 2 h in air | 2.1 μm, shell thickness 180 nm | NR | NR | 40 |
| 95 | MgO hollow spheres | MgCl2·6H2O, urea | Ostwald ripening | Water, EG | 120 °C, 10 h | 450 °C, 1 h in air | 3–4 μm | 130 | 0.414 | 7 |
| 96 | MgO hollow spheres | Mg(Oac)2·4H2O, PVP K-30, NH4OH | Ostwald ripening | EG | 185 °C, 5 h | 500 °C, 1 h in Ar + 1 h in air | 1 μm | 343 | 1.9 | <30 |
| 97 | MnO2 hollow spheres | KMnO4, SiO2 spheres, Pluronic F127 | SiO2 etching | Water | 150 °C, 48 h | None | 210 nm | 233 | NR | NR |
| 98 | MnO2 hollow spheres | KMnO4 | Hollow carbon spheres | Water | 160 °C, 5 h | None | 316 nm, shell thickness 69 nm | 30 | 0.112 | 19.4 |
| 99 | MnO2 hollow spheres | KMnO4, Ce(NO3)3·6H2O, HNO3 | Ostwald ripening | Water | 140 °C, 3 h | None | 3–4 μm | 29 | 0.3 | 2 |
| 100 | C@MnO2 spheres | MnSO4·H2O, (NH4)2S2O8, glucose | Ostwald ripening | Water | 180 °C, 3 h | None | 1.5 μm | 142 | 0.27 | 3–4 |
| 101 | MoO2 hollow spheres | MoO3, diethylenetriamine | Ostwald ripening | Water | 200 °C, 144 h | 700 °C, 4 h in Ar | 3–5 μm | NR | NR | NR |
| 102 | MoO2@MoO2 yolk–shell particles | MoO2(acac)2d, HNO3 | Ostwald ripening | Isopropanol, water | 180 °C, 24 h | 350 °C, 2 h in N2 | 1 μm, shell thickness 80 nm | 31 | NR | 3–4 |
| 103 | NiO hollow spheres | Ni(NO3)2·6H2O, NH3, L-cysteine | Ostwald ripening | Water | 120 °C, 10 h | 600 °C, 1 h in air | 2–3 μm, shell thickness 400 nm | 66 | 0.442 | 10–50 |
| 104 | NiO multi-shelled spheres | Ni(NO3)3·6H2O, NH3, D-glucose | In situ carbon from glucose | Water | 150 °C, 15 h | 500 °C, 6 h in air | 2–3.5 μm, shell thickness 50 nm | 29 | NR | NR |
| 105 | Core-in-double shell NiCo2O4 spheres | Ni–glycerate spheres prepared hydrothermally | Kirkendall mechanism | None | NA | 350 °C, 2 h@1 °C min−1 | 400 nm outer shell, 200 nm inner shell, 40 nm core, 70 nm and 40 nm outer and inner shell thickness | 61 | NR | <10 |
| 81 | SnO2 hollow spheres | SnCl4·5H2O | Ostwald ripening | Water, DMF, TFA | 180 °C, 48 h | None | 2 μm | 108 | NR | 6.04 |
| 106 | SnO2 hollow spheres | SnSO4 | Ostwald ripening | Water | 120 °C, 48 h | None | 100–200 nm | 69 | NR | 4 |
| 107 | SnO2 hollow spheres | SnF2, H2O2 | Ostwald ripening | Water | 180 °C, 12 h | None | 100–200 nm, shell thickness 40–50 nm | 156 | NR | NR |
| 108 | SnO2 hollow spheres | K2SnO3·3H2O, urea | Ostwald ripening | Water, ethanol | 150 °C, 24 h | None | 150–400 nm | 110 | NR | 4 |
| 109 | SnO2 hollow spheres | SnCl2·2H2O, HCl, urea | Hollow polystyrene spheres | Mercaptoacetic acid | 120 °C, 6 h | 400 °C, 2 h in air | 650 nm, shell thickness 100 nm | 62 | NR | 3–8 |
| 73 | Yolk–shell SnO2 | SnCl2·2H2O, HCl | Carbon spheres | DMF, water | 180 °C, 12 h | 600 °C, 3 h in air | 420 nm | 43 | 0.073 | 6.8 |
| 110 | SnO2 multishell spheres | SnCl4·5H2O, sucrose | In situ carbon from sucrose | Water | 190 °C, 24 h | 600 °C, 3 h in air | 0.5–2 μm | 36 | 0.197 | 2.50 |
| 111 | SnO2/C hollow spheres | Sn spheres, glucose | Kirkendall mechanism | Water | 180 °C, 3 h | 500 °C, 3 h in N2 | 100 nm | NR | NR | NR |
| 108 | SnO2/C hollow spheres | SnO2 hollow spheres, glucose | In situ carbon from glucose | Water | 180 °C, 3 h | 550 °C, 3 h in N2 | 150–400 nm | NR | NR | NR |
| 112 | Perovskite SrTiO3 hollow spheres | Anatase TiO2, SrCl2·6H2O, NaOH | Kirkendall mechanism | Water | 180 °C, 6 h | None | 3–5 μm, shell thickness 700 nm | NR | NR | NR |
| 113 | SiO2–Ta2O5 hollow spherese | Tantalum isopropoxide, CTAB, TEOS, NH3 | Ostwald ripening | Water, ethanol | 120 °C, 48 h | 550 °C, 5 h in air | 100–250 nm, shell thickness 50 nm | 249 | 0.48 | 14.8 |
| SiO2–Ta2O5 hollow spheresf | 200 nm, shell thickness 60 nm | 225 | 0.26 | 13.5 | ||||||
| 73 | Yolk–shell Tb4O7 | Tb(NO3)3, NH4Ac·2H2O | Carbon spheres | Ethanol | 180 °C, 12 h | 600 °C, 3 h in air | 200 nm | NR | NR | NR |
| 114 | V2O5 hollow spheres | NH4VO3 | In situ gas bubbles | EG | 180 °C, 24 h | 500 °C, 2 h in air | 3 μm, shell thickness 1.125 μm | 22 | NR | 5–8 |
| 115 | V2O5 hollow spheres | VO(C5H7O2)2, PVP | PVP micelles templated | EG | 140 °C, 12 h | 350 °C, 2 h in air | 800 nm | NR | NR | NR |
| 116 | V2O5@V2O5 yolk–shell spheresg | Vanadium oxytriisopropoxide | Carbon spheres | Isopropanol | 200 °C, 12 h | 350 °C, 2 h in air | 1 μm, shell thickness 200 nm | NR | NR | NR |
| V2O5@V2O5 yolk–shell spheresh | Vanadium oxytriisopropoxide | Carbon spheres | Isopropanol, water | 200 °C, 12 h | 350 °C, 2 h in air | 2 μm, shell thickness 100 nm | NR | NR | NR | |
| 117 | V2O5 yolk–shell spheres | V2O5, oxalic acid | Ostwald ripening | Water, isopropanol | 200 °C, 2.5 h | 350 °C, 2 h in air | 1 μm, shell thickness 100 nm | 28 | 0.15 | NR |
| 118 | ZnO hollow spheres | ZnCl2, glucose | In situ carbon from glucose | Water | 180 °C, 24 h | 500 °C, 4 h | 0.8 μm | 63 | 0.17 | <5, 9–90 |
| 119 | ZnO single shell hollow spheres | ZnSO4·7H2O, glucose | In situ carbon from glucose | Water | 180 °C, 12 h | 550 °C directly, 3 h in air | 1 μm | 10 | 0.04 | NR |
| ZnO double shell hollow spheres | 550 °C, 5 °C min−1, 3 h in air | 1 μm | 19 | 0.07 | NR | |||||
| ZnO triple shell hollow spheres | 550 °C, 2 °C min−1, 3 h in air | 1 μm | 25 | 0.09 | NR | |||||
| 62 | ZrO2 hollow spheres | ZrOCl2·8H2O, HCl, urea | Ostwald ripening | Ethanol, water | 160 °C, 24 h | None | 1–2 μm | 136 | 0.1 | 2–105 |
| 120 | Yolk–shell ZnCo2O4 | ZnAc2·2H2O, CoAc2·4H2O | Carbon spheres | EG | 180 °C, 12 h | 600 °C, 3 h in air | 300–500 nm | 16 | 0.063 | 20 |
Tables 3 and 4 provide a summary of metal oxide particles obtained by a solution precipitation method together with relevant references.13,16–20,120–159 However, the range of metal oxide particles that can be made by this method is not as extensive as in the case of hydrothermal synthesis, probably due to the lower solubility of metal oxide precursors at lower temperatures and the smaller flexibility at the temperatures used. Nevertheless, such mild conditions are attractive in terms of green technology and cost effectiveness.
| Ref. | Particle type | Reactants | Solvents | Reaction conditions | Calcination conditions | Particle size | BET SA (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|---|---|---|---|---|---|
|
a Sonicated.
b When the ratio of Al2(SO4)3·16H2O:Al(NO)3·9H2O is between 0.33 and 0.167.
c To obtain V2O3 phase.
d To obtain V2O5 phase.
e Depending on water or pyridine concentration.
f Mol ratio of Na2WO4·2H2O:concentrated HCl = 1:50.
g Removed by NaOH etching.
h By varying the Ce/HMT ratio.
i Assumed room temperature. NR: not reported. |
|||||||||
| 19 | Ag2O–MnO2 spheres | MnSO4, (NH4)2S2O8, Ag nanoparticles | Water | 50 °C, 1 ha | None | 2.2 μm | NR | NR | NR |
| 121 | α-Al2O3 spheres | Al2(SO4)3·16H2O, Al(NO3)3·9H2O, urea | Water | 98 °C, 1.5 h | 1100 °C, 1 h in air | 125–430 nmb | 76b | NR | 2–20 |
| γ-Al2O3 spheres | Al2(SO4)3·16H2O, Al(NO3)3·9H2O, urea | Water | 98 °C, 1.5 h | 900 °C, 1 h in air | NR | 102b | NR | 2–20 | |
| 18 | CuO spheres | Cu powder, NaOH, (NH4)2S2O8 | Water | 25i °C, 20 h | None | 1–2 μm | 8 | NR | NR |
| 122 | CuO spheres | Cu(NO3)2·H2O, NH3, NaOH | Water, glycol | 100 °C, 2 h | 300 °C, 4 h in air | 1–3 μm | 88 | NR | NR |
| 17 | MnO2 spheres | (CH3COO)2Mn·4H2O, AgNO3, H2SO4, oxone monopersulfate | Water | 25i °C, 36 h | None | 1–3 μm | 163 | NR | 65 |
| 21 | MnO2 spheres | MnSO4, (NH4)2S2O8, FeSO4 | Water | 50 °C, 1.5 ha | None | 700 nm | NR | NR | NR |
| 20 | α-MnO2 spheres | MnSO4·H2O, K2S2O8, K2SO4, H2SO4, AgNO3 | Water | 40 °C, 12 h | None | 2 μm | 150 | NR | 2, 10–20 |
| 60 °C, 12 h | None | 2 μm | 106 | NR | 2, 10–20 | ||||
| 80 °C, 12 h | None | 2 μm | 83 | NR | 2, 10–20 | ||||
| 123 | Nb2O5 spheres | NbCl5, HNO3, resol, PEO-b-PS diblock copolymer | THF | 50 °C, 24 h + 100 °C, 24 h | 350 °C, 3 h and 550 °C, 2 h in N2 + 400 °C, 3 h in air | 0.2–1 μm | 131 | 0.26 | 11.4 |
| 124 | NiO spheres | Ni(NO3)2·6H2O, NH3 | Water | 97 °C, 1 h | 300 °C, 2 h in air | 5 μm | 216 | 0.38 | 64.3 |
| 125 | SnO2 spheres | Na2SnO3·3H2O, D-glucose monohydrate | Water | 50 °C, 12 h | None | 50 nm | 160 | 0.196 | 2.55 |
| 150 °C in air | 50 nm | 146 | NR | NR | |||||
| 300 °C in air | 50 nm | 103 | NR | NR | |||||
| 500 °C in air | 50 nm | 75 | NR | NR | |||||
| 126 | SnO2 spheres | SnSO4 | Water, ethanol | 25i °C, 1 h | 500 °C, 2.5 h in air | 100–800 nm | 29 | NR | 4 |
| 127 | V2O5 spheres | Vanadium isopropoxide | Acetone, pyridine, water | 25i °C, 0.5 h | 400 °C, 2 h in H2c + 300 °C, 1 h in aird | 150–1000 nme | 31 | NR | <30 |
| 128 | V2O5 spheres | NH4VO3, HCl, hydrazine | Water | 25i °C, 0.5 h | 350 °C, 2 h in air | 400 nm | 12 | NR | <50 |
| 129 | WO3 spheres | Na2WO4·2H2O, HClf | Water, EG | 75 °C, 12 h | 450 °C, 2 h in air | 3–5 μm | 13 | NR | 3.3–5.4 |
| 130 | WO3 spheres | Na2WO4, HCl, oxalic acid | Water | 25i °C, 1 ha | 500 °C in air | 1–3 μm | 13 | NR | 28.1 |
| 131 | WO3·H2O spheres | Na2WO4·2H2O, HCl | Water | 70 °C, 10 h | 400 °C, 2 h in air | 2–3 μm | 11 | NR | 1.7–30 |
| 132 | Y2O3:Er spheres | Mesoporous SiO2 spheresg, Y(NO3)3, Er(NO3)3, urea | Water | 90 °C, 2 h | 700 °C, 3 h in air | 560 nm | 85 | 0.196 | 5.7 |
| 133 | ZnCo2O4@CeO2 core–shell spheres | ZnCo2O4, Ce(NO3)3, hexamethylenetetramine | Water, ethanol | 60 °C, 2 h | None | 1.55–1.68 μmh | 34–57h | NR | NR |
| 134 | ZnO spheres | Zn(CH3COO)2, TEA | Water | 25i °C, 2 ha | None | 520 nm | 17 | NR | 25, 180 |
| 135 | ZnO spheres | Zn(CH3COO)2·2H2O, hexamine, sodium citrate | Water | 90 °C, 6 h | 600 °C in air | 2.5 μm | NR | NR | NR |
| 136 | ZrO2 spheres | ZrOCl2·8H2O, porous polymer spheres | Water, ethanol | 25i °C, 0.17 ha | 600 °C, 6 h in air | 2.6 μm | 22 | 0.17 | 31 |
| Ref. | Particle type | Reactants (mol/mass ratios) | Hollowing mechanism | Solvents | Reaction conditions | Calcination conditions | Particle and shell dimensions | BET SA (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Hollow-microporous organic network (H-MON).
b By varying the solvent ratio or the SiO2 amount.
c For H-MON prepared with the ratio of toluene:triethylamine = 1:1.
d
Micrococcus lylae.
e Sonicated.
f Mol ratio of Na2WO4·2H2O:concentrated HCl = 1:15.
g Polyvinylalcohol@glucose derived carbon rich polysaccharide spheres.
h Poly(styrene-acrylic acid).
i When the molar ratio of ZrOCl2·8H2O:ethanol is between 0.011 and 0.032.
