DOI:
10.1039/C6CE01668E
(Paper)
CrystEngComm, 2016,
18, 8291-8300
Solvatomorphism in (Z)-4-fluoro-N′-(3-fluorophenyl)benzimidamide: the role of intermolecular O–H⋯F interaction†
Received
29th July 2016
, Accepted 15th September 2016
First published on 16th September 2016
Abstract
The synthesized compound (Z)-4-fluoro-N′-(3-fluorophenyl)benzimidamide exhibits solvatomorphism in the solid state. The anhydrous and the hydrate forms were obtained at room temperature from slow evaporation of cyclohexane and hexane solvents, respectively. Both forms crystallize in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two symmetry-independent molecules in the asymmetric unit. The crystal packing of the anhydrous form is stabilized via a strong N–H⋯N chain, whereas the hydrate form is stabilized via a strong (N–H⋯N)–(N–H⋯O)–(O–H⋯N) chain. The water molecule plays an important role in the crystal packing by the formation of “extremely short” N–H⋯O and O–H⋯N hydrogen bonds, which are further supported by weak O–H⋯F–C and C–H⋯O interactions. The basic structural building motifs were structurally and energetically equivalent in both forms. PIXEL and QTAIM approaches provide quantitative insights into the nature and energetics of strong as well as weak intermolecular interactions. The NCI descriptor shows the “attractive” nature of the rarely observed O–H⋯F interaction. The thermal stabilities of the solvatomorphs were characterized via differential scanning calorimetry, hot stage microscopy, and thermogravimetric analysis. Hirshfeld surface analysis and 2D fingerprint plots of the individual molecules present in both forms differentiate the trends in crystal packing as well as the contribution from the different intermolecular interactions.
with two symmetry-independent molecules in the asymmetric unit. The crystal packing of the anhydrous form is stabilized via a strong N–H⋯N chain, whereas the hydrate form is stabilized via a strong (N–H⋯N)–(N–H⋯O)–(O–H⋯N) chain. The water molecule plays an important role in the crystal packing by the formation of “extremely short” N–H⋯O and O–H⋯N hydrogen bonds, which are further supported by weak O–H⋯F–C and C–H⋯O interactions. The basic structural building motifs were structurally and energetically equivalent in both forms. PIXEL and QTAIM approaches provide quantitative insights into the nature and energetics of strong as well as weak intermolecular interactions. The NCI descriptor shows the “attractive” nature of the rarely observed O–H⋯F interaction. The thermal stabilities of the solvatomorphs were characterized via differential scanning calorimetry, hot stage microscopy, and thermogravimetric analysis. Hirshfeld surface analysis and 2D fingerprint plots of the individual molecules present in both forms differentiate the trends in crystal packing as well as the contribution from the different intermolecular interactions.
Introduction
Solvatomorphism,1–9 which is also called pseudopolymorphism,10 is of interest in crystal engineering as well as in the drug and pharmaceutical industry for its various properties. It has the ability to co-crystallize a compound with different stoichiometries of solvent molecules.11 The role of solvatomorphism in crystal engineering has been discussed in a recent review by Aakeroy et al.12 Currently, the challenge in crystal engineering is to develop our understanding of both strong/weak intermolecular interactions and their control in the precise formation of various supramolecular architectures.12–15 The solvent molecules present in solvatomorphs affect the molecular conformation as well as the molecular arrangements via the strong/weak intermolecular interactions. The compound amlodipine besylate exists in anhydrous, monohydrate and dihydrate forms.16 Amongst all of them, the dihydrate is the most stable form in the aqueous environment compared with the other two. This is because the anhydrous and monohydrate forms always undergo phase transformation to the dihydrate form. The commercially available indapamide compound always exists in the hydrate form,12 although the compound produced solvatomorphs with solvents like cyclohexane, carbon tetrachloride and diethyl ether.17 It is reported that the solvent has a strong role in the desolvation characteristics of each solvatomorph of the compound naproxen sodium18 with different solvents (ethanol, n-propanol, isopropanol, n-butanol, and isobutanol).
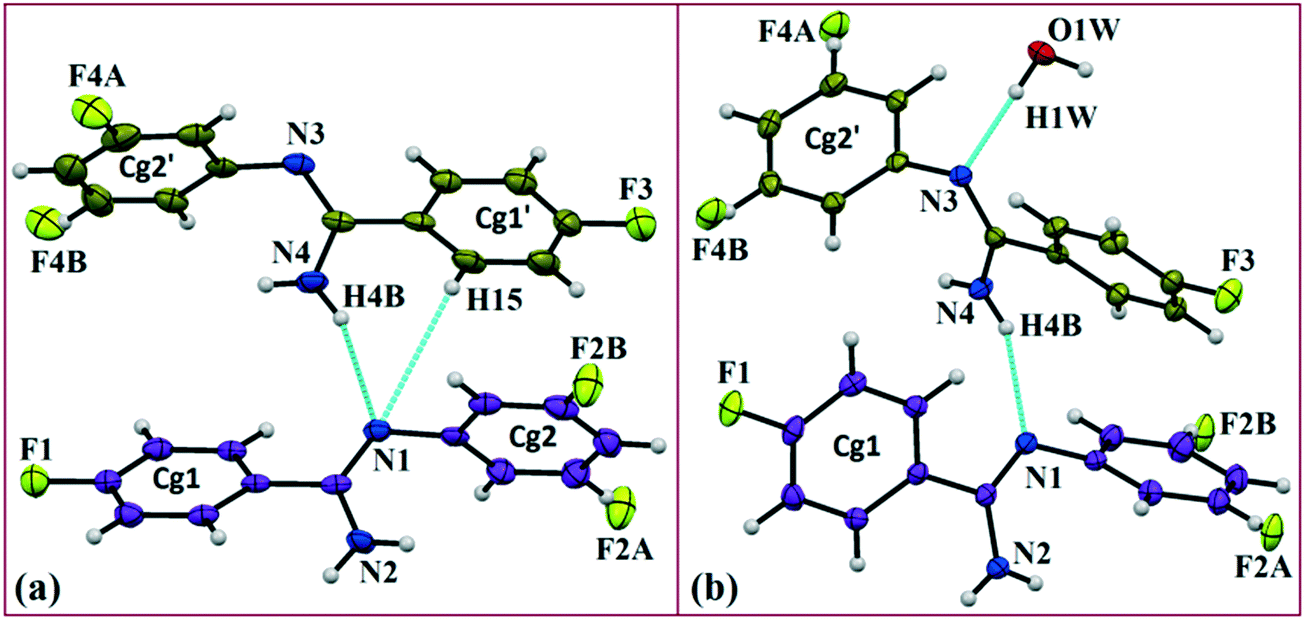
In the current study, as part of our investigation into the polymorphic characteristics of fluorinated phenyl benzimidamides, we have observed the solvatomorph formation in (Z)-4-fluoro-N′-(3-fluorophenyl)benzimidamide (code DB32) and characterized the same via1H-NMR and FTIR spectroscopic methods (Scheme S1, Fig. S1 and S2†). The crystal growth of a similar family of compounds using a variety of growth conditions leads to the formation of an E/Z isomer,19 polymorphs19,20 and the observation of isostructurality.20 The compound was sparingly soluble in solvents hexane and cyclohexane at room temperature and completely soluble after warming at 60 °C. DB32 (anhydrous: plate) (Fig. 1a) was obtained from the slow evaporation of cyclohexane solvent at room temperature whereas DB32W (hydrated: plate) (Fig. 1b) was obtained from the slow evaporation of hexane at room temperature. The solvatomorphs of this compound were characterized via single crystal X-ray diffraction and powder X-ray diffraction. Both forms crystallize in the centrosymmetric P space group with two crystallographically independent molecules in the asymmetric unit (Z′ = 2 and 2 + 1H2O molecule, respectively) with different unit cell parameters (Table S1†). The two symmetry independent molecules (1 and 2) are represented in different color codes; molecule 1 with indigo color and molecule 2 with olive color (Fig. 2).
