
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling electron emission from the photoactive yellow protein chromophore by substitution at the coumaric acid group†
Michael A.
Parkes
,
Ciara
Phillips
,
Michael J.
Porter
and
Helen H.
Fielding
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.h.fielding@ucl.ac.uk
First published on 24th March 2016
Abstract
Understanding how the interactions between a chromophore and its surrounding protein control the function of a photoactive protein remains a challenge. Here, we present the results of photoelectron spectroscopy measurements and quantum chemistry calculations aimed at investigating how substitution at the coumaryl tail of the photoactive yellow protein chromophore controls competing relaxation pathways following photoexcitation of isolated chromophores in the gas phase with ultraviolet light in the range 350–315 nm. The photoelectron spectra are dominated by electrons resulting from direct detachment and fast detachment from the 21ππ* state but also have a low electron kinetic energy component arising from autodetachment from lower lying electronically excited states or thermionic emission from the electronic ground state. We find that substituting the hydrogen atom of the carboxylic acid group with a methyl group lowers the threshold for electron detachment but has very little effect on the competition between the different relaxation pathways, whereas substituting with a thioester group raises the threshold for electron detachment and appears to ‘turn off’ the competing electron emission processes from lower lying electronically excited states. This has potential implications in terms of tuning the light-induced electron donor properties of photoactive yellow protein.
1 Introduction
Photoactive proteins are exploited widely in nature and technology to make systems responsive to light. At the heart of a photoactive protein is a small molecule chromophore that absorbs the light. One photoactive protein that continues to attract a great deal of attention is photoactive yellow protein (PYP), the photoreceptor that is responsible for the negative phototaxis of the Halorhodospira halophila bacterium.1–4 The small molecule chromophore in PYP is a deprotonated para-coumaric acid anion (pCA−) and it is linked to the protein by a thioester covalent bond to a cysteine residue, Cys69 (Fig. 1). In PYP, the negative charge on the chromophore is stabilised by a network of hydrogen-bonds involving Cys69, Glu46, Tyr42 and Thr50 residues and a positively charged Arg52 residue. Following absorption of a photon, the chromophore undergoes rapid trans–cis isomerisation, the hydrogen-bond network then reorganises to accommodate the cis conformation and the protein unfolds and protonates.5–8 Subsequent cis–trans isomerisation of the chromophore, refolding of the protein and deprotonation, completes the photocycle. In the absence of chromophore-protein interactions, in solution and in the denatured protein, the chromophore does not form a stable cis intermediate but relaxes back to the ground state from a twisted trans conformation.9 Understanding how the interactions between the chromophore and the surrounding protein control the dynamical pathway of the chromophore, and thus the protein function, is a major challenge in photochemistry and photobiology.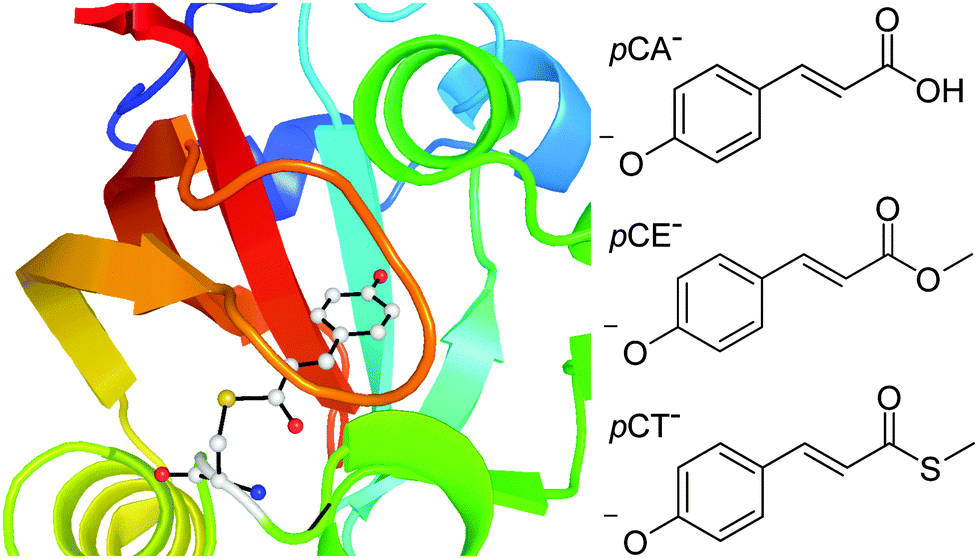 | ||
| Fig. 1 Left: PYP chromophore in its protein environment (Protein Data Bank). Right: Structures of the deprotonated PYP chromophores employed in this work. | ||
There have been numerous experimental investigations of the spectroscopy and excited state dynamics of isolated model PYP chromophores, in the gas-phase and in solution, aimed at disentangling the roles of the chromophore and the protein.9–27 Action spectra of the deprotonated coumaric acid anion in the gas-phase have been found to have a maximum around 430 nm, which is reasonably close to the absorption maximum of PYP (446 nm)21 and it is now generally accepted that this similarity is the combined effect of numerous chromophore-protein interactions. Although the absorption spectra of the gas-phase chromophore and PYP are similar, time-resolved measurements of a deprotonated methyl terminated ketone analogue of the PYP chromophore in the gas-phase, excited at 400 nm (3.1 eV), revealed that 80% of the excited state population relaxed back to the ground state on a timescale of 52 ps via a twisted intermediate, similar to isolated chromophores in solution, and that the remaining 20% of the excited state population underwent autodetachment (electron emission).16 From this, the authors concluded that one of the roles of the protein environment was to funnel the excited state population through the S1/S0 conical intersection seam to the cis isomer and to suppress electron emission. It is worth noting that electron emission has also been observed in isolated chromophores in aqueous solution12 and in PYP28 at higher photoexcitation energies. Interestingly, the spectrum of the electron emitted from the chromophore in PYP was found to be similar to that of a solvated electron, suggesting that the protein pocket provides a local environment for an electron that is similar to water.
