
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular tectonics: homochiral coordination polymers based on pyridyl-substituted cyclic tetrapeptides†
Alexander
Ganß
a,
Chaojie
Xu
b,
Aurélie
Guenet
*b,
Harald
Kelm
c,
Nathalie
Kyritsakas
b,
Jean-Marc
Planeix
*b,
Stefan
Kubik
 *a and
Mir Wais
Hosseini
*b
*a and
Mir Wais
Hosseini
*b
aFachbereich Chemie - Organische Chemie Erwin-Schrödinger-Straße, Technische Universität Kaiserslautern, 67663 Kaiserslautern, Germany. E-mail: kubik@chemie.uni-kl.de
bMolecular Tectonics Laboratory, UMR UDS-CNRS 7140, icFRC, University of Strasbourg, F-67000 Strasbourg, France. E-mail: aguenet@unistra.fr; planeix@unistra.fr; hosseini@unistra.fr
cFachbereich Chemie - Anorganische Chemie Erwin-Schrödinger-Straße, Technische Universität Kaiserslautern, 67663 Kaiserslautern, Germany
First published on 16th September 2016
Abstract
Upon combining enantiopure bis-4-pyridylphenyl-substituted cyclotetrapeptides with HgCl2 and CdCl2, homochiral 1D zigzag coordination polymers were obtained, while the use of a bis-4-pyridyl-substituted cyclotetrapeptide with HgCl2 led to the formation of a different type of 1D coordination network.
Applying the concepts of molecular tectonics,1 combinations of organic coordinating tectons with metal ions, complexes or clusters acting as metallic nodes by self-assembly processes lead to the formation of coordination networks (CNs), also known as coordination polymers (CPs), of different dimensionalities (1D, 2D, 3D). This type of architectures in the crystalline phase has attracted considerable interest over the last two decades because of their possible applications in, for example, gas storage, catalysis or artificial photosynthesis.2 Among a very large variety of CPs and CNs reported to date, homochiral architectures3 based on enantiopure tectons are of interest for asymmetric catalysis or as stationary phases for enantiomeric separation. Chiral amino acids and peptide derivatives are backbones of choice for the design of chiral tectons as, in addition to amino and carboxylate groups, they may be further equipped with peripheral coordinating groups.4,5 However, chiral tectons based on amino acids and acyclic peptides are often flexible. Although this feature may be exploited for the design of adaptable and/or responsive porous materials,6 it may be detrimental in terms of structural stability.7 In order to increase the predictability of the design in terms of connectivity, dimensionality and geometry, one may rigidify this type of frameworks using secondary interactions such as intra- and intermolecular H-bonding7a or reducing the conformational space of the backbone using cyclic peptides for example. Surprisingly, cyclopeptides have scarcely been explored8 for the design of peptide-based coordination polymers, while they have been exploited in various applications such as anion binding or the formation of artificial nanotubular materials.9 To the best of our knowledge, only two examples of cyclic peptide-based coordination polymers characterised by X-ray diffraction techniques have been reported to date. The two examples are either based on a naturally occurring cyclopeptide8c or a synthetic tris-pyridyl-appended cyclopeptide.8b However, in both cases, the peripheral coordinating sites are not directly connected to the cyclic backbone, imparting thus some flexibility through C–C bond rotations.
Herein we report on the design, synthesis and structural characterisation of two cyclotetrapeptide-based tectons 1 and 2 (Scheme 1) and their use for the formation of homochiral coordination polymers in the crystalline phase. Both tectons 1 and 2 are based on L-proline as the chiral centre. In both cases, the two L-proline units are interconnected into cyclic structures thanks to bis-amide junctions using either two 3-amino-5-(pyridine-4-yl)benzoate (1) or 5-aminopyridine-3-carboxylate (2) moieties as peripheral coordinating sites.