 |
| | Fig. 1 Optical images of the solvatomorphs: (a) DB32 obtained from cyclohexane at room temperature and (b) DB32W obtained from hexane at room temperature. | |
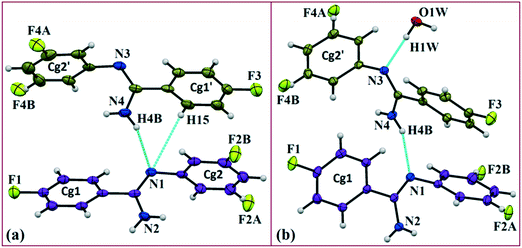 |
| | Fig. 2
ORTEP of the solvatomorphs (a) DB32 and (b) DB32W drawn with 50% ellipsoidal probability. Molecules 1 (indigo color) and 2 (olive color) are the symmetry independent molecules present in the asymmetric unit. | |
Results and discussion
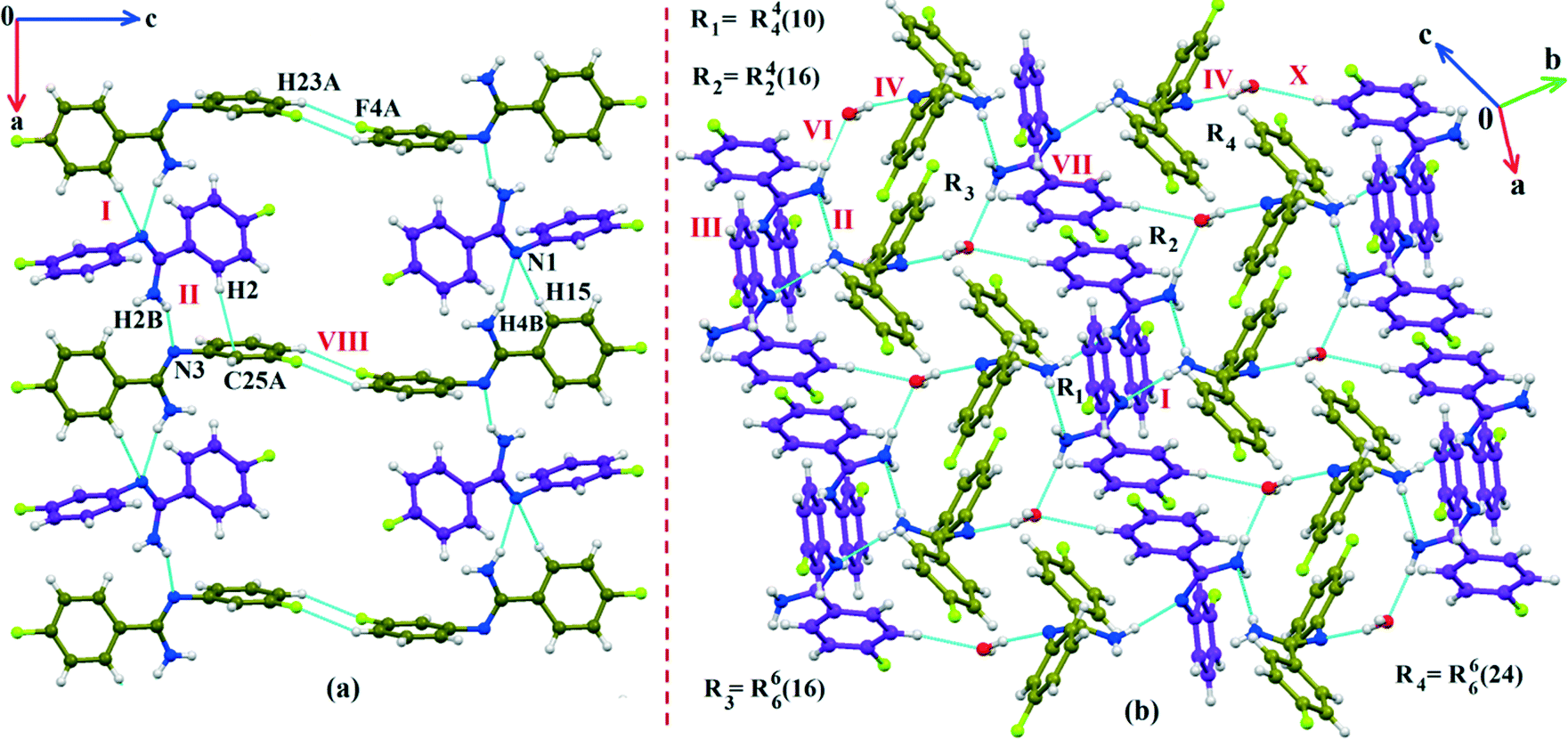
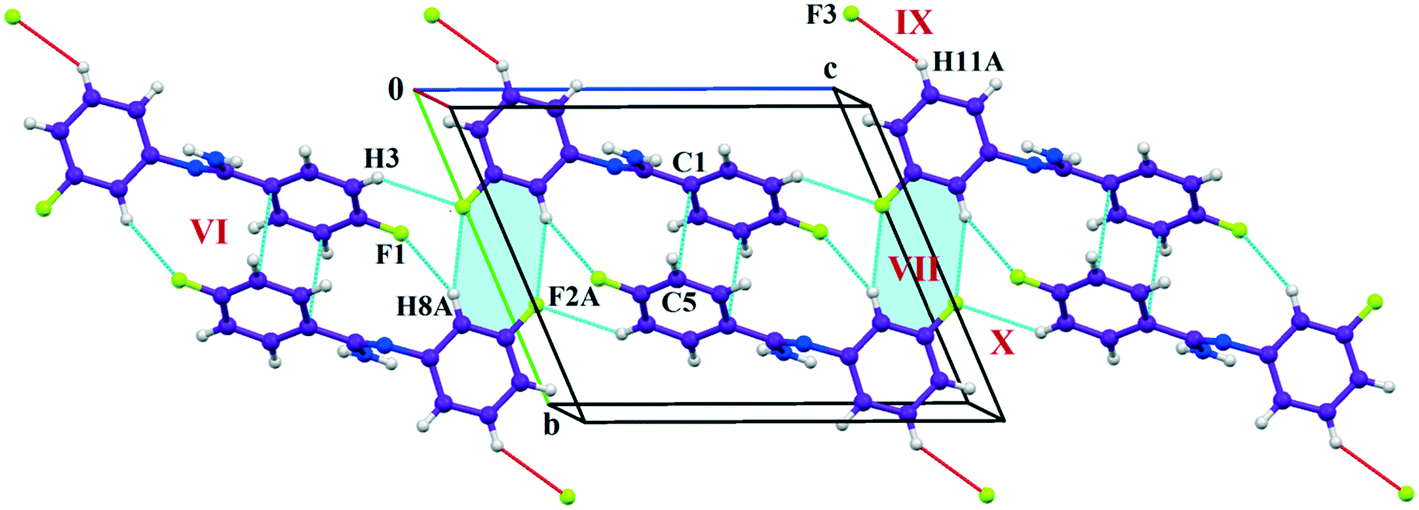
The fluorine atoms (F2 and F4) attached at the meta position of the phenyl rings of molecules 1 and 2, are positionally disordered (93–97% for part A), namely, F2A and F2B; F4A and F4B, respectively. Table 1 lists all the molecular pairs involved in the crystal packing for DB32 and DB32W along with their stabilization energies. In the asymmetric unit (I) of DB32, molecule 1 interacts with molecule 2via a strong N4–H4B⋯N1 hydrogen bond and weak C15–H15⋯N1 interaction (Fig. 2a) with the corresponding stabilization energy (−10.9 kcal mol−1; 61% electrostatic contribution and 39% dispersion contribution). The crystal packing of DB32 (anhydrous) shows that molecules 1 and 2 are connected in an alternative manner via a strong N–H⋯N hydrogen bond [I and II (−10.9 kcal mol−1)] along the a direction leading to the formation of an N–H⋯N chain associated with weak C–H⋯N and C–H⋯π intermolecular interactions (Fig. 3a). Two successive N–H⋯N chains were attached via C–H⋯F dimeric motifs (VIII) associated with molecule 2 along the c direction. Molecule 1 also formed C–H⋯F dimeric motifs (VII) which were connected via the C–H⋯F chain (X) and the centrosymmetric dimer VI associated with the formation of intermolecular C–H⋯F interaction and π⋯π stacking (Fig. 4). Furthermore, the molecules 1 and 2 were together connected via a weak C–H⋯F motif (IX) along the b direction involving H11A with F3 (Fig. 4) having the stabilization energy of −1.6 kcal mol−1 and the molecular pair V (−5.2 kcal mol−1) associated with C19–H19⋯F1 and C5–H5⋯N3 interactions (41% electrostatic contribution) (Fig. S3a†).
Table 1 List of the molecular pairs with their stabilization energies (in kcal mol−1) and related possible intermolecular interactions with their geometrical parameters (D⋯A, H⋯A and ∠D–H⋯A)
| Motifs |
Symmetry |
Distance (Å) |
E
Coul
|
E
Pol
|
E
Disp
|
E
Rep
|
E
Tot
|
Possible interactions |
Geometry (Å/Å/°) |
| DB32 (P) |
| I(1-2) |
x, y, z |
5.208 |
−9.7 |
−4.7 |
−9.1 |
12.0 |
−10.9 |
N4–H4B⋯N1, C15–H15⋯N1 |
3.059(2), 2.07, 159; 3.629(3), 2.83, 131 |
| II(1-2) |
1 + x, y, z |
5.082 |
−9.0 |
−3.9 |
−8.7 |
10.7 |
−10.9 |
N2–H2B⋯N3, C2–H2⋯π(C25A) |
3.032(3), 2.03, 162; 3.560(3), 2.68, 139 |
| III(1-1) |
1 − x, 2 − y, 1 − z |
5.291 |
−2.4 |
−0.9 |
−7.5 |
4.2 |
−6.5 |
C12A–H12A⋯C1, C11A–H11A⋯C4 |
2.921(2), 2.82, 142; 3.105(2), 3.00, 140 |
| IV(2-2) |
2 − x, 2 − y, 1 − z |
4.693 |
−2.1 |
−0.9 |
−7.4 |
5.2 |
−5.3 |
C24A–F4A⋯C17, C25A–H25A⋯C14 |
2.963(1); 3.847(3), 2.92, 144 |
| V(1-2) |
2 − x, 1 − y, 1 − z |
7.175 |
−2.7 |
−1.1 |
−5.5 |
4.1 |
−5.2 |
C19–H19⋯F1, C5–H5⋯N3 |
3.531(2), 2.60, 144; 3.613(2), 2.59, 158 |
| VI(1-1) |
1 − x, 1 − y, 1 − z |
6.493 |
−0.9 |
−0.7 |
−8.6 |
5.3 |
−4.9 |
C1⋯C5, C8A–H8A⋯F1 |
3.440(1); 3.560(3), 2.68, 139 |
| VII(1-1) |
1 − x, 1 − y, 2 − z |
9.019 |
−0.6 |
−0.2 |
−1.5 |
0.6 |
−1.8 |
C8A–H8A⋯F2A |
3.614(2), 2.66, 146 |
| VIII(2-2) |
2 − x, 2 − y, −z |
12.710 |
−1.2 |
−0.3 |
−2.0 |
1.9 |
−1.7 |
C23A–H23A⋯F4A |
3.327(3), 2.42, 141 |
| IX(1-2) |
1 − x, 2 − y, 2 − z |
12.446 |
−0.5 |
−0.1 |
−1.5 |
0.5 |
−1.6 |
C11A–H11A⋯F3 |
3.510(3), 2.68, 133 |
| X(1-1) |
x, y, −1 + z |
12.346 |
0.2 |
−0.3 |
−1.7 |
0.8 |
−1.0 |
C3–H3⋯F2A |
3.514(3), 2.63, 138 |
| |
| DB32W (P) |
| I(1-2) |
x, y, z |
4.882 |
−10.0 |
−4.6 |
−10.9 |
14.5 |
−11.2 |
N4–H4B⋯N1, C19–H19⋯C12A |
2.985(2), 1.96, 175; 3.878(3), 2.91, 149 |
| II(1-2) |
−x + 2, −y + 2, −z + 1 |
5.345 |
−5.6 |
−2.8 |
−10.5 |
8.2 |
−10.7 |
N4–H4A⋯N2, N2–H2A⋯C24A, C24A–H24A⋯C10A |
3.382(2), 2.42, 155; 3.581(2), 2.68, 146 3.779(2), 2.84, 146 |
| III(2-2) |
−x + 1, −y + 1, −z + 1 |
5.073 |
−4.8 |
−2.2 |
−10.1 |
9.1 |
−8.1 |
C15–H15⋯N3, C16–H16⋯Cg2′ |
2.868(2), 2.75, 148; 3.491(2), 2.47, 157 |
| IV (2-W) |
x, y, z |
2.968 |
−15.3 |
−8.6 |
−5.2 |
22.6 |
−6.5 |
O1W–H1W⋯N3 |
2.754(1), 1.83, 170 |
| V(1-1) |
−x + 2, −y + 2, −z + 1 |
5.209 |
−2.8 |
−1.6 |
−9.9 |
9.1 |
−5.2 |
C11A–H11A⋯F1 C12A–H12A⋯Cg1 |
3.770(2), 2.80, 150; 3.511(1), 2.55, 148 |
| VI(1-W) |
x, y + 1, z |
4.334 |
−6.1 |
−2.1 |
−2.2 |
5.8 |
−4.7 |
N2–H2B⋯O1W |
2.892(2), 1.89, 162 |
| VII(1-1) |
−x + 1, −y + 2, −z + 1 |
6.498 |
−1.8 |
−0.9 |
−8.9 |
7.1 |
−4.5 |
C8A–H8A⋯F1, C1⋯C3 |
3.487(2), 2.45, 160; 3.374(2) |
| VIII(1-1) |
−x + 1, −y + 2, −z |
8.302 |
−2.1 |
−0.5 |
−3.2 |
1.6 |
−4.1 |
C8A–H8A⋯F2A, F2A⋯N2 |
3.414(2), 2.74, 120; 3.109(2) |
| IX(2-2) |
−x + 2, −y + 1, −z + 1 |
6.502 |
−0.9 |
−0.6 |
−5.3 |
2.8 |
−4.0 |
C22A–F4A⋯C19, C22A–F4A⋯C26 |
3.328(2); 3.266(2) |
| X(1-W) |
−x + 1, −y + 1, −z + 1 |