Studies of numerous chromophore analogues in aqueous solution have found that the substituent on the coumaryl tail of the chromophore plays a key role in the excited state dynamics, highlighting the significance of the thioester link to the Cys69 amino acid residue. These studies found that derivatives with strong electron accepting groups, such as thioesters, return to the initial trans configuration on timescales of ∼1 ps, whereas those with weak electron accepting groups, such as amides or carboxylate, have longer excited state lifetimes, up to 10 ps for the carboxylate, and measurable trans–cis isomerisation yields.9,11,12,18
From a theoretical perspective, there have been numerous studies of PYP and the PYP chromophore, in the gas-phase and in solution, aimed at investigating the role of the environment on its electronic structure and dynamics.29–37 Various strategies have been employed, ranging from studies of a few solvent molecules or the closest protein residues to quantum-mechanical/molecular mechanics and averaged solvent electrostatic potential/molecular dynamics methods. Such studies have highlighted the importance of torsions around the C–C single bond between the phenoxide ring and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in controlling the trans–cis isomerisation process in the protein.29 They have also rationalised the blue solvatochromic shift of pCA− and its derivatives in terms of a transfer of electron density from the phenolate end of the chromophore to the rest of the chromophore, resulting in a decrease in dipole moment between the ground and first electronically excited states.34 García-Prieto and coworkers have recently reported a detailed theoretical study of the absorption spectra of different chromophore analogues in the gas-phase and solution and found that the presence of a sulfur atom on the coumaryl tail modulates the solvent effect for the first few excited electronic states37 which could explain the differences observed in the excited state dynamics in aqueous solution discussed above.9,11,12,18
C double bond in controlling the trans–cis isomerisation process in the protein.29 They have also rationalised the blue solvatochromic shift of pCA− and its derivatives in terms of a transfer of electron density from the phenolate end of the chromophore to the rest of the chromophore, resulting in a decrease in dipole moment between the ground and first electronically excited states.34 García-Prieto and coworkers have recently reported a detailed theoretical study of the absorption spectra of different chromophore analogues in the gas-phase and solution and found that the presence of a sulfur atom on the coumaryl tail modulates the solvent effect for the first few excited electronic states37 which could explain the differences observed in the excited state dynamics in aqueous solution discussed above.9,11,12,18
In a previous anion photoelectron spectroscopy study of gas-phase pCA− and its ortho- and meta-isomers, we found that moving the position of the O− group on the phenoxide group of the chromophore controlled the competition between electron emission and internal conversion.27 The focus of this paper is to determine whether the thioester link also plays a role in controlling the competition between electron emission and internal conversion in the gas-phase. We employ a combination of anion photoelectron spectroscopy and quantum chemistry calculations to investigate the effect of substituting the hydrogen atom of the carboxylic acid group with a methyl group and substituting the oxygen atom of the methoxy group with a sulfur atom. Specifically, we excite pCA−, its methyl ester, pCE−, and its methyl thioester, pCT−, (Fig. 1) with ultraviolet light in the range 350–315 nm (3.54–3.94 eV), within the 21ππ* ← S0 absorption band. Improving our understanding of the role of the thioester link in controlling electron emission in PYP may provide a basis for practical applications such as the rational design of photoactive materials with specific redox properties.
2 Experimental and computational methods
2.1 Chromophores
pCA was purchased from Sigma-Aldrich and pCE was purchased from Tokyo Chemical Industry, both were used without further purification. pCT was synthesised by a modification of the method of Naseem et al.26 (ESI†).2.2 Photoelectron spectroscopy
Photoelectron spectra were recorded using our photoelectron imaging apparatus that has been described elsewhere.27,38–40 Briefly, deprotonated anions of pCA, pCE or pCT were generated by electrospray ionisation of ∼1 mM solutions of the chromophores in methanol, with a few drops of aqueous ammonia. The pCA−, pCE− or pCT− anions were mass-selected by a quadrupole and passed into a collision cell doubling as a hexapole ion-trap. The anions were released from the trap at a frequency of 20 Hz, to match the repetition rate of the laser system, and transported to the interaction region of collinear velocity-map imaging optics. In the interaction region, the ion packet was crossed with nanosecond laser pulses of wavelength 315, 320 and 350 nm, generated by frequency doubling the output of a Nd:YAG pumped dye laser. Electrons generated by the laser pulse were then accelerated towards a position sensitive detector and imaged using a phosphor screen and CCD camera. Laser-only images were subtracted from images recorded following the interaction of the laser light with the anions, to eliminate background counts arising from scattered laser light and ionisation of residual gas. The photoelectron images were inverted using the pBASEX method.41 The wavelength of the laser light was measured using a wavemeter and the energy scale of the detector was calibrated by recording the photoelectron spectrum of I−. The energy resolution is ∼5% and the error in electron kinetic energy is around ±0.05 eV.2.3 Calculations
All electronic structure calculations were performed using the Gaussian 09 suite of programmes.42 The geometric structures of the phenolate form of pCE−, pCT− and pCA−, and corresponding neutral radicals, were optimised using the B3LYP hybrid functional43–46 and a 6-311++G(3df,3pd) basis set.47–49 Frequency calculations were performed to ensure that the optimised structures were true minima.Vertical excitation energies (VEEs) of the singlet excited electronic states of the phenolate deprotonated coumaric acid chromophores were calculated using the CAM-B3LYP/6-311++G(3df,3pd) method. The long-range corrected version of B3LYP using the Coulomb Attenuating Method (CAM)50 was chosen for its potential to describe excited states with charge-transfer character and because it has been shown to yield reasonable results for the phenolate form of the pCA− anion.22
Vertical detachment energies (VDEs) were determined using electron propagator theory (EPT)51,52 and a 6-311++G(3df,3pd) basis set. We have already benchmarked this approach against the high-resolution photoelectron spectrum of the phenoxide anion and shown that it yields good results for the deprotonated green fluorescent protein chromophore anion.40 Adiabatic detachment energies (ADEs) were determined using the B3LYP/6-311++G(3df,3pd) method, ADE = Eradicalmin + EradicalZPE − [Eanionmin + EanionZPE], where subscripts ‘min’ and ‘ZPE’ represent the minimum energy and zero-point energies, respectively. Higher VDEs were determined by calculating the VEEs of the first electronically excited doublet states of the neutral radicals at the optimised geometries of the corresponding anions using the CAM-B3LYP/6-311++G(3df,3pd) method and adding these to the VDEs calculated using the EPT method.
3 Results and discussion
Photoelectron spectra of the deprotonated coumaric acid anion (pCA−), an ester analogue (pCE−) and a thioester analogue (pCT−) (Fig. 1) were recorded as a function of electron kinetic energy (eKE) and are presented as a function of electron binding energy, eBE = hν − eKE in Fig. 2.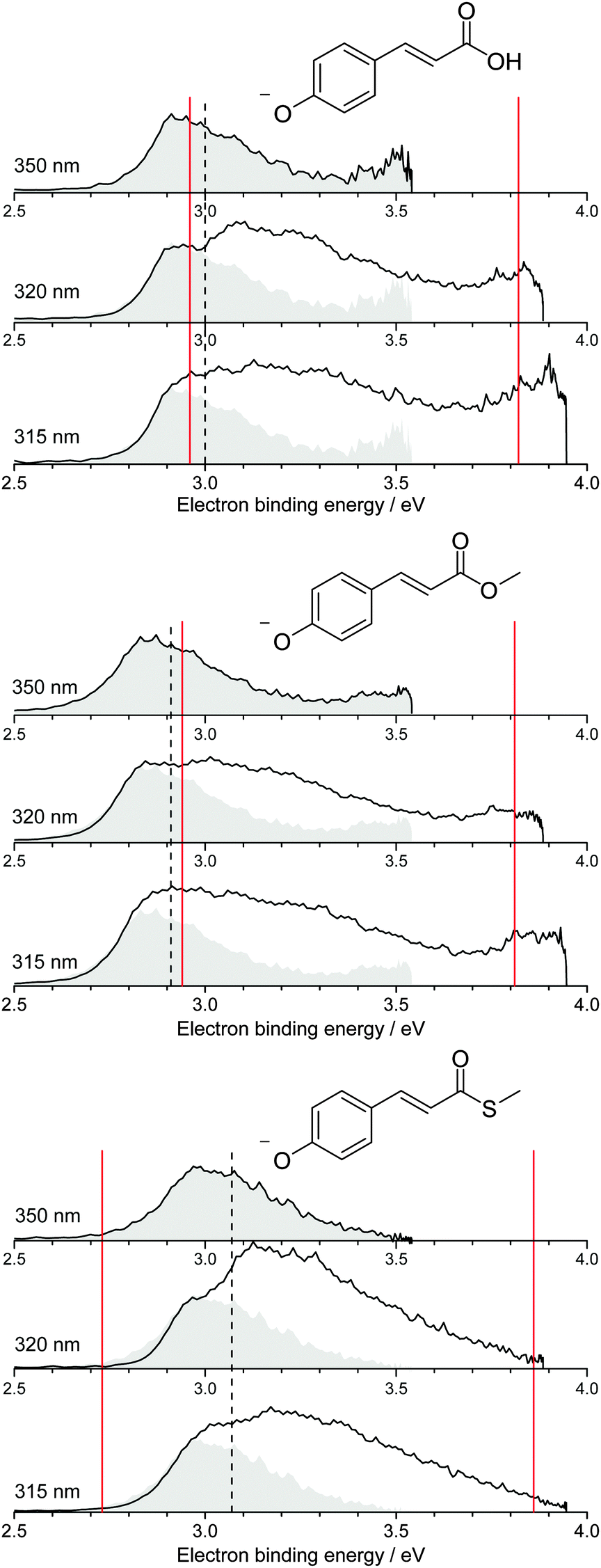 | ||
| Fig. 2 Photoelectron spectra of pCA−, pCE− and pCT− recorded at 350 nm (3.54 eV), 320 nm (3.87 eV) and 315 nm (3.94 eV) (solid black lines). The 350 nm photoelectron spectra (shaded grey) are superimposed on photoelectron spectra recorded at shorter wavelengths, whose intensities have been scaled so the rising edges are aligned. Dashed black lines mark the calculated VDEs (Table 1) and solid red lines mark the calculated VEEs determined by García-Prieto and coworkers for the two lowest energy singlet ππ* states,37 listed in Table 2. | ||
All the photoelectron spectra are dominated by broad, unresolved features at low eBEs. The rising edges of these broad features remain at constant eBE for all photon energies and are therefore attributed to direct photodetachment (PD). The maxima of the 350 nm (3.54 eV) spectra are in good agreement (∼0.1 eV) with the calculated vertical detachment energies (VDEs) and adiabatic detachment energies (ADEs), which are presented in Table 1. It is worth noting that there is significant photoelectron emission at lower eBEs than the calculated VDEs and ADEs in all the photoelectron spectra. In pCA−, and one of its isomers oCA−, this has been attributed to rotation around the single bond between the CC double bond and the phenoxide group, so we assume these torsional motions play a similar role in pCE− and pCT−. The fact that the calculated VDEs and ADEs are so close to one another suggests that the minimum energy geometries of the anion and the radical are very similar, in agreement with the results of our combined photoelectron spectroscopy and computational study of gas-phase pCA− and its ortho- and meta-isomers.27 Our calculated VDEs are also in good agreement with the EOM-IP-CCSD/6-311+G(df,pd) value reported by Zuev et al. for pCA− (2.92 eV),53 the OVGF/aug-cc-pVDZ value reported by Gromov et al. for pCT− (2.90 eV)30 and the experimental VDE reported for the methyl ketone analogue (2.9 eV).16
| Chromophore | VDE | ADE (0–0) | Expt |
|---|---|---|---|
| pCA− | 3.00 (0.879) | 2.99 | 2.91 ± 0.05 |
| pCE− | 2.91 (0.875) | 2.91 | 2.83 ± 0.05 |
| pCT− | 3.07 (0.879) | 3.03 | 2.96 ± 0.05 |
It is worth noting that pCA− can be formed during electrospray ionisation as a carboxylate anion or as a phenolate anion, whereas pCE− and pCT− have only one deprotonation site and can only be formed as phenolate anions. It is clear from the photoelectron spectra (Fig. 2) that all three chromophores have similar VDEs (around 3 eV), which supports our earlier suggestion that pCA− is formed in its phenolate form in our instrument when using methanol with a few drops of aqueous ammonia as a solvent.27 This contrasts with the observations of Almasian et al.,25 but perhaps emphasises the importance of instrumental parameters, such as the position of the electrospray head, as well as choice of solvent in determining the deprotonation site.54 The VDE for the carboxylate form of pCA− (4.68 eV)27 is significantly higher than for the phenolate form so it is also possible that both phenolate and carboxylate forms are present in our instrument but that we are only sensitive to the phenolate form in experiments with photons <4 eV.