 | ||
| Scheme 1 Chemical structures of tectons 1 and 2 (top) and the synthetic strategy for the preparation of compound 1 (bottom). See the ESI† for the synthesis of 2. | ||
Both cyclopeptides were prepared by conventional peptide synthesis from the respective dipeptides in solution. Scheme 1 shows the synthesis of 1 as an example (see the ESI† for the synthesis of 2). The dipeptide subunit of 1 was obtained in two steps by first coupling Boc-L-proline and butyl 3-amino-5-bromobenzoate 310 in the presence of chlorotripyrrolidinophosphonium hexafluorophosphate (PyCloP) to give 4 followed by a Suzuki Miyaura reaction to afford 5. The butyl ester 3 was chosen to avoid transesterification during the Suzuki–Miyaura reaction.11 Synthesis of 2 started by PyCloP-mediated coupling of Boc-L-proline and methyl 5-aminopyridine-3-carboxylate. To facilitate workup of the carboxy-deprotected tetrapeptides at a later step of the syntheses, both dipeptides were converted into the respective benzyl esters. To this end, they were saponified and the resulting free acids were esterified with benzyl alcohol using O-(1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) as a coupling reagent. Chain elongation was achieved by coupling the Boc deprotected forms of these benzyl esters to the respective Boc protected free acids in the presence of TBTU. The tetrapeptides thus obtained were first hydrogenated and then Boc deprotected. Final cyclization of the corresponding products afforded 1 and 2 with cyclization yields of 16% and 52%, respectively.
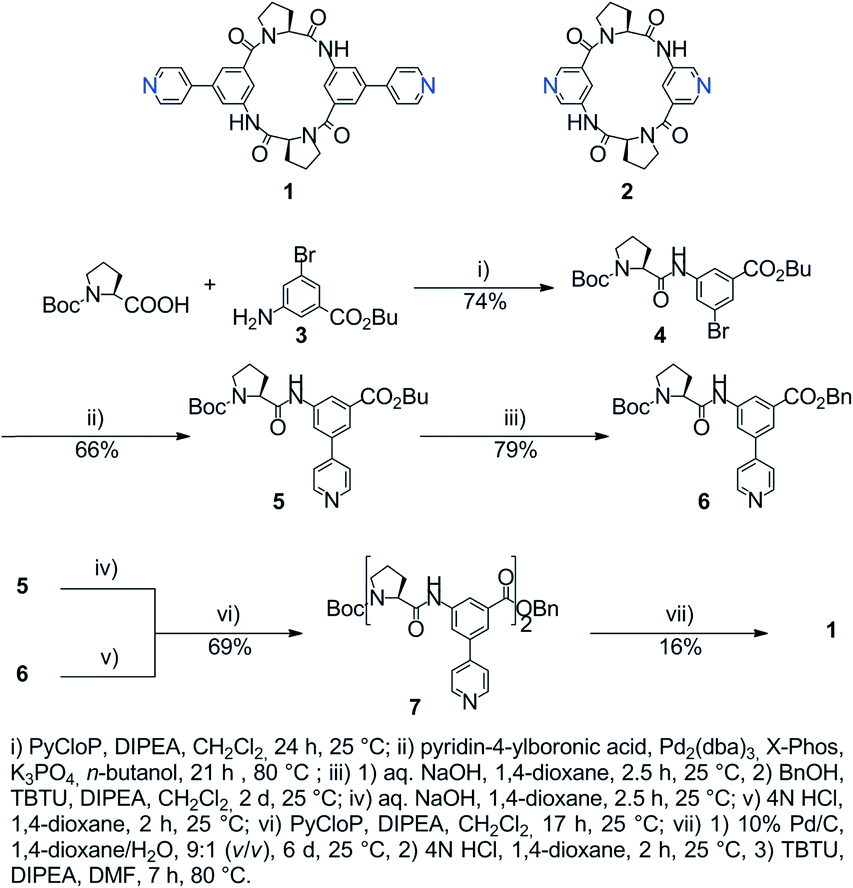
The crystal structures of both tectons 1 and 2 were investigated in the solid state by single-crystal X-ray diffraction (Fig. 1, see the ESI† for crystallographic data).‡ Cyclopeptide 1 crystallises in the P43212 space group (tetragonal system) with two water molecules and one disordered EtOH molecule in the lattice. Tecton 1 adopts a V-shaped conformation with the two pyridyl units pointing to the same side of the cyclic backbone. The angle between the mean planes of the two phenyl units amounts to 52.0°, while the two pyridyl units are tilted with an angle of 38.6° with respect to the phenyl groups (considering the angle between the mean planes of both aromatic moieties). No direct H-bonds between consecutive cyclopeptides are observed. However, a H2O molecule bridges three cyclopeptides through H-bonds between NH groups of the secondary amide, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of the tertiary amide and the N atom of a pyridyl moiety (distances:
O groups of the tertiary amide and the N atom of a pyridyl moiety (distances: 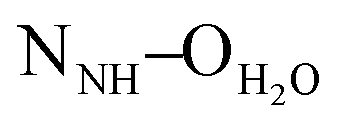 = 2.77 Å;
= 2.77 Å; 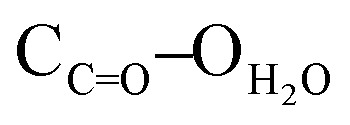 = 2.78 Å;
= 2.78 Å; 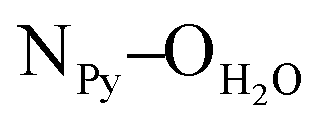 = 2.81 Å, see the ESI†). Weak π–π interactions between pyridyl units are also observed (shortest C–C distance = 3.56 Å, see the ESI†). Tecton 2 crystallises in the P212121 space group (orthorhombic system) with two independent CH3OH molecules. Again, as with tecton 1, compound 2 exhibits a V-shaped conformation with the two pyridyl units pointing towards the same face of the backbone with an angle of 51.2° between the mean planes of the two pyridyl moieties. As expected, the distance between the N atoms of the two pyridyl moieties is shorter (dN–N = 6.84 Å) than that in 1 (dN–N = 10.72 Å). Moderate H-bonding interactions are observed (distances: NPy–NNH = 2.9 Å;
= 2.81 Å, see the ESI†). Weak π–π interactions between pyridyl units are also observed (shortest C–C distance = 3.56 Å, see the ESI†). Tecton 2 crystallises in the P212121 space group (orthorhombic system) with two independent CH3OH molecules. Again, as with tecton 1, compound 2 exhibits a V-shaped conformation with the two pyridyl units pointing towards the same face of the backbone with an angle of 51.2° between the mean planes of the two pyridyl moieties. As expected, the distance between the N atoms of the two pyridyl moieties is shorter (dN–N = 6.84 Å) than that in 1 (dN–N = 10.72 Å). Moderate H-bonding interactions are observed (distances: NPy–NNH = 2.9 Å; 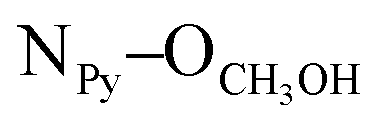 = 2.8 Å and
= 2.8 Å and 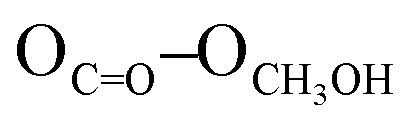 = 2.7 Å). However, when compared to tecton 1, the H-bonds pattern is more intricate involving the different H-bond donor and acceptor sites of 2 and CH3OH molecules (see the ESI†). The structural investigations discussed above showed that both chiral compounds 1 and 2 may be described as enantiopure distorted V-shaped building units.
= 2.7 Å). However, when compared to tecton 1, the H-bonds pattern is more intricate involving the different H-bond donor and acceptor sites of 2 and CH3OH molecules (see the ESI†). The structural investigations discussed above showed that both chiral compounds 1 and 2 may be described as enantiopure distorted V-shaped building units.