6.401 |
−2.7 |
−0.9 |
−1.2 |
2.5 |
−2.4 |
C3–H3⋯O1W |
3.347(2), 2.29, 166 |
| XI (1-W) |
−x + 1, −y + 1, −z |
7.675 |
−2.1 |
−0.9 |
−1.8 |
2.5 |
−2.3 |
O1W–H2W⋯F2A |
2.902(1), 2.15, 136 |
| XII(1-2) |
x + 1, y + 1, z + 1 |
7.618 |
−0.4 |
−0.3 |
−2.4 |
1.1 |
−1.9 |
C5–H5⋯F3 |
3.580(1), 2.79, 130 |
| XIII(1-2) |
−x + 2, −y + 2, −z + 2 |
12.440 |
−0.8 |
−0.2 |
−1.3 |
0.8 |
−1.5 |
C23A–H23A⋯F1 |
3.282(1), 2.51, 127 |
| XIV(1-2) |
x + 1, y, z + 1 |
10.430 |
0.4 |
−0.2 |
−1.8 |
0.7 |
−0.9 |
C18–H18⋯F4A |
3.534(2), 2.67, 120 |
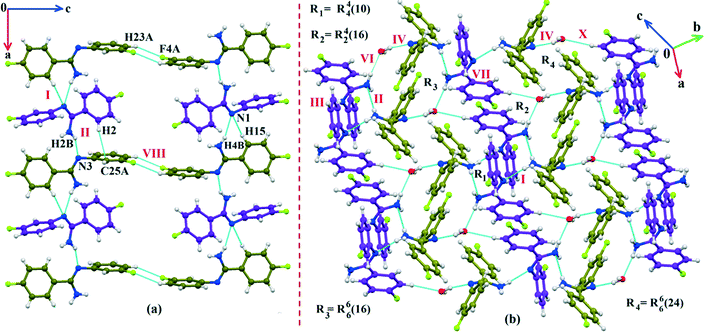 |
| | Fig. 3 (a) The crystal packing of DB32 (molecule 1: indigo colour; molecule 2: olive colour) down the ac plane showing the N–H⋯N and C–H⋯N chains (along the a axis) connected with a C–H⋯F dimer; (b) the crystal packing of DB32W down the ab plane forming the cyclic tetrameric rings R1 & R2 and hexameric rings R3 & R4 associated with strong N–H⋯N, N–H⋯O and O–H⋯N hydrogen bonds. | |
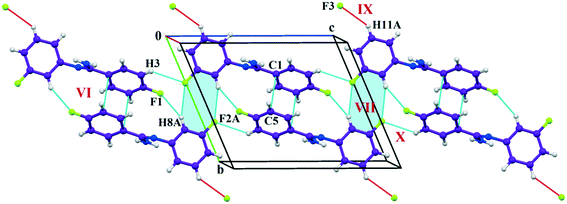 |
| | Fig. 4 Crystal packing arrangement of molecule A (indigo colour) in DB32 down the bc plane depicting C–H⋯F and π⋯π contacts. | |
In the case of DB32W (Fig. 2b), molecules 1 and 2 interact via a strong N4–H4B⋯N1 hydrogen bond (I) in the asymmetric unit with the stabilization energy of −11.2 kcal mol−1 (57% electrostatic contribution and 43% dispersion contribution). Molecule 2 interacts with the water molecule present in the asymmetric unit via a O1W–H1W⋯N3 hydrogen bond (IV; −6.5 kcal mol−1). Fig. 3b shows the molecular arrangement in the hydrate form down the ab plane. Here, the primary structural motifs involving short and highly directional strong hydrogen bonds generate different types of cyclic tetrameric (R1 and R2) and hexameric (R3 and R4) synthons (Desiraju and co-workers21 have discussed about the tetrameric and hexameric supramolecular synthons associated with a strong N–H⋯O hydrogen bond for aniline-phenol co-crystals). In this case, all the cyclic rings are interconnected with each other. Two symmetry independent molecules (1 and 2) and one water molecule form a molecular chain via the strong N4–H4B⋯N1 (I), O1W–H1W⋯N3 (IV) and N2–H2B⋯O1W (VI; −4.7 kcal mol−1) hydrogen bonds along the b axis. Molecules 1 and 2 together form the cyclic tetramer R1, utilizing the strong N4–H4B⋯N1 (I) and N4–H4A⋯N2 (II; −10.7 kcal mol−1; Fig. S3b†) H-bonds. Meanwhile, the cyclic tetramer R2 (involving molecule 2 and a water molecule) is generated with the help of a water molecule which is connected with molecule 2via the strong N2–H2B⋯O1W (VI) hydrogen bond and the short C3–H3⋯O1W (X; −2.4 kcal mol−1) interaction. The molecular pair V (−5.2 kcal mol−1; involving C–H⋯F and C–H⋯π interactions) is formed inside tetramer R1, and VII [−4.5 kcal mol−1; involving π⋯π stacking and C–H⋯F interaction] is formed inside tetramer R2, respectively. The symmetry independent molecules (1 and 2) and the water molecule combined to form a cyclic hexamer R3 involving the molecular pairs II, IV and VI. Meanwhile, a cyclic hexamer R4 (water molecule, molecules 1 and 2) is formed via the molecular pairs I, IV and X. The molecular dimer III [−8.1 kcal mol−1; involving C–H⋯N and C–H⋯π interactions; 41% electrostatic and 59% dispersion contribution] is generated inside hexamer R4.
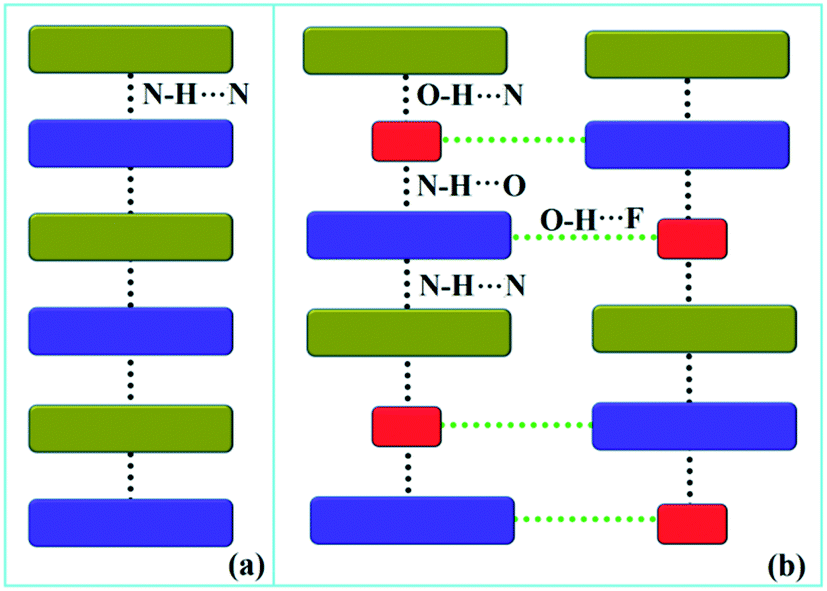
Down the bc plane, the molecular arrangement (Fig. S4†) is completely different due to the participation of O–H⋯F interaction [XI; −2.3 kcal mol−1]. Here, two successive strong parallel molecular chains (along the b axis) are connected with each other either via the molecular pairs III and VII or the dimer VIII [−4.1 kcal mol−1; involving C8A–H8A⋯F2A interaction and F2A⋯N2 contact]. There is also the formation of tetrameric R5 (VI, XI and VIII) and hexameric R6 (I, IV and XI) cyclic rings associated with strong hydrogen bonds and O–H⋯F interaction. In addition, the molecular pairs IX (−4.0 kcal mol−1), XII (−1.9 kcal mol−1), XIII (−1.5 kcal mol−1) and XIV (−0.9 kcal mol−1) also contribute towards the extra stability as supporting motif in the crystal packing. A comparison of the molecular arrangements between the solvatomorphs is shown in Fig. 5. Here, the same color codes (like in the previous crystal packing) are used to design the molecular network. In the case of DB32, the alternate arrangement of crystallographically independent molecules 1 (indigo color) and 2 (olive color) leads to the formation of a strong N–H⋯N chain (Fig. 5a). Although the presence of a water molecule (red color) in DB32W alters the crystal packing, the molecular chain has been preserved by the formation of strong H-bonds forming a (N–H⋯N)–(N–H⋯O)–(O–H⋯N) chain (Fig. 5b) using the water molecule between two more stabilized molecular pairs (DB32W–I). The water molecule forms a strong N–H⋯O hydrogen bond with molecule 1 and a strong O–H⋯N hydrogen bond with molecule 2. Finally, two such chains were connected via O–H⋯F interaction formed between molecule 1 and the water molecule.