Although the VDEs of the chromophores are similar, the maxima in the photoelectron spectra and calculated VDEs (Table 1) increase in the order pCE− < pCA− < pCT−. This trend can be understood in terms of the stabilising effect of electron accepting substituents lowering the energy of the resonantly stabilised anions as the electron affinity increases in the order OMe < OH < SMe.
The calculated vertical excitation energies for the first two 1ππ* states and the first 1nπ* states of pCA−, pCE− and pCT− are listed in Table 2, alongside the results of higher-level SA-CASSCF(14,12)-PT2/cc-pVDZ calculations by García-Prieto and coworkers37 and experimental values reported by Rocha-Rinza and coworkers.21 The molecular orbitals involved in the transitions are plotted in Fig. 3. Our calculated value for pCE− is consistent with that calculated using the CAM-B3LYP/aug-cc-pVTZ method22 and our value for pCA− lies within the range of values calculated by Zuev et al.53 and Uppsten and Durbeej,55 although all the values are ∼0.5 eV higher than the experimental values determined from action spectra21 and those calculated using the SA-CASSCF(14,12)-PT2/cc-pVDZ method.37 Nonetheless, the characters of the excited states calculated using the CAM-B3LYP/6-311++G(3df,3pd) method, and the energies of the excited states with respect to one another, are in good agreement with those calculated using the SA-CASSCF(14,12)-PT2/cc-pVDZ method. To guide our interpretation of the photoelectron spectra, we have marked the more accurate VEEs determined by García-Prieto and coworkers using the SA-CASSCF(14,12)-PT2/cc-pVDZ method37 on our experimental spectra (Fig. 2) and use the configurations obtained from the CAM-B3LYP/6-311++G(3df,3pd) to determine the characters of the excited states with respect to the detachment continua.
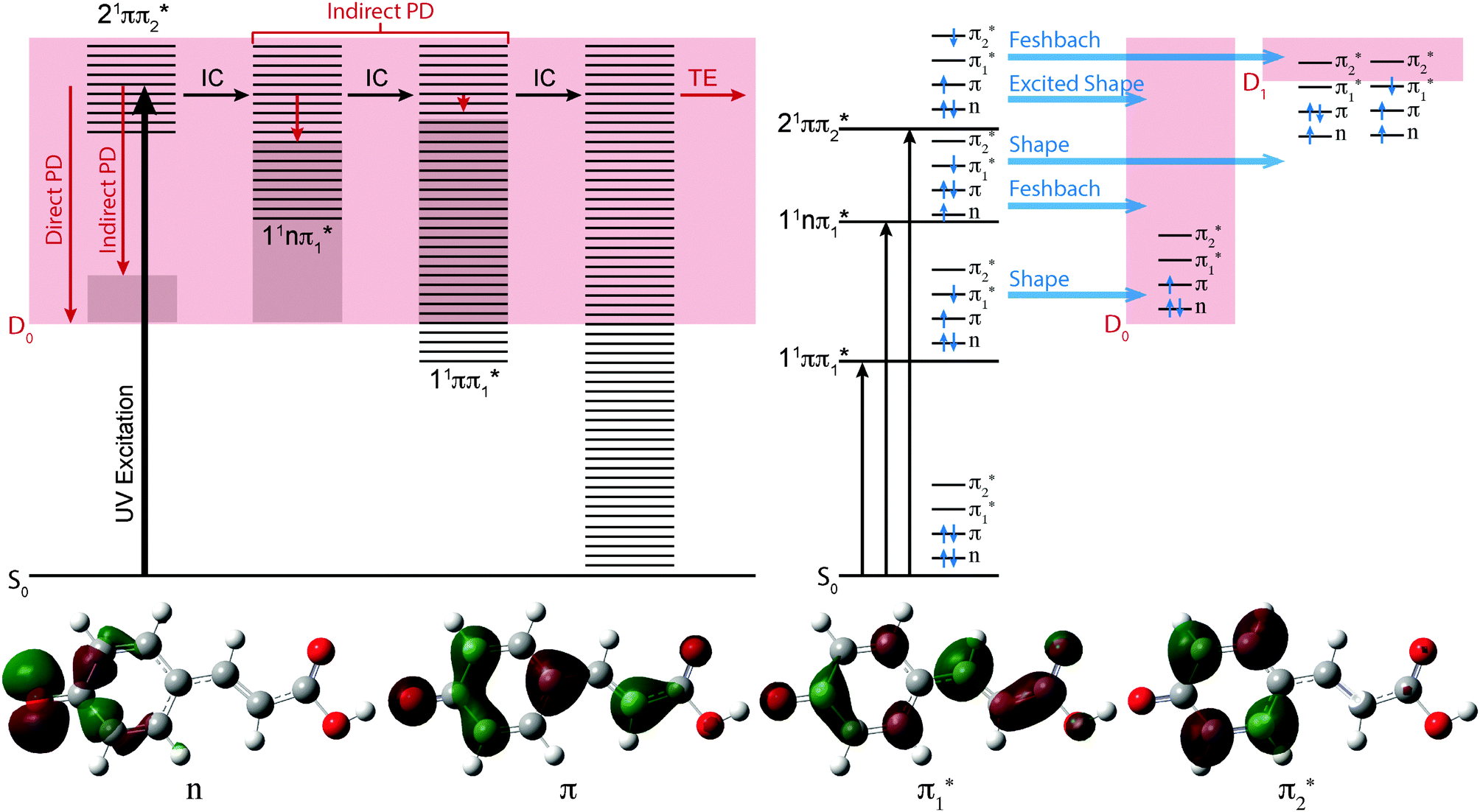 | ||
| Fig. 3 Top left: Jablonski diagram illustrating the electronic relaxation and emission processes following UV photoexcitation of pCA−, pCE− and pCT−. Horizontal black lines represent the vibrational levels of the excited electronic states and the solid pink area represents the electron detachment continuum. Vertical red arrows represent the eKE of direct and indirect electron emission processes and the solid grey areas represent the vibrational energy left in the neutral radical after electron detachment (determined by the propensity for conservation of vibrational energy). The horizontal black arrows represent some of the possible internal conversion (IC) processes and the horizontal red arrow represents electrons with eKE ∼ 0 eV following thermionic emission (TE) from vibrationally hot S0. Top right: Schematic energy level diagram showing the resonance character (horizontal blue arrows) of the first three excited singlet electronic states of the chromophores with respect to the D0 and D1 electronic continua. Bottom: Main CAM-B3LYP/6-311++G(3df,3pd) molecular orbitals involved in the transitions to the first three excited singlet electronic states. | ||
The effect of substituting the hydrogen on the carboxylic acid group in pCA− for a methyl group (pCE−) has very little effect on any of the VEEs. However, substituting the oxygen atom for a sulfur atom (pCT−) causes the 11ππ* and 11nπ* states to red-shift by 0.1–0.2 eV, although it has very little effect on the 21ππ* state. These observations can be understood in terms of the molecular orbitals involved in the transitions (Fig. 3). Transitions to the 11ππ* and 11nπ* states are to the π1* molecular orbital, which is delocalised across the anion and therefore stabilised by the electron accepting methyl thioester group, whereas the 21ππ* transition is to the π2* molecular orbital, which is localised on the phenoxide group and is barely influenced by changing the substituents on the coumaryl tail.
It is clear from Fig. 2 that the 11ππ* and D0 states are very close to one another in pCA− and pCE−, but that the 11ππ* state is 0.2–0.3 eV lower in energy than the D0 state in pCT−. This suggests that the thioester link between the chromophore and the protein plays a role in ensuring that the 11ππ* state is bound with respect to photodetachment following excitation at the maximum of 1ππ* ← S0 absorption band. This is consistent with the fact that there have not been any reports of electron emission from PYP following excitation within the 1ππ* ← S0 absorption maximum at 446 nm (2.78 eV), even though electron emission has been observed following excitation at higher photon energies.28
As the photon energy increases, the broad features in the photoelectron spectra change shape on the high eBE side, characteristic of an indirect PD process following resonant excitation of a higher lying electronically excited state (autodetachment). In pCA−, and one of its isomers mCA−, this broadening has already been rationalised in terms of resonant excitation of higher-lying 21ππ* excited states followed by autodetachment.27 Similar effects have also been observed in photoelectron spectra of the deprotonated green fluorescent protein (GFP) chromophore following resonant excitation of higher lying excited 1ππ* states.40,56,57
The 350 nm (3.54 eV) photoelectron spectra are least influenced by resonant excitation of the 21ππ* states, which have VEEs around 3.8–3.9 eV for all three chromophores (Fig. 2), so the 350 nm spectra have been superimposed on the 320 nm and 315 nm photoelectron spectra to highlight the contributions from resonant excitation of the 21ππ* states in the photoelectron spectra recorded at shorter wavelengths. The difference is most pronounced for the 320 nm photoelectron spectrum of pCT−, when the photon energy (3.87 eV) is resonant with the 2ππ* ← S0 absorption maximum (3.86 eV37).
In Fig. 3, the possible electronic relaxation and electron emission processes following photoexcitation of pCA−, pCE− and pCT− are illustrated on a Jablonski diagram and the resonance characters of the first three singlet excited electronic states with respect to the D0 and D1 electronic continua are also shown. The 21ππ* state responsible for the broadening on the high eBE edges of the photoelectron spectra (Fig. 2) is an excited shape resonance with respect to the D0 continuum, implying a strong coupling between the 21ππ* state and the D0 continuum and fast electron emission.
The photoelectron spectra of pCA− and pCE− also have features at high eBE (low eKE) that shift towards higher eBEs as the photon energy increases, characteristic of indirect PD processes. Interestingly, this feature is not observed in the photoelectron spectra of pCT−. The first question to ask is whether the D1 continua are accessible energetically. The VDEs to the first electronically excited states of the neutral radicals have been calculated to be 4.38, 4.32 and 4.50 eV for pCA−, pCE− and pCT−, respectively. The calculated VDEs are higher than the photon energies used in our experiments; however, it is possible that they are overestimated by ∼0.5 eV, similar to the VEEs calculated for the anion using the same method (Table 2), in which case the D1 continuum would be accessible energetically in pCA− and pCE− at 320 and 315 nm and at 350 nm if the ADEs were 0.3–0.4 eV lower than the VDEs. The next question to ask is if any of the excited states of pCA− and pCE− are coupled strongly to the D1 continuum. The 21ππ* state is a Feshbach resonance with respect to the D1 continuum, implying a weak coupling between the 21ππ* state and the D1 continuum and slow electron emission. Thus, indirect electron emission from the 21ππ* state to the D1 continuum is unlikely to compete with fast electron emission to the D0 continuum. However, ultrafast internal conversion to a lower lying excited electronic state may compete with the fast electron emission to the D0 continuum. Subsequent electron emission from lower lying electronically excited states could generate low eKE electrons, if the displacements between the minima of these states and the D0 state were sufficiently small that the Franck–Condon factors were largest between the high vibrational levels of the electronically excited states and high vibrational levels of D0 (Fig. 3). The 11nπ* state has shape resonance character with respect to the D1 continuum and Feshbach character with respect to the D0 continuum, whereas the 11ππ* state has shape resonance character with respect to the D0 detachment continuum and Feshbach character with respect to the D1 continuum. Consequently, it seems likely that any population relaxing to the 11nπ* state would undergo fast electron emission to the D1 continuum or internal conversion to 11ππ* or S0 states and any population relaxing to the 11ππ* state would undergo fast electron emission to the D0 continuum or internal conversion to S0. Internal conversion processes populating high vibrational levels of the electronic ground state of the anion would also result in low eKE electrons from thermionic emission (Fig. 3).
It is possible that the low eKE (high eBE) electrons observed in the 350 nm photoelectron spectra of pCA− and pCE− arise from a different relaxation process than those in the 320 nm and 315 nm photoelectron spectra. The action absorption spectrum for pCE− shows that 350 nm lies between the 11ππ* and 21ππ* absorption bands,22 so the non-zero absorption could be the result of populating high lying vibrational levels of the 11ππ* state which could then autodetach to the D0 continuum or undergo internal conversion to S0 followed by thermionic emission.
Thus, the presence of high eBE (low eKE) electrons in the 315 nm and 320 nm photoelectron spectra of pCA− and pCE−, that shift towards higher eBEs as the photon energy increases, suggests that IC to lower lying electronic states or the ground electronic state compete with PD. The presence of high eBE (low eKE) electrons in the 350 nm photoelectron spectra of pCA− and pCE− can either be attributed to a similar process or to resonant excitation of the 11ππ* state followed by autodetachment or internal conversion to the ground state and thermionic emission. The absence of high eBE (low eKE) electrons in the 315 nm and 320 nm photoelectron spectra of pCT− could be the result of raising the threshold for detachment into the D1 continuum and ‘turning off’ the 11nπ* → D1 + e− detachment channel. However, if this were the case, subsequent electronic relaxation from the 11nπ* state to the 11ππ* or S0 states would themselves generate low eKE electrons by fast electron detachment into the D0 continuum or thermionic emission. A more likely explanation seems to be that lowering the 11nπ* and 11ππ* states in the Franck–Condon region makes conical intersections between the 21ππ* and 11nπ* or 11ππ* states inaccessible or less accessible, effectively ‘turning off’ internal conversion to these states and the ground electronic state, leaving indirect electron emission from the 21ππ* state to the D0 continuum as the only relaxation pathway. The absence of high eBE (low eKE) electrons in the 350 nm photoelectron spectrum of pCT− can be explained either as ‘turning off’ internal conversion processes from the low vibrational levels of the 21ππ* state or ‘turning off’ autodetachment and internal conversion from high vibrational levels of the 11ππ* state. This opens the interesting possibility of using changes to the link between the chromophore and the protein in PYP to manipulate the UV induced electron donor properties of the protein and isolated chromophores, with the potential to monitor and manipulate redox processes.58
4 Conclusions
In this paper, we have used photoelectron spectroscopy and quantum chemistry calculations to investigate the role of the thioester group in controlling the competition between internal conversion and electron emission in isolated PYP chromophores in the gas phase. Following photoexcitation with ultraviolet light in the range 350–315 nm, we see photoelectrons with high eKEs that arise from direct photodetachment or from excitation of the 21ππ* state followed by indirect photodetachment to the D0 continuum. We also see photoelectrons with low eKEs that appear to arise from an indirect electron emission process. We attribute these low eKE electrons to photodetachment from lower lying 11nπ* or 11ππ* states or to the vibrationally hot electronic ground state and subsequent thermionic emission. We find that substituting the hydrogen atom of the carboxylic acid group with a methyl group lowers the vertical detachment energy but has very little effect on the competition between internal conversion to lower lying electronic states and electron emission, whereas substituting with a thioester group raises the vertical detachment energy and appears to ‘turn off’ competing electron emission processes from lower lying electronically excited states or the electronic ground state. This suggests that one of the roles of the thioester link between the chromophore and the protein is to contribute to impeding electron emission following photoexcitation of the 1ππ* state and another is to ‘turn off’ competing relaxation pathways to allow the chromophore to act as an efficient light-induced electron donor following photoexcitation at shorter wavelengths within the 21ππ* ← S0 absorption band.Acknowledgements
This work was made possible by EPSRC grants (EP/L005646/1 and EP/D054508/1). We acknowledge use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London and computational support from Dr Jörg Saßmannshausen at UCL.References
- T. E. Meyer, Biochim. Biophys. Acta, 1985, 806, 175–183 CrossRef CAS.
