
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Final rotational state distributions from NO(vi = 11) in collisions with Au(111): the magnitude of vibrational energy transfer depends on orientation in molecule–surface collisions†
Bastian C.
Krüger
a,
Nils
Bartels
a,
Alec M.
Wodtke
abc and
Tim
Schäfer
*a
aInstitute of Physical Chemistry, Georg-August University of Göttingen, Tammannstraße 6, 37077 Göttingen, Germany. E-mail: tschaef4@gwdg.de
bDepartment of Dynamics at Surfaces, Max Planck Institute for Biophysical Chemistry, Am Faßberg 11, 37077 Göttingen, Germany
cInternational Center for Advanced Studies of Energy Conversion, Georg-August University of Göttingen, Tammannstrasse 6, 37077 Göttingen, Germany
First published on 19th May 2016
Abstract
When NO molecules collide at a Au(111) surface, their interaction is controlled by several factors; especially important are the molecules' orientation with respect to the surface (N-first vs. O-first) and their distance of closest approach. In fact, the former may control the latter as N-first orientations are attractive and O-first orientations are repulsive. In this work, we employ electric fields to control the molecules' incidence orientation in combination with rotational rainbow scattering detection. Specifically, we report final rotational state distributions of oriented NO(vi = 11) molecules scattered from Au(111) for final vibrational states between vf = 4 and 11. For O-first collisions, the interaction potential is highly repulsive preventing the close approach and scattering results in high-J rainbows. By contrast, these rainbows are not seen for the more intimate collisions possible for attractive N-first orientations. In this way, we reveal the influence of orientation and the distance of closest approach on vibrational relaxation of NO(vi = 11) in collisions with a Au(111) surface. We also elucidate the influence of steering forces which cause the O-first oriented molecules to rotate to an N-first orientation during their approach to the surface. The experiments show that when NO collides at the surface with the N-atom first, on average more than half of the initial vibrational energy is lost; whereas O-first oriented collisions lose much less vibrational energy. These observations qualitatively confirm theoretical predictions of electronically non-adiabatic NO interactions at Au(111).
Introduction
Quantum-state resolved experiments probing molecules colliding at surfaces have revealed important dynamical information about interactions leading to energy transfer.1 A well-studied example is the scattering of NO molecules from a Au(111) surface.2–7 Here, energy transfer is mediated by electron transfer (ET) events forming a transient NO− that result in the coupling of NO vibration to the metal's electronic degrees of freedom.2,8 During this process, many quanta of NO vibration can be lost to the surface within a sub-ps scattering time.2,6 The ET mediated energy transfer process is strongly orientation dependent; the relaxation probability is enhanced for N-first compared to O-first collisions.3 A subtle dependence of the relaxed vibrational distributions was also reported but remained unexplained.4 The orientation influence on ET probability is at least partly due to the fact that N-first oriented molecules may more closely approach the gold surface. Evidence for this is found in ab initio calculations9 as well as in observations of strong rotational rainbows for O-first oriented collisions3,5,10,11 – rotational rainbows are enhanced by repulsive O-first Au interaction.NO scattering from Au(111) has also been investigated by the independent electron surface hopping (IESH) method,9 an algorithm for propagating classical trajectories12 on an electron transfer (Newns–Anderson) Hamiltonian, hybridized to the metal electronic continuum.13 IESH gives good agreement with many experimental results,6 but due to inaccuracies in the interaction potential used for the calculation, it does not always compare favorably with experiment when compared in a one-to-one fashion.14,15 A good example of this problem concerns the influence of NO orientation on vibrational relaxation, which was studied by IESH for the vibrational relaxation of NO(vi = 15) using classical trajectories.9 For N-first trajectories with the NO bond perpendicular to the gold surface, strong multi-quantum vibrational relaxation was seen, whereas for O-first trajectories little or no vibrational relaxation was found. O-first trajectories did result in vibrational relaxation, but only due to dynamical steering; that is, re-orientation of the O-first orientation to N-first orientation when the molecules approach the surface.9
Experiments with oriented molecules do not compare well with IESH theory. One reason is that experiments are not governed by classical mechanics. Instead, the quantum laws of angular momentum enforce that only rather broad initial NO orientation distributions can be produced in the laboratory. Indeed, orientation distributions are so broad that a nominal N-first distribution contains some O-first oriented molecules.3,16 Beyond this, to find good agreement between experiment and theory, the theory would need to accurately describe the weak forces in the entrance channel that govern dynamical steering, which it cannot yet do.17 Hence, we seek an alternative experimental approach to testing the qualitative predictions of the IESH theory as they apply to the vibrational relaxation of highly vibrationally excited NO in collisions with a Au(111) surface. These are: (1) N-first collisions result in the loss of many vibrational quanta, (2) O-first collisions result in little or no vibrational energy loss unless (3) they are dynamically steered to an N-first orientation.9
We accomplish this by combining experimental control of initial NO orientation using externally applied electric fields and detection of rotational scattering rainbows. Specifically, we obtain rotational state distributions accompanying multi-quantum relaxation of NO(vi = 11)/Au(111) surface scattering as a function of initial NO orientation. We derive rotational state population distributions for final scattered vibrational states between vf = 4 and 11. We observe strong rotational rainbows when Δv is small and the rotational rainbow vanishes by the time Δv < −5. For these highly inelastic scattering processes, NO orientation has no detectable influence on the scattering rotational state distribution, a finger-print of dynamical steering. As the high-J rotational rainbow is caused by O-first collisions that experience the repulsive O-Au interaction, we conclude that O-first scattering leads to less vibrational relaxation than N-first scattering unless dynamical steering reorients the NO on its approach to the surface. This allows us to derive vibrational distributions for N-first and O-first surface collisions and yields results that are qualitatively in agreement with IESH theoretical predictions.