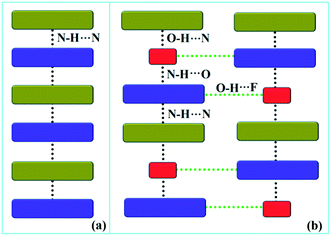 |
| | Fig. 5 The molecular arrangement in (a) DB32 and (b) DB32W [molecule 1, molecule 2, and the water molecule are represented by indigo, olive and red colors, respectively]. | |
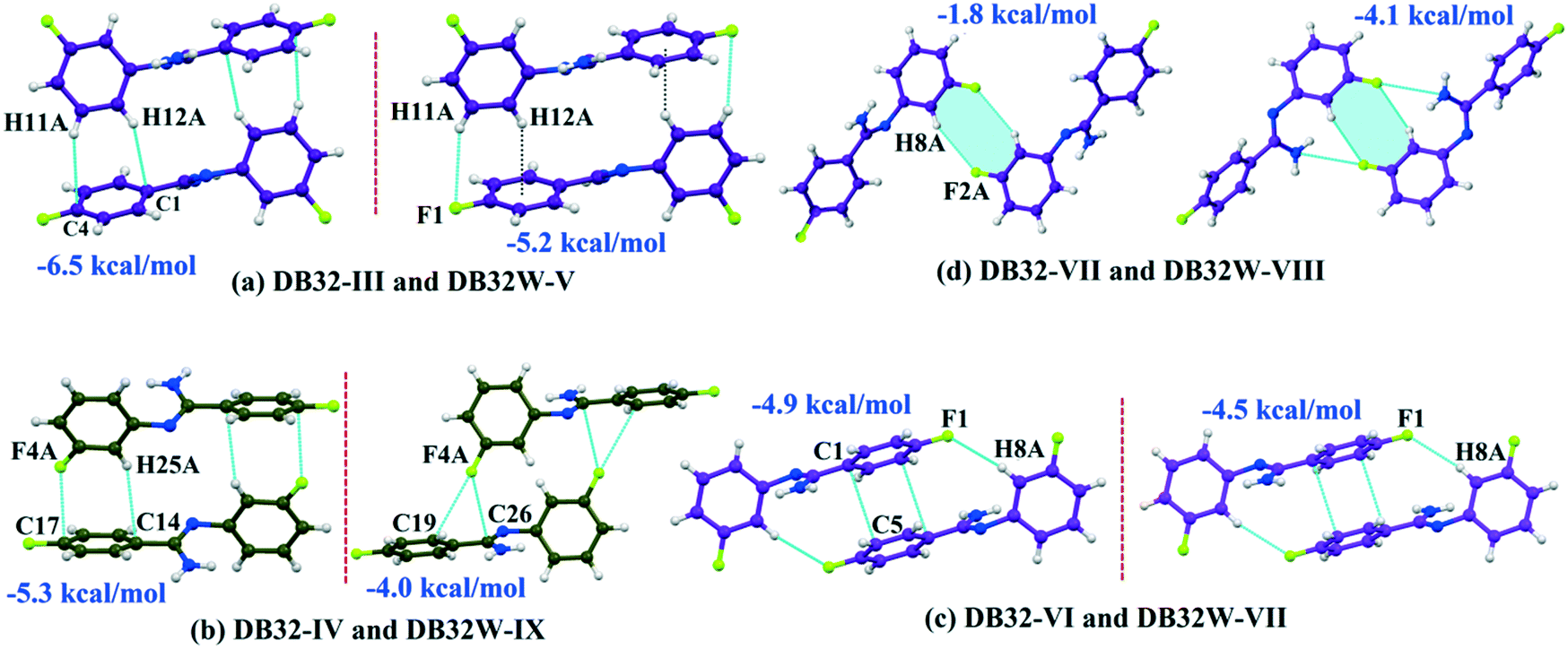
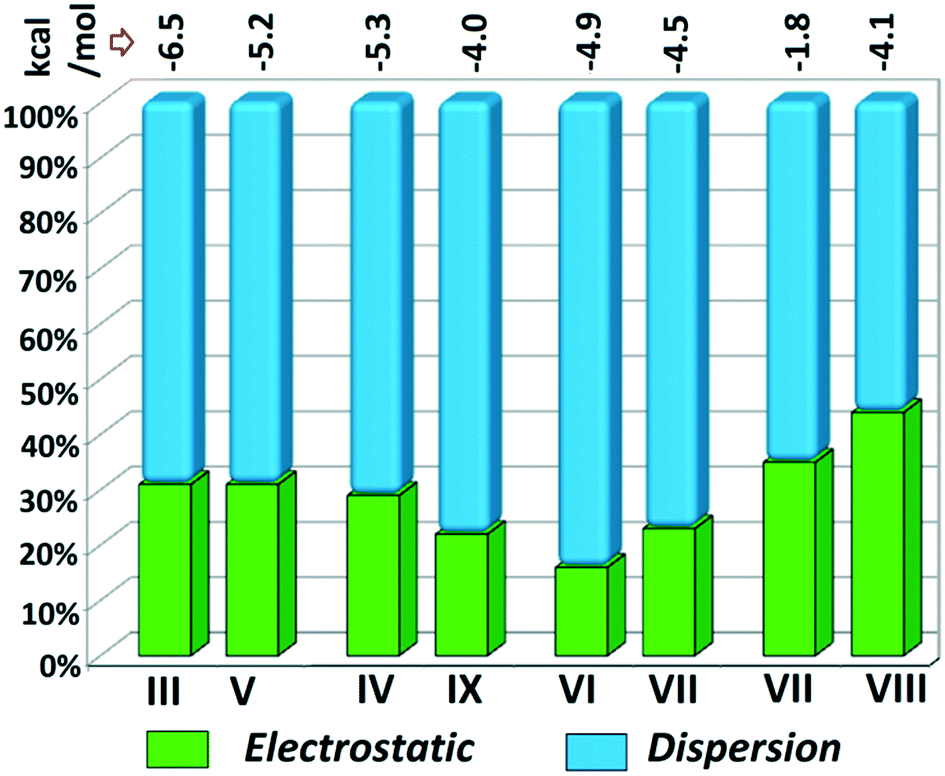
Although the crystal packing of the hydrated one (DB32W) is different compared to the anhydrous one (DB32), both forms have some similarity in packing features. The molecular chain formed via strong hydrogen bonds is a common structural aspect in both forms. In addition to that, there are some motifs which are structurally and energetically equivalent and are present in both forms, as shown in Fig. 6. Between the molecular pairs DB32–III (−6.5 kcal mol−1) and DB32W–V (−5.2 kcal mol−1), the electrostatic and dispersion contributions towards the total stabilization are the same (Fig. 6a and 7). Meanwhile, between the molecular pairs DB32–IV (−5.3 kcal mol−1) and DB32W–IX (−4.0 kcal mol−1), the electrostatic contribution is higher for the former case than for the latter (Fig. 6b and 7). This is due to the translation of one molecule with respect to the other in the case of IX and hence there is no formation of C–H⋯C interaction. The molecular pairs DB32–VI and DB32W–VII have a similar stabilization energy (∼4.7 kcal mol−1). As the intermolecular C–H⋯F is shorter and more directional for DB32W–VII than in the case of DB32–VI, the electrostatic contribution is higher for the former case (Fig. 6c and 7). The molecular pairs DB32–VII (−1.8 kcal mol−1) and DB32W–VIII (−4.1 kcal mol−1) are partially equivalent with respect to the centrosymmetric C–H⋯F dimer. The difference between DB32–VII and DB32W–VIII is the change in orientation of the interacting molecules (Fig. 6d). Because of this, the –NH2 group comes closer to the fluorine atom of the other molecule, leading to the formation of F⋯N contact. In this case, the electrostatic contribution is 10% more for the latter when compared to DB32–VII (Fig. 7).