j Assumed room temperature. NR: not reported. |
||||||||||
| 137 | Al2O3 hollow spheres | Al2O3 spheres, PVP, NaOH | NaOH etching/Kirkendall | Water | 25j °C, few minutes | 400 °C in air | 190 nm, shell thickness 23–30 nm | 292 | 0.442 | 6.3 |
| 138 | Al2O3, ZrO2, ZnO shell | Metal salt | Solid core | Buffer solution | 70 °C, 2 h | 450 °C, 2 h in air | Shell thickness tuneable 1–20 nm | NR | NR | NR |
| 139 | CdO hollow spheres | Cd(CH3COO)2, NaOH | Yeast | Water | 25j °C, 12 h | 500 °C, 4 h in air | 2.3 μm, shell thickness 250–280 nm | 5 | 0.009 | 3–30 |
| 140 | CeO2 hollow spheres | Ce(NO3)3·6H2O, HMT | PS spheres | Water | 75 °C, 2 h | 600 °C, 2 h in air | 190 nm, shell thickness 15 nm | 66 | 0.19 | NR |
| 141 | Co3O4 hollow spheres | Co(NO3)2 | Untreated carbon spheres | Water | 25j °C, 1 h | 450 °C, 2 h in Ar + 450 °C in air | 240 nm, shell thickness 40 nm | 223 | 0.29 | 15.3 |
| Co(NO3)2 | Acid treated carbon spheres | 240 nm, shell thickness 15 nm | 301 | 0.36 | 9.9 | |||||
| Co(NO3)2 | Alkali treated carbon spheres | 240 nm, shell thickness 70 nm | 174 | 0.2 | 23 | |||||
| 142 | Co3O4 hollow spheres | Co2(CO)8 | H-MONa spheres | Toluene | 100 °C, 12 h | 500 °C, 5 h in air | 500 nm, shell thickness 20–80 nmb | 64c | 0.32c | NR |
| 14 | Co3O4 hollow spheres | CoCl2·6H2O, NaBH4 | Bacterial suspensiond | Water | 25j °C, 12 h | None | 1 μm | 149 | 0.26 | 7.7 |
| 143 | CuO hollow spheres | CuSO4, KOH, NH3 | Ostwald ripening | Water | 68 °C, 24 h | None | 3–5 μm, shell thickness 500 nm | NR | NR | 1–2.2, 5–30 |
| 144 | CuO hollow spheres | Cu(CH3COO)2·H2O, urea | In situ gas bubbles | Water | 80 °C, 2 he | None | 400–500 nm, shell thickness 45 nm | 60 | 0.104 | 3.6 |
| 145 | Gd2O3 hollow spheres | Gd(NO3)3, urea | Carbon spheres | Water, ethanol | 90 °C, 6 h | 800 °C, 2 h in air | 200–250 nm, shell thickness 20 nm | 33 | 0.17 | 10.9 |
| 146 | In2O3 hollow spheres | InCl3 | Polymer spheres | C2Cl4 | 55 °C, 6 h | 600 °C, in air | 720 nm, shell thickness 110 nm | 329 | NR | 3 |
| 75 °C, 6 h | 600 °C, in air | 950 nm, shell thickness 140 nm | 28 | NR | 3 | |||||
| 95 °C, 6 h | 600 °C, in air | 1180 nm, shell thickness 220 nm | 27 | NR | 3 | |||||
| 147 | MnO2 hollow spheres | MnSO4 | CH2Cl2/H2O interface | Water, CH2Cl2 | 25j °C, 48 h | 300 °C, 2 h in air | 200–500 nm | 219 | 0.451 | 5.9 |
| 148 | NiO hollow spheres | Ni(NO3)2·6H2O, urea | Sulfonated polystyrene hollow spheres | Water, ethanol | 80 °C, 12 h | 450 °C, 2 h in air | 500 nm, shell thickness 100 nm | 62 | NR | 2–4 |
| 149 | NiO hollow spheres | NiCl2·6H2O, (NH4)2C2O4 | Calcination of organic species | Water | 25j °C, 0.67 he | 500 °C, 1 h in air | 1.7 μm | 32 | NR | 3–20 |
| 150 | SnO2 hollow spheres | SnCl2, HCl | Ostwald ripening | Water | 90 °C, 12 h | None | 100–300 nm, shell thickness 10 nm | 89 | NR | NR |
| 151 | SnO2 hollow spheres | Tin butoxide | Microemulsion template | CTAB, hexanol, n-dodecane, methanol, water | 20 °C, 12 h | None | 15–25 nm, shell thickness 3–5 nm | 417 | NR | NR |
| 152 | SnO2 hollow spheres | SnCl2·2H2O | Hollow SiO2 spheres | None | 80 °C, 24 h | 700 °C, 2 h in air | 340 nm, shell thickness 50 nm | 46 | NR | 2–5 |
| 129 | WO3 hollow spheres | Na2WO4·2H2O, HClf | Ostwald ripening | Water, EG | 75 °C, 12 h | 450 °C, 2 h in air | 3–4 μm | 16 | NR | 5.4–89.6 |
| 153 | WO3 hollow spheres with multiple shells | WCl6 | PVA@GCPg | Ethanol | 0 °C, 12 h | 450 °C, 1 h in air | 500 nm | 124 | 0.14 | 4.3 |
| 154 | WO3/WO3·H2O hollow spheres | Na2WO4·2H2O, HCl, oxalic acid | Ostwald ripening | Water, isopropyl alcohol | 80 °C, 12 h | 200 °C, 2 h in air | 2 μm, shell thickness 200 nm | 22 | 0.079 | 14.2 |
| 155 | Y2O3 hollow spheres | Y(NO3)3, urea | Melamine formaldehyde spheres | Water | 85 °C, 3 h | 800 °C, 2 h in air | 1.8 μm, shell thickness 100 nm | NR | NR | NR |
| 156 | Y2O3:Ln3+ hollow spheres | Y(NO3)3, Eu(NO3)3, urea | PS spheres | Water | 90 °C, 4 h | 800 °C, 2 h in air | 2.1 μm, shell thickness 70 nm | 62 | 0.313 | 20.7 |
| 157 | Y2O3:Tb3+ hollow spheres | Y(NO3)3, Tb(NO3)3, urea | PS spheres | Water, ethyl alcohol | 85 °C, 3 h | 800 °C, 2 h in air | 1.3 μm, shell thickness 50 nm | NR | NR | NR |
| 158 | ZnO hollow spheres | Zn(NO3)2·6H2O, (CH2)6N4, sodium citrate | Ostwald ripening | Water | 95 °C, 5 h | 400 °C, 2 h in air | 2 μm | 42 | NR | 5–8 |
| 159 | ZnO hollow spheres | Zn(CH3COO)2, HMT, sodium citrate | Calcination of organic species | Water | 95 °C, 3 h | 400 °C, 2 h in air | 2–3 μm | 138 | NR | NR |
| 160 | ZrO2 hollow spheres | ZrOCl2·8H2O, NH3 vapour | PSAh spheres | Ethanol | 50 °C, NR | 700 °C, 4 h in air | 3.2–3.4 μm, shell thickness 80–200 nmi | NR | NR | NR |
In general, metal salts are used for the synthesis of metal oxide spheres, often supplemented by complexing/structure directing agents and basic reactants in the case of precipitation method. A reducing or oxidizing agent can also be used. For instance, NaBH4 was used to reduce Co2+ into Co nanoparticles, which subsequently were spontaneously oxidized in air onto bacterial templates to form Co3O4,14 ascorbic acid was employed to reduce Cu(OH)2 to form composite Cu/Cu2O spheres under microwave hydrothermal conditions,15 and N2H4·H2O was used to reduce Cu2+ in solution to Cu2O in the reduction-induced precipitation method,16 while oxidizing agents such as oxone monopersulfate17 or (NH4)2S2O818,19 were employed to increase the valence states of metal cations. Moreover in some cases, metal ions were used as catalysts. For the synthesis of MnO2 spheres, Ag+ was used as a catalyst to help the reaction proceed at low temperature,17,20 while in another work, Fe2+ was used to oxidize Mn2+ to MnO2 crystals.21
The hydrothermal and solution precipitation syntheses are general methods that include hard templating, soft templating, hydrolysis, Ostwald ripening and the Kirkendall effect, which are briefly discussed in the following subsections.
2.1.1.1 Shell structures. Hard templating is the most common method for production of hollow spheres. In hard templating, nanoparticles are attached to the surface of a solid sphere and they aggregate to form a shell. A decade ago, the commonly used hard templates for the creation of spherical shells were carbon,161–163 silica164 or polymer spheres.165 As can be seen from Tables 2 and 4 these templates are still very popular, while bio-organisms have also been used. The aforementioned templates have a large number of reactive oxygen functional groups that are electron donors and therefore attract positively charged metal cations, as depicted in Fig. 3. The size of the inner hollow space can be tailored by selecting solid templates with appropriate sizes, while the thickness of the shell can be adjusted by changing the concentration of the reactants160 or by changing the hydrophilicity of the template's surface by alkaline or acid treatment.141 Just recently, hollow TiO2 nanospheres with a thin single layer of TiO2 nanoparticles were synthesized by using quasi-nanosized carbonaceous spheres as a template.166 In addition, hollow mesoporous organic networks (H-MONs), a new class of functional materials prepared through various carbon–carbon coupling reactions between organic building blocks, were prepared with varying shell thickness and used as templates to generate Co3O4 shells of different thicknesses and having surface areas between 60 and 67 m2 g−1.142
 | ||
| Fig. 3 Hard templating method for synthesis of hollow metal oxide spheres. | ||
However, the use of templates does not restrict the final shape to hollow spheres. Lou and co-workers116 found that the deposition of vanadium species on carbon sphere (CS) templates via a non-hydrolytic hydrothermal reaction between vanadium oxytriisopropoxide and isopropanol produced core–shell CS@V particles with shells made from interconnected nanosheets. Interestingly, when a small amount of water was added to the process, yolk–shell structures with rough surfaces were obtained. The VO2 species formed by sol–gel reaction on the carbon spheres underwent an Ostwald ripening process to form a well-defined gap between the core and the shell. Yolk–shell particles of V2O5@V2O5 were also created from C@V2O5 core–shell particles116 whereby the vanadium species bound to the carbon core surface would shrink into a core during annealing. Similarly, yolk–shell ZnCo2O4,120 SnO2, CeO2 and Tb4O773 were generated by hydrothermal loading of the metal precursor into the carbon template pores followed by calcination in air. TGA studies revealed that the removal of the template occurred in two steps to give the shell first and then the core. Zeng et al.167 reported the synthesis of multi-shell ZnO with single, double or triple shells by simply using carbon templates of different diameters for loading of the ZnO precursor.
Elsewhere, a penetration–solidification–annealing method was used to synthesize multi-shell spheres of CoxMn3−xO4 by using carbon spheres as templates and controlling the molar ratios of Co and Mn oxide precursors.168 Based on the anion-adsorption mechanism and usage of carbon spheres as templates, Wang and co-workers developed a sequential templating approach for production of multi-shell hollow spheres with different composition including ZnO, TiO2, SnO2, Co3O4, α-Fe2O3, Mn2O3, V2O5, etc.166,169–176 For example, multi-shell V2O5 hollow spheres were synthesized by a novel method involving competitive anion adsorption on carbon sphere templates followed by a Trojan catalytic combustion procedure (Fig. 4).176 The carbon spheres were pre-treated to create a negative charge, which could facilitate adsorption of metal anions from a solution of ammonium salt. The NH4+ cations also penetrated the templates, neutralizing the negative charges and stimulating further anion adsorption. As a result, single- or multi-shell structures were formed either by using different precursor concentrations or by performing multiple adsorption processes. The method was shown to be flexible and could be extended to the synthesis of MnO2, MoO3, Cr2O3 and WO3 multi-shell hollow spheres.
 | ||
| Fig. 4 Multi-shell metal oxides prepared via an anion-adsorption mechanism: (A) schematic representation of two synthesis routes to obtain multi-shell hollow microspheres. (i) Cation-adsorption process. (ii) Anion-adsorption process. (B) Effects of synthesis conditions on the morphology of products. (C) Morphological and structural analysis of V2O5 spheres: (a–f) TEM images of the as-prepared samples. (g–i) SEM images of the as-prepared samples. Reproduced with permission from ref. 176. Copyright © 2016, Nature Publishing Group. | ||
However, hard templating has a few disadvantages. An additional synthesis step is required to remove the templates, which is by either calcination in air for carbon-rich templates or alkaline or HF etching for SiO2 templates. The use and subsequent removal of the solid templates represents a waste in resources, which goes against “green” processing. Furthermore, calcination can lead to the partial or full collapse of the shell architectures, while alkaline etching can implicate formation of unwanted crystalline phases, such as sodium titanate (by reacting with TiO2) or impurities,152,177 whereas HF is a very toxic chemical to deal with. Nevertheless, a recent study led to the successful synthesis of hollow spheres with sandwich-type heterostructured shells via SiO2 templating, whereby hydrothermal treatment resulted in crystallization of metal oxide and simultaneous etching of SiO2 in the superhot water.178
Carbon templates can also be produced in situ during hydrothermal synthesis by adding glucose, sucrose or other organics to the metal oxide precursors during the one-pot hydrothermal process. For instance, this method afforded carbon-supported amorphous and crystalline V2O3 microspheres consisting of assembled ultrathin nanosheets of ca. 10 nm thickness by using NH4VO3 and citric acid in a mixed water and ethanediol solvent, followed by calcination in a N2 atmosphere.50 The carbon was generated by carbonization of citric acid and ethanediol, which resulted in its uniform distribution in the resulting composite having 10.6% and 8.1% of carbon in the amorphous and crystalline V2O3 respectively. Hollow spheres and multiple shells of α-Fe2O3, Cr2O3, Co3O4, NiO and ZnO were also successfully prepared by hydrothermal heating of metal chlorides in a solution of fructose at a moderate temperature of 135 °C for 6 h, followed by calcination in air.179 The carbonaceous core of partially dehydrated fructose contains functional groups such as –CO and –OH that can attract positive metal ions forming a metal oxide–carbonaceous composite. In another work,180 composite carbonaceous and Y2O3 spheres produced under hydrothermal conditions were used to generate hollow Y2O3 spheres with 1 to 4 shells by controlled calcination at different heating rates. Other multi-shell structures were successfully produced via this method such as ZnO,119 NiO,104 SnO2110 and CoFe2O4.77 However, the metal oxide shells produced this way sometimes tend to aggregate and may be non-uniform in size.75,110,181
Nevertheless, hard templates could be environmentally friendly through the use of biotemplates such as yeast139 and bacteria.14 The utilization of bacteria (Micrococcus lylae) as a template to synthesize flower-like hierarchical Co3O4 hollow spheres with a uniform size of 1 μm, a high surface area of 149 m2 g−1 and a pore volume of 0.26 cm3 g−1 was achieved via a one-pot reduction/oxidation reaction at room temperature.14 The bacterial surface is naturally covered with carboxyl, ester, amine and hydroxyl groups, which could readily capture cations. The bio-templating method is very attractive as it is facile, scalable and cost effective and could be extended to fabricate other materials and composites.
The choice of the template is important in determining the quality of the produced metal oxide nanoparticles. This has been shown when TiO2 shells were prepared by coating a core with a TiO2 layer followed by adding an outer protective layer around the TiO2.182 The core and the protective layer were made of either SiO2 or a resorcinol–formaldehyde resin-derived carbon, which was then removed by either etching or calcination in air. It was found that better crystallization occurred with the use of resorcinol–formaldehyde resin as a template due to enhanced flexibility and volume shrinkage of the resin during carbonization, while the growth of TiO2 crystals was inhibited by impregnated silicate species.
2.1.1.2 Solid spheres. The solid templates have been mainly used in the synthesis of metal oxide spheres for the creation of large mesopores. In this case, the metal species were infiltrated into mesoporous templates, which upon template removal created a porous spherical structure. Some reported examples are porous ZrO2 spheres obtained by using the EDA-functionalized poly(GMA-co-EGDMA) microspherical templates, Y2O3:Er mesoporous spheres fabricated by using mesoporous SiO2 spheres as templates, and porous WO3 spheres created by employing carbon microsphere templates.53,132,136 In another work, Chen et al.74 prepared multi-core–shell Pd@SiO2@mesoporous–CeO2 by assembling CeO2 nanoparticles around multiple Pd@SiO2 cores via a hydrothermal method. Once the silica was removed with concentrated NaOH, a multi-yolk–shell Pd@mesoporous–CeO2 structure was obtained with a size of 150–200 nm and a BET surface area of ∼103 m2 g−1.
2.1.2.1 Shell structures. Soft templating is also a commonly used method for production of hollow spheres and yolk–shell spheres. In this method the gas–liquid, liquid–liquid or dissolved long chained organic micelles are used as soft templates for the formation and crystallization of metal oxide nanoparticles. Gas–liquid interfaces are in situ formed around gas bubbles that are generated during chemical reactions; for example, CO2 bubbles are formed from urea, NH4HCO3183 and ferric potassium oxalate82 during decomposition at high temperatures or N2 bubbles are formed from the reaction of NH4VO3 with EG.114 Nevertheless, the hollowing of structures on gas bubbles is difficult to prove and it could be more likely that Ostwald ripening is responsible for the generation of hollow spheres.
A liquid–liquid interface occurs between immiscible liquids such as water and oil. Hierarchical mesoporous MnO2 spheres were formed at the droplet interface between CH2Cl2 and H2O.147 Similarly, SnO2 hollow spheres were prepared in a water-in-oil microemulsion of CTAB–hexanol-n-dodecane–methanol–water.151 The calcined SnO2 particles had a surface area of 119 m2 g−1. In another work, a mixture of TBOT and PS in toluene was emulsified in formamide containing the triblock polymer Pluronic P123.184 After removal of toluene and PS spheres via heat treatment, macroporous TiO2 spheres of sizes between 500 nm and 2000 nm were obtained. Finally, a glycerol, water and Fe oxide precursor mixture was hydrothermally heated at 145 °C to obtain mesoporous α-Fe2O3via a quasi-emulsion templating mechanism. The product had a surface area of 103 m2 g−1 when using a glycerol/water volume ratio of 1:7;83 however a different phase of α-FeOOH90 was produced at 120 °C.
A long-chained polymer such as PVP K30 (Mw ∼ 40000) can form micelles in solution with a hydrophobic core and a hydrophilic shell. These micellar structures were used as soft templates to attach vanadium species and upon calcination to form uniform V2O5 hollow spheres with sizes of ∼800 nm115 and with exposed [110] facets, as shown in Fig. 5. During the formation of hollow spheres, VO(acac)2 accumulated on the hydrophilic PVP micelles in the EG solvent to form VO(acac)2@PVP core–shell particles. The C5H8O2 ligand subsequently was replaced by EG to form VEG, which oligomerized via the LaMer process into a tight layer on the surface of micelles. Upon calcination, V2O5 hollow spheres were formed. On the other hand, α-Fe2O3 with multi-shell morphology of ∼3 μm in size and with a surface area and pore volume of 14 m2 g−1 and 0.07 cm3 g−1, respectively, was obtained with the L-histidine amino acid as a template.87 The Fe(OH)3–L-histidine–H+–NO3– complex obtained under hydrothermal conditions underwent size shrinkage and phase separation during the calcination stage to form porous multi-shell particles. In another work,97 a double templating method was employed by decorating soft micelles of the F127 copolymer onto SiO2 spheres to obtain MnO2 hollow particles following hydrothermal treatment of the template with KMnO4. The morphology and surface area could be varied from urchin-like (233 m2 g−1) to flower-like (201 m2 g−1) and non-hierarchical (120 m2 g−1) by varying the F127/SiO2 mass ratio to 0.2, 0.4 and 0.6 respectively.
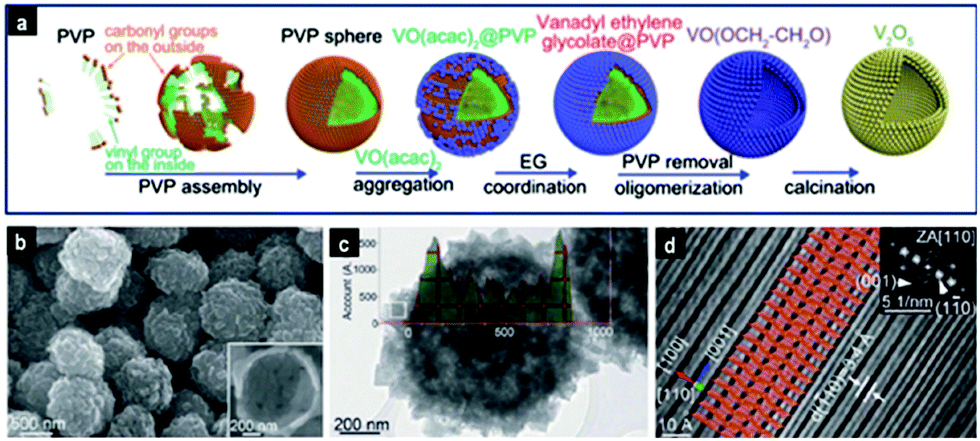 | ||
| Fig. 5 Hierarchical orthorhombic V2O5 hollow nanospheres prepared via soft-templating: (a) schematic illustration of the evolution of V2O5 hollow nanospheres. (b) FESEM and (c) TEM images of V2O5 nanospheres; (d) atomic resolution HRTEM image, from which the interlayer structure of V2O5 was directly observed. Reproduced with permission from ref. 115. Copyright © 2014, Royal Society of Chemistry. | ||
2.1.2.2 Solid spheres. In the synthesis of metal oxide spheres, long chained organics are mostly used as soft templates for surface stabilization of building blocks. These organics accumulate on the nanocrystallites favoring their growth in certain directions or planes, which affects the spatial orientation of crystal nanoparticles and growth of hierarchical structures. Eventually, the removal of these organics by calcination frees hidden pores in the assembled metal oxide structures. Some examples of the organic compounds used as structure directing agents are amino acids such as L-asparagine56 and glycine,41 sugars such as D-glucose monohydrate,125 carbohydrates such as starch185 and polymers such as sodium alginate,33,45 polyethyleneimine2 and also PVP, which has been commonly employed.22–24,47,48 To avoid unnecessary use of polymers and surfactants, alcohols and carboxylic acids have been proved to be very competent capping/structure directing agents by chelating with metal ions. Moreover when mixed together, they react at elevated temperatures to produce esters, which can further influence the growth rate and self-assembly process of nanocrystallites.26,69
Some researchers have used micelles to create mesoporous structures. Luo et al.123 examined a resol-assisted solvent evaporation method in the presence of THF, PEO-b-PS block copolymer and NbCl5. Upon evaporation of THF, the block copolymer aggregated into cylindrical micelles covered by the resol/Nb5+ composite. Upon further evaporation, the micelles tended to bend and aggregate into spherical particles, while resol was acting as a binder. Subsequent pyrolysis and calcination produced Nb2O5 spheres of diameter 0.2–1 μm having uniform mesopores with an average size of 11.4 nm, a high surface area of 131 m2 g−1, and a pore volume of 0.26 cm3 g−1. This solvent evaporation-driven self-assembly was also recently used to create mesoporous TiO2 microspheres with [101] exposed facets from spherical composite micelles consisting of PEO–PPO–PEO and titania oligomers.186 Similarly, surfactants such as Pluronic P123, due to its long hydrophobic chains, can be used to create large mesopores between 12 and 15 nm in the WO3/TiO2 composite spheres.54 Elsewhere, Wang's group187 used a water in oil system in the presence of acrylamide and azobisisobutyronitrile to synthesize hierarchically mesoporous hematite microspheres with high surface area and bimodal structure with mesopores of 2.5 nm and 9 nm.
 | ||
| Fig. 6 Schematic illustration of the formation of metal oxide spheres by a sol–gel process. | ||
Controlled hydrolysis has been used to prepare TiO2 spheres,188–196 core–shell,197 yolk–shell,198,199 and hollow177,183,200–204 particles. A more detailed discussion on the synthesis of TiO2 has been presented elsewhere; the reader is encouraged to refer to a comprehensive review on the synthesis of spherical TiO2 nanostructures by Chen et al.5
Nevertheless, other types of metal alkoxides have also been used to produce the respective oxides, such as vanadium oxiisopropoxide,51,116 vanadium(V) oxytriisopropoxide127 and tin tert-butoxide.151 As shown in Fig. 7, V2O5 mesoporous spheres were synthesized at room temperature by reacting vanadium isopropoxide in a mixture of acetone, pyridine and water at a volume ratio of 983:500:1.127 The average size of the particles was tuned between ∼1 μm and ∼150 nm by increasing the amount of water while maintaining the ratio of pyridine/acetone. The reduction of particle size with increasing water content was attributed to the increased number of sites for nucleation of particles.
 | ||
| Fig. 7 SEM image of the V2O5 porous microspheres; the inset shows the porous structure of a single sphere. Reproduced with permission from ref. 127. Copyright © 2011, Royal Society of Chemistry. | ||
Silica supported Ta2O5 (SiO2–Ta2O5) composite shells were produced by sol–gel synthesis using TEOS, tantalum isopropoxide, CTAB, H2O, NH3 and ethanol.113 NH3 catalyzed the reaction but also assisted in the dissolution of cores at higher temperature. The diameter and shell thickness were tuned by changing the molar ratio of Si:Ta. The BET surface area of the calcined particles increased from 225 to 610 m2 g−1 with increasing Ta content.