Experimental
The molecular beam surface scattering apparatus has been described previously.5,16 Briefly, we expand 10% NO/H2 into a vacuum chamber through a piezoelectric valve (1 mm diameter nozzle, 10 Hz, 3 atm stagnation pressure). This mixture yields an incidence translational energy of 0.51 eV. After passing through two differential pumping chambers, highly vibrationally excited NO X2Π1/2(vi = 11 and Ji = 0.5) is produced using the Pump–Dump–Sweep approach.18 The molecules then pass an electrode used to generate a strong electric field (|E| = 33 kV cm−1) normal to the room temperature Au(111) surface, which orients the vibrationally excited NO molecules prior to the collision. The NO orientation can be reversed by switching the polarity of the orientation field.5 The scattered NO molecules are detected using (1 + 1) resonance enhanced multi-photon ionization (REMPI) spectroscopy via the A2Σ+(v = 0–7) state and subsequent detection of the ions with micro-channel plates (Tectra MCP 050 in chevron assembly). The Au(111) crystal is cleaned by Ar-ion sputtering (LK Technologies; NGI3000) and subsequent annealing for 20 min at 950 K. The cleanliness of the surface is verified by means of Auger electron spectroscopy (Staib, ESA 100).For Pump–Dump–Sweep,18 we require three laser pulses. For the Pump step, the 887 nm output of a frequency doubled Nd:YAG laser (Spectra Physics, Quanta Ray Lab 170-10, 10 Hz, 8–12 ns pulse width (FWHM) of the fundamental) pumped home-built optical parametric oscillator (OPO)19 is mixed with the fourth harmonic of the Nd:YAG to obtain radiation resonant with the NO A2Σ+(v = 2, J = 0.5) ← X2Π1/2(v = 0, J = 0.5) transition at 204.708 nm. The same frequency doubled Nd:YAG laser pumps a second home-built OPO, whose output is mixed with the residual Nd:YAG output at 532 nm producing laser pulses at 336.10 nm suitable for the DUMP step transferring population from A2Σ+(v = 2, J = 0.5) → X2Π1/2(vI = 11, JI = 0.5).
For the Sweep step, the 450.87 nm radiation supplied by a frequency tripled Nd:YAG laser (Spectra Physics, Quanta Ray PRO-270-10) pumped dye laser (Sirah, Precision Scan, PRSCDA-24) removes residual A state population by further excitation to a dissociative state. This prevents the undesired population of various vibrational states in the ground electronic state via fluorescence. We use the 245–315 nm output of a commercially available OPO laser system (Continuum Sunlite Ex, 3 GHz bandwidth, 2 mJ per pulse@255 nm) to record rovibrationally resolved REMPI spectra of scattered molecules. These spectra contain all necessary information to derive rotational and vibrational distributions of ground electronic state NO with vibrational quantum numbers ranging from 4 to 11. Table 1 lists the employed REMPI transitions. In order to derive the rotational state distributions we analyze the REMPI data by fitting simulated spectra to the experiment as explained in more detail in the ESI.†
| v in X2Π1/2 | v in A2Σ+ |
|---|---|
| 11 | 3 |
| 9 | 2 |
| 8 | 1 |
| 7 | 1 |
| 6 | 1 |
| 6 | 0 |
| 5 | 1 |
| 4 | 1 |
Results and discussion
Fig. 1 shows the key observations of this work: rotational state population distributions for several vibrational states produced after scattering NO X2Π1/2(vi = 11 and Ji = 0.5) with 0.51 eV incidence translational energy from Au(111). An example of recorded REMPI spectra and a spectral fit, from which the population distributions are derived, can be found in the ESI.† Several of the rotational distributions exhibit a peak near J ∼ 35. This peak is a previously reported rotational rainbow that results from the repulsive interaction in collisions of the NO molecule with the Au(111) surface where the O-atom strikes the gold surface.3,5 We see that the J ∼ 35 rainbows are present when the vibrational energy loss is small (Δv > −5) and when an O-first incidence orientation is employed. Weak high-J rainbows can be seen for N-first orientations; we attribute this to the small fraction of O-first oriented molecules present in the broad orientation distributions.3,5,16 Particularly striking is the observation that as the vibrational energy loss increases, the J ∼ 35 rainbow diminishes and low J-states become increasingly populated. Furthermore, for a high vibrational energy loss, incidence orientation does not influence the rotational distribution of the scattered molecules – a fingerprint of dynamical steering.5 | ||
| Fig. 1 Final rotational state population distributions are influenced by incidence orientation. NO X2Π1/2 (vI = 11 and JI = 0.5) approaches the surface with 0.51 eV incidence translational energy. Rotational state distributions of scattered NO molecules in X2Π (vS = 11, 9, 8, 7, 6, 5, 4) are shown. Three orientation cases are shown: N-first (blue), O-first (red) and unoriented (green). The peaks near J ∼ 35 are due to a rotational rainbow arising from NO collisions where the O-atom strikes the gold surface. Solid lines are drawn to guide the reader's eye. The errors for vS = 6 are calculated based on two datasets for the populations of the rotational states derived from the γ(0,6) and γ(1,6) band. We expect similar relative errors for the population of rotational states belonging to other vibrational states. | ||
These observations suggest a simple interpretation, the key points of which we now emphasize to the reader.
• When NO(vi = 11) molecules are incident with an O-first orientation, they may:
∘ collide with the surface with the O-atom first and produce a J ∼ 35 rainbow or
∘ be dynamically steered to an N-first orientation on their approach to the surface, in which case no rainbow is seen.
• For NO(vi = 11) molecules that are incident with an O-first orientation and do not suffer dynamical steering, the vibrational energy loss is low.
• For NO(vi = 11) molecules that are incident with or which are dynamically steered to an N-first orientation, the vibrational energy loss is much larger.
Previously, we reported a steric influence on the relaxed vibrational distribution for NO(vi = 11) colliding with Au(111) – see Fig. 3b of ref. 4. Here, also information about the data analysis of vibrational state distributions can be found. In light of the rotationally resolved scattering population distributions presented in this work, the explanation for the steric influence is now clear. Indeed, we can use these insights along with the results from Fig. 3b of ref. 4 to understand the orientation influence on the relaxed vibrational distributions for NO(vi = 11) collisions at Au(111). See Fig. 2, in which the asymmetry parameter R(v) for each vibrational state is plotted using data from ref. 4.
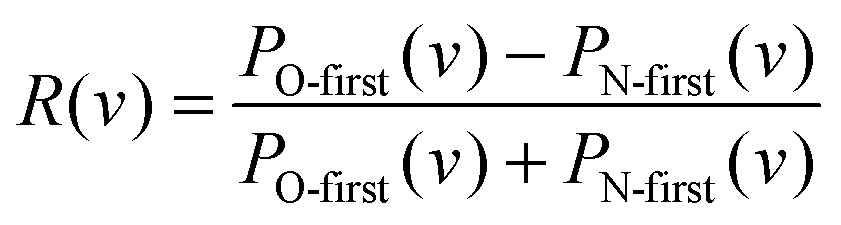 | (1) |
 | ||
| Fig. 2 Upper panel: The probability to find the molecule in a certain final vibrational state after surface scattering for two reverse orientations, taken from Fig. 3b of ref. 4. Lower panel: Asymmetry parameter R(v) = (PO-first(v) − PN-first(v))/(PO-first(v) + PN-first(v)) for each final vibrational state. The dashed line denotes an asymmetry parameter, for which the orientation effect vanishes. The positive asymmetry parameter for final vibrational states vf > 6 shows that these states are predominantly populated when NO hits the surface with the O-atom first. For final vibrational states vf < 6 the orientation effect vanishes due to dynamical steering as indicated by an asymmetry parameter close to zero. | ||
Earlier observations have shown that NO's incidence orientation strongly influences the probability of vibrational energy transfer.3–5 This has been explained by a facile electron transfer event for N-first collisions that is not as likely for O-first collisions.9 The results presented here show that – not only is there orientation dependence on the vibrational relaxation probability – there is clear incidence orientation dependence on the final vibrational state population distribution. That is, the dynamics of energy transfer and the magnitude of the energy transferred are dependent on incidence orientation.
A possible explanation for this behavior is that the magnitude of electronically non-adiabatic coupling is strongly dependent on the distance of closest approach – an N-first collision may approach more closely due to the attractive bonding interaction. We also point out that the qualitative aspects of the orientation behavior observed here were previously predicted from IESH theory.9 Specifically, these predictions are: (1) that collisions with O-first orientation are approximately vibrationally elastic whereas collisions with N-first orientations transfer large amounts of vibrational energy and (2) that dynamical steering of O-first oriented NO to N-first orientations is an important element of the electronically non-adiabatic energy transfer process. Furthermore, for IESH trajectories with N-first collisions multiple ET events are possible (leading to more vibrational energy loss) whereas for O-first trajectories fewer ET events can occur.
Conclusions
We report final rotational state distributions of oriented NO(vi = 11) molecules scattered from Au(111) for a large number of final vibrational states. A pronounced high-J rotational rainbow is observed in rotational state distributions for final vibrational states vf = 11–7. This rainbow vanishes for relaxation leading to lower final vibrational states. This allows us to extract the dynamics of O-first and N-first collisions while accounting for the influence of dynamical steering.Acknowledgements
We gratefully acknowledge support from the Alexander von Humboldt foundation. We acknowledge support from the CRC1073 under project A04, from the Deutsche Forschungsgemeinschaft (DFG), the Ministerium für Wissenschaft und Kultur (MWK) Niedersachsen, and the Volkswagenstiftung under Grant No. INST 186/902-1.Notes and references
- K. Golibrzuch, N. Bartels, D. J. Auerbach and A. M. Wodtke, Annu. Rev. Phys. Chem., 2015, 66, 399–425 CrossRef CAS PubMed.
- Y. Huang, C. T. Rettner, D. J. Auerbach and A. M. Wodtke, Science, 2000, 290, 111–114 CrossRef CAS PubMed.
- N. Bartels, K. Golibrzuch, C. Bartels, L. Chen, D. J. Auerbach, A. M. Wodtke and T. Schäfer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17738–17743 CrossRef CAS PubMed.
- N. Bartels, B. C. Krüger, D. J. Auerbach, A. M. Wodtke and T. Schäfer, Angew. Chem., Int. Ed., 2014, 53, 13690–13694 CrossRef CAS PubMed.
- N. Bartels, K. Golibrzuch, C. Bartels, L. Chen, D. J. Auerbach, A. M. Wodtke and T. Schäfer, J. Chem. Phys., 2014, 140, 054710 CrossRef PubMed.
- R. Cooper, C. Bartels, A. Kandratsenka, I. Rahinov, N. Shenvi, K. Golibrzuch, Z. Li, D. J. Auerbach, J. C. Tully and A. M. Wodtke, Angew. Chem., 2012, 124, 5038–5042 CrossRef.
- N. Bartels, PhD thesis, Institute of Physical Chemistry, University of Göttingen, Göttingen, 2015, http://https://ediss.uni-goettingen.de/handle/11858/11800-11735-10000-10022-16053-11859.
- J. W. Gadzuk, J. Chem. Phys., 1983, 79, 6341–6348 CrossRef CAS.
- N. Shenvi, S. Roy and J. C. Tully, Science, 2009, 326, 829–832 CrossRef CAS PubMed.
- M. G. Tenner, F. H. Geuzebroek, E. W. Kuipers, A. E. Wiskerke, A. W. Kleyn, S. Stolte and A. Namiki, Chem. Phys. Lett., 1990, 168, 45–50 CrossRef CAS.
- M. G. Tenner, E. W. Kuipers, A. W. Kleyn and S. Stolte, Surf. Sci., 1991, 242, 376–385 CrossRef CAS.
- N. Shenvi, S. Roy and J. C. Tully, J. Chem. Phys., 2009, 130, 174107 CrossRef PubMed.
- S. Roy, N. A. Shenvi and J. C. Tully, J. Chem. Phys., 2009, 130, 174716 CrossRef PubMed.
- B. C. Krüger, N. Bartels, C. Bartels, A. Kandratsenka, J. C. Tully, A. M. Wodtke and T. Schäfer, J. Phys. Chem. C, 2015, 119, 3268–3272 Search PubMed.
- A. Kandratsenka, J. Altschäffel, Private Communication, 2015.
- T. Schäfer, N. Bartels, N. Hocke, X. Yang and A. M. Wodtke, Chem. Phys. Lett., 2012, 535, 1–11 CrossRef.
- K. Golibrzuch, P. R. Shirhatti, I. Rahinov, A. Kandratsenka, D. J. Auerbach, A. M. Wodtke and C. Bartels, J. Chem. Phys., 2014, 140, 044701 CrossRef PubMed.
- N. Bartels, B. C. Krüger, S. Meyer, A. M. Wodtke and T. Schäfer, J. Phys. Chem. Lett., 2013, 4, 2367–2370 CrossRef CAS.
- L. Velarde, D. P. Engelhart, D. Matsiev, J. LaRue, D. J. Auerbach and A. M. Wodtke, Rev. Sci. Instrum., 2010, 81, 063106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02100j |
| This journal is © the Owner Societies 2016 |