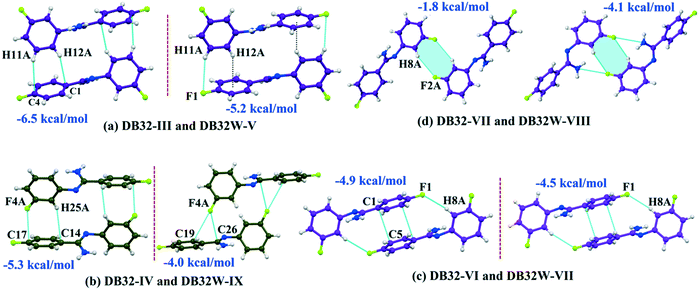 |
| | Fig. 6 The equivalent molecular pairs (a)–(d) for the solvatomorphs DB32 and DB32W with their stabilization energies. | |
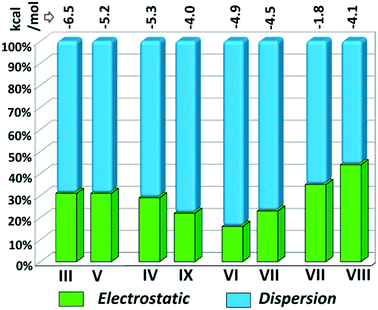 |
| | Fig. 7 Comparison of the energetic contribution (%) to the total stabilization for the equivalent molecular pairs [the upper values indicate their individual stabilization energies in kcal mol−1]. | |
QTAIM22,23 analysis has been performed to obtain quantitative insights into the topological characteristics of the intermolecular interaction for some selected molecular pairs extracted from the crystal packings of DB32 and DB32W. The molecular graphs indicate the bond critical points [BCP (3, −1)], as shown in Fig. S5 and S6,† thereby depicting the bonding character of these interactions. The topological parameters [the electron density (ρBCP), the Laplacian (∇2ρBCP), the local potential energy (Vb), the kinetic energy density (Gb) and the dissociation energy DEV(int)] at the BCP for the intermolecular interactions are given in Table S2.† The value of the electron density (ρBCP) and the Laplacian (∇2ρBCP) at the BCP for the weak C–H⋯F interactions are observed in the range of 0.0238–0.0513 e Å−3 and 0.379–0.749 e Å−5, respectively. The corresponding bond dissociation energies are in the range of 0.77 to 1.77 kcal mol−1. The values of the electron density and the Laplacian for the short C3–H3⋯O1W and O1W–H2W⋯F2A interactions are 0.0820 e Å−3 and 1.032 e Å−5; 0.0992 e Å−3 and 1.441 e Å−5, respectively. The corresponding bond dissociation energies are 2.43 and 3.79 kcal mol−1, respectively.
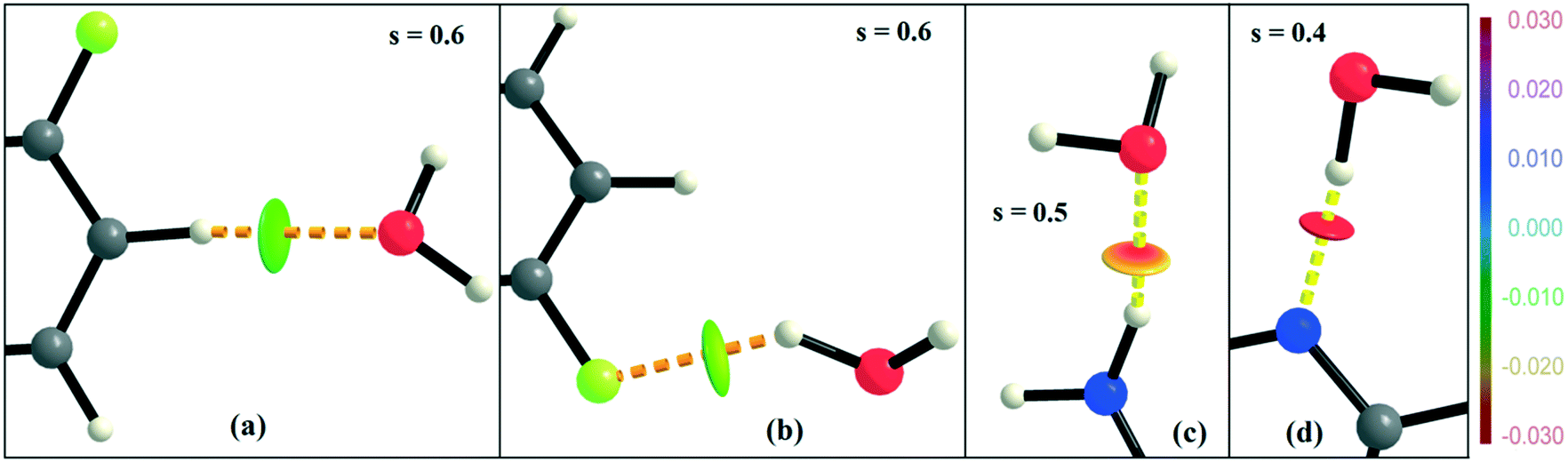
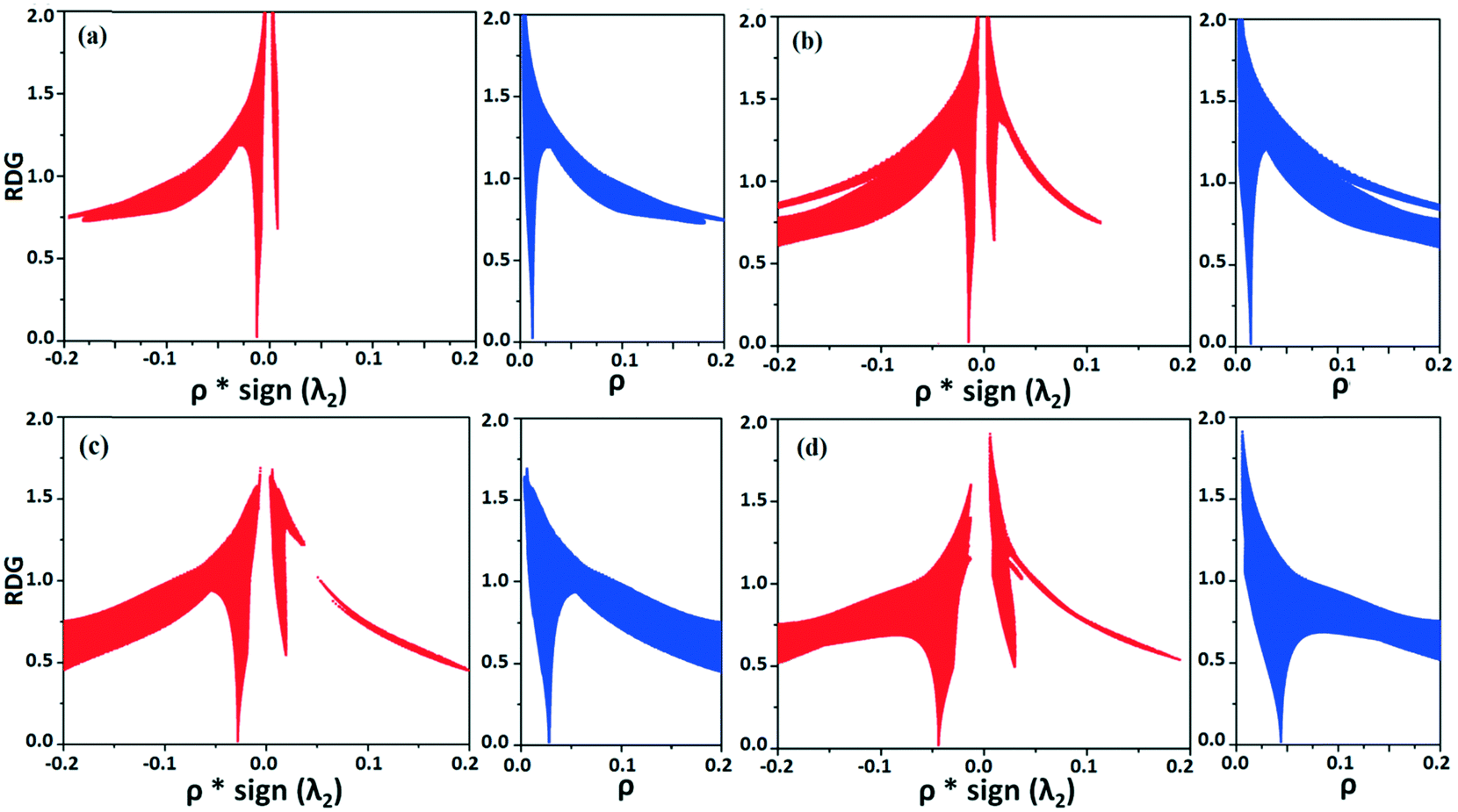
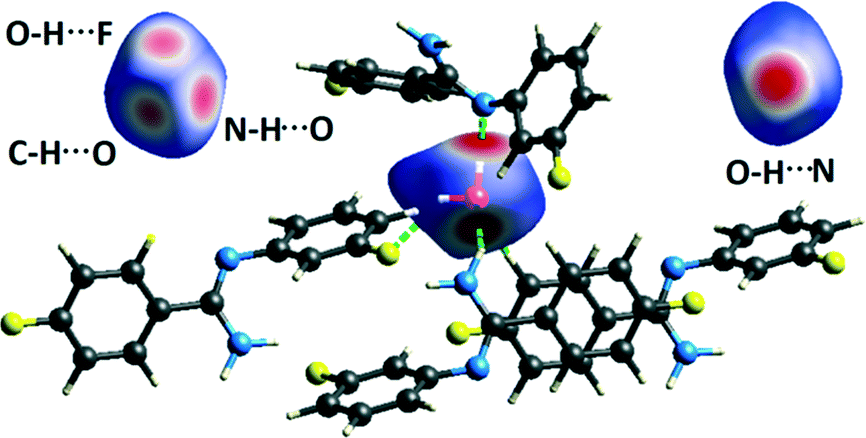
NCI analysis24,25 has been performed using the program NCImilano26,27 to investigate the interactions based on the reduced density gradient (RDG) (s = 1/(2(3π2)1/3)|∇ρ|/ρ4/3).24 Around the bond critical point (3, −1), it is a graphical representation of the region in real space where the non-covalent interaction occurs.28 This analysis was carried out for the intermolecular interactions involving the water molecule present in DB32W. All the RDG isosurfaces were plotted (Fig. 8) with the color range −0.03 < ρ * sign (λ2) < 0.03 au, where λ2 is the second eigenvalue of the Hessian matrix. The sign of the second eigenvalue of the Hessian matrix differentiates whether any non-covalent interaction is stabilized (λ2 < 0) or destabilized (λ2 > 0). The 2D plots (Fig. 9) associated with the RDG vs. ρ * sign (λ2) gives the corresponding value of the spike for a particular interaction. In the case of the short and highly directional C–H⋯O interaction (dO⋯H = 2.29 Å), a green colored disc-shaped (Fig. 8a) RDG isosurface with the less negative (Fig. 9a) value (−0.0123 au) of ρ * sign (λ2) indicates the attractive nature of the C–H⋯O interaction. A light-green colored disc-shaped (Fig. 8b) RDG isosurface was observed for the short O–H⋯F (dF⋯H = 2.15 Å) interaction which has a very sharp peak (Fig. 9b) with the value −0.0167 au of ρ * sign (λ2). It signifies the “attractive” nature of the O–H⋯F interaction. Meanwhile. the strong N–H⋯O (dO⋯H = 1.89 Å) hydrogen bond gives a reddish-yellow colored disc-shaped RDG isosurface (Fig. 8c) with the value of −0.03 au (Fig. 9c), which indicates its more attractive nature compared to the two previous weak intermolecular interactions. However, the short and highly directional O–H⋯N hydrogen bond (dO⋯H = 1.83 Å) gives a deep red colored disc-shaped (Fig. 8d) RDG isosurface (s = 0.4) having a broad peak with a very high negative (Fig. 9d) value (−0.04 au) of ρ * sign (λ2) signifying the strong hydrogen bond.