The sol–gel method can be also used to manufacture templates for the synthesis of hollow and porous spheres. The one-pot sol–gel polymerization of formamide–resorcinol was employed to create vesicle templates for the synthesis of hollow In2O3 spheres,205 while the porosity in SnO2 spheres was created upon removal of the carbon template from composite Sn–resorcinol–formaldehyde resin particles.206
 | ||
| Fig. 8 Ostwald ripening initiated by dissolution of the middle core (panel a), and localized Ostwald ripening (panel b). | ||
In some cases, due to the localized Ostwald ripening around a dense core, an intermediate yolk–shell architecture is formed (Fig. 8b), as in the case of MoO2@MoO2,102 CeO2@CeO2,70 SnO2@SnO2,106 TiO2@TiO2208 and V2O5@V2O5,117 which subsequently is converted to a multi-shell structure having 2–3 shells after a prolonged hydrothermal process, and finally to hollow spheres. Similarly, double-shell CoO and Co3O4,209 double-shell α-Fe2O3,84 as well as perovskite-type BaZrO3@BaZrO364 and LaFeO3@LaFeO391 structures were obtained. In a different work, Li et al.86 found that the two interfaces created by hydrothermal etching of silica from Fe3O4@SiO2@TiO2 in 1 M NaOH at 150 °C for 24 h allowed for the dissolution of TiO2 crystals and their subsequent growth in an opposite direction to eventually form Fe3O4@TiO2 double-shell spheres with flower-like morphology and with a uniform size of ∼560 nm and a high surface area of 150 m2 g−1. Ostwald ripening involving selective etching of crystals is favored in basic and acidic solutions and it is apparent from Tables 2 and 4 that bases such as urea, KOH, NaOH and amines or acids such as HCl, HNO3 and other organic acids facilitate this process.
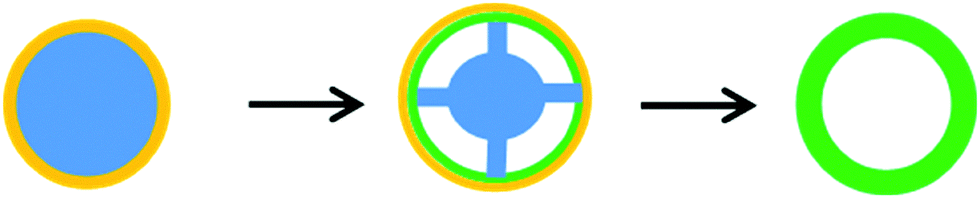 | ||
| Fig. 9 Hollowing by the Kirkendall effect. | ||
The thermal treatment in air of a film decorated with the Cu(II) complex showed that the bulk diffusion of atoms/ions at the Cu–O interface gave Cu2O-rich and CuO-poor spheres at 200 °C.212 As the temperature increases, the Cu from the core moves outwards through the oxide shell to react with oxygen, leaving a hollow space (since the outward diffusion of Cu ions is much faster than the inward diffusion of O ions) until a hollow sphere of pure CuO is formed at 400 °C. Core-in-double-shell hollow NiCo2O4 spheres were obtained by slow annealing of NiCo–glycerate spheres in air due to a combination of the Kirkendall effect and the contraction and adhesion forces during the oxidative degradation of organic species.105 This method can also be extended to the synthesis of ZnCo2O4 and CoMn2O4 with complex interior structures.
The Kirkendall effect can also occur during the hydrothermal reaction stage. The reaction of TiO2 microspheres with a solution of strontium chloride hexahydrate at 180 °C for 6 h generated perovskite-type SrTiO3 hollow spheres having a size of 3–5 μm and a shell thickness of ∼700 nm.112 With the assistance of NaOH, the Ti–O–Ti bonds can be broken to form Ti–O–Na on the surface of the sphere. Then, the Sr2+ ions can react with the sodium titanate to form a thin layer of SrTiO3 shell, separating the inner TiO32− ions from the Sr2+ ions in solution. Hence, the concentration gradient between these two types of ions permitted TiO32− to diffuse out and the Sr2+ ions to diffuse in through the shell, resulting in hollow SrTiO3 spheres. Additionally, composite SnO2–C hollow spheres were prepared by Wu et al.111 under hydrothermal conditions by reacting Sn spheres in a solution of glucose at 180 °C.
Interestingly, a simple solution route by mixing hydrothermal carbon spheres in a solution of KMnO4 at room temperature produced MnO2 spheres of different morphologies.214 Solid MnO2 spheres were produced with 100 ml of 25 g L−1 KMnO4, C@MnO2 yolk–shell spheres were obtained with 100 ml of 2.5 g L−1 KMnO4 and finally MnO2 hollow shell spheres were obtained with 200 ml of 2.5 g L−1 KMnO4 solution. The formation of different morphologies was achieved due to different stages of the Kirkendall effect occurring through the soft surface of the hydrothermally synthesized carbon spheres by varying MnO4− concentration.
The Kirkendall mechanism provides a pathway for the selective etching of the surface-protected metal oxides to produce hollow structures. For instance, the PVP-protected TiO2 solid spheres were selectively etched by fluoride ions to form hollow or yolk–shell TiO2.215 Similarly, it was also reported that NaOH and HCl were used to etch the PVP-protected colloidal Al2O3 and ZnO into hollow spheres, respectively.137
It is apparent that during the Kirkendall mechanism, a solid core acts as a sacrificial template by reacting with its surrounding environment to form different hollow structures. This method could therefore be extremely useful to synthesize a variety of complex hollow compounds and composites from various solid templates.
2.2 Spray method
Spray methods include electrospray ionization216–219 and gas phase processes such as aerosol220–225 and flame or ultrasonic spray pyrolysis (USP).226–229 These methods use high temperatures to evaporate the liquid from the colloidal precursor solution released by the spray nozzle to form solid or hollow spherical structures. Several types of metal oxides such as CeO2 spheres,216 TiO2 spheres,217 WO3 spheres,228 ZnO spheres,221,225 α-Fe2O3 microspheres,230 CuO hollow spheres,222 Mn3O4 hollow spheres,224 WO3 hollow spheres,226 hollow TiO2 and ZrO2 spheres,223 TiO2 core–shell particles,218 Bi2WO6 spheres,229 Fe3O4–carbon composite spheres,220 Li2O–CuO–SnO2 multi-deck cage-type spherical composites219 and α-Fe2O3 multi-shell hollow spheres231 have been produced via the spray method. Some morphologies including porous spheres,221 hollow spheres,222,223 yolk–shell spheres,232 yolk–multi-shell spheres,233 and “ant-cave” spherical structure234 are shown in Fig. 10. | ||
| Fig. 10 Different types of metal oxide structures prepared by the aerosol method: (a) porous sphere (reproduced with permission from ref. 221; Copyright © 2014, Royal Chemical Society); (b) hollow sphere loaded with nanometals (reproduced with permission from ref. 223. Copyright © 2013, Wiley); (c) hollow shell (reproduced with permission from ref. 222. Copyright © 2013, Wiley); (d) yolk–shell sphere (reproduced with permission from ref. 232. Copyright © 2013, Wiley); (e) yolk–multi-shell sphere (reproduced with permission from ref. 233. Copyright © 2013, Wiley); and (f) “ant-cave” spherical structure (reproduced with permission from ref. 234. Copyright © 2013, American Chemical Society). | ||
In the electrospray method, the liquid is evaporated via the potential difference between the nozzle and the metal receptor, while in the gas phase processes, the colloids pass through a flame or horizontal furnace (Fig. 11). Kang and co-workers have published numerous works on spray pyrolysis for the synthesis of hollow spheres, yolk–shell particles, multi-shell spheres and porous microspheres. Hollow WO3 spheres with thin and porous shells were produced by USP using citric acid as the carbon source.226 Multi-shell structures were created from the precursor dissolved in sucrose solution. During decomposition, a dense carbon–metal oxide composite was formed, which upon further heating resulted in contraction and combustion of the carbon to form the multi-shell structures. This method was used to prepare yolk–shell TiO2 and composite multi-component systems (composed of up to 5 components including TiO2, Al2O3, ZrO2, CeO2 and Y2O3),232 double-shell LiNi0.5Mn1.5O4 particles,235 Pd loaded double-shell SnO2 particles233 and double-shell SnO2 spheres.234 Alternatively, the spray-pyrolysis method has been extended to the synthesis of yolk–shell structured metal oxide with 10 kinds of metal components in one step as shown in Fig. 11b.236 The method could also be modified to produce metal sulfide multi-shell spheres. SnO2 yolk–double shell spheres were indeed treated with H2S gas to produce SnS yolk–double shell spheres.237 Another work reported the synthesis of a new structured material named “ant-cave microball”, where polystyrene nanobeads were used as templates to create MoO3–C composite spheres.234 The decomposition of these nanobeads resulted in unique morphology of porous composite spheres with nanochannels, effectively resembling an ant-cave.
 | ||
| Fig. 11 Spray pyrolysis for the formation of yolk–shell-structured LiNi0.5Mn1.5O4 spheres (panel a; reproduced with permission from ref. 235. Copyright © 2013, Royal Chemical Society) and yolk–shell ten-component transition metal oxide powder (panel b; reproduced with permission from ref. 236. Copyright © 2014, Royal Chemical Society). | ||
ZnO spheres were synthesized via an aerosol method using an organometallic precursor dissolved in toluene and Brij 58 as the structure directing agent.221 The ZnO spheres had a BET surface area, crystal size and maximum pore volume of 61 m2 g−1, 8.6 nm and 13 nm, respectively. Al and S could be easily incorporated into the ZnO matrix by adding similar organometals into the precursor solution. Unfortunately, the dopants reduced the crystal size and hence the maximum pore size of the resulting ZnO spheres but this could be counteracted by using the triblock copolymer P123. Recently, very high surface area α-Fe2O3 microspheres with an average size of 560 nm, a BET surface area of 301 m2 g−1 and an average pore size of 2.1 nm were synthesized by USP using Fe(NO3)3 and Na2CO3 as precursors.230 The average particle size could be tuned by changing the concentration of the precursors.
Hollow or macroporous structures could be synthesized by using hard templates,223,228 furnace synthesis at elevated temperatures,225in situ bubble reactions222,224 or non-equilibrium air calcination.231 Au nanorods, Pd nanocubes and Au core/Pd shell nanorods were successfully introduced into hollow TiO2 and ZrO2 spheres by initially embedding these nanometals in PS nanospheres. The PS spheres were dispersed in solution containing metal alkoxides and then sprayed by using N2 through a tube furnace. Subsequently, calcination was performed to remove the PS template, leaving hollow TiO2 and ZrO2 spheres of average diameters of 0.8 μm and 0.6 μm, respectively, and containing nanometals inside the hollow space. In another work,225 ZnO spheres of various shapes were produced simply by changing the furnace temperature. Amorphous porous spheres were obtained between 40 °C and 100 °C, solid spheres at 400 °C, yolk–shell spheres at 600 °C and hollow spheres with different crystallite sizes between 700 °C and 1200 °C. The hollowing process was induced by the Kirkendall effect. An interesting in situ bubble hollowing method was devised by Jian et al.222 to prepare hollow CuO spheres by adding sucrose and H2O2 to Cu(NO3)2 solution. The decomposition of the sucrose into CO2 and H2O (with H2O2 acting as a catalyst) within the aerosol at high temperature inflated the spheres like balloons to produce particles with an average size of ∼85 nm and very thin walls of 5–10 nm. The same strategy was used to prepare hollow Mn3O4 spheres.224 α-Fe2O3 multi-shell hollow spheres231 were synthesized by spray drying a mixture of Fe(III) citrate and sucrose. The obtained Fe(III)–sucrose composite was then calcined in air to remove the carbon template. From the effect of non-equilibrium heating in air, the number of shells could be varied between 2 and 4 by simply changing the Fe(III) citrate/glucose ratio between 0.25 and 1.5.
The spray method is a simple and continuous process with a short residence time (a few seconds) of particles at a high temperature, which produces high purity products and can be easily implemented on an industrial scale. Moreover, other constituents can be included in the precursor solution allowing the preparation of composite or doped metal oxide particles. However, due to the low residence time of the particles at high temperature, further annealing may be required to improve the crystallinity of the products. Furthermore, the method has not yet been able to create hierarchical structures and the surface areas of the particles are often in the low to moderate range.
2.3 Other methods
Besides the methods presented above, there are a variety of other routes for the preparation of colloidal metal oxide structures such as template- and solvent-free methods, ultrasonic irradiation- and microwave-assisted syntheses, electrodeposition, direct printing, and methods involving lasers or taking advantage of gas–liquid diffusion.A template- and solvent-free method was devised by Wang et al.238 for the synthesis of hierarchical metal oxide spheres (HMOS) of TiO2, Fe2O3, ZrO2 and their composites. This method involves grind milling of the metal oxides in the presence of PEG and some water to create a paste, which was spread into a film and annealed. The process generated microspheres via PEG modification and self-assembly and has great potential for large scale production of HMOS.
During ultrasonic irradiation, the formation and collapse of bubbles in the aqueous phase results in localized extremely high temperatures (>5000 K), high pressures (>20 MPa) and very high cooling rates (1010 K s−1), which can supply enough energy to drive the formation of spherical metal oxide structures.144 Various metal oxide spherical particles such as ZnO hollow nanospheres of size ∼80 nm,239 mesoporous NiO hollow spheres,149 MnO2 spheres made of interconnected nanoflakes,21 ZnO spheres with bimodal pores at 25 nm and 180 nm,134 CuO hollow spheres,144 composite Ag2O–MnO2 spheres with Ag2O residing at the end of MnO2 nanowires,19 and WO3 spheres130 were synthesized that way.
The electrodeposition method was used to prepare uniformly distributed 100–500 nm sized MnO2 spheres with a very high surface area of 129 m2 g−1 and mesopores in the range of 5–12 nm, which were composed of randomly oriented nanorod-like structures.240 This synthesis is inexpensive, operates at room temperature and the deposition potential is an extra parameter that can be varied to achieve different morphologies. However, the synthesized oxides have low crystallinity and require further annealing. Other examples of particles prepared via electrodeposition are Y(OH)3 and Y2O3 nanospheres,241 Co3O4 hollow spheres deposited on PS spheres and organized into a close-packed monolayer array242 and SnO2 spheres.243
The microwave-assisted synthesis is analogous to the hydrothermal method but offers a much faster heating rate of the solution. An enormous advantage of this method is the very short time which is reduced to minutes instead of hours (as shown in Tables 1 and 2) as compared to the hydrothermal method. Some examples of metal oxides prepared by this method are Fe3O4 and γ-Fe2O3 hollow spheres,85 NiO spheres41 and hollow spheres244 with very high surface areas reaching 200 m2 g−1, TiO2 spheres195,245 and Bi2O3 spheres.22
Laser irradiation is another powerful and versatile way to obtain CuO246 and ZrO2 spheres.247 This method was even used to make hollow spheres of metals and semiconductors such as Fe, Co, Ni, TiO2, Co3O4, NiO, WO3 and Fe2O3.248 The method offers control over the size of particles and high crystallinity, and therefore no annealing step is required. The high energy dispersed during laser heating was successful in producing single-crystalline rutile TiO2 at room temperature with an average size of 540 nm248 from commercial anatase TiO2 nanoparticles dispersed in acetone. The hollowing was attributed to the Kirkendall effect. The size of spheres could be tuned by controlling the laser beam and irradiation time; however the size of the void space could not be controlled. In another light-driven approach, UV irradiation was used to decompose titanium glycolate spheres into highly uniform mesoporous TiO2 spheres with amorphous structure.249
Some other methods include thermal decomposition of various precursors such as Ni(CH3COO)2·4H2O at 500 °C for 10 h to produce NiO mesoporous spheres,250 synthesis of hollow CuO spheres212 involving direct printing of metal–ion complex ink on a substrate followed by thermal heating, and a gas–liquid diffusion method to prepare Co3O4 hollow spheres by controlled precipitation from a solution of Co(NO3)2 in the presence of a vapor from crushed ammonium carbonate.251
2.4 Methods for controlling the precipitation process
To prevent the uncontrolled precipitation of the metal oxide into large aggregates, the release of reactants in solution can be regulated. The commonly used procedures to control the precipitation process include metal complexation, pH control and controlled release of other reactants. Very low concentrations of metal precursors (usually in mmol amounts) are used as the first preventive measure.| [M(H2O)6]2+ + OH− → [M(H2O)5(OH)]+ + H2O | (1) |
| [M(H2O)5(OH)]+ + OH− → [M(H2O)4(OH)2] + H2O | (2) |
47,48 and W6+52,53 can precipitate from the solution via forced hydrolysis at elevated temperatures without the need of a basic additive. The soluble hydroxyl complexes created by hydrolysis reaction (as per eqn (3)) form precursors for the nucleation of crystallites.| [M(H2O)n]z+ ⇌ [M(H2O)n−y(OH)y](z−y)+ + yH+ | (3) |
To successfully coat a homogeneous and very thin layer of metal oxide on a substrate can be a very challenging task. This subject has been studied by Zhang et al.,138 who reported a simple but interesting method of controlling the amount of OH− ions available for precipitation of metal hydroxides, which could be converted to metal oxides upon calcination. For controlled precipitation, the ionic product of the metal hydroxide precursor (Kmp) has to be equal to or slightly higher than its solubility constant (Ksp). For a metal hydroxide M(OH)n, Kmp can be related to pH as expressed by eqn (4) and (5):
| Kmp = [Mn+aq][OHaq−]n | (4) |
| Kmp = [Mn+aq][Kw × 10pH]n | (5) |
| Kmp ≥ Ksp | (6) |
The deposition of a very thin layer of TiO2via the modified Stöber sol–gel method is normally difficult. However, it was successfully achieved by controlling the hydrolysis rate of the TiO2 precursor by varying the amount of ammonia, which has a crucial role in controlling the reaction kinetics for the formation of TiO2 shells.204 Accordingly, the thickness of the TiO2 shell could be changed from 25 nm to 70 nm by varying the ammonia concentration from 0.25 vol% to 0.4 vol%.