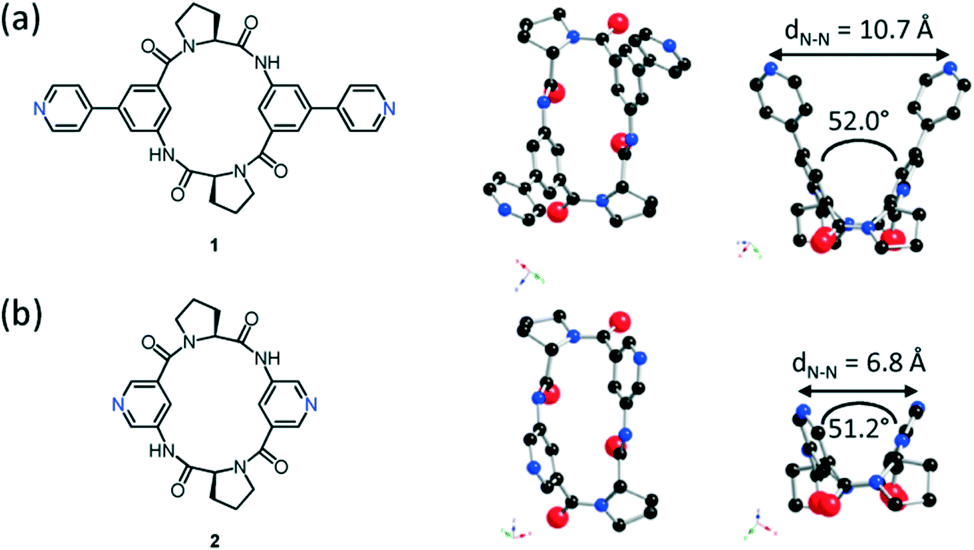 | ||
| Fig. 1 Chemical and crystal structures of 1 (a) and 2 (b) along two directions showing the solid state conformations. H atoms and solvent molecules are omitted for clarity. | ||
As connecting metallatectons, both neutral CdCl2 and HgCl2 complexes have been chosen. The rationale behind this choice was their neutral nature, on the one hand, which should lead to neutral coordination polymers upon their combination with the neutral coordinating tectons 1 and 2. On the other hand, in principle, both CdCl2 and HgCl2 could behave as two connecting achiral V-shaped nodes. Thus, it was expected that combination of coordinating tectons 1 and 2 with either CdCl2 or HgCl2 should lead to enantiomerically pure 1D coordination polymers. Tecton 1 was combined with either CdCl2 or HgCl2.§ Crystals thus obtained were structurally investigated by X-ray diffraction on single crystals (Fig. 2 and the ESI†). For both cases (1·CdCl2 and 1·HgCl2), crystals (C2 space group, monoclinic system) are composed of tecton 1 and the metallic node MCl2 (M = Hg or Cd). No solvent molecules are present in the lattice. Both combinations lead to the formation of isostructural 1D coordination polymers (Fig. 2 and the ESI†). For 1·CdCl2 (Fig. 2), the Cd atom adopts a slightly distorted tetrahedral geometry with its coordination sphere occupied by two pyridyl moieties of two consecutive tectons 1 (dCd–N = 2.258(5) Å) and two chloride ions (dCd–Cl = 2.437(3) Å). The N–Cd–N, Cl–Cd–Cl and N–Cd–Cl angles are 109.7(3)°, 139.20(19)°, 100.72(15)° and 102.43(16)°, respectively. Thus, as predicted, the CdCl2 moiety behaves as a two-connecting node bridging two consecutive tectons 1 (Fig. 2a). Within the 1D polymer, two consecutive Cd atoms are separated by ca. 18.6 Å. Similarly, for 1·HgCl2, the Hg atom also adopts a distorted tetrahedral coordination geometry with slightly different geometrical parameters (dHg–N = 2.388(7) Å, dHg–Cl = 2.377(3) Å; N–Hg–N, Cl–Hg–Cl and N–Hg–Cl angles of 98.3(4)°, 152.65(15)°, 95.7(2)° and 102.1(2)°, respectively) (see the ESI†). The distance of 18.3 Å between two Hg nodes in the chain is similar to the one observed for 1·CdCl2 (18.6 Å). For both structures, 1 adopts overall a similar V-shaped conformation to the free form (Fig. 1), exhibiting, however, larger angles between the mean planes of the phenyl units of 89.8° and 89.4° for 1·CdCl2 and 1·HgCl2, respectively. The tilt angle between the pyridyl and phenyl groups is slightly smaller (16.8° for 1·CdCl2 and 13.8° for 1·HgCl2). Because of the coordination geometry adopted by the M2+ metal ion and the conformation of tecton 1, both 1D polymers are of the zigzag type (Fig. 2b and the ESI†). Furthermore, the presence of the NH groups and CO moieties of the