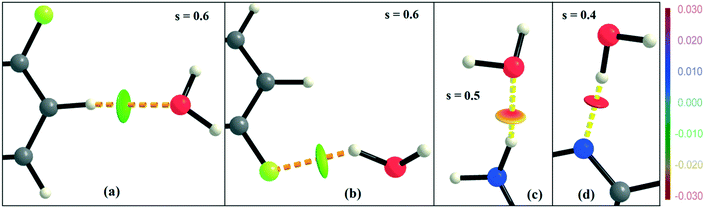 |
| | Fig. 8 NCI-based RDG isosurfaces for intermolecular (a) C–H⋯O and (b) O–H⋯F interactions and (c) N–H⋯O and (d) O–H⋯N hydrogen bonds in DB32W. | |
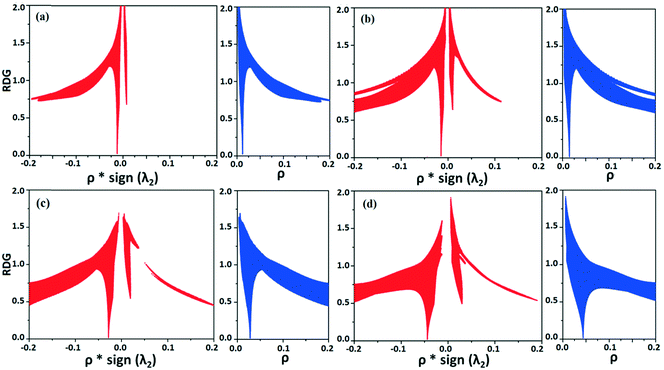 |
| | Fig. 9 The plot of the reduced density gradient (RDG) versus electron density multiplied by the sign of the second Hessian eigenvalue [ρ * sign (λ2)] and electron density (ρ) for intermolecular (a) C–H⋯O and (b) O–H⋯F interactions and (c) N–H⋯O and (d) O–H⋯N hydrogen bonds. | |
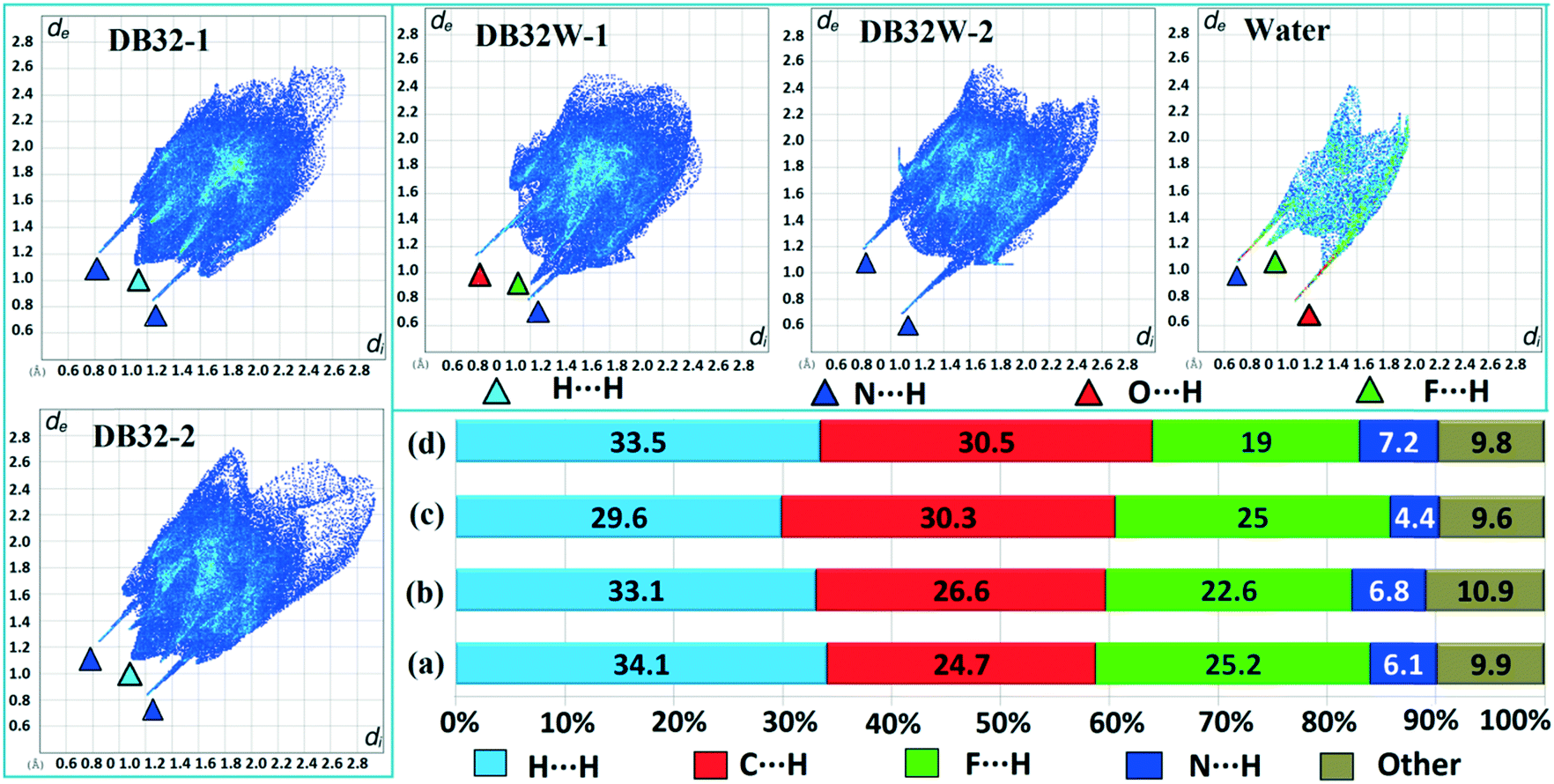
Hirshfeld surface analysis29 for the two solvatomorphs was performed using the program CrystalExplorer 3.1.30 It helps to distinguish the similarities and differences between the two crystal structures as well as the symmetry independent molecules present in the asymmetric unit in the crystal environment. It is a unique approach to obtain information about the trends in crystal packing.31 The 2D fingerprint plots32,33 (Fig. 10 and Fig. S7†) obtained from the Hirshfeld surface analysis provide quantitative information for the individual intermolecular atom⋯atom contacts of a molecule in the crystal environment. In the case of DB32, the fingerprint plots for the two crystallographically independent molecules 1 and 2 are shown in the left side of Fig. 10. The sharp spikes observed due to the different atom⋯atom contacts were represented with the different colored (cyan, blue, red and green) triangles. In both fingerprints plots for molecules 1 and 2, the two sharp spikes responsible for the strong N–H⋯N hydrogen bond formation and the wings regions responsible for the H⋯H contact were observed. However, the plot for molecule 2 exhibits two small spikes which correspond to F⋯H contact (C19–H19⋯F1 and C23–H23⋯F4A) which is absent in the case of molecule 1. The fingerprint plots for DB32W (molecule 1, molecule 2 and water) are shown at the top of the right side (Fig. 10). It was observed that the fingerprint plot of molecule 1 has three different sharp spikes i.e.; the upper one (red triangle) is for the strong N–H⋯O hydrogen bond and weak C–H⋯O interaction, the middle one (green triangle) is for the short O–H⋯F interaction and the lower one (blue triangle) is for the strong N–H⋯N and O–H⋯N hydrogen bonds. On the other hand, the fingerprint plot of molecule 2 has two sharp spikes (blue triangle) which correspond to the strong N–H⋯N hydrogen bonds. The Hirshfeld surface was mapped with dnorm for the water molecule (Fig. 11) present in DB32W. In dnorm, a total of four red spots were observed; the deep red is for the short O–H⋯N hydrogen bond, and the other three (light red color) are for N–H⋯O, O–H⋯F and C–H⋯O interactions, respectively. Subsequently, three different spikes (blue, green and red colored triangles) were also observed in the fingerprint plot for the water molecule. These spikes correspond to the above-mentioned contacts related to the water molecule. The relative contributions from the different atom⋯atom contacts in DB32 [(a) and (b)] and DB32W [(c) and (d)] have been plotted in Fig. 10. In DB32, the contribution from the F⋯H contact is 3% more for molecule 1 compared to molecule 2, whereas the contribution from the C⋯H contact is 2% more for molecule 2 in comparison with molecule 1. The N⋯H contribution is almost similar for both molecules (1 and 2). For DB32W, due to the presence of a water molecule, the contributions from N⋯H and F⋯H contacts are different in molecules 1 and 2. The difference in F⋯H and N⋯H contributions between the two molecules is 6% and 3%, respectively. As molecule 1 forms H-bonds with the water molecule leading to the formation of N–H⋯O, C–H⋯O and O–H⋯F interactions, the N⋯H contribution only comes from the N–H⋯N hydrogen bond. In the case of molecule 2, the N⋯H contribution comes from the N–H⋯N hydrogen bond as well as the O–H⋯N hydrogen bond. That is why the N⋯H contribution is greater for molecule 2 than for molecule 1.