Some researchers used the in situ generated water during chemical reactions for the synthesis of metal oxide particles. Liu et al.254 used the aldol condensation reaction between titanium isopropoxide and acetone to generate water molecules for the formation of TiO2 spheres. Similarly, the water produced during the reaction of vanadium(V) oxytriisopropoxide with acetic acid catalyzed the hydrolysis–condensation process that led to the formation of vanadium oxide spheres.51 Guo et al. synthesized TiO2 spheres by using water generated during esterification reaction of ethanol and acetic acid.255 In another example, TiO2 shells were prepared on hydrated sulfate templates of ZnSO4·7H2O in ethanol.256 The spherical templates of ZnSO4·7H2O in ethanol that were formed acted as sites for deposition of titanium species. Moreover, water present in hydrated crystals acted as a supplier of water molecules for hydrolysis of titania precursors.
3. Effect of synthesis parameters
In this section, we examine the effects of the different experimental parameters such as reaction time, reaction temperature, calcination, pH and reactant and solvent types on the structure and phase of metal oxide spherical particles.3.1 Reaction time
During initial stages of the reaction, nanocrystallites are formed and start aggregating into spheres to reduce their surface energy. The spheres are amorphous in nature but their size, long range ordering and the degree of crystallinity improve with increasing synthesis time.Solution phase reactions at high temperatures favor hollowing via Ostwald ripening as time increases. The hollowing can be initiated at the central core whereby the relatively small crystallites dissolve and migrate to recrystallize on larger crystals on the surface of spheres. This mechanism has been used to obtain hollow spheres of different metal oxides, such as CeO2,67,69,72 CuO,33,78,79,143 CuO/CuO2,80 ZnO,158 MgO,95 ZrO2,62 SnO2,150 NiO,244 TiO2257 and In2O3.94 Some examples are presented in Tables 2 and 4. The shell thickness67 or hollow core size94 could be increased by increasing the reaction time. However, the synthesis time should be carefully adjusted because an excessive reaction time can corrode and collapse the formed hollow structures.33,158
Another hollowing Ostwald ripening mechanism occurs at localized spots within the spheres, at fracture points or low crystal densities. Such cases can result in the formation of yolk–shell particles such as MoO2@MoO2,102 TiO2@TiO2,81,203 CeO2@CeO2,70 γFe2O3@γFe2O3,88 BaZrO3@BaZrO364 and LnFeO3@LnFeO3 (Ln = La, Pr–Tb),91 multi-shell particles such as Fe3O4@TiO2 double-shell structure86 and V2O5 with up to three shells117 or porous In2O3 particles.37 However these structures are, in general, eventually transformed into hollow shells due to further dissolution of the core if reaction time is further increased.
3.2 Reaction temperature
The reaction temperature is an important parameter, which affects the rate of crystal nucleation and therefore the size, morphology and polymorph of metal oxide particles.Temperature affects the crystallization rate, which limits the quantity of crystals formed. This in turn affects the shape32,36,64,79 and size21,32,146 of the synthesized particles. As the nanocrystals continuously form, they aggregate into hierarchical structures. During hydrothermal growth of CuO spheres, the particle shape evolved from irregular CuO nanoparticles at 80 °C to uniform spindle-shaped CuO nanorods with sharp ends at 100 °C, small amounts of nanospheres at 140 °C and large scale monodisperse nanospheres with a wave-like surface at 160 °C.32 On the other hand, hollow flower-like spheres of α-GaOOH changed into microspheres and finally into rods as the temperature increased from 175 °C to 225 °C.36 Therefore, the optimum temperature for achieving a maximum amount of the required structures needs to be experimentally determined. The diameters of solid spheres21 or shells32,146 can also increase with temperature. The ultrasound-assisted solution precipitation of MnO2 produced spherical particles with an average size that could be tuned between 0.4 and 1.28 μm by varying the temperature from 30 °C to 70 °C.21 High temperature was shown to favor the hollowing process of solid spheres via Ostwald ripening.64,79,244 Conversely, the synthesis of hollow structures via bubble82 and emulsion templating83 is not recommended at high temperatures due to reduced stability of the liquid medium.
Changes in temperature can also affect the growth patterns of the crystals, giving rise to different crystal phases. As an example, the Cu2O content in CuO spheres increased with temperature79,80 while α-Fe2O3 was favored at elevated temperatures instead of FeOOH.35,90
3.3 Calcination
Calcination improves the crystallinity of metal oxides by densifying the crystallites and also removes organic impurities bound to the precursor during the hydrothermal process. As a result, the grain size of the crystals increases and the removal of water and CO2 creates additional pores and interspaces. The sum of these effects generally decreases the surface area of particles (although an increase in the surface area has been observed in some cases56) and increases the pore volume.Calcination in an oxygen-containing atmosphere is required to remove carbon templates in the synthesis of porous or hollow spheres. Sometimes, multi-step calcination involving a pyrolysis step followed by oxidative calcination is required. During the synthesis of mesoporous Nb2O5,123 the PEO-b-PS copolymer was used as a pore forming template and resol as a “glue” for the niobia composite following high temperature polymerization. The composite was pyrolyzed first at 350 °C to selectively decompose PEO-b-PS, then at 550 °C to crystallize Nb2O5 crystals without collapsing the polymer skeleton. Finally, the carbon skeleton was removed by calcination in air at 400 °C to obtain mesoporous crystalline Nb2O5 particles.
Some researchers reported the formation of yolk–shell structures of ZnCo2O4,120 SnO2, CeO2 and Tb4O773via simple calcination of metal oxide precursors at 600 °C (using a rate of 5 °C min−1) that were hydrothermally loaded into the pores of carbon spheres. The template removal occurred in two steps with the burning off of the first layer closest to the surface, separating the oxide shell and the composite core. Further calcination removed the carbon template from the core to produce the yolk–shell metal oxide structures.
In some circumstances, the heating rate was found to have some interesting effects on the final structure of calcined particles. A zinc oxide precursor–carbon composite was calcined in air via three methods.119 The first sample was calcined for 3 h in a preheated furnace at 550 °C, the second sample was calcined at a heating rate of 5 °C min−1 to 550 °C and maintained for 3 h and the third sample was calcined at a heating rate of 2 °C min−1 to 550 °C and maintained for 3 h. Single-shell hollow spheres were obtained from the first sample, while double- and triple-shell spheres were formed from the second and third samples, respectively. The formation of multi-shell particles was explained by the occurrence of a temperature gradient between the exterior and interior of the precursors, giving rise to an inside out Ostwald ripening process. Dong et al.170 went a step further by proposing an easy way to control the number of shells and inter-shell spacing in hollow ZnO microspheres by controlling the heating processes (heating rate and final temperature) and precursor concentration on the carbon sphere templates. In another work, a gradual removal of the carbon core from C@V composite microspheres was successfully used to obtain V2O5@V2O5 yolk–shell particles.116 The core was much larger when the C@V sample was annealed at 350 °C at a rate of 3 °C min−1 as compared to a rate of 1 °C min−1, due to the relatively fast removal of the carbon core at the highest rate, leaving behind a V2O5 core formed from the vanadium species that were initially bound to the carbon core. However, annealing at 400 °C at a rate of 1 °C min−1 gave porous single-shell spheres. In a similar fashion, the control of heating rate could generate core-in-double-shell NiCo2O4 spheres105 and multi-shell Y2O3 spheres.180
The calcination temperature can also govern the metal oxide polymorphism. For example, anatase TiO2 forms between 300 °C and 600 °C and the more crystalline rutile phase starts to form at temperatures >600 °C,182,191,258 the perovskite LaFeO3 would not form below an annealing temperature of 750 °C,91 β-Bi2O3 was obtained at 350 °C while α-Bi2O3 was formed at 450 °C,259 the change of phase from γ-Ga2O3 to β-Ga2O3 occurred at T > 700 °C92 and γ-Al2O3 and α-Al2O3 were formed at 900 °C and 1100 °C, respectively.121
3.4 pH
We have already mentioned how pH alters the degree of precipitation and induces Ostwald ripening in the particles. Nevertheless, the solution pH can affect particle growth in various ways, for instance, by controlling adsorption of the precursor on carbon templates,175 changing the degree of ionization of the ligands,67 modifying the electric charge of the surface of nanoparticles253 and altering the degree of complexation of the metal ions in solution.104In the synthesis of Mn2O3, multi-shell structures were formed with the help of carbon sphere templates; the amount of metal cations that can be adsorbed on the carbon spheres was varied by using different solution pH (since high precursor concentration results in its accumulation on the template surface rather than in its infiltration into template pores).175 This strategy was shown to be well suited to alter the zeta potential of carbon spheres and hence their electrostatic interaction with metal ions. As a result, depending on pH, single-, double- or triple-shell Mn2O3 structures were formed upon calcination.
CeO2 particles of different shapes were respectively obtained at pH of 1, 2 and 3.5 in the presence of citric acid used as a ligand, namely, solid spheres, hollow spheres and microplates.67 Due to the three levels of ionization of citric acid (pKa = 3.13, 4.76 and 6.4), different ligands were formed at different pH. At pH below 1, most of the citric acid was in solution as H3Cit without ionization, which coordinated with Ce3+ to give solid microspheres. At pH = 3.5, H2Cit− concentration was higher than H3Cit concentration, giving rise to microplates. At pH = 2, the ligands were a mix of H3Cit and H2Cit−, which resulted in the formation of coordination polymers (metal organic frameworks) in a metastable stage between solid spheres and microplates, compromising the shape into hollow spheres.
The pH of a microemulsion system was found to alter the surface energy and the electric charge distribution on the surface of In(OH)3 nanoparticles as well as the amount of adsorbed CTAB on the different crystal faces.253 As a result, the particles aggregated and grew in different directions to form bundles of nanorods at pH = 5 and spheres at pH = 3.
The hydrothermal synthesis of NiO in the presence of D-glucose was performed at different ammonia concentrations104 to investigate its effect on the shape of final particles. A high concentration of ammonia decreased the concentration of free Ni2+ by forming stable [Ni(NH3)x]2+ complexes in solution and thereby controlling the rate of precipitation of Ni(OH)2. Therefore, aggregated porous structures were obtained at pH < 10.5, and at pH 10.5 a controlled precipitation of the Ni species on the in situ formed carbon spheres followed by calcination afforded hollow microspheres. A further increase of pH to 10.9 reduced the rate of precipitation of nickel hydroxide, allowing formation of carbon layers on the surface of Ni(OH)2 particles, which upon calcination were converted to multi-shell NiO microspheres. Therefore, pH is an important parameter to consider for controlling the morphology of particles.
3.5 Solvents and reactants
The type and composition of solvents is also an important factor that can be used to manipulate the morphology and surface area of metal oxide particles.55,83,90,95,122,251,260A mixture of water and ethanol was used to synthesize multi-shell Co3O4 when using carbon spheres as templates as opposed to single shells when only water was used.171 The effect of ethanol was used to decrease the number of aqua groups coordinated to Co ions, hence to decrease the size of the hydrated Co ions. This strategy was used to control the diffusion of ions through the carbon template, allowing the production of spherical structures, which upon calcination gave multi-shell structures. The same principle was applied to synthesize α-Fe2O3 multi-shell hollow microspheres.172
The quasi-emulsion template was used for the formation of α-Fe2O3 hollow spheres;83 the concentration of the soft template was varied by changing the glycerol/water ratio at 145 °C. As a result, the morphology was evolved from solid flower-like particles consisting of densely packed needle-like subunits to hollow spheres and finally solid spheres made up of nanosheets at the solvent ratios of 1:19, 1:7 and 1:4, respectively. The effect of the EG/water ratio was investigated during hydrothermal synthesis of MgO microspheres.95 It was found that at a high EG volume, spheres were obtained, while at a high water volume, nanoplates were formed.
In another work, the glycol concentration had no effect on the morphology but rather affected the surface area of the synthesized particles.122 The BET surface area of CuO spheres (before calcination) decreased from 157 m2 g−1 to 104 m2 g−1 as the glycol content in water increased.
The solvent ratio can also influence the particle size. The solution phase precipitation at room temperature of V2O5 spheres in a mixed solvent of acetone, pyridine and water gave particles of different sizes (between ∼150 nm and ∼1000 nm) simply by varying the water or pyridine concentration.127
An increase in the ratio of the structure directing agent (SDA) to the metal precursor improved the sphericity of particles such as ZnO,56,185 SnO2,125 and Bi2WO6 spheres24 as well as α-Fe2O3 multi-shell hollow spheres87 because SDA acted as a capping agent and prevented uncontrolled aggregation, although an excess of SDA can be detrimental to the crystallinity118 or development of the structures.24 Moreover, this ratio has also been used to control the size and morphology of particles such as NiO,43 Y2O3,261 ZnO,134 ZnCo2O4@CeO2,133 Bi2O3,23 SnO248 and Co3O4.28 Conversely, the effect of the change in the precursor concentration while keeping the overall ratio constant has been examined in the low temperature precipitation of ZnO hollow spheres using HMT at a 1:1 ratio.159 Hollow fluffy-like spheres having a size of 2–3 μm and a surface area of 138 m2 g−1 were produced; however a decrease in the concentration of the reactants (but keeping the same ratio) gave solid spheres with a net-like surface and a much higher surface area of ∼368 m2 g−1.
The type of reactant can also impact the shape or phase of the final products. CuO/Cu2O hollow spheres were obtained via a hydrothermal method using ethanolamine but CuO flower-like spheres were obtained when ammonia was used.80 In the hydrothermal synthesis of NiO spheres, the β-Ni(OH)2 phase was formed when NaOH or ethanolamine was used while α-Ni(OH)2 was formed with urea instead.43 This study indicates that the phase and structure of the final products depend on the special properties and structure of the reactants used.
4. Applications of spherical metal oxide particles
Metal oxides are used in diverse and multidisciplinary areas ranging from industrially relevant sensing and catalytic uses to biomedical, environmental and energy-related applications. This section shows how the characteristics of different metal oxides affect their respective applications.4.1 Sensing and catalytic applications
Basically sensors operate in two ways. Biosensors make use of immobilized enzymes that can break down a biomolecule to give an electrochemical signal. On the other hand, n-type oxide semiconductors such as SnO2, ZnO, In2O3, TiO2 and WO3 operate by the chemoresistive detection of reducing gases at their surface, involving the diffusion of the analyte gas towards the sensor surface and its electrochemical oxidation with a negatively charged adsorbed oxygen.94 Sensors should have a high sensitivity (at the ppb or ppm level), very fast response (a few seconds), good selectivity towards similar substances, stability, a wide response range, a linear dependency with respect to gas concentration, good repeatability and reusability. These qualities are imparted by the proper choice of sensor material with a microstructure that has been tailored to have specific properties such as a high surface area, porosity, thermal and chemical stability, a short diffusion length for efficient mass transfer, non-toxicity and biocompatibility (for biosensors). From morphology viewpoint, porous or hollow particles have been found to be well suited as sensing materials since their pores allow for rapid mass diffusion and can accommodate numerous active sites, while hollow particles offer short diffusion lengths and an empty structure that can act as a reservoir to continuously replenish active sites with the monitored substance. To improve their performance, some sensors have been loaded with precious metals such as Au,47,56,262 Er93 and Rh,94 made as composites, for example α-Fe2O3/In2O3 hollow spheres263 or as hierarchical structures with high surface area such as nanosheet-assembled WO3 microspheres130 or Co3O4 microspheres composed of large and thin nanoplatelets.27
Kim et al.94 observed that 1.67 atomic% Rh loaded into In2O3 hollow spheres showed an excellent response to 2–100 ppm ethanol, which was up to 180 times higher than in the case of unloaded In2O3 hollow spheres. Also, Rh additionally decreased the optimum operating temperature from 475 °C to 371 °C and enhanced the selectivity to ethanol 15–25 times. The sensing time was very short (0.4 s) but recovery time was relatively large (∼200 s) due to thermal promotion of the surface reactions. A higher Rh loading could decrease the recovery time at the expense of a lower response, which was attributed to the formation of unwanted rhodium and indium phases.
Hierarchical MnO2 spheres consisting of nanorod subunits were synthesized via electrodeposition and loaded with the glucose oxidase enzyme for the mediator-less detection of glucose.240 The particles exhibited a high surface area of 129 m2 g−1 and pore size between 5 and 12 nm, which could easily accommodate the enzyme molecules. The sensor had a high sensitivity of 31.6 μA mM cm−2, a large linear range up to 3.15 mM and a low detection limit of 0.35 μM. Moreover, no interference was observed with species that coexist with glucose in blood, such as ascorbic acid, uric acid and acetaminophen, indicating good selectivity of the sensor.