secondary amide groups, oriented in opposite directions, leads to the formation of two hydrogen bonds per cyclopeptide unit. Thus, consecutive zigzag chains are interconnected along the c axis (Cd–Cd axis) through hydrogen bonds between the two NH units of a cyclopeptide belonging to one zigzag chain and the two CO moieties belonging to consecutive parallel chains (distances for 1·CdCl2: HNH–OCO 2.00 Å, NNH–OCO 2.80 Å; distances for 1·HgCl2: HNH–OCO 1.98 Å, NNH–OCO 2.80 Å) (Fig. 2b and the ESI†). The consecutive zigzag chains are separated along the c axis by ca. 5.0 Å corresponding to the shortest Cd–Cd or Hg–Hg distance (Fig. 2b and the ESI†). Finally, consecutive 1D polymers are packed in a staggered manner along the b axis (Fig. 2a and the ESI†). Taking into account both types of interactions, i.e. coordination and hydrogen bonds, the overall architecture is a 2D hybrid H-bonding/coordination network. For both polymers, the purity of the phase was established by powder X-ray diffraction (PXRD) on microcrystalline batches which revealed a good match between the observed and simulated patterns from the XRD data (see the ESI†).
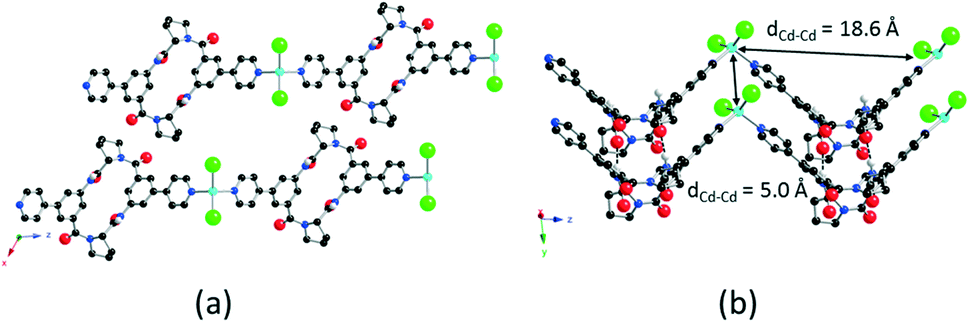 | ||
| Fig. 2 Portions of the crystal structure of the 1D polymer 1·CdCl2 along (a) the b axis, and (b) perpendicular to the Cd–Cd axis highlighting the staggered packing of parallel 1D zigzag networks and showing the H-bonds between two cyclopeptides belonging to two distinct polymers. H-bonds are depicted as dashed lines. Only the H atoms involved in H-bonding interactions are presented. | ||
Independent of the 2/HgCl2 molar ratio, the combination of cyclopeptide 2 with HgCl2 afforded crystalline materials§ composed of a different 1D coordination network 2·HgCl2·CH3OH (Fig. 3). In contrast with 1·HgCl2, the crystal (space group C2221, orthorhombic system) contains, in addition to the organic tecton 2, HgCl2 and CH3OH molecules. Within the lattice, two non-equivalent Hg centres (Hg1 and Hg2) are present. They differ by their coordination number, sphere and geometry. Indeed, whereas Hg1 is found to be 5-coordinated, Hg2 is 3-coordinated (Fig. 3b). The Hg atom labelled as Hg2 is coordinated to two chloride ions and one nitrogen atom belonging to one pyridyl unit of a cyclopeptide. It adopts a distorted T-shaped geometry (dHg–Cl = 2.22(3) Å and 2.32(2) Å, dHg–N = 2.38(3) Å, Cl–Hg–Cl angle of 165.7(9)° and Cl–Hg–N angles of 88.7(10)° and 104.1(11)°) (Fig. 3b). It should be noted that the site occupancy for Hg2 is 0.5. The latter acts as a terminal metallic centre, and thus not as a node of the network. In marked contrast, Hg1 behaves as a three-connecting node leading to the formation of the extended periodic network. The pentacoordinated Hg1 adopts a distorted trigonal bipyramidal geometry. The trigonal base is occupied by two chloride ions and one Npy atom belonging to one pyridyl moiety of 2 (dHg–Cl = 2.323(6) Å and 2.337(7) Å, dHg–N = 2.43(2) Å and Cl–Hg–Cl angle of 151.3(4)°, Cl–Hg–N angles of 100.7(6)° and 107.7(6)°). The remaining two axial positions are occupied by a Npy atom belonging to another tecton 2 and an O atom belonging to the CO unit of a third cyclopeptide 2 (dHg–Nax = 2.68(3) Å, dHg–Oax = 2.81 Å and Nax–Hg–N angle of 81.6(8)°, Cl–Hg–Nax angles of 95.9(5)° and 91.9(5)°, Oax–Hg–N angle of 78.87° and Oax–Hg–Cl angles of 88.90° and 92.91°). It is worth noting that, although the Hg–O distance of 2.81 Å is rather long, it is less than the sum of the O and Hg van der Waals radii and thus may be considered as a bonding interaction. The overall connectivity of the network 2·HgCl2·CH3OH is rather complex. In order to simplify its description, we shall first consider only the Hg–N connectivity. Based on this type of bonding, a trimeric unit composed of one central and two peripheral tectons 2 is spotted. The central cyclic peptide is connected to the two peripheral tectons by two 5-coordinated Hg1 atoms. The two peripheral tectons are capped by the 3-coordinated Hg2 atom with an occupancy of 0.5 (Fig. 3c and d). The two Npy atoms of the central cyclopeptide are oriented divergently and both are located at the trigonal base of two distinct Hg1 atoms. The two pyridyl moieties of the peripheral tectons 2 behave differently since one of them occupies the axial position of the 5-coordinated Hg1, whereas the second one is coordinated to the terminal 3-coordinated Hg2. The two peripheral cyclopeptides exhibit angles between the mean planes of the aromatic rings of ca. 57.5° comparable with the one observed for the free tecton 2. However, for the central cyclopeptide, a smaller angle of ca. 38.3° is observed. Within the trimeric units, the three cyclopeptides are not coplanar, preventing thus the formation of a zigzag architecture as observed for 1·HgCl2. The bridging of consecutive trimeric units through Hg–O(C) bonds leads to the formation of a 1D coordination network. Indeed, consecutive trimeric units are interconnected through Hg1–O(C) bonds between one peripheral tecton 2 and Hg1 (Fig. 3a). As the two 5-coordinated Hg1 atoms of one trimeric unit are coordinated to two different CO groups belonging to two different trimeric units in a divergent orientation, a 1D coordination network is generated. Consecutive 1D networks are packed in a parallel fashion with MeOH solvent molecules occupying the empty space (see the ESI†). The purity of the microcrystalline phase obtained was again checked by PXRD which revealed a good match between the recorded and simulated patterns using the single crystal data (see the ESI†).
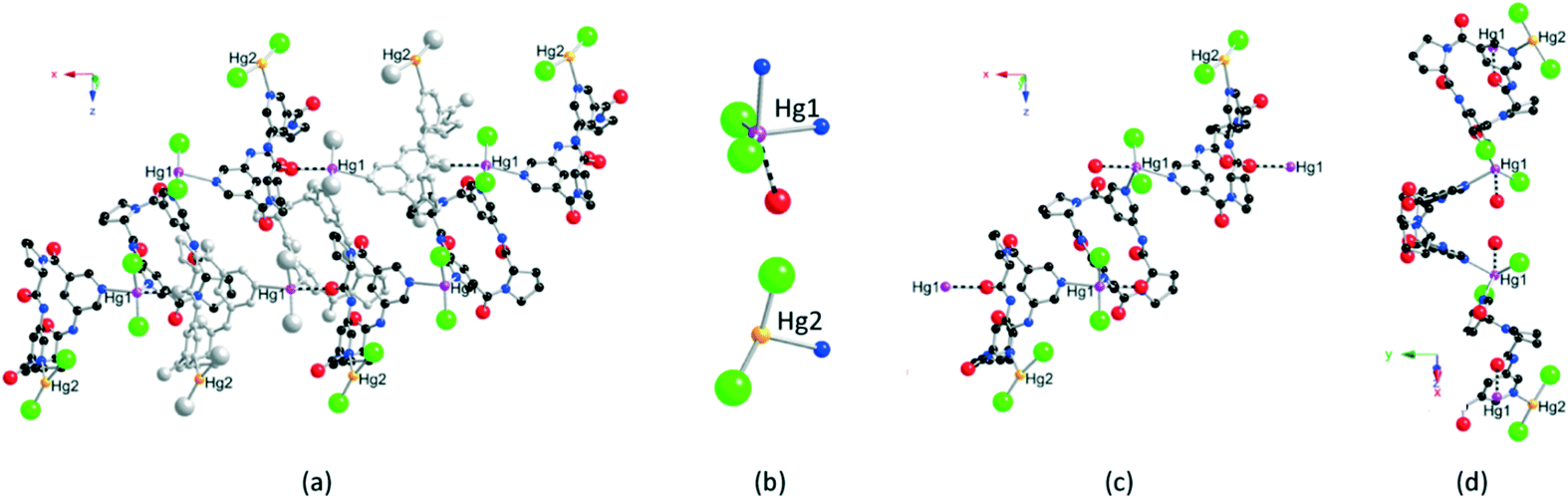 | ||
| Fig. 3 Portions of the crystal structure of the 1D network 2·HgCl2·CH3OH along the 1D axis of the network (a) showing the coordination sphere of the two distinct Hg atoms (b) as well as the trimeric unit along the axis of the 1D network (c) and along another axis highlighting the V-shaped conformation of the central cyclopeptide unit (d). The two non-equivalent Hg atoms are depicted in orange (Hg2) and purple (Hg1). One of the trimeric units is coloured in grey in Fig. 3a to differentiate adjacent trimeric units. Hg2 is represented in each trimeric unit even though its site occupancy is 0.5. The Hg–O bond is depicted as a dotted line. H atoms and solvent molecules are not shown for clarity. | ||
In conclusion, two new enantiopure L-proline based cyclopeptides bearing either 4-pyridyl or 4-phenylpyridyl units have been synthesised. Combinations of tectons 1 or 2 behaving as chiral V-shaped coordinating tectons with CdCl2 or HgCl2 metallic nodes afforded three homochiral coordination polymers. Although both tectons are rather rigid, owing to the cyclic nature of their backbone and depending on the distance between the two pyridyl units, either a 2D hybrid H-bonded/coordination network or a 1D coordination architecture is obtained. Efforts towards the formation of other coordination networks using cyclic peptide-based tectons and other metal centres are currently under way.
This contribution is part of the Chiranet project funded by the European Regional Development Fund (ERDF) under the INTERREG IV Upper Rhine Programme and as part of the Science Offensive of the Trinational Upper Rhine Metropolitan Region. Financial support from the University of Strasbourg, the International Centre for Frontier Research in Chemistry (icFRC), Strasbourg, the Institut Universitaire de France, and the CNRS is acknowledged.
Notes and references
- (a) M. Simard, D. Su and J. D. Wuest, J. Am. Chem. Soc., 1991, 113, 4696 CrossRef CAS; (b) S. Mann, Nature, 1993, 365, 499 CrossRef CAS; (c) M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313 CrossRef CAS PubMed.
- (a) Chem. Rev., 2012, 112, MOFs special issue Search PubMed; (b) Chem. Soc. Rev., 2014, 43, themed issue on MOFs Search PubMed.
- (a) K. Kim, M. Banerjee, M. Yoon and S. Das, Top. Curr. Chem., ed. M. Schröder, Springer Berlin Heidelberg, 2010, vol. 293, p. 115 Search PubMed; (b) W. Lin, Top. Catal., 2010, 53, 869 CrossRef CAS; (c) G. Nickerl, A. Henschel, R. Grünker, K. Gedrich and S. Kaskel, Chem. Ing. Tech., 2011, 83, 90 CrossRef CAS; (d) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed; (e) K. K. Bisht, B. Parmar, Y. Rachuri, A. C. Kathalikattil and E. Suresh, CrystEngComm, 2015, 17, 5341 RSC; (f) A. Jouaiti, M. W. Hosseini and N. Kyritsakas, Chem. Commun., 2002, 1898 RSC; (g) P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2013, 49, 4468 RSC; (h) N. Marets, V. Bulach and M. W. Hosseini, New J. Chem., 2013, 37, 3549 RSC; (i) P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Dalton Trans., 2014, 43, 2000 RSC; (j) P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Dalton Trans., 2014, 43, 166 RSC.
- T. Takayama, S. Ohuchida, Y. Koike, M. Watanabe, D. Hashizume and Y. Ohashi, Bull. Chem. Soc. Jpn., 1996, 69, 1579 CrossRef CAS.
- (a) I. Imaz, M. Rubio-Martinez, J. An, I. Sole-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287 RSC; (b) X.-L. Yang and C.-D. Wu, CrystEngComm, 2014, 16, 4907 RSC.
- J. Rabone, Y.-F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053 CrossRef CAS PubMed.
- (a) C. Martí-Gastaldo, J. E. Warren, K. C. Stylianou, N. L. O. Flack and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 11044 CrossRef PubMed; (b) C. Martí-Gastaldo, J. E. Warren, M. E. Briggs, J. A. Armstrong, K. M. Thomas and M. J. Rosseinsky, Chem. – Eur. J., 2015, 21, 16027 CrossRef PubMed.
- (a) F. Fujimura and S. Kimura, Org. Lett., 2007, 9, 793 CrossRef CAS PubMed; (b) Y. Dong, D. T. J. Loong, A. K. L. Yuen, R. J. Black, S. O'Malley, J. K. Clegg, L. F. Lindoy and K. A. Jolliffe, Supramol. Chem., 2012, 24, 508 CrossRef CAS; (c) S. Chakraborty, P. Tyagi, D.-F. Tai, G.-H. Lee and S.-M. Peng, Molecules, 2013, 18, 4972 CrossRef CAS PubMed.
- (a) R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448 RSC; (b) S. Kubik, Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd, 2012, vol. 3, p. 1179 Search PubMed; (c) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS PubMed; (d) A. K. Yudin, Chem. Sci., 2015, 6, 30 RSC.
- (a) N. Dales, J. Fonarev, J. Fu and Z. Zhang, WO2010112520A1, 2010; (b) R. Reuter and H. A. Wegner, Chem. Commun., 2013, 49, 146 RSC.
- K. L. Billingsley, K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 3484 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical (NMR, mass spectra) and crystallographic data. CCDC 1473103–1473107. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01944g |
| ‡ 1: single crystals obtained by slow evaporation of a solution of 1 in a 2/2/1 mixture of CH2Cl2/MeOH/EtOH; 2: single crystals of 2 grown upon cooling a hot saturated MeOH solution containing the cyclopeptide 2 (see the ESI†). |
| § 1·CdCl2: single crystals obtained by slow diffusion of a solution of CdCl2·2.5H2O in MeOH into a solution of 1 in a mixture of CH2Cl2/MeOH followed by solvent evaporation; 1·HgCl2: single crystals obtained by slow diffusion of a HgCl2 solution in MeOH into a solution of 1 in a mixture of CH2Cl2/MeOH; 2·HgCl2·CH3OH: single crystals obtained by slow diffusion of a HgCl2 solution in MeOH into a solution of 2 in CH2Cl2/MeOH followed by slow solvent evaporation (see the ESI†). |
| This journal is © The Royal Society of Chemistry 2016 |