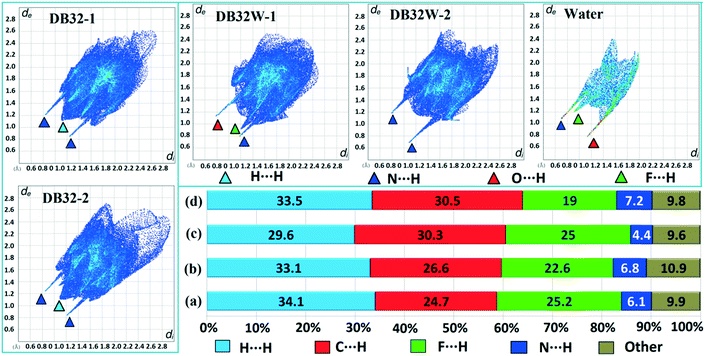 |
| | Fig. 10 The fingerprint plots for DB32 (left side: molecules 1 and 2) and DB32W (upper right side: molecule 1, molecule 2 and water) and the relative contributions from different atom⋯atom contacts for DB32 [(a) molecule 1 and (b) molecule 2] and DB32W [(c) molecule 1 and (d) molecule 2]. | |
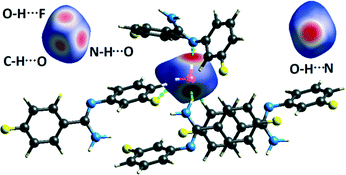 |
| | Fig. 11 Hirshfeld surface mapped with dnorm (front and back views) for the water molecule in DB32W, showing the four red spots associated with the intermolecular O–H⋯N, N–H⋯O O–H⋯F and C–H⋯O interactions. | |
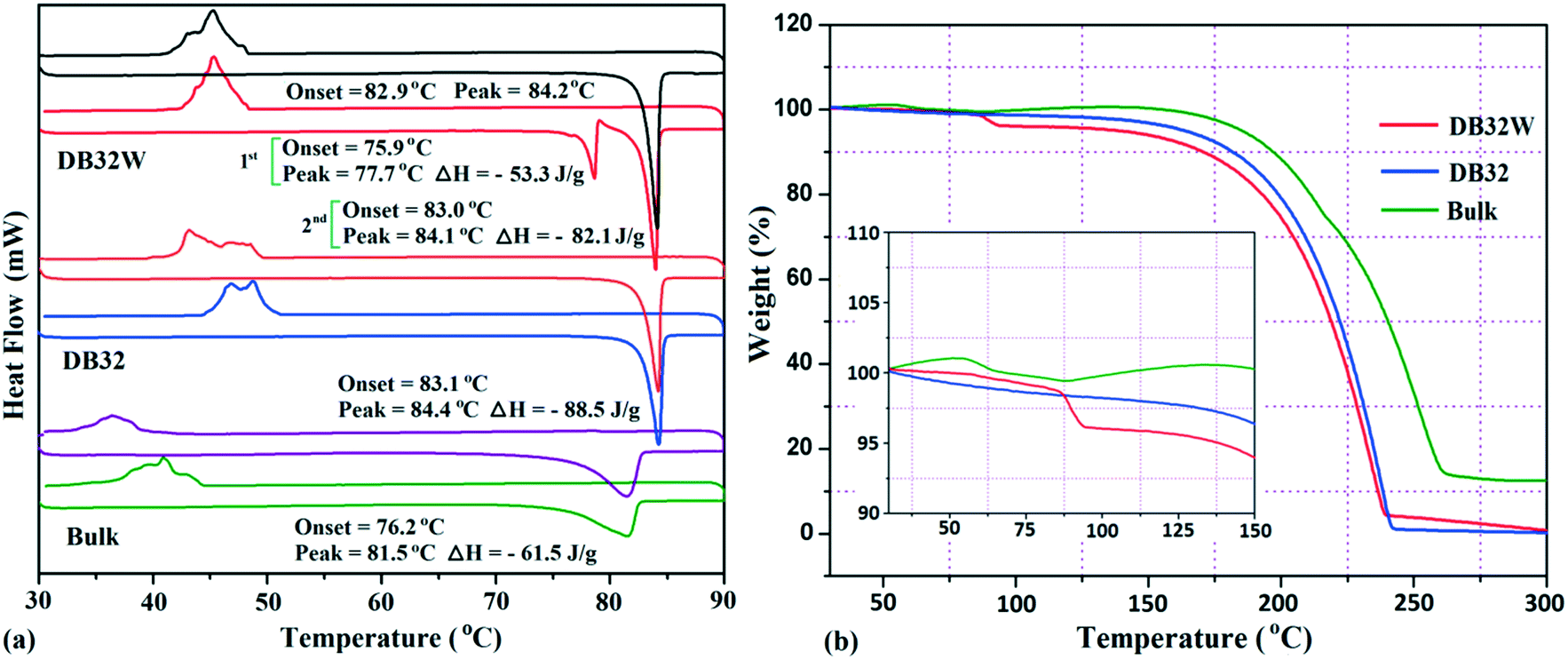
The thermal stability of these solvatomorphs has been investigated via differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The DSC measurements were performed using a Perkin-Elmer DSC 6000 instrument under a nitrogen gas atmosphere. Firstly, all the samples (∼2.5 mg) were prepared in a covered aluminum pan and then the experiment was performed under vacuum in the covered aluminum pan. The sample was heated from 30 °C to 90 °C with a heating rate of 2 °C min−1 and again cooled up to 30 °C. In Fig. 12a, the DSC curves are shown in different colors; the bulk compound in green color, DB32 in blue color and DB32W in red color. For the bulk compound, only one broad endothermic peak, at the onset value of 76.2 °C (ΔH = −61.5 J g−1), was observed during the first heating cycle followed by solidification by cooling at a temperature range of 35–40 °C. Meanwhile, DB32 (anhydrous) melted, indicated by a sharp peak at the onset value of 83.1 °C (ΔH = −88.5 J g−1), and again solidified (exothermic) at ∼47 °C in the cooling cycle. In the second heating/cooling cycle, there was no change in the endothermic peaks for the bulk compound (purple color) and the anhydrous DB32 (orange color). An interesting feature was observed in the case of DB32W (hydrate form) in the first heating cycle. During heating, a small sharp endothermic peak at the onset value of 75.9 °C which corresponds to the melting of DB32W was observed; then, it completely melted at 77.7 °C (ΔH = −53.3 J g−1) and again solidified at ∼79 °C in the same heating cycle. The new solid obtained from melted DB32W started melting in the same heating cycle at the onset value of 83.0 °C (ΔH = −82.1 J g−1) and then completely melted, indicated by a sharp peak at 84.1 °C. During cooling, an exothermic peak was observed at the same temperature range like in the solidification (first and second cycles) of the anhydrous form DB32. It is clear that the melted DB32W was crystallized without the water molecule during heating (first cycle) i.e., the new solid form was the anhydrous DB32.
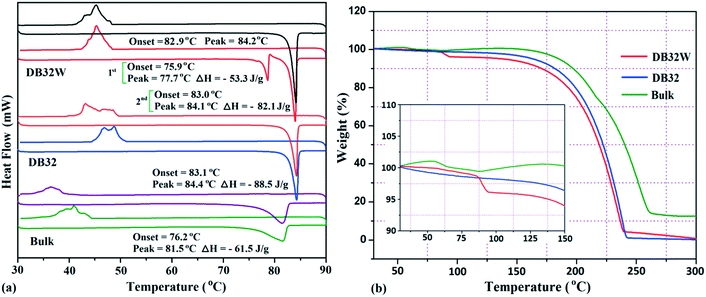 |
| | Fig. 12 (a) DSC heating/cooling curves (1st and 2nd cycles) for the solvatomorphs DB32 and DB32W, and the bulk compound @ 2 °C min−1 and (b) TGA curves for the solvatomorphs and the bulk compound @ 5 °C min−1. | |
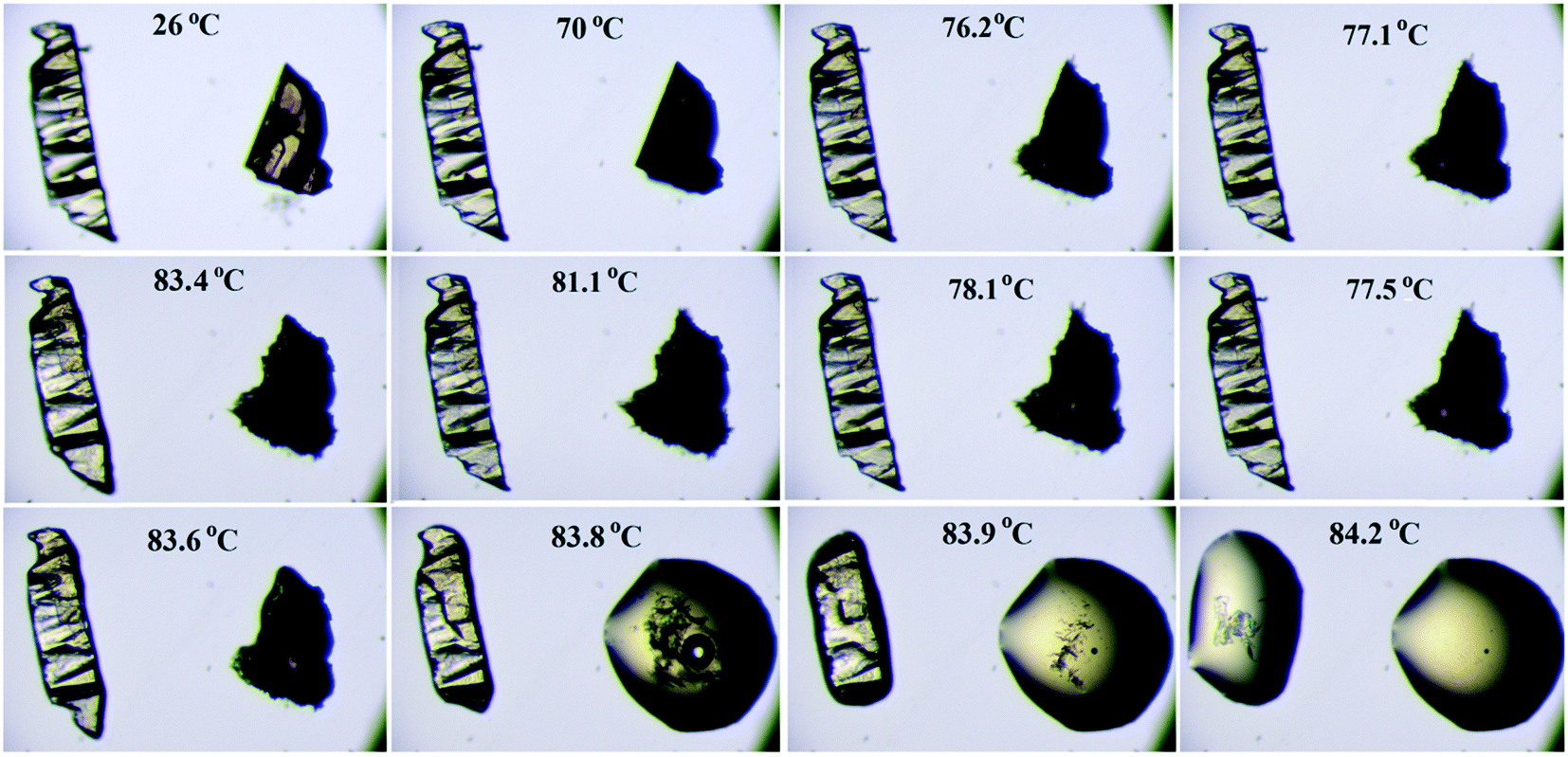
Thermogravimetric analysis shows that the nature of the curves for the hydrate form is totally different from that of the anhydrous form and the bulk compound (Fig. 12b). There is a drastic change in the weight loss for the hydrate form i.e. it supports the presence of a solvent (water) molecule in DB32W. It is observed that after 100 °C, the nature of both curves (blue and red colors) is similar. To visualize the thermal stability and the changes due to the increase in temperature, hot stage microscopy measurements were performed using a hot stage apparatus (Linkam LTS420) equipped with a stereomicroscope. For this experiment, small single crystals of DB32 (left side) and DB32W (right side) were used, as shown in Fig. 13. The crystal images were obtained using a Leica EC3 camera connected to the microscope. The single crystals of the solvatomorphs were placed on a glass slide and focused under the microscope, and then heated from 25 °C to 90 °C with a heating rate of 0.5 °C min−1. The HSM study shows that at 70 °C only the crystal of DB32W has changed in color from light orange to black on heating. From 76 °C, the crystal of DB32W started melting, and small spikes appeared in the vicinity of the compound. Above 79 °C, it again started to solidify to a new form (DB32) but at the same time (at the same temperature) the crystal of DB32 remained unchanged. After that, both crystals (DB32 and the new form) started melting at 83.4 °C and completely melted at 84.2 °C. The solvatomorphs DB32 and DB32W were also characterized via powder X-ray diffraction patterns. The overlay diagram of the powder X-ray diffraction patterns (PXRD) for the solvatomorphs is shown in Fig. S8.† The PXRD pattern (experimental) for the hydrate form (DB32W) fully matches with the calculated PXRD pattern. Meanwhile, the PXRD patterns for the bulk material (experimental) and the anhydrous one (calculated, DB32) are similar. Hence, it can be concluded that the bulk material is the anhydrous form.
 |
| | Fig. 13 Hot stage microscopy images for the solvatomorphs (DB32 in the left side and DB32W in the right side) heated @ 0.5 °C min−1. | |