With the advent of more and more chemicals that are classified as toxic and the implementation of more stringent laws for their detection, the development of more versatile and sensitive sensors will be required in the future and metal oxides can effectively offer a solution.
 | ||
| Fig. 12 (A) Operating principle of a metal oxide support loaded with nanoparticles of a metal catalyst. (B) Catalytic reduction of 4-NP to 4-AP. (a) Reaction equation. (b) Time-dependent absorption spectra of the reaction solution in the presence of the Pd nanocube-embedded hollow mesoporous TiO2 microspheres. (c) Plot of ln[C(t)/C(0)] against the reaction time. The R2 = 0.9956 is the coefficient of determination obtained from the linear fitting. (d) Plot of C(t)/C(0) against the reaction time for five successive cycles of the reduction reaction catalyzed by Pd nanocube-embedded hollow mesoporous ZrO2 microspheres. (e) TEM image of the ZrO2 microspheres after five cycles of the reduction reaction (panel B reproduced with permission from ref. 223. Copyright © 2013, Wiley-VCH). | ||
The environmentally friendly room temperature degradation of HCHO to CO2 and H2O was achieved by using Pt loaded γ-Al2O3 hollow spheres.63 As shown in Fig. 12B, the hierarchical macro–mesoporous structure allowed for high dispersion of Pt and accessible pores facilitated diffusion of reactive molecules and products to/from reaction sites, respectively. In another work,223 the embedded Pd nanoparticles inside the hollow space of mesoporous TiO2 and ZrO2 spheres synthesized via the PS nanospherical template-assisted aerosol method prevented the aggregation of metal nanoparticles and reduced the loss of catalyst during recycling, enhancing the reduction reaction of 4-nitrophenol. Similarly, high CO oxidation at low temperatures was possible via the use of nanosized Au in Au@TiO2 yolk–shell particles due to the unique synergy between Au and TiO2 and the protection of nanometal by the shell.199
Porous ZrO2 microspheres have strong Lewis acidity and poor Brønsted sites but their treatment with phosphoric acid could improve the acidity of the Brønsted sites.136 The synthesized catalyst was tested for the catalytic Friedel–Crafts alkylation of indoles with chalcones, giving 98% yield in 6 h and showed a negligible loss in its activity even after 22 reuses. Similarly, Friedel–Crafts reactions were carried out with mesoporous Nb2O5 spheres40 and Fe3O4@TiO2 double-shell yolk–shell particles86 once the Lewis and Brønsted sites were improved by acidification with sulfuric acid and hydrothermal treatment in basic solution, respectively.
CeO2 on the other hand shows strong oxygen storage and release capacity via facile conversion between Ce3+ and Ce4+. This makes ceria a model catalyst for oxidation reactions. Hollow CeO2 spheres converted 43% CO at 295 °C,70 while 10% Cu doped CeO2 spheres showed >98% CO conversion at 210 °C68 and a complete oxidation was achieved on the core–shell ZnCo2O4@CeO2 particles at less than 200 °C.133 However, loading CeO2 with noble nanometals such as Pd,74 Ag26 and Au69 drastically improved its performance at much lower temperatures. For instance, Pd@CeO2 multi-yolk–shell particles achieved 100% CO oxidation at 110 °C.74 The catalyst also performed excellently in the case of aerobic oxidation of cinnamyl alcohol into cinnamaldehyde with >99.9 conversion after 1.5 h. In another case, CeO2 doped into MnO2 hollow spheres could catalytically oxidize benzene with 90% oxidation at 252 °C and completely oxidize it at 340 °C.99
Mesoporous NiO spheres and flowers having a size of 500 nm and mesopores in the range of 10–30 nm showed high catalytic activity for the transformation of toxic phenolic pollutants.2 Furthermore, the NiO nanostructures with large scale nanocrystal domains and well-shaped morphologies imparted magnetic properties to an otherwise antiferromagnetic system due to the quantum confinement effect, allowing easy magnet-assisted separation.
Other types of metal oxides and their combinations have been investigated for several other reactions. Perovskite LnFeO3 (Ln = La, Pr–Tb) hollow spheres showed an excellent catalytic performance for NO + CO reaction at high temperatures between 200 °C and 500 °C due to their outstanding thermal and chemical stability.91 MgO spheres and flowers performed much better than bulk MgO in the Claisen condensation of benzaldehyde and acetophenone under solvent-free conditions.265 Co3O4 hollow spheres were used for methane conversion141 and H2O2 oxidation.142 Fe3O4@SiO2@void@TiO2 particles were employed as a catalyst for epoxidation of styrene,198 giving a high conversion and selectivity of 90.2% and 88.5% respectively. In the latter work, the Fe3O4 core was well protected by the SiO2 layer and the void space was successfully loaded with Au nanometals, which respectively provided magnetic separability and high reactivity and reusability.
4.2 Biomedical applications
Some semiconductors were shown to have photoluminescence capabilities. This arises from oxygen vacancy defects that can accommodate photogenerated electrons, which release photons upon recombination with free photogenerated holes. Some examples of semiconductors that have been found to possess photoluminescence properties are perovskite BaZrO3 hollow spheres,64 In2O3 spheres37,253 and γ-Ga2O3 hollow nanoflowers,92 with the latter also showing excellent solar blind detection performance.
 | ||
| Fig. 13 (A) Schematic illustration of loading and unloading a drug to/from the PEG-coated hollow metal oxide particles. (B) (a) HRSEM image, (b) TEM image of hematite HNS, (c) HRSEM image, (d) TEM image of magnetite HNS, (e) HRTEM images of hematite HNS showing lattice fringes of the (006) plane, and (f) magnetite HNS showing lattice fringes of the (311) plane. (C) (a) Dynamics of extracellular dissolution of total Fe throughout the course of microbial growth in the absence and presence of bacteria and (b) intracellular content of Fe in the bacteria after 4 h incubation with nanoparticles of 500 μg ml−1 concentration in LB broth (the inset illustrates the enhancement of the antimicrobial process). (Panels B and C reproduced with permission from ref. 269. Copyright © 2016, Wiley-VCH.) | ||
Nor et al. reported hematite hollow nanospheres (HNS) synthesized by the hard templating method; reduction of hematite HNS by H2 led to magnetite HNS (Fig. 13B). It was reported that magnetite HNS (hematite HNS and C-magnetite) shows superior antibacterial performance towards both E. coli and S. epidermidis (Fig. 13C). In comparison to hematite HNS, magnetite HNS allows for a multiple-fold increase in the generated soluble iron ions, showing that the control over both the composition and nanostructure is crucial for tuning the antimicrobial activity of iron oxides.
4.3 Environmental applications
The morphology of the material is of utmost importance for designing a photocatalyst. A large surface area affords numerous reaction sites and a high crystallinity favors photocatalytic activity due to effective charge migration. Indeed, the annealing process improves the crystallinity of the material but at the expense of the total surface area. Therefore, a compromise needs to be reached for the best crystal size and the largest possible surface area for optimum photocatalytic performance. SnO2 spheres calcined at 150 °C had the best activity under UV light for the degradation of methyl orange as compared to uncalcined samples and samples calcined at 200 °C, 300 °C and 500 °C respectively.125 Similarly, Bi2O3 spheres calcined at 350 °C degraded 99% of methyl orange under visible light after 3.5 hours as compared to only 25% degradation for samples calcined at 500 °C. It was also shown that hollow structures performed better than solid ones due to multiple light reflection and absorption within the structure cavities (Fig. 14A) and stability of the photogenerated charge carriers.
 | ||
| Fig. 14 (A) Schematic illustration of multiple reflection and absorption of UV light in a hollow shell photocatalyst. (B) SEM image and schematic illustration of photoexcitation electrons and holes migrating to the opposite sides of the interface in hollow ZnO spheres (panel B reproduced with permission from ref. 225. Copyright © 2014, American Chemical Society). | ||
Dilger et al.225 studied the effect of nanoarchitecture on the photoconductivity of ZnO particles (Fig. 14B). They found that the time needed for the current from photogenerated charge carriers to drop down to 10% of its maximum value (T0.1) was <60 s for bulk and porous ZnO, whereas for yolk–shell and hollow spheres, T0.1 was substantially higher reaching 521 s and 1150 s, respectively. This structural effect is duly reflected by photocatalytic experiments using various materials. Porous CeO2 hollow spheres degraded 92% of gas phase acetaldehyde within 24 h, four times better than in the case of CeO2 nanoparticles, despite the fact that the latter had a smaller band gap (2.88 eV as compared to 3.01 eV for hollow CeO2 spheres) and better crystallinity.71 WO3 hollow spheres had better performance than flower-like spheres, with first order kinetics of 0.056 min−1 as compared to 0.0151 min−1 for the removal of rhodamine B under UV light.129 While both structures possessed similar surface areas (13 m2 g−1 and 16 m2 g−1 for the microspheres and hollow spheres, respectively), the improvement in the catalytic activity of the hollow spheres was attributed to the large textural porosity between 3.3 nm and 89.6 nm. Increasing the number of shells also resulted in better photoactivity. For example, ZnO hollow spheres with 1, 2 and 3 shells degraded 84.1%, 88.3% and 99% of rhodamine B respectively,119 while the required time for complete degradation of the same pollutant under visible light was 1.5 h for WO3 with multiple shells as compared to 2.5 h for single shell particles.153
The crystal phase, due to its geometric structure, is another important aspect that is related to photoactivity. Hou et al.273 showed that the β-phase of Ga2O3 exhibited superior photocatalytic activity towards the gas phase degradation of aromatic compounds as compared to α- and γ-Ga2O3. Also, studies of Fe2O3 as catalyst showed that α-Fe2O3 spheres could completely degrade rhodamine 6G after 1 h under visible light irradiation,34 while F doped γ-Fe2O3 needed UV light to degrade rhodamine B.88
TiO2 has been the most extensively studied semiconductor for photocatalytic purposes due to its abundance, low cost, non-toxicity and high photoactivity accompanied by a high oxidation and reduction potential. TiO2 can oxidize a large variety of organic compounds in water or air182,189,193,200,203,254–257,274–278 and has also been successful for reducing metal ions such as Cr6+193 and for H2 production.254 The major factors affecting the degree of photoactivity are surface area, porosity, crystallinity and morphology. A high crystallinity is desired rather than a large surface area to increase the separation between the photogenerated electrons and holes, and reduce their instantaneous recombination. However, the rutile phase despite its higher crystallinity and lower band gap energy has a lower activity than the anatase phase because of the lower surface area of the rutile particles. As a result, the surface area and the crystal size of particles have to be finely tuned due to their inverse role in photocatalysis, which is the reason why a wide discrepancy has been observed for the optimum calcination temperatures of the synthesized TiO2 particles (between 450 °C and 800 °C for grain sizes varying between ca. 12 and 30 nm), depending on their sizes, morphologies and treatment methods.177,182,200,278 The activity of TiO2 under visible light has been enhanced by doping with Pt,254 CO32−,254 N,193,279 metal oxides,256 CdS184 and even trace organics from the solvent or organic precursor,255 since the inclusion of a dopant within the TiO2 matrix provides a sink for photogenerated electrons, thereby reducing the degree of electron–hole recombination. Similar to other semiconductors discussed in this section, the hollow shell morphology TiO2 performed better than its other counterparts203 because of multi-reflection of light.
95 and 569.7 mg g−1 respectively.96 ZrO2 solid and hollow spheres,62 CuO spheres and Cu2O hollow spheres,32 NiO spheres,42,43 perovskite BaZrO3 hollow spheres64 and CeO2 hollow spheres70 were used for the removal of anionic dyes, while urchin-like α-FeOOH hollow spheres90 had high removal capacities towards both organic dyes and heavy metal ions. MnO2 spheres with a hierarchical dandelion-like surface structure, a high surface area of 163 m2 g−1 and main mesopores of about 65 nm showed both oxidation and adsorption properties over lethal As(III) species.17 The As(III) species could be effectively oxidized by MnO2 particles to the less toxic As(V) species, followed by their adsorption.
4.4 Energy storage and conversion applications
 | ||
| Fig. 15 (A) Charging and discharging of Li ions in a yolk–shell type Li-ion battery; (B) schematic illustration of the formation process of yolk–shell MoO2 microspheres; (C) (a) discharge–charge voltage profiles of the MoO2 electrode at different current densities of 50, 500 and 2000 mA g−1; and (b) cycling performance of the as-synthesized MoO2 microspheres at 50, 500 and 2000 mA g−1. (Panels B and C reproduced with permission from ref. 102. Copyright © 2013, Royal Chemical Society.) | ||
V2O5 microspheres synthesized by an additive-free hydrothermal method involving a hierarchical assembly of nanoporous fibers possessed a moderate surface area of 42 m2 g−1.51 The structure endowed the material with shorter diffusion pathways for easier Li and electron transport hence enhancing electrochemical performance. The microspheres displayed a very stable capacity retention of 130 mA h g−1 over 100 cycles at a current rate of 0.5C and showed an excellent rate capability with a capacity of 105 mA h g−1, even at a high rate of 30C. Also, TiO2 anatase spheres consisting of ultrathin nanosheets formed via crystals growing in the [001] direction188 performed well at low and high rates. In another work,250 NiO spheres with loosely connected crystals, a surface area of 30 m2 g−1 and pore size distribution in the range of 10–60 nm provided suitable pathways for efficient transport of electrolyte ions, large surface to volume ratios, and good structural stability to deliver a reversible capacity of 800 mA h g−1 after 100 cycles at a current density of 500 mA g−1. The material was also tolerant to various charge and discharge currents, indicating high rate performance for high power applications. Aside from the morphology, crystal polymorphs can also be a deciding factor in the fabrication of metal oxide-based Li-ion batteries. It was shown that the anatase phase exhibited higher capacity than either brookite or rutile due to the more favorable Li ion insertion mechanism.188,191,281,282
Hollow structures have also attracted great attention due to the increased contact area between the electrode material and electrolyte as well as their mechanical flexibility. Hierarchical orthorhombic V2O5 hollow spheres showed good performance in Na-ion batteries (an alternative to Li-ion batteries) due to the predominantly exposed [110] crystal planes, which provided channels for easy Na+ insertion and extraction as well as high tolerance to the deformation imparted by voids in the shells of hollow spheres.115
Mn3O4 hollow spheres synthesized by an aerosol method featured a high surface area of 96 m2 g−1 and thin shells of 5–10 nm. These hollow spheres exhibited good stability with a high capacity retention of 980 mA h g−1 over 140 cycles and exceptional rate capability by retaining a capacity of 300 mA h g−1 at an ultra-high current density of 10000 mA g−1.224 These values represent the best electrochemical performance for Mn3O4 anode materials to date and were attributed to the unique thin wall hollow structure, which provided considerably reduced diffusion paths for electrons and Li ions. On the other hand, multi-shell hollow α-Fe2O3 showed a superior capability of 1203 mA h g−1 at a current density of 100 mA g−1 after 128 cycles and an excellent cycling stability of 870 mA h g−1 at a current density of 400 mA g−1 after 300 cycles due to the material's hierarchical porosity and structure, which assured shorter diffusion pathways for efficient transport of electrons and Li ions.87 An excellent performance of multi-shell hollow α-Fe2O3 spheres was indeed recently confirmed by another work,172 reporting up to 1702 mA h g−1 at a current density of 50 mA g−1, which was due to the enhanced volumetric capacity of the structure that allows for maximum lithium storage. Recently, multi-shell V2O5 hollow microspheres, synthesized by a simple method involving adsorption of anions on carbon templates, were found to exhibit an exceptionally high specific capacity of 447.9 mA h g−1 (at a high current density of 1000 mA g−1), exceptional rate capability and cycling stability, due to the ample charge storage sites, short transport paths and good structural stability of the material.176 These attributes also belong to yolk–shell structures such as MoO2@MoO2102 and V2O5@V2O5.116,117 The superiority of the yolk–shell structure over solid spheres was demonstrated for the V2O5@V2O5 yolk–shell particles retaining 89% of its specific discharge capacity after 50 cycles as compared to around 60% only for solid V2O5 spheres.117
Composites or multi-component structures are attractive for studies due to the synergetic effect between the different components. Carbon-supported amorphous and crystalline V2O3 microspheres both showed excellent high rate and electrochemical performance due to the uniform distribution of partly graphitized carbon within the framework and also due to the stable structure of spheres, which assured a low charge resistance, fast electronic transport, a large surface area and excellent stability.50 The amorphous spheres showed 95% retention in the discharge capacity after 7000 cycles at a high current density of 2 A g−1, while the crystalline ones retained 98% after 9000 cycles. Ant-cave structured MoO3–C composite microspheres234 synthesized by USP exhibited high initial discharge and charge capacities of 1109 and 724 mA h g−1 and still delivered a discharge capacity of 733 mA h g−1 after 300 cycles. The high performance was attributed to the combination of ant-cave channel structure and conductivity of the carbon in the composite. A yolk–shell structure of CuO@NiO spheres exhibited much higher capacity than the theoretical value of 1061 mA h g−1 after 200 cycles due to the unique multilayer hollow structure, which provided a large electrochemically active surface, more active sites for Li ion storage and facilitated Li diffusion.283 In another example, the carbon coated triple-shell hollow spheres of CoMn2O4 possessed a high specific capacity of 726.7 mA h g−1 and nearly 100% capacity retention after 200 cycles.168
Numerous other metal oxides and composites of various shapes and accommodating space for lithium have been successfully used in Li-ion batteries. The list of metal oxides used in Li-ion batteries is quite long and includes the following: CuO spheres and hollow spheres,18,78,79,122 Fe2O3 hollow spheres,83,88 α-Fe2O3 multi-shell hollow spheres,172,231 Co3O4 spheres,28 Co3O4 multiple shells,171 Gd2O3 hollow spheres,145 NiO spheres and hollow spheres,41,103,149,284 TiO2 hollow spheres,285 TiO2 yolk–shell spheres,281 multi-shell TiO2 hollow microspheres,174 Fe2O3@TiO2 core–shell spheres,204 V2O5 spheres and hollow spheres,114,127,128 SnO2 spheres and hollow spheres,106,109,206,264 SnO2 multi-shell spheres,110 Li2O–CuO–SnO2 multi-deck cage spheres,219 CoFe2O4 and other metal ferrite spheres,29,77 core-in-double-shell NiCo2O4 particles,105 multi-shell LiMn2O4 hollow microspheres,286 Cr2O3–C core–shell spheres,31 SnO2@C spheres,49 SnO2/C composite hollow spheres108,111 and Fe3O4–C composite spheres.220
The surface area of MnO2 hollow spheres prepared by a double templating method was varied by changing the ratio of the F127 surfactant and SiO2 spheres used as soft and hard templates, respectively.97 The urchin-like hollow spheres showed the best capacitance of 266.6 F g−1 within the potential range of 1 V at a current density of 0.1 A g−1 due to their high surface area of 233 m2 g−1 relative to the other hollow structures. Co3O4 hollow spheres with thin shells of 130 nm, a moderate surface area of 60 m2 g−1 and mesopores centered at 7.8 nm, prepared hydrothermally using sucrose as a precursor for carbon sphere templates, performed well giving a specific capacitance of 470 F g−1 at 1 A g−1 with no obvious capacitance decrease observed over 1000 cycles.75 On the other hand, bacteria-templated Co3O4 hollow spheres prepared by a one-pot mineralization method at room temperature featured a similar average pore size of 7.7 nm but a larger surface area of 149 m2 g−1 due to their fluffy-like surface.14 These particles showed a high capacitance of 214 F g−1 at 2 A g−1, a Coulombic efficiency averaging over 95% and excellent cycling stability that showed a capacitance retention of about 95% after 4000 cycles. Mn2O3 triple-shell hollow microspheres with thin porous shells and a large pore volume of 0.52 cm3 g−1 but a moderate surface area of 37 m2 g−1 showed a record high specific capacitance up to 1651 F g−1 at 0.5 A g−1, an excellent rate capability of 1422 F g−1 at 10 A g−1 and a cycling stability retention of 92% after 2000 cycles.175 These findings show the importance of porosity, surface texture and high surface area for the performance of metal oxide particles as supercapacitors.
However, the particle morphology may surpass the effect of surface area in some cases, as it was shown in the case of NiO spheres prepared by a hydrothermal method.43 It was found that NiO spheres made of ultrathin nanowires performed much better than urchin-like spheres with nearly 100% capacity retention after 200 cycles at a current density of 10 A g−1, despite the higher surface area of the latter (243 m2 g−1vs. 215 m2 g−1). The high capacity was attributed to the bimodal pore size distribution of the ultrathin nanowire-assembled spheres at 3.2 nm and 8.9 nm as opposed to the monomodal 4 nm pores in the urchin-like spheres as well as to their network-like surface texture, which assured efficient diffusion paths for OH− ions and greatly enhanced intercalation of the electrolyte ions, ensuring sufficient Faradic reactions.
Doped or composite structures can also perform well as supercapacitors as it was shown for Fe3O4 doped MnO2 microspheres,21 Ag2O–MnO2 composite spheres19 and MnO2/C composite spheres.100 Core–shell C@MnO2 had a high specific capacitance of 583 F g−1 at a current density of 1 A g−1 in 0.1 M Na2SO4 electrolyte,214 which was attributed to the inner graphitized carbon core coupled with porous interconnected MnO2 nanorods for enhanced electrolyte accessibility, short ion diffusion length and charge transfer pathways. Ternary metal oxides with two different metal cations also exhibited high electrochemical performance as it was demonstrated for core-in-double-shell hollow NiCo2O4 spheres105 with a complex interior, porous shells and consisting of small nanocrystalline particles. This material delivered high pseudocapacitance values of 1141, 1048, 965, 862 and 784 F g−1 at current densities of 1, 2, 5, 10 and 15 A g−1. Furthermore, after 4000 cycles at 5 A g−1, 94.7% of the specific capacitance was retained, proving the high stability of the spheres.
Other examples of metal oxides that have been used as supercapacitors are NiO spheres and hollow spheres,41,124,148,244,287 La doped NiO spheres,44 Co3O4 hollow spheres242 and MnO2 hollow spheres.98
To address this issue, an additional layer of metal oxide particles has been deposited to enhance the light to current capability of DSSCs. The engineered particles have increased dye loading, interconnected crystals to improve the current transport, improved crystallization to intensify electron–hole separation and more importantly, enhanced back-scattering of light of longer wavelength (visible and near infrared region) onto the primary layer of DSSCs, which augments absorption of light, as shown in Fig. 16. There are numerous reports on the use of TiO2 particles of varying architecture in DSSCs such as spheres,184,190,192,195,217,245,288–290 hollow spheres,227,248,291 and yolk–shell structures.208
 | ||
| Fig. 16 Scattering of long wavelength light (red light) due to the layer of larger particles on the top of TiO2 film in a DSSC. | ||
Macroporous TiO2 spheres were prepared by an emulsion templating method using PS particles as templates for the macropores,184 and were formed via calcination of the PS–TiO2 composite spheres. These spheres had size in the range of 500–2000 nm, pore size in the range of 200–300 nm, a surface area of 76 m2 g−1 and were composed of crystals with sizes of 6–8 nm. The high surface area assured a high amount of dye loading, which was about 1.5 times higher than that adsorbed on a commercial nano-TiO2, and the proper size of spheres improved the scattering of light in the wavelength range of 500–800 nm. Hence, the geometry of particles assured a significant improvement in light scattering, and increased dye loading as well as interconnected pores (fewer grain boundaries) enhanced light to current efficiency as compared to conventional TiO2 materials.
TiO2 spheres of size 2.1 μm consisting of nanorods were prepared by a hydrothermal method.289 The large surface area (64 m2 g−1) and size of the particles resulted in a high power conversion efficiency of 10.34%. The high performance was also due to the crystal size and structure, which increased the electron transport rate and slowed down the recombination rate of electrons and holes. On the other hand, TiO2 mesoporous microspheres of size ca. 800 nm, created by an evaporation-driven self-assembly method,186 possessed a large surface area of 112 m2 g−1, a large pore volume of 0.164 cm3 g−1, and highly crystalline walls with [101] exposed facets. These attributes resulted in DSSCs with high energy conversion efficiency of up to 12.1%.
The wavelength of the scattered light is closely related to the size of particles. Xu et al.202 showed that TiO2 particles with a size of 380 nm exhibited strong resonance with light of wavelength 366 nm. However, Yu et al.288 studied the effect of light scattering by the particles with sizes in the range of 260–800 nm and found that particles with a size of 450 nm showed the highest scattering of light in the range 600–750 nm and therefore the best photon to current efficiency; this was attributed to the size uniformity and long range ordering when the particles were applied as a light scattering layer in DSSCs.
Other semiconductors such as SnO2,126,243 quintuple shell SnO2173 and Nb2O539 have also been used in DSSCs. However, the power conversion efficiencies did not reach those of TiO2-based electrodes.
5. Conclusions and perspectives
We have reviewed the synthesis methods and potential applications of metal oxide spherical nanoparticles. These particles were engineered to have a high surface area to volume ratio, a high pore volume, and high mechanical strength, which endorse them for a variety of applications in the catalytic, environmental, biomedical and energy fields. They can show synergistic interactions with noble metals when used as supports, making them potentially attractive catalysts. Furthermore, for specific crystal size, the quantum effects can impart semiconducting or magnetic properties to the particles, which can be beneficial in the field of photocatalysis and separation. The most common methods currently used for the synthesis of metal oxide spheres are the high temperature hydrothermal and low temperature solution precipitation methods. However, these methods are time consuming and suitable for batch production only. Thus, the emerging methods such as aerosol and spray drying or spray pyrolysis methods are well suited for continuous large scale production, and can be used to synthesize porous spheres and shells, allowing high loading of noble metals. However, further studies are required toward controlling the uniformity and size of spheres and the thickness of shells. For multi-shell hollow spheres, the finely controlled synthesis of concentric hollow spheres is still a challenge and more research in this direction is needed. Further studies on the design and synthesis of metal oxide structures, especially core–shell and yolk–shell particles with cores and shells of tailored surface properties, porosity and chemical composition for advanced catalytic, biomedical and energy-related applications, are highly desirable. Since chemistry of metal oxide nanoparticles is a rapidly growing research area, new discoveries in the synthesis and applications of these materials are anticipated.In perspective, synergetic interactions of metal oxide composites need to be further investigated to possibly avoid the use of precious metals as active agents in catalysis, and enhance the energy storage capacities of the materials used in the energy field. Multiple functionalization within the same structure could generate catalysts that can cope with tandem reactions, hence avoiding the use of two or more catalysts. Furthermore, more work on the synthesis of hollow metal oxide spheres in the nanometer regime needs to be carried out for use as nanoreactors in colloidal solutions. The new generation of porous non-silica metal oxide submicrospheres with precisely controlled functionality can be envisioned to be created by taking advantage of prosperous nanochemistry synthesis methods under mild conditions. A further development of the computationally assisted design and fabrication of complex porous non-silica metal oxide particles is needed for structure prediction and reduction of the number of synthesized samples. In the past decade, there has been a remarkable evolution in porous non-silica metal oxide submicrospheres with unique properties as platforms for emerging applications, which envisages a bright and exciting future for these unique materials.
Abbreviations
| BET | Brunauer–Emmett–Teller |
| CS | Carbon spheres |
| CTAB | Cetyltrimethylammonium bromide |
| DEX | Dexamethasone |
| DMF | N,N-Dimethylformamide |
| EDA | Ethylenediamine |
| EG | Ethylene glycol |
| ERY | Erythromycin |
| GMA-co-EGDMA | Glycidyl methacrylate-co-ethylene glycol dimethacrylate |
| HMT | Hexamethylenetetramine |
| IBU | Ibuprofen |
| IUPAC | International Union of Pure and Applied Chemistry |
| MEA | Monoethanolamine |
| PEG | Poly-ethylene glycol |
| PEO-b-PS | Polyethylene oxide-block-polystyrene |
| PEO–PPO–PEO | Poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) |
| PS | Polystyrene |
| PVP | Polyvinyl pyrrolidone |
| SDBS | Sodium dodecylbenzenesulfonate |
| TBOT | Titanium butoxide |
| TEA | Triethanolamine |
| TEOS | Tetraethyl orthosilicate |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
Acknowledgements
The authors would like to thank Prof. Shaobin Wang for fruitful discussions.Notes and references
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- M. Khairy, S. A. El-Safty, M. Ismael and H. Kawarada, Appl. Catal., B, 2012, 127, 1–10 CrossRef CAS.
- H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- P. Zhang, A. Li and J. Gong, Particuology, 2015, 22, 13–23 CrossRef CAS.
- D. Chen and R. A. Caruso, Adv. Funct. Mater., 2013, 23, 1356–1374 CrossRef CAS.
- W. Wu, Z. Wu, T. Yu, C. Jiang and W.-S. Kim, Sci. Technol. Adv. Mater., 2015, 16 DOI:10.1088/1468-6996/16/2/023501.
- J.-H. Lee, Sens. Actuators, B, 2009, 140, 319–336 CrossRef CAS.
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 CAS.
- Z. Wang and L. Zhou, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- J. Hu, M. Chen, X. Fang and L. Wu, Chem. Soc. Rev., 2011, 40, 5472–5491 RSC.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- K. Byrappa and T. Adschiri, Prog. Cryst. Growth Charact. Mater., 2007, 53, 117–166 CrossRef CAS.
- H.-W. Shim, A.-H. Lim, J.-C. Kim, E. Jang, S.-D. Seo, G.-H. Lee, T. D. Kim and D.-W. Kim, Sci. Rep., 2013, 3, 2325, DOI:10.1038/srep02325.
- X. Zou, H. Fan, Y. Tian, M. Zhang and X. Yan, Dalton Trans., 2015, 44, 7811–7821 RSC.
- H. Cao, A. Yang, H. Li, L. Wang, S. Li, J. Kong, X. Bao and R. Yang, Sens. Actuators, B, 2015, 214, 169–173 CrossRef CAS.
- T. Zhang and D. D. Sun, Chem. Eng. J., 2013, 225, 271–279 CrossRef CAS.
- Z. Yuan, Y. Wang and Y. Qian, RSC Adv., 2012, 2, 8602–8605 RSC.
- Y. Dai, S. Tang, S. Vongehr and X. Meng, ACS Sustainable Chem. Eng., 2014, 2, 692–698 CrossRef CAS.
- T. Lin, L. Yu, M. Sun, G. Cheng, B. Lan and Z. Fu, Chem. Eng. J., 2016, 286, 114–121 CrossRef CAS.
- J. Zhu, S. Tang, H. Xie, Y. Dai and X. Meng, ACS Appl. Mater. Interfaces, 2014, 6, 17637–17646 CAS.
- M.-G. Ma, J.-F. Zhu, R.-C. Sun and Y.-J. Zhu, Mater. Lett., 2010, 64, 1524–1527 CrossRef CAS.
- F. Qin, H. Zhao, G. Li, H. Yang, J. Li, R. Wang, Y. Liu, J. Hu, H. Sun and R. Chen, Nanoscale, 2014, 6, 5402–5409 RSC.
- Y. Li, J. Liu, X. Huang and G. Li, Cryst. Growth Des., 2007, 7, 1350–1355 CAS.
- D. Ma, S. Huang, W. Chen, S. Hu, F. Shi and K. Fan, J. Phys. Chem. C, 2009, 113, 4369–4374 CAS.
- X. Liang, J. Xiao, B. Chen and Y. Li, Inorg. Chem., 2010, 49, 8188–8190 CrossRef CAS PubMed.
- C. Sun, S. Rajasekhara, Y. Chen and J. B. Goodenough, Chem. Commun., 2011, 47, 12852–12854 RSC.
- W. Hao, S. Chen, Y. Cai, L. Zhang, Z. Li and S. Zhang, J. Mater. Chem. A, 2014, 2, 13801–13804 CAS.
- S. Yoon, J. Appl. Electrochem., 2014, 44, 1069–1074 CrossRef CAS.
- H. Ma, Y. Xu, Z. Rong, X. Cheng, S. Gao, X. Zhang, H. Zhao and L. Huo, Sens. Actuators, B, 2012, 174, 325–331 CrossRef CAS.
- L.-Y. Jiang, S. Xin, X.-L. Wu, H. Li, Y.-G. Guo and L.-J. Wan, J. Mater. Chem., 2010, 20, 7565–7569 RSC.
- S. Yang, S. Zhang, H. Wang, H. Yu, Y. Fang and F. Peng, RSC Adv., 2014, 4, 43024–43028 RSC.
- R. Liu, J. Yin, W. Du, F. Gao, Y. Fan and Q. Lu, Eur. J. Inorg. Chem., 2013, 1358–1362 CrossRef CAS.
- G. Liu, Q. Deng, H. Wang, D. H. Ng, M. Kong, W. Cai and G. Wang, J. Mater. Chem., 2012, 22, 9704–9713 RSC.
- C. Wang, Y. Cui and K. Tang, Nanoscale Res. Lett., 2013, 8, 1–4 CrossRef PubMed.
- M. Muruganandham, R. Amutha, M. S. A. Wahed, B. Ahmmad, Y. Kuroda, R. P. Suri, J. J. Wu and M. E. Sillanpaa, J. Phys. Chem. C, 2011, 116, 44–53 Search PubMed.
- H. Yang, L. Liu, H. Liang, J. Wei and Y. Yang, CrystEngComm, 2011, 13, 5011–5016 RSC.
- B. Tao, Y. Zhang, D. Han, Y. Li and Z. Yan, J. Mater. Chem. A, 2014, 2, 5455–5461 CAS.
- X. Jin, C. Liu, J. Xu, Q. Wang and D. Chen, RSC Adv., 2014, 4, 35546–35553 RSC.
- C. C. Li, J. Dou, L. Chen, J. Lin and H. C. Zeng, ChemCatChem, 2012, 4, 1675–1682 CrossRef CAS.
- A. K. Mondal, D. Su, Y. Wang, S. Chen, Q. Liu and G. Wang, J. Alloys Compd., 2014, 582, 522–527 CrossRef CAS.
- T. Zhu, J. S. Chen and X. W. Lou, J. Phys. Chem. C, 2012, 116, 6873–6878 CAS.
- X. Li, S. Xiong, J. Li, J. Bai and Y. Qian, J. Mater. Chem., 2012, 22, 14276–14283 RSC.
- D. Han, X. Jing, J. Wang, P. Yang, D. Song and J. Liu, J. Electroanal. Chem., 2012, 682, 37–44 CrossRef CAS.
- P.-P. Jin, X. Zou, L.-J. Zhou, J. Zhao, H. Chen, Y. Tian and G.-D. Li, Sens. Actuators, B, 2014, 204, 142–148 CrossRef CAS.
- Z. Li, Q. Zhao, W. Fan and J. Zhan, Nanoscale, 2011, 3, 1646–1652 RSC.
- X. Wang, S. Qiu, C. He, G. Lu, W. Liu and J. Liu, RSC Adv., 2013, 3, 19002–19008 RSC.
- X. Wang, S. Qiu, J. Liu, C. He, G. Lu and W. Liu, Eur. J. Inorg. Chem., 2014, 863–869 CrossRef CAS.
- X. W. Lou, J. S. Chen, P. Chen and L. A. Archer, Chem. Mater., 2009, 21, 2868–2874 CrossRef CAS.
- C. Niu, M. Huang, P. Wang, J. Meng, X. Liu, X. Wang, K. Zhao, Y. Yu, Y. Wu and C. Lin, Nano Res., 2015, 1–11 CAS.
- C. Zhang, Z. Chen, Z. Guo and X. W. D. Lou, Energy Environ. Sci., 2013, 6, 974–978 CAS.
- S. Jeon and K. Yong, J. Mater. Chem., 2010, 20, 10146–10151 RSC.
- W. Zeng, C. Dong, B. Miao, H. Zhang, S. Xu, X. Ding and S. Hussain, Mater. Lett., 2014, 117, 41–44 CrossRef CAS.
- S. A. K. Leghari, S. Sajjad and J. Zhang, RSC Adv., 2013, 3, 15354–15361 RSC.
- M. K. Devaraju, S. Yin and T. Sato, Inorg. Chem., 2011, 50, 4698–4704 CrossRef CAS PubMed.
- X. Liu, J. Zhang, L. Wang, T. Yang, X. Guo, S. Wu and S. Wang, J. Mater. Chem., 2011, 21, 349–356 RSC.
- W. Wang, Y. Tian, X. Wang, H. He, Y. Xu, C. He and X. Li, J. Mater. Sci., 2013, 48, 3232–3238 CrossRef CAS.
- A. Lei, B. Qu, W. Zhou, Y. Wang, Q. Zhang and B. Zou, Mater. Lett., 2012, 66, 72–75 CrossRef CAS.
- W. Guo, T. Liu, J. Wang, W. Yu, R. Sun, Y. Chen, S. Hussain, X. Peng and Z. Wang, Ceram. Int., 2013, 39, 5919–5924 CrossRef CAS.
- Y. Lai, M. Meng and Y. Yu, Appl. Catal., B, 2010, 100, 491–501 CrossRef CAS.
- X. Fu, X. Yang, Z. Qiu, F. Zhao, J. Zhuang, A. He, L. Chen, C. Wu, X. Duan and C. Liang, CrystEngComm, 2013, 15, 3334–3340 RSC.
- C. Wang, Y. Le and B. Cheng, Ceram. Int., 2014, 40, 10847–10856 CrossRef CAS.
- L. Nie, A. Meng, J. Yu and M. Jaroniec, Sci. Rep., 2013, 3, 3215, DOI:10.1038/srep03215.
- Z. Dong, T. Ye, Y. Zhao, J. Yu, F. Wang, L. Zhang, X. Wang and S. Guo, J. Mater. Chem., 2011, 21, 5978–5984 RSC.
- S.-H. Hsieh, G.-J. Lee, C.-Y. Chen, J.-H. Chen, S.-H. Ma, T.-L. Horng, K.-H. Chen and J. J. Wu, Top. Catal., 2013, 56, 623–629 CrossRef CAS.
- Y. Huo, Y. Jin and Y. Zhang, J. Mol. Catal. A: Chem., 2010, 331, 15–20 CrossRef CAS.
- Z. Shen, J. Liu, F. Hu, S. Liu, N. Cao, Y. Sui, Q. Zeng and Y. Shen, CrystEngComm, 2014, 16, 3387–3394 RSC.
- D. Zhang, Y. Qian, L. Shi, H. Mai, R. Gao, J. Zhang, W. Yu and W. Cao, Catal. Commun., 2012, 26, 164–168 CrossRef CAS.
- Y. Jiao, F. Wang, X. Ma, Q. Tang, K. Wang, Y. Guo and L. Yang, Microporous Mesoporous Mater., 2013, 176, 1–7 CrossRef CAS.
- Z. Yang, J. Wei, H. Yang, L. Liu, H. Liang and Y. Yang, Eur. J. Inorg. Chem., 2010, 3354–3359 CrossRef CAS.
- S. Yuan, Q. Zhang, B. Xu, Z. Jin, Y. Zhang, Y. Yang, M. Zhang and T. Ohno, RSC Adv., 2014, 4, 62255–62261 CAS.
- G. Shen, H. Liu, Q. Wang, Z. Wang and Y. Chen, J. Nanopart. Res., 2012, 14, 1–8 CrossRef.
- H. Jiu, Y. Sun, L. Zhang, C. Zhang, J. Zhang and J. Liu, Ceram. Int., 2014, 40, 3149–3154 CrossRef CAS.
- C. Chen, X. Fang, B. Wu, L. Huang and N. Zheng, ChemCatChem, 2012, 4, 1578–1586 CrossRef CAS.
- H. Du, L. Jiao, Q. Wang, J. Yang, L. Guo, Y. Si, Y. Wang and H. Yuan, Nano Res., 2013, 6, 87–98 CrossRef CAS.
- J. Zhao, Y. C. Zou, X. X. Zou, T. Y. Bai, Y. P. Liu, R. Q. Gao, D. J. Wang and G. D. Li, Nanoscale, 2014, 6, 7255–7262 RSC.
- S. Li, A. Li, R. Zhang, Y. He, Y. Zhai and L. Xu, Nano Res., 2014, 7, 1116–1127 CrossRef CAS.
- C. Wang, Q. Li, F. Wang, G. Xia, R. Liu, D. Li, N. Li, J. S. Spendelow and G. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 1243–1250 CAS.
- X. Guan, L. Li, G. Li, Z. Fu, J. Zheng and T. Yan, J. Alloys Compd., 2011, 509, 3367–3374 CrossRef CAS.
- S. Wang, P. Li, H. Zhu and W. Tang, Powder Technol., 2012, 230, 48–53 CrossRef CAS.
- D. Li, Q. Qin, X. Duan, J. Yang, W. Guo and W. Zheng, ACS Appl. Mater. Interfaces, 2013, 5, 9095–9100 CAS.
- Z. Wu, K. Yu, S. Zhang and Y. Xie, J. Phys. Chem. C, 2008, 112, 11307–11313 CAS.
- B. Wang, J. S. Chen, H. B. Wu, Z. Wang and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 17146–17148 CrossRef CAS PubMed.
- L. Wang, Z. Lou, J. Deng, R. Zhang and T. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 13098–13104 CAS.
- S.-W. Cao, Y.-J. Zhu, M.-Y. Ma, L. Li and L. Zhang, J. Phys. Chem. C, 2008, 112, 1851–1856 CAS.
- W. Li, Y. Deng, Z. Wu, X. Qian, J. Yang, Y. Wang, D. Gu, F. Zhang, B. Tu and D. Zhao, J. Am. Chem. Soc., 2011, 133, 15830–15833 CrossRef CAS PubMed.
- Z.-G. Wu, Y.-J. Zhong, J.-T. Li, X.-D. Guo, L. Huang, B. Zhong and S.-G. Sun, J. Mater. Chem. A, 2014, 2, 12361–12367 CAS.
- L.-P. Zhu, L.-L. Wang, N.-C. Bing, C. Huang, L.-J. Wang and G.-H. Liao, ACS Appl. Mater. Interfaces, 2013, 5, 12478–12487 CAS.
- F. X. Ma, H. Hu, H. B. Wu, C. Y. Xu, Z. Xu and L. Zhen, Adv. Mater., 2015, 27, 4097–4101 CrossRef CAS PubMed.
- B. Wang, H. Wu, L. Yu, R. Xu and T. T. Lim, Adv. Mater., 2012, 24, 1111–1116 CrossRef CAS PubMed.
- X. Li, C. Tang, M. Ai, L. Dong and Z. Xu, Chem. Mater., 2010, 22, 4879–4889 CrossRef CAS.
- Y. Teng, L. X. Song, A. Ponchel, Z. K. Yang and J. Xia, Adv. Mater., 2014, 26, 6238–6243 CrossRef CAS PubMed.
- T. Zhang, M. Chen, F. Gu, D. Han, Z. Wang and G. Guo, Integr. Ferroelectr., 2012, 138, 117–122 CrossRef CAS.
- S.-J. Kim, I.-S. Hwang, C. W. Na, I.-D. Kim, Y. C. Kang and J.-H. Lee, J. Mater. Chem., 2011, 21, 18560–18567 RSC.
- L.-X. Li, D. Xu, X.-Q. Li, W.-C. Liu and Y. Jia, New J. Chem., 2014, 38, 5445–5452 RSC.
- S. Yang, P. Huang, L. Peng, C. Cao, Y. Zhu, F. Wei, Y. Sun and W. Song, J. Mater. Chem. A, 2016, 4, 400–406 CAS.
- J. Ma, Q. Cheng, V. Pavlinek, P. Saha and C. Li, New J. Chem., 2013, 37, 722–728 RSC.
- S.-W. Bian, Y.-P. Zhao and C.-Y. Xian, Mater. Lett., 2013, 111, 75–77 CrossRef CAS.
- D. Li, X. Wu and Y. Chen, J. Phys. Chem. C, 2013, 117, 11040–11046 CAS.
- G. Wang, H. Xu, L. Lu and H. Zhao, J. Mater. Chem. A, 2015, 3, 1127–1132 CAS.
- H. Zhang, Y. Li, Z. Hong and M. Wei, Mater. Lett., 2012, 79, 148–151 CrossRef CAS.
- X. Zhang, X. Song, S. Gao, Y. Xu, X. Cheng, H. Zhao and L. Huo, J. Mater. Chem. A, 2013, 1, 6858–6864 CAS.
- D. Xie, Q. Su, Z. Dong, J. Zhang and G. Du, CrystEngComm, 2013, 15, 8314–8319 RSC.
- L. Chu, M. Li, Z. Wan, L. Ding, D. Song, S. Dou, J. Chen and Y. Wang, CrystEngComm, 2014, 16, 11096–11101 RSC.
- L. Shen, L. Yu, X. Y. Yu, X. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2015, 54, 1868–1872 CrossRef CAS PubMed.
- X. M. Yin, C. C. Li, M. Zhang, Q. Y. Hao, S. Liu, L. B. Chen and T. H. Wang, J. Phys. Chem. C, 2010, 114, 8084–8088 CAS.
- S. Liu, G. Huang, J. Yu, T. W. Ng, H. Y. Yip and P. K. Wong, ACS Appl. Mater. Interfaces, 2014, 6, 2407–2414 CAS.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, Chem. Mater., 2008, 20, 6562–6566 CrossRef CAS.
- S. Ding and X. W. D. Lou, Nanoscale, 2011, 3, 3586–3588 RSC.
- H. X. Yang, J. F. Qian, Z. X. Chen, X. P. Ai and Y. L. Cao, J. Phys. Chem. C, 2007, 111, 14067–14071 CAS.
- P. Wu, N. Du, H. Zhang, C. Zhai and D. Yang, ACS Appl. Mater. Interfaces, 2011, 3, 1946–1952 CAS.
- Z. Zheng, B. Huang, X. Qin, X. Zhang and Y. Dai, J. Colloid Interface Sci., 2011, 358, 68–72 CrossRef CAS PubMed.
- M. Sharma, D. Das, A. Baruah, A. Jain and A. K. Ganguli, Langmuir, 2014, 30, 3199–3208 CrossRef CAS PubMed.
- E. Uchaker, N. Zhou, Y. Li and G. Cao, J. Phys. Chem. C, 2013, 117, 1621–1626 CAS.
- D. Su, S. X. Dou and G. Wang, J. Mater. Chem. A, 2014, 2, 11185–11194 CAS.
- H. B. Wu, A. Pan, H. H. Hng and X. W. D. Lou, Adv. Funct. Mater., 2013, 23, 5669–5674 CrossRef CAS.
- A. Pan, H. B. Wu, L. Yu and X. W. D. Lou, Angew. Chem., 2013, 125, 2282–2286 CrossRef.
- J. Yu and X. Yu, Environ. Sci. Technol., 2008, 42, 4902–4907 CrossRef CAS PubMed.
- X. Ma, X. Zhang, L. Yang, K. Wang, K. Jiang, Z. Wei and Y. Guo, CrystEngComm, 2014, 16, 7933–7941 RSC.
- Y. Sun, L. Zhang, J. Zhang, P. Chen, S. Xin, Z. Li and J. Liu, Ceram. Int., 2014, 40, 1599–1603 CrossRef CAS.
- H.-S. Roh, G. K. Choi, J.-S. An, C. M. Cho, D. H. Kim, I. J. Park, T. H. Noh, D.-W. Kim and K. S. Hong, Dalton Trans., 2011, 40, 6901–6905 RSC.
- Z. Zhang, H. Chen, H. Che, Y. Wang and F. Su, Mater. Chem. Phys., 2013, 138, 593–600 CrossRef CAS.
- W. Luo, Y. Li, J. Dong, J. Wei, J. Xu, Y. Deng and D. Zhao, Angew. Chem., Int. Ed., 2013, 52, 10505–10510 CrossRef CAS PubMed.
- C. Yuan, X. Zhang, L. Su, B. Gao and L. Shen, J. Mater. Chem., 2009, 19, 5772–5777 RSC.
- P. Manjula, R. Boppella and S. V. Manorama, ACS Appl. Mater. Interfaces, 2012, 4, 6252–6260 CAS.
- G. Shang, J. Wu, M. Huang, J. Lin, Z. Lan, Y. Huang and L. Fan, J. Phys. Chem. C, 2012, 116, 20140–20145 CAS.
- S. Wang, Z. Lu, D. Wang, C. Li, C. Chen and Y. Yin, J. Mater. Chem., 2011, 21, 6365–6369 RSC.
- J. Shao, X. Li, Z. Wan, L. Zhang, Y. Ding, L. Zhang, Q. Qu and H. Zheng, ACS Appl. Mater. Interfaces, 2013, 5, 7671–7675 CAS.
- J. Huang, X. Xu, C. Gu, G. Fu, W. Wang and J. Liu, Mater. Res. Bull., 2012, 47, 3224–3232 CrossRef CAS.
- S. Bai, K. Zhang, L. Wang, J. Sun, R. Luo, D. Li and A. Chen, J. Mater. Chem. A, 2014, 2, 7927–7934 CAS.
- J. Huang, X. Xu, C. Gu, M. Yang, M. Yang and J. Liu, J. Mater. Chem., 2011, 21, 13283–13289 RSC.
- P. Zhao, Y. Zhu, X. Yang, K. Fan, J. Shen and C. Li, RSC Adv., 2012, 2, 10592–10597 RSC.
- F. Wang, X. Wang, D. Liu, J. Zhen, J. Li, Y. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 22216–22223 CAS.
- H. Liu, G. Shao, W. Jia, Z. Zhang, Y. Zhang, J. Liang, X. Liu, H. Jia and B. Xu, CrystEngComm, 2013, 15, 3615–3622 RSC.
- P. Ma, Y. Wu, Z. Fu and W. Wang, J. Alloys Compd., 2011, 509, 3576–3581 CrossRef CAS.
- J. He, J. Chen, L. Ren, Y. Wang, C. Teng, M. Hong, J. Zhao and B. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 2718–2725 CAS.
- J. H. Pan, Y. Bai and Q. Wang, Langmuir, 2015, 31, 4566–4572 CrossRef CAS PubMed.
- W. Zhang, Z. X. Chi, W. X. Mao, R. W. Lv, A. M. Cao and L. J. Wan, Angew. Chem., 2014, 126, 12990–12994 CrossRef.
- B. Bai, W. Guan, Z. Li and G. Li Puma, Mater. Res. Bull., 2011, 46, 26–31 CrossRef CAS.
- Y. Chen and J. Lu, J. Porous Mater., 2012, 19, 289–294 CrossRef CAS.
- C.-A. Wang, S. Li and L. An, Chem. Commun., 2013, 49, 7427–7429 RSC.
- N. Kang, J. H. Park, M. Jin, N. Park, S. M. Lee, H. J. Kim, J. M. Kim and S. U. Son, J. Am. Chem. Soc., 2013, 135, 19115–19118 CrossRef CAS PubMed.
- Y. Qin, F. Zhang, Y. Chen, Y. Zhou, J. Li, A. Zhu, Y. Luo, Y. Tian and J. Yang, J. Phys. Chem. C, 2012, 116, 11994–12000 CAS.
- C. Deng, H. Hu, X. Ge, C. Han, D. Zhao and G. Shao, Ultrason. Sonochem., 2011, 18, 932–937 CrossRef CAS PubMed.
- G. Tian, Z. Gu, X. Liu, L. Zhou, W. Yin, L. Yan, S. Jin, W. Ren, G. Xing and S. Li, J. Phys. Chem. C, 2011, 115, 23790–23796 CAS.
- W. J. Tseng, T. T. Tseng, H. M. Wu, Y. C. Her and T. J. Yang, J. Am. Ceram. Soc., 2013, 96, 719–725 CrossRef CAS.
- Y. Liu, Z. Chen, C.-H. Shek, C.-M. L. Wu and J. K.-L. Lai, ACS Appl. Mater. Interfaces, 2014, 6, 9776–9784 CAS.
- S. Ding, T. Zhu, J. S. Chen, Z. Wang, C. Yuan and X. W. D. Lou, J. Mater. Chem., 2011, 21, 6602–6606 RSC.
- D. Xie, W. Yuan, Z. Dong, Q. Su, J. Zhang and G. Du, Electrochim. Acta, 2013, 92, 87–92 CrossRef CAS.
- L. Shi and H. Lin, Langmuir, 2010, 26, 18718–18722 CrossRef CAS PubMed.
- F. Gyger, M. Hubner, C. Feldmann, N. Barsan and U. Weimar, Chem. Mater., 2010, 22, 4821–4827 CrossRef CAS.
- S. Ding, J. S. Chen, G. Qi, X. Duan, Z. Wang, E. P. Giannelis, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 133, 21–23 CrossRef PubMed.
- G. Xi, Y. Yan, Q. Ma, J. Li, H. Yang, X. Lu and C. Wang, Chem. – Eur. J., 2012, 18, 13949–13953 CrossRef CAS PubMed.
- Y. Liu, Q. Li, S. Gao and J. Shang, CrystEngComm, 2014, 16, 7493–7501 RSC.
- G. Jia, H. You, Y. Song, Y. Huang, M. Yang and H. Zhang, Inorg. Chem., 2010, 49, 7721–7725 CrossRef CAS PubMed.
- Z. Xu, Y. Gao, T. Liu, L. Wang, S. Bian and J. Lin, J. Mater. Chem., 2012, 22, 21695–21703 RSC.
- H. Jiu, Y. Fu, L. Zhang, Y. Sun, Y. Wang and T. Han, Micro Nano Lett., 2012, 7, 947–950 Search PubMed.
- F. Meng, J. Yin, Y.-Q. Duan, Z.-H. Yuan and L.-J. Bie, Sens. Actuators, B, 2011, 156, 703–708 CrossRef CAS.
- Y. Wei, Y. Huang, J. Wu, M. Wang, C. Guo, D. Qiang, S. Yin and T. Sato, J. Hazard. Mater., 2013, 248, 202–210 CrossRef PubMed.
- H. Yao, D. Jia and H. Zhang, Ceram. Int., 2015, 41, 1531–1534 CrossRef CAS.
- Y. Xia and R. Mokaya, J. Mater. Chem., 2005, 15, 3126–3131 RSC.
- X. Sun, J. Liu and Y. Li, Chem. – Eur. J., 2006, 12, 2039–2047 CrossRef CAS PubMed.
- M.-M. Titirici, M. Antonietti and A. Thomas, Chem. Mater., 2006, 18, 3808–3812 CrossRef CAS.
- P. M. Arnal, C. Weidenthaler and F. Schüth, Chem. Mater., 2006, 18, 2733–2739 CrossRef CAS.
- D. G. Shchukin and R. A. Caruso, Chem. Mater., 2004, 16, 2287–2292 CrossRef CAS.
- H. Ren, J. J. Sun, R. B. Yu, M. Yang, L. Gu, P. R. Liu, H. J. Zhao, D. Kisailus and D. Wang, Chem. Sci., 2016, 7, 793–798 RSC.
- X. Zeng, J. Yang, L. Shi, L. Li and M. Gao, Nanoscale Res. Lett., 2014, 9, 1–10 CrossRef CAS PubMed.
- G. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2014, 53, 9041–9044 CrossRef CAS PubMed.
- X. Lai, J. Li, B. A. Korgel, Z. Dong, Z. Li, F. Su, J. Du and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 2738–2741 CrossRef CAS PubMed.
- Z. H. Dong, X. Y. Lai, J. E. Halpert, N. L. Yang, L. X. Yi, J. Zhai, D. Wang, Z. Y. Tang and L. Jiang, Adv. Mater., 2012, 24, 1046–1049 CrossRef CAS PubMed.
- J. Y. Wang, N. L. Yang, H. J. Tang, Z. H. Dong, Q. Jin, M. Yang, D. Kisailus, H. J. Zhao, Z. Y. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417–6420 CrossRef CAS PubMed.
- S. M. Xu, C. M. Hessel, H. Ren, R. B. Yu, Q. Jin, M. Yang, H. J. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 CAS.
- Z. Dong, H. Ren, C. M. Hessel, J. Wang, R. Yu, Q. Jin, M. Yang, Z. Hu, Y. Chen, Z. Tang, H. Zhao and D. Wang, Adv. Mater., 2014, 26, 905–909 CrossRef CAS PubMed.
- H. Ren, R. Yu, J. Wang, Q. Jin, M. Yang, D. Mao, D. Kisailus, H. Zhao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- J. Y. Wang, H. J. Tang, H. Ren, R. B. Yu, J. Qi, D. Mao, H. J. Zhao and D. Wang, Adv. Sci., 2014, 1 DOI:10.1002/advs.201400011.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef.
- J. B. Joo, I. Lee, M. Dahl, G. D. Moon, F. Zaera and Y. Yin, Adv. Funct. Mater., 2013, 23, 4246–4254 CrossRef CAS.
- J. Chen, D. W. Wang, J. Qi, G. D. Li, F. Y. Zheng, S. X. Li, H. J. Zhao and Z. Y. Tang, Small, 2015, 11, 420–425 CrossRef CAS PubMed.
- H. M. Abdelaal and B. Harbrecht, C. R. Chim., 2015, 18, 379–384 CrossRef CAS.
- L. Zong, P. Xu, Y. Ding, K. Zhao, Z. Wang, X. Yan, R. Yu, J. Chen and X. Xing, Small, 2015, 11, 2768–2773 CrossRef CAS PubMed.
- X. Li, F. Chen, X. Lu, C. Ni, X. Zhao and Z. Chen, J. Porous Mater., 2010, 17, 297–303 CrossRef CAS.
- H. Liu, J. B. Joo, M. Dahl, L. Fu, Z. Zeng and Y. Yin, Energy Environ. Sci., 2015, 8, 286–296 CAS.
- G. Yang, P. Hu, Y. Cao, F. Yuan and R. Xu, Nanoscale Res. Lett., 2010, 5, 1437–1441 CrossRef CAS PubMed.
- G. Veerappan, D.-W. Jung, J. Kwon, J. M. Choi, N. Heo, G.-R. Yi and J. H. Park, Langmuir, 2014, 30, 3010–3018 CrossRef CAS PubMed.
- G. Zhang, X. Shen and Y. Yang, J. Phys. Chem. C, 2011, 115, 7145–7152 CAS.
- Y. Liu, R. Che, G. Chen, J. Fan, Z. Sun, Z. Wu, M. Wang, B. Li, J. Wei and Y. Wei, Sci. Adv., 2015, 1, e1500166 Search PubMed.
- D. Mao, J. X. Yao, X. Y. Lai, M. Yang, J. A. Du and D. Wang, Small, 2011, 7, 578–582 CrossRef CAS PubMed.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef CAS PubMed.
- J. H. Pan, Z. Cai, Y. Yu and X. Zhao, J. Mater. Chem., 2011, 21, 11430–11438 RSC.
- W.-G. Yang, F.-R. Wan, Q.-W. Chen, J.-J. Li and D.-S. Xu, J. Mater. Chem., 2010, 20, 2870–2876 RSC.
- Z. Lin, M. Zheng, B. Zhao, G. Wang, L. Pu and Y. Shi, J. Solid State Electrochem., 2014, 18, 1673–1681 CrossRef CAS.
- D. Chen, F. Huang, Y. B. Cheng and R. A. Caruso, Adv. Mater., 2009, 21, 2206–2210 CrossRef CAS.
- H. Ming, Z. Ma, H. Huang, S. Lian, H. Li, X. He, H. Yu, K. Pan, Y. Liu and Z. Kang, Chem. Commun., 2011, 47, 8025–8027 RSC.
- K. Del Ángel-Sánchez, O. Vázquez-Cuchillo, M. Salazar-Villanueva, J. Sánchez-Ramirez, A. Cruz-López and A. Aguilar-Elguezabal, J. Sol–Gel Sci. Technol., 2011, 58, 360–365 CrossRef.
- H.-E. Wang, L.-X. Zheng, C.-P. Liu, Y.-K. Liu, C.-Y. Luan, H. Cheng, Y. Y. Li, L. Martinu, J. A. Zapien and I. Bello, J. Phys. Chem. C, 2011, 115, 10419–10425 CAS.
- D. Chen, L. Cao, F. Huang, P. Imperia, Y.-B. Cheng and R. A. Caruso, J. Am. Chem. Soc., 2010, 132, 4438–4444 CrossRef CAS PubMed.
- Y. Cui, L. Liu, B. Li, X. Zhou and N. Xu, J. Phys. Chem. C, 2010, 114, 2434–2439 CAS.
- C. Wang, J. Chen, X. Zhou, W. Li, Y. Liu, Q. Yue, Z. Xue, Y. Li, A. A. Elzatahry and Y. Deng, Nano Res., 2014, 1–8 Search PubMed.
- I. Lee, J. B. Joo, Y. Yin and F. Zaera, Angew. Chem., 2011, 123, 10390–10393 CrossRef.
- J. B. Joo, Q. Zhang, M. Dahl, I. Lee, J. Goebl, F. Zaera and Y. Yin, Energy Environ. Sci., 2012, 5, 6321–6327 CAS.
- X. Cheng, M. Chen, L. Wu and G. Gu, Langmuir, 2006, 22, 3858–3863 CrossRef CAS PubMed.
- H. Xu, X. Chen, S. Ouyang, T. Kako and J. Ye, J. Phys. Chem. C, 2012, 116, 3833–3839 CAS.
- L. Cao, D. Chen and R. A. Caruso, Angew. Chem., Int. Ed., 2013, 52, 10986–10991 CrossRef CAS PubMed.
- W. Li, J. Yang, Z. Wu, J. Wang, B. Li, S. Feng, Y. Deng, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2012, 134, 11864–11867 CrossRef CAS PubMed.
- B. Li, Y. Xie, M. Jing, G. Rong, Y. Tang and G. Zhang, Langmuir, 2006, 22, 9380–9385 CrossRef CAS PubMed.
- P. Gurunathan, P. M. Ette and K. Ramesha, ACS Appl. Mater. Interfaces, 2014, 6, 16556–16564 CAS.
- H. Chun Zeng, Curr. Nanosci., 2007, 3, 177–181 CrossRef.
- W. Q. Fang, X. H. Yang, H. Zhu, Z. Li, H. Zhao, X. Yao and H. G. Yang, J. Mater. Chem., 2012, 22, 22082–22089 RSC.
- D. Zhang, J. Zhu, N. Zhang, T. Liu, L. Chen, X. Liu, R. Ma, H. Zhang and G. Qiu, Sci. Rep., 2015, 5, 8737, DOI:10.1038/srep08737.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- J. Liu, S. Z. Qiao, J. S. Chen, X. W. D. Lou, X. Xing and G. Q. M. Lu, Chem. Commun., 2011, 47, 12578–12591 RSC.
- Y.-H. Choi, D.-H. Kim, H. S. Han, S. Shin, S.-H. Hong and K. S. Hong, Langmuir, 2014, 30, 700–709 CrossRef CAS PubMed.
- M. Kong, W. Zhang, Z. Yang, S. Weng and Z. Chen, Appl. Surf. Sci., 2011, 258, 1317–1321 CrossRef CAS.
- S. Li and C.-A. Wang, J. Colloid Interface Sci., 2015, 438, 61–67 CrossRef CAS PubMed.
- J. H. Pan, X. Z. Wang, Q. Huang, C. Shen, Z. Y. Koh, Q. Wang, A. Engel and D. W. Bahnemann, Adv. Funct. Mater., 2014, 24, 95–104 CrossRef CAS.
- W. Yu, Q. Ma, C. Wang, X. Dong, J. Wang and G. Liu, Mater. Express, 2014, 4, 435–440 CrossRef CAS.
- D. Hwang, H. Lee, S.-Y. Jang, S. M. Jo, D. Kim, Y. Seo and D. Y. Kim, ACS Appl. Mater. Interfaces, 2011, 3, 2719–2725 CAS.
- B. Liu, K. Nakata, M. Sakai, H. Saito, T. Ochiai, T. Murakami, K. Takagi and A. Fujishima, Langmuir, 2011, 27, 8500–8508 CrossRef CAS PubMed.
- Y. Yu, C. H. Chen and Y. Shi, Adv. Mater., 2007, 19, 993–997 CrossRef CAS.
- X. Jia, Z. Chen, X. Cui, Y. Peng, X. Wang, G. Wang, F. Wei and Y. Lu, ACS Nano, 2012, 6, 9911–9919 CrossRef CAS PubMed.
- D. Lehr, D. Großmann, W. Grünert and S. Polarz, Nanoscale, 2014, 6, 1698–1706 RSC.
- G. Jian, L. Liu and M. R. Zachariah, Adv. Funct. Mater., 2013, 23, 1341–1346 CrossRef CAS.
- Z. Jin, F. Wang, J. Wang, J. C. Yu and J. Wang, Adv. Funct. Mater., 2013, 23, 2137–2144 CrossRef CAS.
- G. Jian, Y. Xu, L.-C. Lai, C. Wang and M. R. Zachariah, J. Mater. Chem. A, 2014, 2, 4627–4632 CAS.
- S. Dilger, M. Wessig, M. R. Wagner, J. S. Reparaz, C. M. Sotomayor Torres, L. Qijun, T. Dekorsy and S. Polarz, Cryst. Growth Des., 2014, 14, 4593–4601 CAS.
- Y. H. Cho, Y. C. Kang and J.-H. Lee, Sens. Actuators, B, 2013, 176, 971–977 CrossRef CAS.
- J. Huo, Y. Hu, H. Jiang, W. Huang, Y. Li, W. Shao and C. Li, Ind. Eng. Chem. Res., 2013, 52, 11029–11035 CrossRef CAS.
- A. B. D. Nandiyanto, O. Arutanti, T. Ogi, F. Iskandar, T. O. Kim and K. Okuyama, Chem. Eng. Sci., 2013, 101, 523–532 CrossRef CAS.
- Y. Huang, Z. Ai, W. Ho, M. Chen and S. Lee, J. Phys. Chem. C, 2010, 114, 6342–6349 CAS.
- J. W. Overcash and K. S. Suslick, Chem. Mater., 2015, 27, 3564–3567 CrossRef CAS.
- Z. Padashbarmchi, A. H. Hamidian, H. Zhang, L. Zhou, N. Khorasani, M. Kazemzad and C. Yu, RSC Adv., 2015, 5, 10304–10309 RSC.
- Y. J. Hong, M. Y. Son, B. K. Park and Y. C. Kang, Small, 2013, 9, 2224–2227 CrossRef CAS PubMed.
- Y. J. Hong, M. Y. Son and Y. C. Kang, Adv. Mater., 2013, 25, 2279–2283 CrossRef CAS PubMed.
- Y. N. Ko, S. B. Park, K. Y. Jung and Y. C. Kang, Nano Lett., 2013, 13, 5462–5466 CrossRef CAS PubMed.
- S. H. Choi, Y. J. Hong and Y. C. Kang, Nanoscale, 2013, 5, 7867–7871 RSC.
- S. H. Choi, J.-K. Lee and Y. C. Kang, Nanoscale, 2014, 6, 12421–12425 RSC.
- S. H. Choi and Y. C. Kang, Small, 2014, 10, 474–478 CrossRef CAS PubMed.
- C. Wang, M. Zhu, H. Liu, Y. Cui and Y. Chen, RSC Adv., 2014, 4, 24176–24182 RSC.
- B. Fang, C. Zhang, G. Wang, M. Wang and Y. Ji, Sens. Actuators, B, 2011, 155, 304–310 CrossRef CAS.
- P. Si, P. Chen and D.-H. Kim, J. Mater. Chem. B, 2013, 1, 2696–2700 RSC.
- M. Aghazadeh, A.-A. M. Barmi and H. M. Shiri, Russ. J. Electrochem., 2013, 49, 344–353 CrossRef CAS.
- X.-H. Xia, J.-P. Tu, X.-L. Wang, C.-D. Gu and X.-B. Zhao, Chem. Commun., 2011, 47, 5786–5788 RSC.
- M. A. Hossain, G. Yang, M. Parameswaran, J. R. Jennings and Q. Wang, J. Phys. Chem. C, 2010, 114, 21878–21884 CAS.
- C.-Y. Cao, W. Guo, Z.-M. Cui, W.-G. Song and W. Cai, J. Mater. Chem., 2011, 21, 3204–3209 RSC.
- P. Chen, J.-D. Peng, C.-H. Liao, P.-S. Shen and P.-L. Kuo, J. Nanopart. Res., 2013, 15, 1–11 Search PubMed.
- H. Wang, A. Pyatenko, K. Kawaguchi, X. Li, Z. Swiatkowska-Warkocka and N. Koshizaki, Angew. Chem., 2010, 122, 6505–6508 CrossRef.
- X. Li, Y. Shimizu, A. Pyatenko, H. Wang and N. Koshizaki, Nanotechnology, 2012, 23, 115602 CrossRef PubMed.
- H. Wang, M. Miyauchi, Y. Ishikawa, A. Pyatenko, N. Koshizaki, Y. Li, L. Li, X. Li, Y. Bando and D. Golberg, J. Am. Chem. Soc., 2011, 133, 19102–19109 CrossRef CAS PubMed.
- X.-X. Zou, G.-D. Li, Y.-N. Wang, J. Zhao, C. Yan, M.-Y. Guo, L. Li and J.-S. Chen, Chem. Commun., 2011, 47, 1066–1068 RSC.
- Z. Ju, C. Guo, Y. Qian, B. Tang and S. Xiong, Nanoscale, 2014, 6, 3268–3273 RSC.
- Q. Jiao, M. Fu, C. You, Y. Zhao and H. Li, Inorg. Chem., 2012, 51, 11513–11520 CrossRef CAS PubMed.
- R. D. Hancock and A. E. Martell, Chem. Rev., 1989, 89, 1875–1914 CrossRef CAS.
- J. Yang, C. Lin, Z. Wang and J. Lin, Inorg. Chem., 2006, 45, 8973–8979 CrossRef CAS PubMed.
- B. Liu, L.-M. Liu, X.-F. Lang, H.-Y. Wang, X. W. D. Lou and E. S. Aydil, Energy Environ. Sci., 2014, 7, 2592–2597 CAS.
- C. Guo, X. Wu, M. Yan, Q. Dong, S. Yin, T. Sato and S. Liu, Nanoscale, 2013, 5, 8184–8191 RSC.
- X. Lü, F. Huang, J. Wu, S. Ding and F. Xu, ACS Appl. Mater. Interfaces, 2011, 3, 566–572 Search PubMed.
- S. Shang, X. Jiao and D. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 860–865 CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. Dunlop, J. W. Hamilton, J. A. Byrne and K. O'Shea, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- G. Zhu, J. Lian, M. Hojamberdiev and W. Que, J. Cluster Sci., 2013, 24, 829–841 CrossRef CAS.
- Y. Jia, X.-Y. Yu, T. Luo, J.-H. Liu and X.-J. Huang, RSC Adv., 2012, 2, 10251–10254 RSC.
- Q. Zhu, J.-G. Li, X. Li, X. Sun and Y. Sakka, Sci. Technol. Adv. Mater., 2011, 12, 055001 CrossRef.
- J. Zhang, X. Liu, S. Wu, M. Xu, X. Guo and S. Wang, J. Mater. Chem., 2010, 20, 6453–6459 RSC.
- G. Han, Q. Lu, G. Liu, X. Ye, S. Lin, Y. Song, B. Liu, X. Yang and G. Li, J. Mater. Sci.: Mater. Electron., 2012, 23, 1616–1620 CrossRef CAS.
- H. Wang, Q. Liang, W. Wang, Y. An, J. Li and L. Guo, Cryst. Growth Des., 2011, 11, 2942–2947 CAS.
- N. Sutradhar, A. Sinhamahapatra, S. K. Pahari, P. Pal, H. C. Bajaj, I. Mukhopadhyay and A. B. Panda, J. Phys. Chem. C, 2011, 115, 12308–12316 CAS.
- P. Yang, S. Gai, Y. Liu, W. Wang, C. Li and J. Lin, Inorg. Chem., 2011, 50, 2182–2190 CrossRef CAS PubMed.
- R. Lv, P. Yang, F. He, S. Gai, G. Yang and J. Lin, Chem. Mater., 2015, 27, 483–496 CrossRef CAS.
- X. Wang, D. Chen, L. Cao, Y. Li, B. J. Boyd and R. A. Caruso, ACS Appl. Mater. Interfaces, 2013, 5, 10926–10932 CAS.
- Y. A. Nor, L. Zhou, A. K. Meka, C. Xu, Y. Niu, H. Zhang, N. Mitter, D. Mahony and C. Yu, Adv. Funct. Mater., 2016, 26, 5408–5418 CrossRef CAS.
- X. Wu, K. Li and H. Wang, J. Hazard. Mater., 2010, 174, 573–580 CrossRef CAS PubMed.
- D. Xiang, F. Qu, X. Chen, Z. Yu, L. Cui, X. Zhang, J. Jiang and H. Lin, J. Sol-Gel Sci. Technol., 2014, 69, 370–377 CrossRef CAS.
- Q. Wang, S. Yu, Z. Tan, R. Zhang, Z. Li, X. Gao, B. Shen and H. Su, CrystEngComm, 2015, 17, 671–677 RSC.
- Y. Hou, L. Wu, X. Wang, Z. Ding, Z. Li and X. Fu, J. Catal., 2007, 250, 12–18 CrossRef CAS.
- G. Li, H. Zhang, J. Lan, J. Li, Q. Chen, J. Liu and G. Jiang, Dalton Trans., 2013, 42, 8541–8544 RSC.
- B. Liu, K. Nakata, M. Sakai, H. Saito, T. Ochiai, T. Murakami, K. Takagi and A. Fujishima, Catal. Sci. Technol., 2012, 2, 1933–1939 CAS.
- H. Bai, Z. Liu, L. Liu and D. D. Sun, Chem. – Eur. J., 2013, 19, 3061–3070 CrossRef CAS PubMed.
- S. Ding, X. Yin, X. Lü, Y. Wang, F. Huang and D. Wan, ACS Appl. Mater. Interfaces, 2011, 4, 306–311 Search PubMed.
- H. Zhang, G. Du, W. Lu, L. Cheng, X. Zhu and Z. Jiao, CrystEngComm, 2012, 14, 3793–3801 RSC.
- B. Chi, L. Zhao and T. Jin, J. Phys. Chem. C, 2007, 111, 6189–6193 CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- S. Yoon and A. Manthiram, J. Phys. Chem. C, 2011, 115, 9410–9416 CAS.
- S. Yoon, C. A. Bridges, R. R. Unocic and M. P. Paranthaman, J. Mater. Sci., 2013, 48, 5125–5131 CrossRef CAS.
- W. Guo, W. Sun and Y. Wang, ACS Nano, 2015, 9, 11462–11471 CrossRef CAS PubMed.
- X. Yan, X. Tong, J. Wang, C. Gong, M. Zhang and L. Liang, Mater. Lett., 2013, 95, 1–4 CrossRef CAS.
- S. Ding, J. S. Chen, Z. Wang, Y. L. Cheah, S. Madhavi, X. Hu and X. W. Lou, J. Mater. Chem., 2011, 21, 1677–1680 RSC.
- F. Wang, J. Y. Wang, H. Ren, H. J. Tang, R. B. Yu and D. Wang, Inorg. Chem. Front., 2016, 3, 365–369 RSC.
- W. Yu, X. Jiang, S. Ding and B. Q. Li, J. Power Sources, 2014, 256, 440–448 CrossRef CAS.
- I. G. Yu, Y. J. Kim, H. J. Kim, C. Lee and W. I. Lee, J. Mater. Chem., 2011, 21, 532–538 RSC.
- J.-Y. Liao, B.-X. Lei, D.-B. Kuang and C.-Y. Su, Energy Environ. Sci., 2011, 4, 4079–4085 CAS.
- P. Cheng, S. Du, Y. Cai, F. Liu, P. Sun, J. Zheng and G. Lu, J. Phys. Chem. C, 2013, 117, 24150–24156 CAS.
- S. Dadgostar, F. Tajabadi and N. Taghavinia, ACS Appl. Mater. Interfaces, 2012, 4, 2964–2968 CAS.
| This journal is © The Royal Society of Chemistry 2016 |