
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple guiding principle for the temperature dependence of the solubility of light gases in imidazolium-based ionic liquids derived from molecular simulations
Daniela
Kerlé
*a,
Majid
Namayandeh Jorabchi
b,
Ralf
Ludwig
*bc,
Sebastian
Wohlrab
c and
Dietmar
Paschek
*b
aUniversität Bremen, Fachbereich Produktionstechnik, Technische Thermodynamik, Badgasteiner Str. 1, D-28359 Bremen, Germany. E-mail: dkerle@uni-bremen.de; Fax: +49 421 218 64771; Tel: +49 421 218 64757
bUniversität Rostock, Institut für Chemie, Physikalische und Theoretische Chemie, Dr.-Lorenz-Weg 1, D-18059 Rostock, Germany
cLeibniz-Institut für Katalyse an der Universität Rostock, Albert-Einstein-Str. 29a, D-18059 Rostock, Germany
First published on 8th November 2016
Abstract
We have determined the temperature dependence of the solvation behavior of a large collection of important light gases in imidazolium-based ionic liquids with the help of extensive molecular dynamics simulations. The motivation of our study is to unravel common features of the temperature dependent solvation under well controlled conditions, and to provide a guidance for cases, where experimental data from different sources disagree significantly. The solubility of molecular hydrogen, oxygen, nitrogen, methane, krypton, argon, neon and carbon dioxide in the imidazolium based ionic liquids of type 1-n-alkyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Cnmim][NTf2]) with varying alkyl side chain lengths n = 2, 4, 6, 8 is computed for a temperature range between 300 K and 500 K at 1 bar. By applying Widom's particle insertion technique and Bennet's overlapping distribution method, we are able to determine the temperature dependent solvation free energies of those selected light gases in simulated imidazolium based ionic liquids with high statistical accuracy. Our simulations demonstrate that the magnitude of the solvation free energy of a gas molecule at a chosen reference temperature and that of its temperature-derivatives are intimately related to one another. We conclude that this “universal” behavior is rooted in a solvation entropy–enthalpy compensation effect, which seems to be a defining feature of the solvation of small molecules in ionic liquids. The observations lead to simple analytical relations, determining the temperature dependence of the solubility data based on the absolute solubility at a certain reference temperature. By comparing our results with available experimental data from many sources, we can show that our approach is particularly helpful for providing reliable estimates for the solvation behavior of very light gases, such as hydrogen, where conflicting experimental data exist.
1 Introduction
Salts with melting points below 100 °C are commonly referred to as ionic liquids (ILs). These liquids have several unique properties,1–4 and are used for a wide range of potential applications.5,6 For the application of ILs in gas separation processes (e.g. flue gas decontamination) it is important to have access to accurate solubility data.7–10 Even more so, the recently introduced supported ionic liquid membranes11–14 are promising new tools for separating various mixtures of gases. Of particular importance, of course, is the ability to separate H2 and CO2 from gas-streams.Till now, a wealth of experimental measurements of the infinite dilution properties of a large number of gases in various ILs have been reported.15 However, the experimental determination of solubilities is particularly difficult for gases with a low molecular weight.1 As a result, the reported solubility data of hydrogen in ILs16–26 are highly inconsistent.
In addition, theoretical methods to determine solubilities of gases in imidazolium-based ILs have been reported in the literature: data were determined from COSMO based methods,27–30 equations of state approaches,31–34 group theory,35 quantitative structure–property relationship (QSPR) and neutral network models,36 as well as molecular dynamics (MD) and Monte Carlo (MC) simulations.37–54 We would like to point out that MD and MC simulations have the advantage that they offer the possibility of both a (semi-)quantitative prediction of the solubility and gaining a fundamental understanding of the molecular mechanism for the solvation process.
For the simulation of imidazolium-based ILs there are different atomic-detailed molecular force fields available.55 Many of those force-fields are capable of reproducing essentially “static” properties, such as thermodynamic properties and structural features quite well. However, most of them are lacking the ability of describing transport properties, such as diffusion coefficients and viscosities satisfactorily. In this study we used the non-polarizable all-atom force field originally introduced by Lopes,56 using the refined parameters from the study by Köddermann et al.57 to simulate the imidazolium-based ILs of type [Cnmim][NTf2] (see Fig. 1). We have shown earlier that a wealth of both thermodynamical and dynamical properties of the pure IL could be described in excellent agreement with experimental data40,57 by using this model. In addition, this modified force field was also capable of describing the solvation behavior of noble gases58 and carbon dioxide59 very satisfactorily. By accurately determining temperature dependent solvation properties, we could demonstrate that the entropy contribution to the solvation free energy plays an important role in the solvation process, not unlike the hydrophobic hydration of small apolar particles in liquid water.60–63 Moreover, also an entropy-driven “solvophobic interaction” of apolar particles could be observed in ILs,58 indicating that specific solvent-mediated interactions could play an important role in ILs.