A study of the Cambridge Structural Database (CSD) for the occurrence of short O–H⋯F intermolecular interactions was carried out using CSD version 5.37. The search was conducted with some constraints, namely: dH⋯F = 2.0 to 2.67 Å, ∠O–H⋯F = 130 to 180°, R factor ≤ 0.1, not disordered, not polymeric, no errors, no ions, no powder structures, and only organic compounds. There was a total of 93 hits (Fig. S9a†), where an –OH group (alcohol or phenol or hydrates) interacts with the fluorine atom of a molecule. There was a total of 31 hits (Fig. S9b†), where the hydrates or the coordinated water molecules interact with the fluorine atom of a molecule, leading to the formation of O–H⋯F interaction. If the fluorine atom is attached to any phenyl ring, then the number of hits is decreased to 12 (Fig. S9c†) from 31. Therefore, the CSD study reveals that the intermolecular O–H⋯F interaction is not very common in organic solids.
Summary
In this study, we have shown the existence of the compound (Z)-4-fluoro-N′-(3-fluorophenyl) benzimidamide as a solvatomorph (hydrate). From the powder X-ray diffraction patterns and thermogravimetric analysis, it is clear that the bulk compound is equivalent to the anhydrous form (DB32). The thermal characterization studies (DSC and HSM) also confirm the transformation of the hydrate form to the anhydrous form, on heating. The supramolecular architectures for both the anhydrous and hydrate forms are mainly stabilized through the strong hydrogen bonded chain along with the weak intermolecular interactions. The hydrate forms stable tetrameric and hexameric supramolecular synthons down the ab and bc planes. The intermolecular O–H⋯F interaction plays an important role in the crystal packing of the hydrate form. NCI analysis shows the nature of the O–H⋯F interaction, which is different in terms of the reduced density gradient compared to the other strong hydrogen bonds. Hirshfeld surface analysis also helps to understand the differences and the similarities between the two crystal structures of the solvatomorphs, mainly due to the presence of a water molecule in the crystal packing.
Acknowledgements
D. D. thanks IISER Bhopal for the research fellowship. D. C. and D. D. thank IISER Bhopal for the research facilities and infrastructure, and DST-SERB for research funding.
References
- B. Stoger, P. Kautny, D. Lumpi, E. Zobetza and J. Frohlich, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 667–676 Search PubMed.
- S. K. Dey and G. Das, Chem. Commun., 2011, 47, 4983–4985 RSC.
- R. Kaur, S. Cherukuvada, P. B. Managutti and T. N. G. Row, CrystEngComm, 2016, 18, 3191–3203 RSC.
- D. Chopra and T. N. G. Row, Cryst. Growth Des., 2006, 6, 1267–1270 CAS.
- K. Iwata, T. Kojima and Y. Ikeda, Cryst. Growth Des., 2014, 14, 3335–3342 CAS.
- K. Iwata, M. Karashima and Y. Ikeda, Cryst. Growth Des., 2016, 16, 4599–4606 CAS.
- H. Lemmer, N. Stieger, W. Liebenberg and M. R. Caira, Cryst. Growth Des., 2012, 12, 1683–1692 CAS.
- D. M. Kaminski, A. A. Hoser, M. Gagos, A. Matwijczuk, M. Arczewska, A. Niewiadomy and K. Wozniak, Cryst. Growth Des., 2010, 10, 3880–3888 Search PubMed.
- R. Shukla, T. P. Mohan, B. Vishalakshi and D. Chopra, CrystEngComm, 2014, 16, 1702–1713 RSC.
-
J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed.
- H. G. Brittain, J. Pharm. Sci., 2010, 101, 464–468 CrossRef PubMed.
- C. B. Aakeroy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC.
- P. Manna, S. K. Seth, M. Mitra, A. Das, N. J. Singh, S. R. Choudhury, T. Kar and S. Mukhopadhyay, CrystEngComm, 2013, 15, 7879–7886 RSC.
- P. Manna, S. K. Seth, M. Mitra, S. R. Choudhury, A. Bauza, A. Frontera and S. Mukhopadhyay, Cryst. Growth Des., 2014, 14, 747–755 CAS.
- P. Manna, S. K. Seth, M. Mitra, S. R. Choudhury, A. Bauza, A. Frontera and S. Mukhopadhyay, Cryst. Growth Des., 2014, 14, 5812–5821 CAS.
- V. Koradia, H. L. de Diego, K. Frydenvang, M. Ringkjobing-Elema, A. Mullertz, A. D. Bond and J. Rantanen, Cryst. Growth Des., 2010, 10, 5279–5290 CAS.
- P. Ghugare, V. Dongre, P. Karmuse, R. Rana, D. Singh, A. Kumar and Z. Filmwala, J. Pharm. Biomed. Anal., 2010, 51, 532–540 CrossRef CAS PubMed.
- K. J. Chavez, M. Guevara and R. W. Rousseau, Cryst. Growth Des., 2010, 10, 3372–3377 CAS.
- D. Dey and D. Chopra, CrystEngComm, 2015, 17, 5288–5298 RSC.
- D. Dey, S. P. Thomas, M. A. Spackman and D. Chopra, Chem. Commun., 2016, 52, 2141–2144 RSC.
- A. Mukherjee, K. Dixit, S. P. Sarma and G. R. Desiraju, IUCrJ, 2014, 1, 228–239 CAS.
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, U.K., 1990 Search PubMed.
-
V. G. Tsirelson, The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, ed. C. Matta and R. Boyd, Wiley-VCH, Weinheim, Germany, 2007, ch. 10, p. 45 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- G. Saleh, L. L. Presti, C. Gatti and D. Ceresoli, J. Appl. Crystallogr., 2013, 46, 1513–1517 CAS.
- G. Saleh, C. Gatti, L. L. Presti and J. Contreras-Garc, Chem. – Eur. J., 2012, 18, 15523–15536 CrossRef CAS PubMed.
- M. P. Johansson and M. Swart, Phys. Chem. Chem. Phys., 2013, 15, 11543–11553 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer, version 3.1, University of Western Australia, Crawley, Australia, 2012 Search PubMed.
- S. K. Seth, I. Saha, C. Estarellas, A. Frontera, T. Kar and S. Mukhopadhyay, Cryst. Growth Des., 2011, 11, 3250–3265 CAS.
- M. A. Spackman and J. J. Mckinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. Mckinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 9820841496291. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01668e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence