
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic nitroxyl formation in the ammonia oxidation on platinum via Eley–Rideal reactions†
Yunxi
Yao
 and
Konstantinos P.
Giapis
*
and
Konstantinos P.
Giapis
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: giapis@cheme.caltech.edu
First published on 14th October 2016
Abstract
For over 90 years, nitroxyl (HNO) has been postulated to be an important reaction intermediate in the catalytic oxidation of ammonia to NO and its by-products (N2, N2O), but never proven to form or exist on catalytic surfaces. Here we show evidence from reactive ion beam experiments that HNO can form directly on the surface of polycrystalline Pt exposed to NH3via Eley–Rideal abstraction reactions of adsorbed NH by energetic O+ and O2+ projectiles. The dynamic formation of HNO in a single collision followed up by prompt rebound from the surface prevents subsequent reactive interactions with other surface adsorbates and enables its detection. In addition to HNO, NO and OH are also detected as direct products in what constitutes the concurrent abstraction of three surface adsorbates, namely NH, N, and H, by O+ projectiles with entirely predictable kinematics. While its relation to thermal catalysis may be tenuous, dynamic HNO formation could be important on grain surfaces of interstellar or cometary matter under astrophysical conditions.
Introduction
The catalytic oxidation of ammonia (NH3) to nitric oxide (NO) on Pt–Rh gauze (Ostwald process) is one of the oldest industrial reactions still in use today for manufacturing nitric acid.1 Surface science techniques and computational approaches over several decades have made great strides towards understanding the ammonia oxidation process.2–7 Yet, a broadly accepted molecular-level description of its mechanism is still lacking. It is stunning that three classical ammonia oxidation mechanisms, proposed between 1926–1935, continue to be debated today. Detection of NH on the Pt surface has favored the imide mechanism of Raschig8 and Zawadzki.9 The hydroxylamine (NH2OH) mechanism of Bodenstein10 and the nitroxyl (HNO) mechanism of Andrussow11 have lost appeal, as the postulated surface intermediates remain undetected.6,12,13 However, detection may be elusive because the intermediates are extremely short-lived—consistent with the very fast kinetics of the oxidation reaction. Nitroxyl, in particular, has been implicated even in recent times in the formation not only of NO,6 but also of the main by-product N2O,14–16 an unregulated but serious atmospheric pollutant.17,18 Curiously, nitroxyl has been detected in astrophysical environments (e.g., interstellar clouds),19–21 and hydroxylamine has been hypothesized to be a precursor for the formation of complex prebiotic molecules on the surface of interstellar dust grains.22,23 Elucidating the surface formation mechanisms of nitroxyl and hydroxylamine may therefore be important not only for catalysis but also for astrochemistry.Detection of short-lived reaction intermediates is challenging but critical to revealing the correct elementary steps in heterogeneous catalytic reactions. Both, in situ spectroscopic7,12 and beam scattering techniques2–4,24 have been applied to the search for reactive intermediates on catalytic surfaces used in the ammonia oxidation process. Beam studies can provide direct mass-spectrometric detection of surface species but they must be performed under collision-free conditions (<10−6 Torr), necessitating a way to bridge the “pressure gap”. Regardless of the technique, the conjectured HNO or NH2OH intermediates have not been detected in spite of extensive efforts. Several other stable species were observed, including: NH2, NH, N, O, OH, and H, providing support for the direct oxidation of NH3 by O or O2 on the Pt surface.2,3 Unless they can be stabilized, transient intermediates produced by forced ejection from the surface must survive the collisional interaction and live long enough to reach the mass spectrometer. The shorter the lifetime, the larger the exit energy required. This prerequisite necessitates the use of projectiles with kinetic energies on the high end of the hyperthermal energy regime (10–100 eV). Collisional interactions at these energies include surface sputtering (i.e., energetic ejection of surface species) but also Eley–Rideal (ER) reactions between projectiles and surface adsorbates. Stable species pre-existing on the surface may be detected intact due to sputtering, or as new molecules when they bond with incident projectiles via Eley–Rideal (ER) reactions. The kinematics of the scattered products can help distinguish unambiguously between these two situations. Of course, ER reactions may provide an alternate pathway to the desired intermediate.
Experimental
The scattering apparatus and associated ion beam-line have been described in detail elsewhere.25–27 Isotopically-pure reactive beams of O+ and O2+, extracted from an inductively-coupled plasma and mass-filtered, are directed onto a polycrystalline Pt surface (4N purity, ESPI) which is pre-cleaned by sputtering in situ with an Ar+ gun. Beam energy is controlled by varying externally the plasma bias with respect to ground. Ion beam currents between 3–5 μA for O+ and 8–20 μA for O2+ (Fig. S1, ESI†) are delivered to a grounded surface over a ∼3 mm spot, with tunable energy between 30 and 150 eV. Once plasma conditions are selected, the O+ beam current is relatively constant over the entire energy range. On the contrary, the O2+ beam current varies considerably with beam energy, though it is stable at any given energy. All product intensity data collected are normalized by the corresponding incident current. The energy width of both incident beams is constant at ∼5 eV (FWHM), determined by the electron temperature of the inductively-coupled plasma. The ion beams are delivered to the sample surface at 45° angle of incidence with respect to the surface normal, and products are detected at 45° angle of exit. Gaseous ammonia is dosed in situ through a tube (2 cm away), using a leak valve to adjust background system pressure. The exposure pressure at the sample is not measured but it could be an order of magnitude higher than the background pressure due to direct dosing. The energetic O+ and O2+ projectiles serve as gas-phase reagent and surface probe. They are typically neutralized efficiently on the incoming path and impact the surface as neutral species.28 The energetic collision with the metal surface causes hyperthermal surface ionization,29 yielding charged products without resorting to electron-impact- or photo-ionization schemes. Finally, the scattered products are energy- and mass-resolved using an electrostatic energy analyzer followed up by a quadrupole mass spectrometer with mass resolution less than 0.5 amu to prevent signal interference between adjacent mass numbers (Fig. S2, ESI†).Results and discussion
Here we perform hyperthermal reactive beam scattering not only to probe surface species but also to study the dynamic formation of intermediates via Eley–Rideal reactions in the ammonia oxidation on polycrystalline Pt at room temperature under collision-free conditions. In the presence of surface oxygen, NH3 adsorbs dissociatively on the Pt surface to form dehydrogenated ammonia species, which are believed to react directly with surface O or O2 to form NO.16 Our experiment simulates this situation by exposing the Pt surface to thermal NH3 molecules while simultaneously bombarding the surface with O+ or O2+ projectiles. Partial oxidation intermediates pre-existing on the surface may be forcibly ejected by the projectiles (collision-induced desorption). Reactive beam projectiles can also abstract directly surface species in single-collision events, thus forming a new molecule whose detection can uniquely identify the adsorbate. During such scattering experiments, we detected the elusive HNO reaction intermediate, along with other important species.Since energetic O+ projectiles serve both as a probe and a reagent, it is important to first establish how they interact with a clean Pt surface: scattered O− and O2− are produced in this experiment (Fig. 1a and b). At O+ incidence energy of 78 eV, the scattered O− signal is bimodal with a broad peak at ∼25 eV from sputtering of adsorbed O atoms, and a narrower dynamic peak at ∼56 eV from single-bounce scattering off of Pt. Only a single dynamic peak is seen for scattered O2− due to ER reactions between O+ projectiles and adsorbed O atoms.30 As soon as the Pt surface is exposed to ammonia at 2 × 10−8 Torr, the dynamic O− peak intensity increases by more than 200× while sputtering contributions diminish (Fig. 1a). The dramatic intensity enhancement is primarily a result of the significant lowering of the Pt surface work function26 due to the large dipole moment of ammonia: indeed, the work function of Pt(111) has been shown to decrease by ∼3 eV upon ammonia adsorption, albeit at 100 K.31 A low work function favours negative ion formation during ion-surface charge exchange.26 The competition for surface adsorption sites upon NH3 exposure reduces O-atom coverage and suppresses O2− formation. Interestingly, the dynamic O2− peak at an exit energy of ∼50 eV virtually disappears upon exposure to NH3, while a slow peak appears at an exit energy of ∼10 eV (Fig. 1b). This slow peak cannot be sputtered O2. A possible alternate assignment of mass 32 amu e−1 is hydrazine N2H4, which however cannot be resolved experimentally from O2. Upon NH3 exposure, NH2 must form on the Pt surface in the presence of oxygen, as reported in the literature. It is conjectured that NH2 can dimerize to form N2H4, which is sputtered by the O+ projectiles to give rise to the observed slow peak at 32 amu in Fig. 1b.
 | ||
| Fig. 1 NH3 exposure effect on scattered products. Energy distributions of: (a) O−, (b) O2−, (c) OH−, (d) NO−, (e) HNO−, (f) NH−, and (g) H− from O+ bombardment of Pt at incidence energy of E0 = 78 eV and at various NH3 exposure pressures as annotated. The curves at 0.0 Torr represent scattering on a clean Pt surface (in the absence of NH3). For all but the O2− product, the scattered peak position remains invariant but signal intensity increases upon ammonia exposure. | ||
Continuing onto the observation of reaction products, three new dynamic molecular-ion exits are observed simultaneously: OH−, NO− and HNO−, shown in Fig. 1c, d and e, respectively. As explained below, these species stem from concurrent direct reactions between O+ projectiles and the respective surface-adsorbed H, N and NH species. In addition, H− and NH− dynamic peaks are also seen, albeit with much lower exit energies (Fig. 1). Though NH2 is not observed, it cannot be ruled out.
Given the significance of observing a peak at 31 amu, some assurance is needed that it is correctly assigned to HNO. First, signal spill over from adjacent peaks at 30 or 32 amu has been discounted by operating the mass spectrometer at very high resolution. Second, contamination from 17ON is not possible given the mass-selected, isotopically pure 16O+ incident beam and the very minor natural abundance of 17O. The only remaining source of contamination may be from 15NO, where the 15N isotope originates in ammonia. The observed peak intensity of mass 31 (“HNO−”) is on the order of 4% of mass 30 (“NO−”), at least one order of magnitude larger than the natural abundance of 15N at 0.37%. Assuming that it is valid to compare the intensities of these peaks, this analysis suggests negligible 15NO contribution to the peak at 31 amu, thus supporting its assignment to HNO.
The dynamics for the observed scattered products is revealed in Fig. 2, by varying the O+ incidence energy; during these experiments, the surface is kept at room temperature while the background pressure of NH3 is raised to 1 × 10−7 Torr. The O−, OH−, NO−, HNO−, NH−, and H− ion exit peak positions vary monotonically with O+ incidence energy, which suggests that their formation is driven by the O+ projectiles.30 The low-energy shoulder in the NO− energy spectra is probably due to sputtering of adsorbed NO, formed likely by surface reactions. Close inspection of the OH− and HNO− peaks reveals a low-energy tail—asymmetric peaks.
 | ||
| Fig. 2 Energy distributions of scattered products: (a) O−, (b) OH−, (c) NO−, (d) HNO−, (e) NH− and (f) H− from O+ bombardment of a polycrystalline Pt surface, held at room temperature and exposed to NH3 at a pressure 1 × 10−7 Torr. Results are shown for various O+ incidence energies, as annotated. | ||
The observation of HNO− is truly surprising. HNO is highly reactive and thus extremely difficult to detect. In 1926, Andrussow postulated the existence of the nitroxyl (HNO) radical as surface intermediate in the formation of both NO and N2 during ammonia oxidation. HNO has remained undetected to date in traditional adsorption studies,6,16,32 as well as beam studies,2–4,24 and this has cast serious doubt to Andrussow's mechanism. Why are we able to finally detect this short-lived intermediate? A notable difference with other beam studies is the use of O+ projectiles with hyperthermal energy large enough to allow for: (i) a small distance-of-closest-approach (apsis) to a Pt surface atom so that a transient state can form between the O, Pt, and adsorbed NH, (ii) an extremely short collision time that ejects an energetic O–NH before it can react further with other surface species, (iii) HNO formation as anion (with concomitant stabilization) by resonant electron transfer from the Pt surface.26 Even with these differentiating advantages, the scattered HNO− signal is weak versus OH− and NO− ion exits, and it is the first to vanish at O+ incidence energies above 100 eV.
Kinematic analysis can help pinpoint the origin of these scattering products. The O− signal is produced by single-bounce scattering of incident projectiles, explainable in full by binary collision theory (BCT).33 The fast OH−, NO− and HNO− products can be described by an “atom-in, molecule-out” mechanism,30 a true ER abstraction reaction of adsorbed H, N, and NH, respectively, by O+ projectiles, as depicted schematically in Fig. 3a. As such, this is the first case ever shown of three ER reactions taking place concurrently on the same surface. We have described elsewhere30 that the ER reaction can be visualized as a three-step process: first, a projectile collides with a substrate atom linked to an adsorbate; second, a triatomic transient state forms at the apsis point between the projectile, the substrate atom and the adsorbate; and third, the transient state disintegrates on the rebound ejecting a projectile-adsorbate molecule from the surface. BCT can predict the kinematic factors of all newly formed molecules.30
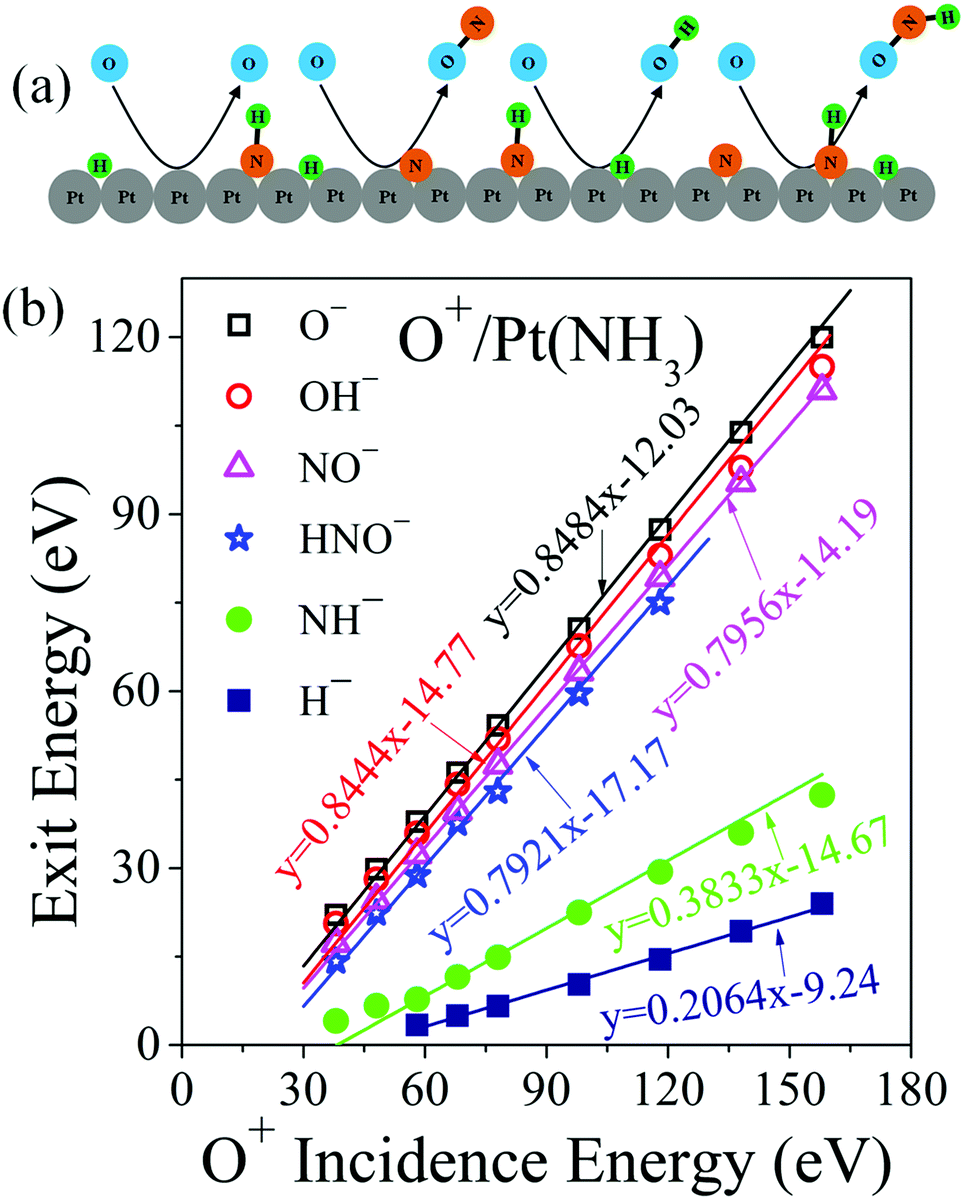 | ||
| Fig. 3 Eley–Rideal reaction mechanisms and associated kinematics. (a) Schematic depiction of scattering and abstraction reactions occurring between incident O+ projectiles and surface adsorbates (H, N, NH). (b) Peak ion exit energies of O−, OH−, NO−, HNO−, NH− and H− as a function of O+ incidence energy. The data points represent the energy of the corresponding dynamic peak in Fig. 1. The solid line for O− is a linear fitting with its slope calculated from standard binary collision theory. Solid lines for the direct reaction products OH−, NO− and HNO− are linear fittings with the slopes predicted from a modified binary collision theory (ref. 30). The NH− data were fitted by assuming NH− forms from dissociation of excited HNO with a mass-weighted kinematic factor. The H− data were linearly fitted with two parameters. | ||
The exit energy data for O−, OH−, NO− and HNO−, plotted in Fig. 3b, exhibit linear dependence on O+ incidence energy. The O− exit energy is described very well by a BCT-calculated kinematic factor of 0.8484, yielding an intercept of −12.03 eV, which corresponds to the inelastic energy loss occurring during the surface scattering process. The calculated kinematic factor for OH− is 0.8444, which fits the OH− data very well. Given the light mass of H, the kinematic factor of OH− is almost equal to that of O−. The inelastic energy loss for OH− is 14.77 eV, which is 2.74 eV larger than the O− exit, probably due to bond rearrangements during its formation (e.g., Pt–H ruptures, O–H forms). The surface H atoms, whose presence is confirmed by an H+ sputtering peak (Fig. S3, ESI†), most likely originate from the dissociation of NHx, though contributions from UHV background hydrogen cannot be excluded. The NH− energy data is dynamic and thus not from sputtering of adsorbed NH. It can be fitted very well by a mass-weighted kinematic factor from dissociating HNO, which suggests that the latter may be formed in an excited state. The H− energy data cannot yet be predicted.
NO− forms by an ER reaction between O+ projectiles and adsorbed N atoms—similar to the reverse process of N+ projectiles abstracting adsorbed O atoms.30 The kinematic factor for NO−, calculated to be 0.7956, describes the NO− exit energy very well. The inelastic energy loss for NO− is 14.19 eV, very close to that of OH−, suggesting similarities in scattering. Likewise, the HNO− formation can be described by an ER reaction between O+ projectiles and adsorbed NH atoms with a kinematic factor of 0.7921. The inelastic energy loss associated with HNO− formation is 17.17 eV, about 3 eV larger than NO− formation and comparable to the bond energy of N–H (3.5 eV) (32). This difference in energy loss may indicate that there is an N–H bond rapture associated with the formation of HNO−, which raises the possibility that this intermediate may be co-produced from O+ projectiles abstracting adsorbed NH2 from the surface.5,7 Though no O–NH2 species was detected, we cannot rule out the possibility of its formation as an internally excited intermediate, which subsequently dissociates spontaneously, producing H+ and HNO− pair ions. Given the light mass of H+, the kinematics of HNO− produced via this pathway will be indistinguishable from the direct abstraction of NH. This analysis raises the question of whether the observed NO− may likewise be a fragment of HNO− undergoing dehydrogenation. If that were the case, the NO− should exit with slightly lower kinetic energy than HNO−, the opposite of what is observed (Fig. 3b).
Clearly, the observed HNO− forms directly via an ER reaction of energetic O atoms with adsorbed NH and, thus, it cannot be considered a surface intermediate. However, its detection is significant as it proves unambiguously that nitroxyl can form in gas-surface collisions and survive long enough to be detected. While this method of producing HNO− is dynamic, it is still a surface reaction—albeit with the O-atom at the apsis point closer to the surface rather than at the equilibrium chemisorption position. A conspicuous difference from the Langmuir–Hinshelwood mechanism lies in the fact that the ER reaction produces HNO as anion, which is immediately ejected from the surface, thus preventing subsequent reactive interactions with other surface species. Under industrial catalytic reaction conditions, the adsorbed HNO may be rapidly oxidized to form NO and OH.6 More importantly, HNO may undergo rapid dehydrative dimerization on the surface, thus explaining the perplexing formation of N2O.16,34,35
The ER surface reaction process was further tested at elevated surface temperatures. An ordered (2 × 2) structure of N atoms has been reported on Pt(111), formed by annealing a NH3–O overlayer to 400 K.36,37 Above 400 K, the surface NHx species are expected to completely dissociate into N and H. Then, low surface coverage in NH should suppress the formation of HNO. This prediction was confirmed by performing scattering experiments at elevated temperatures. When raising the temperature from 300 to 420 K, several trends are observed as shown in Fig. 4. First, the O− scattering intensity decreases, which may appear counterintuitive as there should be fewer surface species to react with projectiles, thus allowing more to scatter. Indeed, increasing temperature favors desorption and should reduce surface coverage in NHx. However, the reduction in adsorbed NHx has a significant effect on the Pt surface work function, which influences anion formation disproportionately. Second, the OH− and NO− products exhibit the same trend as O− with a more dramatic reduction in signal intensity. Here the work function effect is compounded by the reduction in surface species (H and N) needed to react with the incident projectiles. Third, the HNO− signal is barely discernible at 350 K, and disappears entirely above this temperature (Fig. S4, ESI†). This behavior with temperature agrees with the reported conversion of surface NHx species to surface N atoms in the presence of co-adsorbed O atoms.37 Therefore, the observed NO− signal at 420 K can be attributed solely to the ER reactions between O+ projectiles and surface N atoms. The kinematics of the observed O−, OH−, and NO− at 420 K is very similar to room temperature (Fig. S5 and S6, ESI†).
 | ||
| Fig. 4 Surface temperature effect on NH3 adsorbates and scattered product formation. Energy distributions for: (a) O−, (b) OH−, (c) NO−, and (d) HNO− from O+ bombardment of Pt at incidence energy of 67 eV. The surface is exposed in situ to 1 × 10−7 Torr NH3 and its temperature is varied from 300 K to 420 K. | ||
The final topic of this study pertains to ammonia oxidation with O2+ projectiles. The Pt surface is dosed with NH3 as before. Compared to atomic O+, molecular O2+ projectiles must undergo collisional dissociation before they can react with adsorbed NH3, probably driven by the high momentum carried by the O2+ projectiles.38 O2+ scattering on Pt, exposed to 1 × 10−7 Torr NH3 at room temperature, yields the following anions: O2−, O−, OH−, HO2−, NO− and HNO− (Fig. 5). The O2− ion exit follows BCT, assuming sequential collisions of the constituent atoms of the molecular projectile. The O− ion exits are produced by collision-induced dissociation, with exit energy roughly about 1/2 of the corresponding O2− ion exits. Surprisingly, there is weak but clear signal corresponding to HO2−, which appears with exit energy almost equal to that of O2−. This suggests that the HO2− is produced by an Eley–Rideal reaction between O2+ projectiles and surface adsorbed H. This direct hydrogenation step may serve to activate the O2 bond under industrial ammonia oxidation conditions, in addition to the catalytic splitting of O2 on Pt. The HO2− peak disappears at O2+ incidence energy above 100 eV, probably due to dissociation into OH− and O (or OH and O−). The fast OH−, NO− and HNO− products must originate from the direct interaction between O2+ projectiles and the respective adsorbed H, N and NHx species, with one fast O atom from O2+ projectile remaining in the final product. Kinematic plots (Fig. S7, ESI†) of O2− and HO2− exit energy data show a linear dependence on O2+ incidence energy: both sets are well described by the BCT-calculated kinematic factor of 0.8484 for the O2− exit. The energy loss to form HO2− is 3.8 eV larger as compared to the formation of O2−. For products of O2 dissociation with subsequent surface reaction: O−, OH−, NO− and HNO−, the kinematic data cannot be fitted linearly. But for any given O2+ incidence energy, the ion exit energies follow a trend similar to that seen in O+/Pt(NH3): E(O−) > E(OH−) ∼ E(NO−) > E(HNO−).
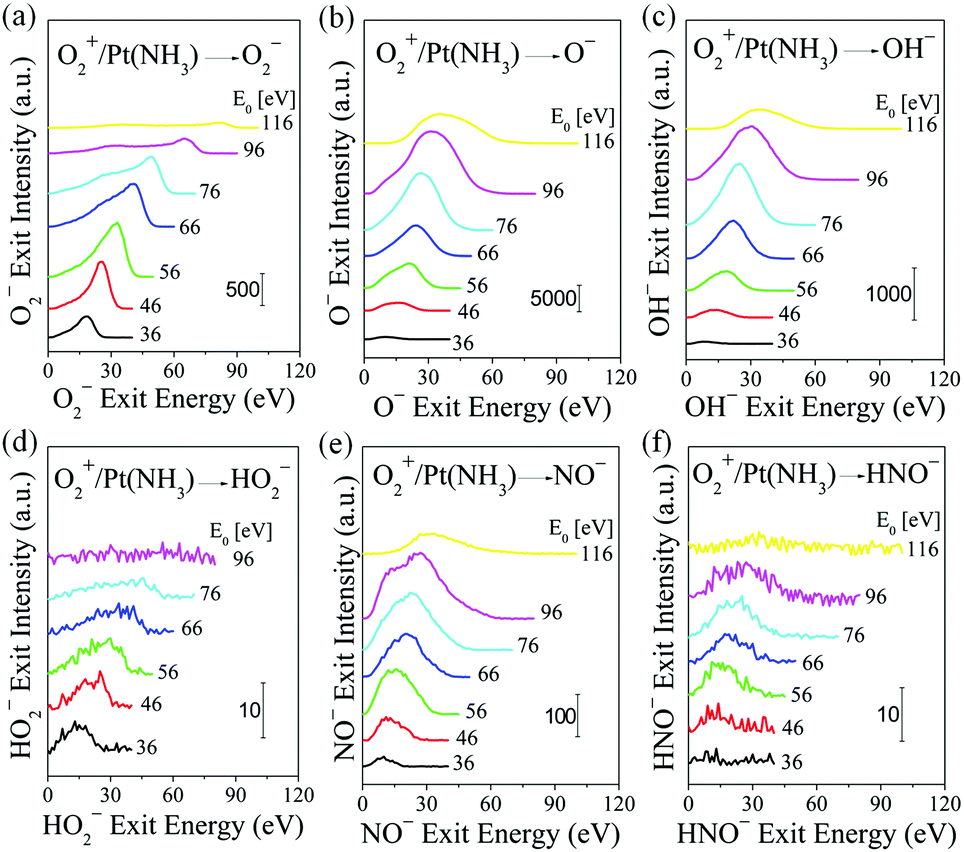 | ||
| Fig. 5 Ammonia oxidation on Pt by energetic molecular O2+. Energy distributions of scattered products: (a) O2−, (b) O−, (c) OH−, (d) HO2−, (e) NO− and (f) HNO− from O2+ bombardment of polycrystalline Pt at room temperature, exposed to 1 × 10−7 Torr NH3 at various O2+ incidence energies as annotated. Only O2− and HO2− form in single collisions with predictable kinematics. The ammonia oxidation products OH−, NO− and HNO− require dissociation of the molecular projectile and, although dynamic, their kinematics cannot be predicted by simple binary collision theory. | ||
Conclusions
The HNO intermediate has been detected in scattering experiments on Pt surfaces exposed to NH3 under simultaneous bombardment by energetic O+ and O2+ projectiles. We argue that its direct formation as anion and its prompt ejection from the surface prevents the rapid reaction of HNO with other surface intermediates, enabling its mass-spectrometric detection. Reactive scattering at hyperthermal energies can serve as a method for detecting transient surface intermediates by directly bonding with them via Eley–Rideal reactions.Acknowledgements
This report was based on work funded by NSF (Award No. 1202567). We appreciate deeply the thorough reading of the manuscript and the extensive suggestions offered by an anonymous reviewer. It is seldom that someone puts so much effort in a review.Notes and references
- G. B. Taylor, T. H. Chilton and S. L. Handforth, Ind. Eng. Chem., 1931, 23, 860–865 CrossRef CAS.
- C. W. Nutt and S. Kapur, Nature, 1968, 220, 697–698 CrossRef CAS.
- C. W. Nutt and S. Kapur, Nature, 1969, 224, 169 CrossRef CAS.
- M. Asscher, W. L. Guthrie, T. H. Lin and G. A. Somorjai, J. Phys. Chem., 1984, 88, 3233–3238 CrossRef CAS.
- W. D. Mieher and W. Ho, Surf. Sci., 1995, 322, 151–167 CrossRef CAS.
- M. Kim, S. J. Pratt and D. A. King, J. Am. Chem. Soc., 2000, 122, 2409–2410 CrossRef CAS.
- S. Günther, A. Scheibe, H. Bluhm, M. Haevecker, E. Kleimenov, A. Knop-Gericke, R. Schlögl and R. Imbihl, J. Phys. Chem. C, 2008, 112, 15382–15393 Search PubMed.
- F. Raschig, Z. Angew. Chem., 1927, 40, 1183–1185 CrossRef CAS.
- J. Zawadzki, Discuss. Faraday Soc., 1950, 8, 466–468 RSC.
- M. Bodenstein, Z. Elektrochem., 1935, 41, 466–468 CAS.
- L. Andrussow, Z. Angew. Chem., 1926, 39, 321–332 CrossRef CAS.
- B. A. Morrow and I. A. Cody, J. Catal., 1976, 45, 151–162 CrossRef CAS.
- G. Novell-Leruth, J. M. Ricart and J. Pérez-Ramírez, J. Phys. Chem. C, 2008, 112, 13554–13562 CAS.
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117–151 CrossRef.
- J. Pérez-Ramírez and E. V. Kondratenko, Chem. Commun., 2004, 376–377 RSC.
- R. Imbihl, A. Scheibe, Y. F. Zeng, S. Günther, R. Kraehnert, V. A. Kondratenko, M. Baerns, W. K. Offermans, A. P. J. Jansen and R. A. van Santen, Phys. Chem. Chem. Phys., 2007, 9, 3522–3540 RSC.
- D. A. Lashof and D. R. Ahuja, Nature, 1990, 344, 529–531 CrossRef CAS.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS PubMed.
- L. M. Ziurys, J. M. Hollis and L. E. Snyder, Astrophys. J., 1994, 430, 706–712 CrossRef CAS.
- T. P. Snow and V. M. Bierbaum, Annu. Rev. Anal. Chem., 2008, 1, 229–259 CrossRef CAS PubMed.
- D. Smith, Chem. Rev., 1992, 92, 1473–1485 CrossRef CAS.
- J. He, G. Vidali, J. L. Lemaire and R. T. Garrod, Astrophys. J., 2015, 799, 49 CrossRef.
- G. Fedosseev, S. Ioppolo, T. Lamberts, J. F. Zhen, H. M. Cuppen and H. Linnartz, J. Chem. Phys., 2012, 137, 054714 CrossRef PubMed.
- Y. M. Fogel, B. T. Nadykto, V. F. Rybalko, V. I. Shvachko and I. E. Korobchaskaya, Kinet. Catal., 1964, 5, 496–504 CAS.
- M. J. Gordon and K. P. Giapis, Rev. Sci. Instrum., 2005, 76, 083302 CrossRef.
- Y. Yao and K. P. Giapis, ChemPhysChem, 2016, 17, 1430–1434 CrossRef CAS PubMed.
- Y. Yao and K. P. Giapis, Angew. Chem., Int. Ed., 2016, 55, 11595–11599 CrossRef CAS PubMed.
- D. Jacobs, Annu. Rev. Phys. Chem., 2002, 53, 379–407 CrossRef CAS PubMed.
- A. Danon and A. Amirav, Int. J. Mass Spectrom. Ion Processes, 1990, 96, 139–167 CrossRef CAS.
- Y. Yao and K. P. Giapis, Phys. Rev. Lett., 2016, 116, 253202 CrossRef PubMed.
- G. B. Fisher, Chem. Phys. Lett., 1981, 79, 452–458 CrossRef CAS.
- L. Gang, B. G. Anderson and R. A. van Santen, Appl. Catal., B, 2003, 40, 101–110 CrossRef CAS.
- D. P. Smith, J. Appl. Phys., 1967, 38, 340–347 CrossRef CAS.
- J. Pérez-Ramírez, E. V. Kondratenko, V. A. Kondratenko and M. Baerns, J. Catal., 2004, 227, 90–100 CrossRef.
- J. Schäffer, V. A. Kondratenko, N. Steinfeldt, M. Sebek and E. V. Kondratenko, J. Catal., 2013, 301, 210–216 CrossRef.
- Z. Liang, H. J. Yang, M. Kim and M. Trenary, J. Chem. Phys., 2014, 140, 114707 CrossRef PubMed.
- Z. Liang, H. J. Yang, J. Oh, J. Jung, Y. Kim and M. Trenary, ACS Nano, 2015, 9, 8303–8311 CrossRef CAS PubMed.
- K. F. Medzihradszky, J. M. Campbell, M. A. Baldwin, A. M. Falick, P. Juhasz, M. L. Vestal and A. L. Burlingame, Anal. Chem., 2000, 72, 552–558 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp06533c |
| This journal is © the Owner Societies 2016 |