 | ||
| Fig. 1 Schematic representation of the studied ionic liquids 1-n-alkyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide [Cnmim][NTf2]. | ||
Here we focus on the infinite dilution properties of “light gases”, like carbon dioxide, oxygen, nitrogen, methane, argon, neon, and hydrogen in imidazolium based ILs. The chosen gases cover a spectrum from very weakly interacting gases, such as hydrogen, to moderately strong interacting molecules, such as carbon dioxide. We apply Widom's particle insertion technique for calculating temperature dependent solvation free energies and solubilities of these gases in imidazolium-based ILs of the type [Cnmim][NTf2] with varying chain lengths n = 2, 4, 6, 8. In addition, to validate these calculations we also use Bennett's overlapping distribution method for selected examples. Calculated Henry's law constants are compared with available experimental data. The temperature behavior of the solubility as well as its dependence of the alkyl chain lengths in the imidazolium cations is determined and discussed. The motivation of our study is to reveal common “universal” features of the temperature dependent solvation under well controlled conditions, and to provide a reasonable guidance for cases, where experimental data from different sources disagree significantly.
2 Experimental section
2.1 Molecular dynamics simulations
We perform constant pressure (NPT) MD simulations of imidazolium based ILs of the type [Cnmim][NTf2] for different chain lengths n = 2, 4, 6, 8 at a pressure of 1 bar, covering a broad temperature range between 300 K and 500 K. All simulated systems are composed of 343 ion pairs, applying the force field from the study by Köddermann et al.57 An additional minor modification from the simulation setup used in previous studies40,57 is that all bond-lengths were kept fixed. As shown before59 this does not have any impact on the studied properties. A cubic simulation box was used, and the system size with 343 ion pairs was chosen, which is large enough that thermodynamical properties do not depend on the system size, as it was reported by Wittich et al.64 for a system of 125 ion pairs of [C4mim][PF6]. For the solutes, various models from the literature without modification of the interacting parameters were employed. For hydrogen, the potential obtained from the study by Potkowski et al.65 was used. Potential modes reported by Guillot et al.66 were employed to describe the noble gases and methane. Nitrogen was described by the potential obtained from the study by Potoff et al.67 and oxygen by the potential obtained from the study by Hansen et al.68 Finally, carbon dioxide the EPM2-model of Harris and Yung69 was used in a modified way as described before.59 All parameters describing the solutes are given in Table 1.All simulations reported here were performed using the Gromacs simulation program.70 The preparation of topology files, as well as the data analysis, was performed using the most recent version of the MOSCITO suite of programs.71 Production runs of 10 ns length were employed for every temperature, starting from previously well equilibrated configurations. To avoid artifacts due to slow dynamics at low temperatures these systems were pre-equilibrated at 500 K. A Nosé–Hoover thermostat72,73 and a Parrinello–Rahman barostat74,75 with coupling times τT = 1.0 ps and τp = 2.0 ps were used to control constant temperature and pressure (1 bar) conditions. The electrostatic interactions were treated by particle mesh Ewald summation.76 A real space cutoff of 1.2 nm was employed, and a mesh spacing of approximately 0.12 nm (4th order interpolation) had been used to determine the reciprocal lattice contribution. The Ewald convergence parameter was set to a relative accuracy of the Ewald sum of 10−5. Lennard-Jones cutoff corrections for energy and pressure were considered. A 2 fs timestep was used in all simulations, and in every 25th step a configuration was saved. Distance constraints were solved by means of the SHAKE procedure.77 The thermodynamic properties of the simulated ILs are essentially identical to the properties reported in ref. 59.
2.2 Infinite dilution properties
The solubility of a solute A in a solvent B is conveniently described by the Ostwald coefficient Ll/g = ρlA/ρgA, where ρlA and ρgA are the number densities of the solute in the liquid and the gas phase of component B, respectively, when both phases are in equilibrium. Alternatively, the solubility of solute A can be expressed in terms of the inverse Henry's law constant kH−1. The relationship between Henry's law constant and the excess chemical potential μlex,A in the liquid phase is given by78| kH−1 = exp[−βμlex,A]/(ρlILRT), | (1) |
According to Widom's potential distribution theorem,79,80 the excess chemical potential μex can be computed as a volume weighted ensemble average
μex = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln〈Vexp(−βΦ)〉/〈V〉. ln〈Vexp(−βΦ)〉/〈V〉. | (2) |
As control, we also determine the excess chemical potential from energy histograms81,82 computed for the energy change ΔU = U(N + 1) − U(N) associated with the insertion p0(ΔU) and removal p1(ΔU) of an (N + 1)th gas molecule from the constant pressure (NPT) simulation. The two distribution functions are related according to
 | (3) |
| lnp1(ΔU) − lnp0(ΔU) = βμex − βΔU. | (4) |
and