- W. W. Sprenger, W. D. Hoff, J. P. Armitage and K. J. Hellingwerf, J. Bacteriol., 1993, 175, 3096–3104 CAS.
- K. J. Hellingwerf, J. Hendriks and T. Gensch, J. Phys. Chem. A, 2003, 107, 1082–1094 CrossRef CAS.
- F. Schotte, H. S. Cho, V. R. I. Kaila, H. Kamikubo, N. Dashdorj, E. R. Henry, T. J. Graber, R. Henning, M. Wulff, G. Hummer, M. Kataoka and P. A. Anfinrud, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19256–19261 CrossRef CAS PubMed.
- R. Kort, H. Vonk, X. Xu, W. D. Hoff, W. Crielaard and K. J. Hellingwerf, FEBS Lett., 1996, 382, 73–78 CrossRef CAS PubMed.
- U. K. Genick, S. M. Soltis, P. Kuhn, I. L. Canestrelli and E. D. Getzoff, Nature, 1998, 392, 206–209 CrossRef CAS PubMed.
- A. Xie, W. D. Hoff, A. R. Kroon and K. J. Hellingwerf, Biochemistry, 1996, 35, 14671–14678 CrossRef CAS PubMed.
- M. Unno, M. Kumauchi, J. Sasaki, F. Tokunaga and S. Yamauchi, J. Am. Chem. Soc., 2000, 12, 4233–4234 CrossRef.
- P. Changenet-Barret, A. Espagne, P. Plaza, K. J. Hellingwerf and M. M. Martin, New J. Chem., 2005, 29, 527 RSC.
- P. Changenet-Barret, P. Plaza and M. M. Martin, Chem. Phys. Lett., 2001, 336, 439–444 CrossRef CAS.
- D. S. Larsen, M. Vengris, I. H. van Stokkum, M. A. van der Horst, R. A. Cordfunke, K. J. Hellingwerf and R. van Grondelle, Chem. Phys. Lett., 2003, 369, 563–569 CrossRef CAS.
- D. S. Larsen, M. Vengris, I. H. M. van Stokkum, M. A. van der Horst, F. L. de Weerd, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2004, 86, 2538–2550 CrossRef CAS PubMed.
- D. S. Larsen, I. H. M. van Stokkum, M. Vengris, M. A. van der Horst, F. L. de Weerd, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2004, 87, 1858–1872 CrossRef CAS PubMed.
- I. B. Nielsen, S. Boye-Peronne, M. O. A. El Ghazaly, M. B. Kristensen, S. Brondsted Nielsen and L. H. Andersen, Biophys. J., 2005, 89, 2597–2604 CrossRef CAS PubMed.
- M. Vengris, D. S. Larsen, M. A. van der Horst, O. F. A. Larsen, K. J. Hellingwerf and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 4197–4208 CrossRef CAS PubMed.
- I.-R. Lee, W. Lee and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 258–262 CrossRef CAS PubMed.
- A. Espagne, D. H. Paik, P. Changenet-Barret, M. M. Martin and A. H. Zewail, ChemPhysChem, 2006, 7, 1717–1726 CrossRef CAS PubMed.
- A. Espagne, P. Changenet-Barret, J. B. Baudin, P. Plaza and M. M. Martin, J. Photochem. Photobiol., A, 2007, 185, 245–252 CrossRef CAS.
- A. Espagne, D. H. Paik, P. Changenet-Barret, P. Plaza, M. M. Martin and A. H. Zewail, Photochem. Photobiol. Sci., 2007, 6, 780–787 CAS.
- L. Lammich, J. Rajput and L. H. Andersen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 051916 CrossRef PubMed.
- T. Rocha-Rinza, O. Christiansen, J. Rajput, A. Gopalan, D. B. Rahbek, L. H. Andersen, A. V. Bochenkova, A. A. Granovsky, K. B. Bravaya, A. V. Nemukhin, K. L. Christiansen and M. Brondsted Nielsen, J. Phys. Chem. A, 2009, 113, 9442–9449 CrossRef CAS PubMed.
- T. Rocha-Rinza, O. Christiansen, D. B. Rahbek, B. Klærke, L. H. Andersen, K. Lincke and M. Brøndsted Nielsen, Chem. – Eur. J., 2010, 16, 11977–11984 CrossRef CAS PubMed.
- J. Rajput, D. B. Rahbek, G. Aravind and L. H. Andersen, Biophys. J., 2010, 98, 488–492 CrossRef CAS PubMed.
- H. Kuramochi, S. Takeuchi and T. Tahara, J. Phys. Chem. Lett., 2012, 3, 2025–2029 CrossRef CAS.
- M. Almasian, J. Grzetic, J. Van Maurik, J. D. Steill, G. Berden, S. Ingemann, W. J. Buma and J. Oomens, J. Phys. Chem. Lett., 2012, 3, 2259–2263 CrossRef CAS PubMed.
- S. Naseem, A. D. Laurent, E. C. Carroll, M. Vengris, M. Kumauchi, W. D. Hoff, A. I. Krylov and D. S. Larsen, J. Photochem. Photobiol., A, 2013, 270, 43–52 CrossRef CAS.
- C. R. S. Mooney, M. A. Parkes, A. Iskra and H. H. Fielding, Angew. Chem., Int. Ed., 2015, 54, 5646–5649 CrossRef CAS PubMed.
- J. Zhu, L. Paparelli, M. Hospes, J. Arents, J. T. M. Kennis, I. H. M. van Stokkum, K. J. Hellingwerf and M. L. Groot, J. Phys. Chem. B, 2013, 117, 11042–11048 CrossRef CAS PubMed.
- G. Groenhof, M. Bouxin-cademartory, B. Hess, S. P. D. Visser, H. J. C. Berendsen, M. Olivucci, A. E. Mark and M. A. Robb, J. Am. Chem. Soc., 2004, 126, 4228–4233 CrossRef CAS PubMed.
- E. V. Gromov, I. Burghardt, J. T. Hynes, H. Köppel and L. S. Cederbaum, J. Photochem. Photobiol., A, 2007, 190, 241–257 CrossRef CAS.
- D. Zuev, K. B. Bravaya, M. V. Makarova and A. I. Krylov, J. Chem. Phys., 2011, 135, 194304 CrossRef PubMed.
- E. V. Gromov, I. Burghardt, H. Köppel and L. S. Cederbaum, J. Phys. Chem. A, 2011, 115, 9237–9248 CrossRef CAS PubMed.
- M. Boggio-Pasqua, M. A. Robb and G. Groenhof, J. Am. Chem. Soc., 2009, 131, 13580–13581 CrossRef CAS PubMed.
- F. F. García-Prieto, I. F. Galván, A. Muñoz Losa, M. A. Aguilar and M. E. Martn, J. Chem. Theory Comput., 2013, 9, 4481–4494 CrossRef PubMed.
- E. V. Gromov, J. Chem. Phys., 2014, 141, 224308 CrossRef PubMed.
- S. Frutos-Puerto, A. Muñoz Losa, M. E. Martn and M. A. Aguilar, Comput. Theor. Chem., 2014, 1040–1041, 287–294 CrossRef CAS.
- F. F. García-Prieto, M. A. Aguilar, F. J. Olivares del Valle, I. Fernández Galván, A. Muñoz Losa, M. L. Sánchez and M. E. Martn, J. Phys. Chem. A, 2015, 119, 5504–5514 CrossRef PubMed.
- A. R. McKay, M. E. Sanz, C. R. S. Mooney, R. S. Minns, E. M. Gill and H. H. Fielding, Rev. Sci. Instrum., 2010, 81, 123101 CrossRef CAS PubMed.
- C. R. S. Mooney, M. E. Sanz, A. R. McKay, R. J. Fitzmaurice, A. E. Aliev, S. Caddick and H. H. Fielding, J. Phys. Chem. A, 2012, 116, 7943–7949 CrossRef CAS PubMed.
- C. R. S. Mooney, M. A. Parkes, L. Zhang, H. C. Hailes, A. Simperler, M. J. Bearpark and H. H. Fielding, J. Chem. Phys., 2014, 140, 205103 CrossRef PubMed.
- G. A. Garcia, L. Nahon and I. Powis, Rev. Sci. Instrum., 2004, 75, 4989 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, H. P. H. X. Li, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. von Ragué Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- T. Yanai, D. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- J. Linderberg and Y. Öhrn, Propagators in Quantum Chemistry, John Wiley and Sons, Hoboken, New Jersey, 2004, p. 79 Search PubMed.
- V. G. Zakrzewski, O. Dolgounitcheva, A. V. Zakjevskii and J. V. Ortiz, Annu. Rep. Comput. Chem., 2010, 6, 79 CAS.
- D. Zuev, K. B. Bravaya, T. D. Crawford, R. Lindh and A. I. Krylov, J. Chem. Phys., 2011, 134, 034310 CrossRef PubMed.
- E. Janusson, A. Hesketh, K. Bamford, K. Hatlelid, R. Higgins and J. S. McIndoe, Int. J. Mass Spectrom., 2015, 388, 1–8 CrossRef CAS.
- M. Uppsten and B. Durbeej, J. Comput. Chem., 2012, 33, 1892–1901 CrossRef CAS PubMed.
- A. V. Bochenkova, B. Klærke, D. B. Rahbek, J. Rajput, Y. Toker and L. H. Andersen, Angew. Chem., Int. Ed., 2014, 53, 9797–9801 CrossRef CAS PubMed.
- C. W. West, J. N. Bull, A. S. Hudson, S. L. Cobb and J. R. R. Verlet, J. Phys. Chem. B, 2015, 119, 3982–3987 CrossRef CAS PubMed.
- A. M. Bogdanov, A. S. Mishin, I. V. Yampolsky, V. V. Belousov, D. M. Chudakov, F. V. Subach, V. V. Verkhusha, S. Lukyanov and K. A. Lukyanov, Nat. Chem. Biol., 2009, 5, 459–461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of S-methyl (E)-3-(4-hydroxyphenyl)prop-2-enethioate (pCT). Coordinates of the optimised geometries of the deprotonared anions. See DOI: 10.1039/c6cp00565a |
| This journal is © the Owner Societies 2016 |