such that

| μex = f1(ΔU) − f0(ΔU). | (5) |
All computed energies are based on the minimum image and include a reaction field correction similar to Roberts and Schnitker.85 Cut-off corrections for the dispersion interactions are included.86 A total of 2 × 105 configurations were analyzed for each IL and for every temperature. Each configuration was sampled by 103 random insertions to determine the f0-functions. The energies computed for those insertions have also been used to determine “Widom-estimates” for the excess chemical potentials. We would like to point out that the values computed from particle insertions are found to lie within the statistical uncertainty of the data from the overlapping distribution theory. Note that the choice of the sampling rate is a critical parameter for successfully computing the chemical potentials via Widom's insertion technique. By reducing the sampling rate significantly, we denote a systematic deviation of the “Widom-estimate” from the data obtained via the overlapping distribution method. This effect was observed by us for sampling rates being about two orders of magnitude lower than the rates reported here. All “converged” computed Henry coefficients are shown in Table 2.
| T/K | k H/bar | ||||
|---|---|---|---|---|---|
| [C2mim] | [C4mim] | [C6mim] | [C8mim] | ||
| CO2 | 300 | 34 ± 3 | 28 ± 3 | 29 ± 3 | 22 ± 3 |
| 350 | 87 ± 6 | 73 ± 5 | 63 ± 4 | 56 ± 6 | |
| 400 | 140 ± 8 | 121 ± 6 | 107 ± 6 | 98 ± 9 | |
| 450 | 207 ± 10 | 171 ± 9 | 152 ± 8 | 139 ± 11 | |
| 500 | 262 ± 12 | 224 ± 11 | 191 ± 9 | 185 ± 13 | |
| Kr | 300 | 188 ± 5 | 143 ± 6 | 119 ± 6 | 89 ± 6 |
| 350 | 266 ± 4 | 210 ± 3 | 175 ± 2 | 139 ± 3 | |
| 400 | 327 ± 3 | 261 ± 3 | 221 ± 2 | 182 ± 3 | |
| 450 | 377 ± 2 | 306 ± 2 | 257 ± 1 | 214 ± 1 | |
| 500 | 411 ± 2 | 337 ± 3 | 284 ± 2 | 240 ± 1 | |
| CH4 | 300 | 300 ± 10 | 224 ± 15 | 214 ± 15 | 142 ± 10 |
| 350 | 381 ± 7 | 300 ± 7 | 277 ± 6 | 203 ± 5 | |
| 400 | 441 ± 3 | 351 ± 4 | 321 ± 4 | 249 ± 3 | |
| 450 | 486 ± 5 | 391 ± 3 | 353 ± 3 | 278 ± 2 | |
| 500 | 509 ± 4 | 415 ± 3 | 373 ± 3 | 299 ± 2 | |
| Ar | 300 | 436 ± 9 | 350 ± 12 | 318 ± 14 | 242 ± 15 |
| 350 | 508 ± 6 | 418 ± 6 | 378 ± 4 | 303 ± 6 | |
| 400 | 548 ± 4 | 455 ± 5 | 410 ± 4 | 340 ± 4 | |
| 450 | 571 ± 3 | 478 ± 3 | 427 ± 2 | 356 ± 2 | |
| 500 | 577 ± 2 | 486 ± 3 | 431 ± 3 | 365 ± 2 | |
| O2 | 300 | 652 ± 22 | 523 ± 21 | 456 ± 23 | 378 ± 26 |
| 350 | 720 ± 7 | 596 ± 8 | 517 ± 5 | 447 ± 10 | |
| 400 | 744 ± 5 | 619 ± 7 | 540 ± 5 | 480 ± 6 | |
| 450 | 750 ± 5 | 629 ± 4 | 545 ± 3 | 482 ± 3 | |
| 500 | 732 ± 4 | 621 ± 5 | 536 ± 3 | 477 ± 3 | |
| N2 | 300 | 1048 ± 47 | 858 ± 42 | 766 ± 46 | 651 ± 53 |
| 350 | 1062 ± 15 | 898 ± 14 | 795 ± 12 | 707 ± 20 | |
| 400 | 1036 ± 7 | 875 ± 11 | 775 ± 9 | 703 ± 11 | |
| 450 | 991 ± 8 | 844 ± 7 | 741 ± 4 | 666 ± 6 | |
| 500 | 942 ± 4 | 802 ± 7 | 699 ± 4 | 629 ± 5 | |
| Ne | 300 | 3418 ± 63 | 3051 ± 88 | 2898 ± 110 | 2752 ± 172 |
| 350 | 2507 ± 23 | 2222 ± 25 | 2088 ± 16 | 1964 ± 36 | |
| 400 | 1943 ± 13 | 1716 ± 10 | 1587 ± 13 | 1480 ± 8 | |
| 450 | 1570 ± 12 | 1388 ± 8 | 1267 ± 5 | 1185 ± 10 | |
| 500 | 1318 ± 6 | 1147 ± 5 | 1047 ± 5 | 977 ± 7 | |
| H2 | 300 | 3875 ± 98 | 3286 ± 121 | 3313 ± 157 | 2718 ± 173 |
| 350 | 2867 ± 42 | 2466 ± 44 | 2415 ± 33 | 2066 ± 42 | |
| 400 | 2216 ± 16 | 1906 ± 23 | 1835 ± 19 | 1610 ± 21 | |
| 450 | 1805 ± 22 | 1552 ± 10 | 1462 ± 7 | 1279 ± 9 | |
| 500 | 1510 ± 5 | 1296 ± 10 | 1190 ± 13 | 1058 ± 8 | |
From the temperature dependence of the computed solvation free energy for infinite dilution, we can comment on the behavior of the first and second derivatives of free energy with respect to temperature. So the solvation entropies, enthalpies, and heat capacities are obtained from fits of the data to a second order expansion of the solvation free energy around the reference state (T° = 298 K at P° = 1 bar) according to
 | (6) |
 and
and  represent the solvation free energy and the solvation entropy in the reference state, respectively, relating the corresponding enthalpy
represent the solvation free energy and the solvation entropy in the reference state, respectively, relating the corresponding enthalpy  and entropy
and entropy  via
via . According to the second order expansion, the solvation heat capacity cP,ex is assumed to be constant over the considered temperature range. The fitted parameters are provided in Table 3.
. According to the second order expansion, the solvation heat capacity cP,ex is assumed to be constant over the considered temperature range. The fitted parameters are provided in Table 3.
| [C2mim] | [C4mim] | [C6mim] | [C8mim] | ||
|---|---|---|---|---|---|
| CO2 |

|
−2.57 | −2.79 | −2.45 | −2.89 |

|
−39.1 | −40.1 | −33.8 | −39.2 | |

|
−14.2 | −14.7 | −12.5 | −14.6 | |
| c P,ex/J K−1 mol−1 | 45 | 48 | 34 | 43 | |
| Kr |

|
1.66 | 1.24 | 1.09 | 0.56 |

|
−20.5 | −21.3 | −20.9 | −23.1 | |

|
−4.45 | −5.09 | −5.13 | −6.32 | |
| c P,ex/J K−1 mol−1 | 19.9 | 21.9 | 21.7 | 26.8 | |
| CH4 |

|
2.78 | 2.36 | 2.55 | 1.73 |

|
−18.5 | −19.7 | −18.4 | −21.8 | |

|
−2.72 | −3.50 | −2.92 | −4.76 | |
| c P,ex/J K−1 mol−1 | 17.6 | 20.8 | 18.6 | 27.0 | |
| Ar |

|
3.76 | 3.48 | 3.54 | 3.06 |

|
−16.2 | −16.7 | −16.6 | −18.4 | |

|
−1.07 | −1.51 | −1.42 | −2.41 | |
| c P,ex/J K−1 mol−1 | 16.9 | 18.4 | 19.3 | 23.5 | |
| O2 |

|
4.76 | 4.48 | 4.43 | 4.17 |

|
−16.8 | −17.2 | −17.1 | −19.0 | |
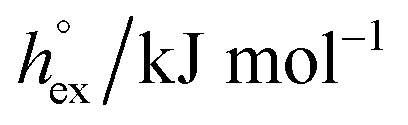
|
−0.23 | −0.66 | −0.67 | −1.50 | |
| c P,ex/J K−1 mol−1 | 19.0 | 20.0 | 20.7 | 26.0 | |
| N2 |

|
5.95 | 5.72 | 5.73 | 5.53 |

|
−15.5 | −16.4 | −16.2 | −18.4 | |

|
1.34 | 0.84 | 0.90 | 0.04 | |
| c P,ex/J K−1 mol−1 | 17.0 | 20.0 | 21.0 | 27.6 | |
| Ne |

|
8.89 | 8.90 | 9.07 | 9.13 |

|
−5.66 | −5.91 | −5.36 | −4.96 | |

|
7.20 | 7.14 | 7.47 | 7.65 | |
| c P,ex/J K−1 mol−1 | 10.1 | 12.3 | 11.7 | 10.8 | |
| H2 |

|
9.19 | 9.09 | 9.40 | 9.11 |

|
−7.29 | −7.72 | −7.29 | −8.99 | |

|
7.02 | 6.79 | 7.23 | 6.43 | |
| c P,ex/J K−1 mol−1 | 11.3 | 13.4 | 14.9 | 19.0 | |
3 Results and discussion
We have computed the solvation free energies μex and the associated Henry coefficients kH of CO2, O2, N2, CH4, Kr, Ar, Ne and H2 at infinite dilution from MD simulations for the four ILs [Cnmim][NTf2] with n = 2, 4, 6, 8, employing the sampling techniques discussed in the previous section. The density data for the simulated IL necessary to inter-convert Henry coefficients kH and solvation free energies μex can be found in ref. 59. All computed Henry coefficients are summarized in Table 2. The temperature dependence of the corresponding solvation free energies μex(T) has been fitted to a second order expansion around a reference state following eqn (6). The parameters obtained for a reference temperature of T° = 298 K are given in Table 3. In Fig. 2 computed solubilities (here given as inverse Henry's law constants) are compared with available experimental and theoretical data. For reasons of clarity, we restrict this comparison to data based on solvation in [C6mim][NTf2], because most experimental datasets are only available for this particular IL, since it had been selected as a reference compound for an IUPAC experimental validation project.88,89 | ||
| Fig. 2 Solubilities kH−1 = exp[−βμlex,gas]/ρlILRT of selected gases in [C6mim][NTf2]. Shown are data for (a) CO2, (b) CH4, (c) Ar, (d) O2, (e) N2, and (f) H2 at 1 bar. The filled symbols represent experimental data given according to the indicated sources. Open symbols specify data from molecular simulations, while crosses represent data obtained from alternative theoretical predictions. All dashed lines show theoretical predictions based on our enthalpy–entropy compensation model. The upper and lower black thin dashed lines are temperature-predictions for solubilities twice and half of the reference-value used for our MD simulation-data (given as black thick dashed lines). The red dashed line represents an enthalpy–entropy compensation model prediction for the experimental solubility data obtained from the study by Kumełan et al. References for the experimental and theoretical data are given in the text. Error bars are shown where they were available and are not smaller than the symbol size. | ||
The remainder of this section is organized as follows: first we will compare our simulated solubility data with available experimental and theoretical data. A short discussion of the available solubility data is given for every gas in detail. The following sections will then focus on a systematic rationalization of the effect of varying the alkyl side chain length, and of changing the temperature based on an enthalpy–entropy compensation behavior obeyed by all the considered gases.
3.1 Comparison with available experimental and theoretical data
3.2 Alkyl side chain length dependence
The computed solubilities as a function of the alkyl side chain length obtained from MD simulation are given as inverse Henry coefficients, and are shown as full symbols in Fig. 3. The data indicate a rather small variation of Henry coefficients for ILs with varying chain lengths. However, there is a tendency towards higher solubilities for gases in ILs with longer alkyl side chains. When comparing the solubility data for [C2mim][NTf2] and [C8mim][NTf2] for all the investigated gases, we consistently observe an increase in solubility of about 30% to 40% for the component with the C8-chain. In ref. 59 we reported the observation that the solvation free energies μex of carbon dioxide showed almost no chain length dependence at a given temperature. By assuming μex to be chain length independent, it follows that the chain length dependence of the solubility data at a given temperature can be solely expressed due to density scaling according to eqn (1), leading to an approximate expression: | (7) |
| ρIL(n) = ρ(0)IL + ρ(1)IL·n + ρ(2)IL·n2 | (8) |
 | ||
| Fig. 3 Comparison of inverse Henry coefficients of all simulated gases in [Cnmim][NTf2] with varying chain lengths n = 2, 4, 6, 8 at 400 K. The dashed lines are predictions of the chain length dependence based on the density scaling procedure described in eqn (7). | ||
3.3 Temperature dependence: enthalpy–entropy compensation effect
From the wealth of experimental and theoretical solubility data presented in Fig. 2 (including our data) we conclude that there apparently exists a systematic relationship between the temperature dependent slope of the solubilities of different gases and their interaction-strength with the solvent: rather “strongly” interacting species, such as CO2, show apparently a strong “anomalous” temperature dependence of the solubility, whereas weakly interacting species such as oxygen and argon show a significantly weaker temperature behavior. Finally, for the case of molecular hydrogen (H2), we observe a change in the sign of the slope, showing a strongly increasing solubility with increasing temperature. The latter finding is apparently supported by the majority of experimental datasets. However, for two cases, namely nitrogen and hydrogen, there are substantially conflicting results from different sources, each suggesting a very different kind of temperature behavior. How could this be resolved? We think that the apparent systematic trend is deeply rooted in the free energy landscape explored by a solvated gas molecule, and that the observed trend can be explained by purely thermodynamic means.By fitting the temperature dependent solvation free energies μex(T) to the second order expansion around a thermodynamic reference state (T° = 298 K and P° = 1 bar) given by eqn (6), we obtain standard solvation free energies  , entropies
, entropies  , enthalpies
, enthalpies  , as well as the solvation heat capacities
, as well as the solvation heat capacities  . Data computed for all studied gases and ILs are collected in Table 3. In addition, we also consider data for a modified CO2 molecule, where we have systematically weakened the interaction with the solvent by switching off the Coulomb interaction, and by scaling the Lennard-Jones interaction using a factor f with εij = (fεiiεjj)1/2. Finally, this prototypical molecule is transformed into a purely repulsive component by modeling the solute–solvent interaction solely via Weeks–Chandler–Andersen-type (WCA) interactions according to Vij(r) = Vij,LJ(r) + εij for r ≤ rLJ,min, and Vij(r) = 0 otherwise. Following the procedure suggested by Simha et al. for the dissolution of gases in polymers,100,101Fig. 4a shows a plot of the standard solvation free energy
. Data computed for all studied gases and ILs are collected in Table 3. In addition, we also consider data for a modified CO2 molecule, where we have systematically weakened the interaction with the solvent by switching off the Coulomb interaction, and by scaling the Lennard-Jones interaction using a factor f with εij = (fεiiεjj)1/2. Finally, this prototypical molecule is transformed into a purely repulsive component by modeling the solute–solvent interaction solely via Weeks–Chandler–Andersen-type (WCA) interactions according to Vij(r) = Vij,LJ(r) + εij for r ≤ rLJ,min, and Vij(r) = 0 otherwise. Following the procedure suggested by Simha et al. for the dissolution of gases in polymers,100,101Fig. 4a shows a plot of the standard solvation free energy  vs. the solvation enthalpy
vs. the solvation enthalpy  for all studied gases in all solvents. It is evident that both properties are linearly related for the entire set of solvation data. This linear relationship can be utilized to predict the temperature dependent solvation data. Furthermore, it can provide us with a quantitative representation of the notion that the absolute solubility of a certain compound and its temperature behavior are somehow related. The thermodynamic definition of the free energy implies that the solvation entropy and enthalpy have to be linearly related as well, as it is demonstrated in Fig. 4b. To quantify the relations shown in Fig. 4 we use the following relation:
for all studied gases in all solvents. It is evident that both properties are linearly related for the entire set of solvation data. This linear relationship can be utilized to predict the temperature dependent solvation data. Furthermore, it can provide us with a quantitative representation of the notion that the absolute solubility of a certain compound and its temperature behavior are somehow related. The thermodynamic definition of the free energy implies that the solvation entropy and enthalpy have to be linearly related as well, as it is demonstrated in Fig. 4b. To quantify the relations shown in Fig. 4 we use the following relation:
 | (9) |
 and
and  can be obtained as
can be obtained as | (10) |
 and
and  over a rather wide range of interaction strengths is apparently intimately related to the process of solvation of small gas molecules. In particular, how a gas molecule and its solvation shell explore the configurational space of the solvated state. The consequence is a positive correlation between the solvation entropy and the solvation enthalpy. We would like to point out that by restricting our study to “small gas molecules”, the variation in the size of the molecules has apparently no big effect, and is being accounted for effectively. For larger solutes this might not necessarily be the case.
over a rather wide range of interaction strengths is apparently intimately related to the process of solvation of small gas molecules. In particular, how a gas molecule and its solvation shell explore the configurational space of the solvated state. The consequence is a positive correlation between the solvation entropy and the solvation enthalpy. We would like to point out that by restricting our study to “small gas molecules”, the variation in the size of the molecules has apparently no big effect, and is being accounted for effectively. For larger solutes this might not necessarily be the case.
 | ||
Fig. 4 (a) Correlation between the solvation free energy  and the solvation enthalpy and the solvation enthalpy  of gases dissolved in [Cnmim][NTf2] in the reference state. (b) Correlation between the solvation entropy of gases dissolved in [Cnmim][NTf2] in the reference state. (b) Correlation between the solvation entropy  and the solvation enthalpy and the solvation enthalpy  . Shown are data for all other examined gases (colored) as well as the scaled potential model variants of CO2 (black squares), taken from ref. 59. . Shown are data for all other examined gases (colored) as well as the scaled potential model variants of CO2 (black squares), taken from ref. 59. | ||
The relationship between  and
and  as outlined above essentially determines the coupling between the absolute solubility and its temperature dependence. However, the second order expansion of μex(T) given in eqn (6) also requires the knowledge of the heat capacity of solvation cP,ex, to fully determine the temperature dependence of all our solubility data. Fortunately, the variation of the computed cP,ex-values, given in Table 3, and shown in Fig. 5, has no big effect. If we neglect the heat capacity contribution completely by setting cP,ex = 0, we arrive at a description, which is qualitatively correct for all investigated gases (not shown). This procedure, however, leads to significant deviations of the predicted data from our simulation data for temperatures above 400 K. A much better description is achieved by using a common value of cP,ex = 20 J K−1 mol−1 for all gases instead, as it is indicated by the predictions represented by thin dashed lines in Fig. 6. However, to improve things further, we make use of a negative correlation between
as outlined above essentially determines the coupling between the absolute solubility and its temperature dependence. However, the second order expansion of μex(T) given in eqn (6) also requires the knowledge of the heat capacity of solvation cP,ex, to fully determine the temperature dependence of all our solubility data. Fortunately, the variation of the computed cP,ex-values, given in Table 3, and shown in Fig. 5, has no big effect. If we neglect the heat capacity contribution completely by setting cP,ex = 0, we arrive at a description, which is qualitatively correct for all investigated gases (not shown). This procedure, however, leads to significant deviations of the predicted data from our simulation data for temperatures above 400 K. A much better description is achieved by using a common value of cP,ex = 20 J K−1 mol−1 for all gases instead, as it is indicated by the predictions represented by thin dashed lines in Fig. 6. However, to improve things further, we make use of a negative correlation between  and cP,ex, suggested in Fig. 5. To complete our model we make use of the linear relationship between
and cP,ex, suggested in Fig. 5. To complete our model we make use of the linear relationship between  and cP,ex:
and cP,ex:
 | (11) |
 | ||
Fig. 5 Correlation between the solvation free energy  and the solvation heat capacity cP,ex dissolved in [Cnmim][NTf2] obtained for the different potential model variants of CO2 (black)59 and for all other examined gases (colored) under standard conditions. and the solvation heat capacity cP,ex dissolved in [Cnmim][NTf2] obtained for the different potential model variants of CO2 (black)59 and for all other examined gases (colored) under standard conditions. | ||
 | ||
| Fig. 6 The symbols denote the temperature dependence of the inverse Henry coefficients for all simulated gases dissolved in [C6mim][NTf2]. The lines indicate the predictions of the enthalpy–entropy compensation model. Thin dashed lines: cP,ex = 20 J K−1 mol−1. Heavy solid lines: cP,ex according to eqn (11). | ||
In addition to the temperature dependent experimental data, we have included predictions according to our enthalpy–entropy compensation model also in Fig. 2. The thick dashed lines in Fig. 2 represent data chosen to match our simulation data. The thin dashed lines are used to illustrate the temperature evolution of the model predictions in the vicinity of one particular solute. Here reference solubilities were chosen to be half and twice the size of the reference-solubility used for matching our MD simulation data. It is quite evident that particularly for N2 and H2, several experimental datasets are incompatible with our model predictions. However, the red dashed lines shown in Fig. 2 demonstrate that the experimental data obtained by the Maurer group in Kaiserslautern (the data obtained from the study by Kumełan et al. are represented by red symbols in Fig. 2a, b, d and f) for a variety of solutes are consistently in very good agreement with the predictions of the enthalpy–entropy compensation model. This includes even the controversial case of molecular hydrogen.
Finally, we would put another argument forward, that a positive slope for the temperature dependence of the hydrogen-solubility data is a very likely scenario. Following the arguments of Hayduk and Laudie,102 which have been reviewed and extended by Beutier and Renon,103 all Henry coefficients and hence all solubilities obtained for a certain solvent should meet at the critical point of that solvent, in our case the ILs. The solubility data shown in Fig. 6 clearly indicate a convergent behavior with increasing temperature. By extrapolating the enthalpy–entropy compensation model to very high temperatures, we find that this convergence even continues. In addition, Fig. 7 demonstrates that by assuming cP,ex to be constant, the model even predicts a single common intersection temperature T* for the solubility of all gases. Eqn (6) and (9) imply that the common temperature T* is defined by
 | (12) |
 | (13) |
 | ||
| Fig. 7 Enthalpy–entropy compensation model predictions for all gases in [C6mim][NTf2] with a constant cP,ex = 20 J K−1 mol−1. Indicated is the intersection temperature T* = 754 K. (a) Predicted solubilities by inverse Henry coefficients. Red squares indicate the simulated values given in Table 2. (b) Predicted solvation free energies μex(T). | ||
4 Conclusion
The systematic behavior of the gas solubility in ionic liquids is studied and described with the help of extensive molecular dynamics simulations. The solubility of hydrogen, oxygen, nitrogen, methane, krypton, argon, neon and carbon dioxide in the ionic liquids of type 1-n-alkyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([Cnmim][NTf2]) with varying chain lengths n = 2, 4, 6, 8 is computed for a large temperature range from 300 K up to 500 K at 1 bar. By applying Widom's particle insertion technique, as well as Bennett's overlapping distribution method, we are able to determine solvation free energies of those selected light gases in imidazolium based ionic liquids with great statistical accuracy. A detailed comparison of the computed solubility data with available experimental and theoretical data is provided.We observe that the chain length dependence of the computed solubility in various solvents can be mostly attributed to the change in the number-density of ion pairs in the solvent, as the computed solvation free energies show almost no chain length dependence.
The data obtained from our MD simulations clearly show that the magnitude of the solvation free energy at a defined reference temperature and its temperature-derivatives are intimately related to one another. This is a consequence of a solvation entropy–enthalpy compensation effect, which seems to be a defining feature of the solvation of small molecules in the investigated ionic liquids. This effect is leading to simple analytical relations quantitatively describing the temperature dependent solubility of gases solely depending on the absolute solubility value at a defined reference temperature.
The enthalpy–entropy compensation model is also predicts that the solubility data all meet at a single temperature, which is in line with the observation made for various fluids that the solubilities for gases meet at the critical temperature. However, this feature of a common temperature exists only, if the model is used with a unique heat capacity of solvation valid for all gases. Since the computed heat capacities of solvation do not vary strongly for different gases, a common value of cP,ex = 20 J K−1 mol−1 is a reasonable approximation for all the investigated solutes.
We would like to point out that the enthalpy–entropy compensation model is particularly helpful for assessing the solvation behavior of very light gases, such as hydrogen, where conflicting experimental data have been reported.
Acknowledgements
This work has been supported by the German Science Foundation (DFG) priority program SPP 1570 with additional support from SFB 652.References
-
P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, VCH-Wiley, Weinheim, 2nd edn, 2007 Search PubMed
.
- C. A. Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC
- F. Endres and S. Z. El Abedin, Phys. Chem. Chem. Phys., 2006, 8, 2101–2116 RSC
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed
- M. Smiglak, A. Metlen and R. D. Rogers, Acc. Chem. Res., 2007, 40, 1182–1192 CrossRef CAS PubMed
- N. V. Plechkova and K. R. Seddon, Chem. Rev., 2008, 37, 123–150 RSC
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef
- D. J. Tempel, P. B. Henderson, J. R. Brzozowski, R. M. Pearlstein and H. Cheng, J. Am. Chem. Soc., 2008, 130, 400–401 CrossRef CAS PubMed
- J. Huang and T. Rüther, Aust. J. Chem., 2009, 62, 298–308 CrossRef CAS
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2010, 43, 152–159 CrossRef CAS PubMed
- S. Raeissi and C. J. Peters, Green Chem., 2009, 11, 185–192 RSC
- C. Myers, H. Pennline, D. Luebke, J. Ilconich, J. K. Dixon, E. J. Maginn and J. F. Brennecke, J. Membr. Sci., 2008, 322, 28–31 CrossRef CAS
- J. Ilconich, C. Myers, H. Pennline and D. Luebke, J. Membr. Sci., 2007, 298, 41–47 CrossRef CAS
- Z. Lei, C. Dai and B. Chen, Chem. Rev., 2013, 114, 1289–1326 CrossRef PubMed
- A. Finotello, J. E. Bara, D. Camper and R. D. Noble, Ind. Eng. Chem. Res., 2008, 47, 3453–3459 CrossRef CAS
- J. Jacquemin, P. Husson, V. Majer and M. F. Costa Gomes, J. Solution Chem., 2007, 36, 967–979 CrossRef CAS
- M. F. Costa Gomes, J. Chem. Eng. Data, 2007, 52, 472–475 CrossRef
- J. Kumełan, A. P. S. Kamps, D. Tuma and G. Maurer, J. Chem. Thermodyn., 2006, 38, 1396–1401 CrossRef
- P. J. Dyson, G. Laurenczy, C. A. Ohlin, J. Vallance and T. Welton, Chem. Commun., 2003, 2418–2419 RSC
- J. Jacquemin, M. F. Costa Gomes, P. Husson and V. Majer, J. Chem. Thermodyn., 2006, 38, 490–502 CrossRef CAS
- J. Jacquemin, P. Husson, V. Majer, A. A. H. Padua and M. F. Costa Gomes, Green Chem., 2008, 10, 944–950 RSC
- J. Jacquemin, P. Husson, V. Majer and M. F. Costa Gomes, Fluid Phase Equilib., 2006, 1, 87–95 CrossRef
- J. Kumełan, A. P. S. Kamps, D. Tuma and G. Maurer, J. Chem. Eng. Data, 2006, 51, 11–14 CrossRef
- J. Kumełan, D. Tuma, A. P. S. Kamps and G. Maurer, J. Chem. Eng. Data, 2010, 55, 165–172 CrossRef
- S. Raeissi, L. J. Florusse and C. J. Peters, J. Chem. Eng. Data, 2011, 56, 1105–1107 CrossRef CAS
- Y. Shimoyama and A. Ito, Fluid Phase Equilib., 2010, 297, 178–182 CrossRef CAS
- N. A. Manan, C. Hardacre, J. Jacquemin, D. W. Rooney and T. G. A. Youngs, J. Chem. Eng. Data, 2009, 54, 2005–2022 CrossRef
- J. Palomar, M. Gonzalez-Miquel, A. Polo and F. Rodriguez, Ind. Eng. Chem. Res., 2011, 50, 3452–3463 CrossRef CAS
- K. Z. Sumon and A. Henni, Fluid Phase Equilib., 2011, 310, 39–55 CrossRef CAS
- J. Abildskov, M. D. Ellegaard and J. P. O'Connell, Fluid Phase Equilib., 2009, 286, 95–106 CrossRef
- J. S. Andreu and L. F. Vega, J. Phys. Chem. B, 2008, 112, 15398–15406 CrossRef CAS PubMed
- X. Zhang, Z. Liu and W. Wang, AIChE J., 2008, 54, 2717–2728 CrossRef CAS
- J. S. Andreu and L. F. Vega, J. Phys. Chem. C, 2007, 111, 16028–16034 CAS
- Y. S. Kim, J. H. Jang, B. D. Lim, J. W. Kang and C. S. Lee, Fluid Phase Equilib., 2007, 256, 70–74 CrossRef CAS
- A. A. Oliferenko, P. V. Oliferenko, K. R. Seddon and J. S. Torrecilla, Phys. Chem. Chem. Phys., 2011, 13, 17262–17272 RSC
- E. J. Maginn, J. Phys.: Condens. Matter, 2009, 21, 373101 CrossRef CAS PubMed
- W. Shi and E. J. Maginn, J. Phys. Chem. B, 2008, 112, 2045–2055 CrossRef CAS PubMed
- W. Shi and E. J. Maginn, J. Phys. Chem. B, 2008, 112, 16710–16720 CrossRef CAS PubMed
- T. Köddermann, D. Paschek and R. Ludwig, ChemPhysChem, 2008, 9, 549–555 CrossRef PubMed
- R. M. Lynden-Bell, N. A. Atamas, A. Vasilyuk and C. G. Hanke, Mol. Phys., 2002, 100, 3225–3229 CrossRef CAS
- C. G. Hanke, A. Johansson, J. B. Harper and R. M. Lynden-Bell, Chem. Phys. Lett., 2003, 374, 85–89 CrossRef CAS
- J. Deschamps, M. F. Costa Gomes and A. A. H. Padua, ChemPhysChem, 2004, 5, 1049–1052 CrossRef CAS PubMed
- J. K. Shah and E. J. Maginn, Fluid Phase Equilib., 2004, 195, 222–223 Search PubMed
- J. K. Shah and E. J. Maginn, J. Phys. Chem. B, 2005, 109, 10395–10405 CrossRef CAS PubMed
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300–5308 CrossRef CAS PubMed
- J. Kumełan, A. P. S. Kamps, I. Urukova, D. Tuma and G. Maurer, J. Chem. Thermodyn., 2005, 37, 595–602 CrossRef
- I. Urukova, J. Vorholz and G. Maurer, J. Phys. Chem. B, 2005, 109, 12154–12159 CrossRef CAS PubMed
- J. Kumełan, A. P. S. Kamps, D. Tuma and G. Maurer, Ind. Eng. Chem. Res., 2007, 46, 8236–8240 CrossRef
- J. Deschamps and A. A. H. Pádua, ACS Symp. Ser., 2005, 901, 150–158 CrossRef CAS
- X. P. Wu, Z. P. Liu and W. C. Wang, Wuli Huaxue Xuebao, 2005, 21, 1138–1142 CAS
- W. Shi, D. C. Sorescu, D. R. Luebke, M. J. Keller and S. Wickramanayake, J. Phys. Chem. B, 2010, 114, 6531–6541 CrossRef CAS PubMed
- A. F. Ghobadi, V. Taghikhani and J. R. Elliott, J. Phys. Chem. B, 2011, 115, 13599–13607 CrossRef CAS PubMed
- R. Singh, E. Marin-Rimoldi and E. J. Maginn, Ind. Eng. Chem. Res., 2015, 54, 4385–4395 CrossRef CAS
- F. Dommert, K. Wendler, R. Berger, L. Delle Site and C. Holm, ChemPhysChem, 2012, 13, 1625–1637 CrossRef CAS PubMed
- J. N. Canongia Lopes, J. Deschamps and A. A. H. Padua, J. Phys. Chem. B, 2004, 108, 2038–2047 CrossRef
- T. Köddermann, D. Paschek and R. Ludwig, ChemPhysChem, 2007, 8, 2464–2470 CrossRef PubMed
- D. Paschek, T. Köddermann and R. Ludwig, Phys. Rev. Lett., 2008, 100, 115901 CrossRef PubMed
- D. Kerlé, R. Ludwig, A. Geiger and D. Paschek, J. Phys. Chem. B, 2009, 113, 12727–12735 CrossRef PubMed
- L. R. Pratt, Annu. Rev. Phys. Chem., 2003, 53, 409–436 CrossRef PubMed
- N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS
- B. Widom, P. Bhimalapuram and K. Koga, Phys. Chem. Chem. Phys., 2003, 5, 3085–3093 RSC
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed
- B. Wittich and U. K. Deiters, J. Phys. Chem. B, 2010, 114, 3452–3463 CrossRef PubMed
- K. Patkowski, W. Cencek, P. Jankowski, K. Szalewicz, J. Mehl, G. Garberoglio and A. H. Harvey, J. Chem. Phys., 2008, 129, 094304 CrossRef PubMed
- B. Guillot and Y. Guissani, J. Chem. Phys., 1993, 99, 8075–8094 CrossRef CAS
- J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS
- N. Hansen, F. A. B. Agbor and F. J. Keil, Fluid Phase Equilib., 2007, 259, 180–188 CrossRef CAS
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CrossRef CAS
-
D. Paschek, MOSCITO 4: MD simulation package, 2008, http://ganter.chemie.uni-dortmund.de/MOSCITO Search PubMed
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS
- S. Nosé and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef
- U. Essmann, L. Perera, M. L. Berkowitz, T. A. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS
- R. P. Kennan and G. L. Pollack, J. Chem. Phys., 1990, 93, 2724–2735 CrossRef CAS
- B. Widom, J. Chem. Phys., 1963, 39, 2808–2812 CrossRef CAS
-
T. L. Beck, M. E. Paulaitis and L. R. Pratt, The Potential Distribution Theorem and Models of Molecular Solutions, Cambridge University Press, Cambrigde, UK, 2006 Search PubMed
- C. H. Bennett, J. Comput. Phys., 1976, 22, 245–268 CrossRef
- K. S. Shing and K. E. Gubbins, Mol. Phys., 1983, 49, 1121–1138 CrossRef CAS
-
D. Frenkel and B. Smit, Understanding Molecular Simulation. From Algorithms to Applications, Academic Press, San Diego, 2nd edn, 2002 Search PubMed
- D. Paschek, J. Chem. Phys., 2004, 120, 6674–6690 CrossRef CAS PubMed
- J. E. Roberts and J. Schnitker, J. Chem. Phys., 1994, 101, 5024–5031 CrossRef CAS
- D. Paschek, J. Chem. Phys., 2004, 120, 10605–10617 CrossRef CAS PubMed
- To interconvert solvation free energies μex and Henry constants kH, we use a second order polynomial fitted to the temperature dependent ion-pair density of the simulated ILs: ρIL(T) = ρ(0)IL + ρ(1)IL·T + ρ(2)IL·T2. [C2mim][NTf2]: ρ(0)IL = 2.985 nm−3, ρ(1)IL = −2.51 × 10−3 nm−3 K−1, and ρ(2)IL = 8.46 × 10−7 nm−3 K−2. [C4mim][NTf2]: ρ(0)IL = 2.639 nm−3, ρ(1)IL = −2.18 × 10−3 nm−3 K−1, and ρ(2)IL = 6.85 × 10−7 nm−3 K−2. [C6mim][NTf2]: ρ(0)IL = 2.368 nm−3, ρ(1)IL = −1.94 × 10−3 nm−3 K−1, and ρ(2)IL = 5.90 × 10−7 nm−3 K−2. [C8mim][NTf2]: ρ(0)IL = 2.127 nm−3, ρ(1)IL = −1.56 × 10−3 nm−3 K−1, and ρ(2)IL = 2.92 × 10−7 nm−3 K−2.
- K. N. Marsh, J. F. Brennecke, R. D. Chirico, M. Frenkel, A. Heintz, J. W. Magee, C. J. Peters, L. P. N. Rebelo and K. R. Seddon, Pure Appl. Chem., 2009, 81, 781–790 CrossRef CAS
- R. D. Chirico, V. Diky, J. W. Magee, M. Frenkel and K. N. Marsh, Pure Appl. Chem., 2009, 81, 791–828 CrossRef CAS
- J. Kumełan, A. P. S. Kamps, D. Tuma and G. Maurer, J. Chem. Thermodyn., 2006, 38, 1396–1401 CrossRef
- S. S. Moganty and R. E. Baltus, Ind. Eng. Chem. Res., 2010, 49, 5846–5853 CrossRef CAS
- D. Kerlé, R. Ludwig, A. Geiger and D. Paschek, Z. Phys. Chem., 2013, 227, 167–176 CrossRef
- D. Camper, J. Bara, C. Koval and R. Noble, Ind. Eng. Chem. Res., 2006, 45, 6279–6283 CrossRef CAS
- J. L. Anderson, J. K. Dixon and J. F. Brennecke, Acc. Chem. Res., 2007, 40, 1208–1216 CrossRef CAS PubMed
- J. Blath, M. Christ, N. Deubler, T. Hirth and T. Schiestel, Chem. Eng. J., 2011, 172, 167–176 CrossRef CAS
- W. Afzal, X. Liu and J. M. Prausnitz, J. Chem. Thermodyn., 2013, 63, 88–94 CrossRef CAS
- J. Kumełan, A. P. S. Kamps, D. Tuma and G. Maurer, J. Chem. Eng. Data, 2009, 54, 966–971 CrossRef
- J. L. Anthony, J. L. Anderson, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2005, 109, 6366–6374 CrossRef CAS PubMed
- J. Kumełan, A. P. S. Kamps, D. Tuma and G. Maurer, J. Chem. Eng. Data, 2006, 51, 1364–1367 CrossRef
- R. Simha and T. Somcynsky, Macromolecules, 1969, 2, 342–350 CrossRef CAS
- H. Xie and R. Simha, Polym. Int., 1997, 44, 348–355 CrossRef CAS
- W. Hayduk and H. Laudie, AIChE J., 1973, 19, 1233–1238 CrossRef CAS
- D. Beutier and H. Renon, AIChE J., 1978, 24, 1122–1125 CrossRef
- L. P. N. Rebelo, J. N. Canongia Lopes, J. M. S. S. Esperança and E. Filipe, J. Phys. Chem. B, 2005, 109, 6040–6043 CrossRef CAS PubMed
- M. G. Freire, P. J. Carvalho, A. M. Fernandes, I. M. Marrucho, A. J. Queimada and J. A. P. Coutinho, J. Colloid Interface Sci., 2007, 314, 621–630 CrossRef CAS PubMed
- N. Rai and E. J. Maginn, Faraday Discuss., 2012, 154, 53–69 RSC
- K. S. Rane and J. R. Errington, J. Phys. Chem. B, 2014, 118, 8734–8743 CrossRef CAS PubMed
- A. Yokozeki, M. B. Shiflett, C. P. Junk, L. M. Grieco and T. Foo, J. Phys. Chem. B, 2008, 112, 16654–16663 CrossRef CAS PubMed
- E.-K. Shin, B.-C. Lee and J. S. Lim, J. Supercrit. Fluids, 2008, 45, 282–292 CrossRef CAS
- J. O. Valderrama and P. A. Robles, Ind. Eng. Chem. Res., 2007, 46, 1338–1344 CrossRef CAS
| This journal is © the Owner Societies 2017 |