
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination polymers of 5-substituted isophthalic acid†
Laura J.
McCormick
*a,
Samuel A.
Morris
a,
Alexandra M. Z.
Slawin
a,
Simon J.
Teat
b and
Russell E.
Morris
a
aEaSTCHEM School of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife, Scotland KY16 9ST, UK. E-mail: ljm22@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 463776
bAdvanced Light Source, Berkeley Laboratory, 1 Cyclotron Road, Berkeley, California 94720, USA
First published on 10th December 2015
Abstract
The synthesis and characterisation of five coordination polymers – Ni2(mip)2(H2O)8·2H2O (1), Zn6(mip)5(OH)2(H2O)4·7.4H2O (2), Zn6(mip)5(OH)2(H2O)2·4H2O (3), Mn(HMeOip)2 (4), and Mn3(tbip)2(Htbip)2(EtOH)2 (5) – are reported. Preliminary nitric oxide release data on compounds 2 and 3 are also given.
Introduction
Design and synthesis of new coordination polymers is currently a burgeoning field, due to the ability to tailor the properties of these materials by adjusting their structure.1 One such property that is presently garnering interest is the use of porous coordination polymers (also known as metal–organic frameworks or MOFs) as gas storage and delivery materials, exploiting the large surface areas of these materials coupled, in many cases, with binding gas molecules to coordination sites from which a coordinated solvent molecule has been removed. The use of these materials as gas delivery systems is advantageous for both energy storage applications2 (adsorbed gas molecules do not contribute to storage vessel pressure, allowing large volumes of gas to be stored without the complications of a high pressure container) and medical applications3 (coordinated gas molecules may be displaced by water, allowing for targeted gas delivery without the systemic exposure necessitated by use of delivery of these gases via inhalation or ingestion4). A medicinal gas of particular interest is nitric oxide (NO), which has been shown to play a role in a variety of biological processes including wound healing and thrombosis prevention, as well as being an anti-bacterial agent.5Coordination polymers based on 5-substituted isophthalate ligands have been shown to exhibit catalytic,6 magnetic,7 gas storage and flexible framework8 properties. Despite being very closely related to the ligands used in some of the best known and widely studied MOFs such as terephthalic acid (MOF-5,9 UiO-66,10 MIL-10111) and trimesic acid (HKUST-1,12 MIL-10013), surprisingly few coordination polymers derived from the commercially available 5-methyl14 and tert-butyl15 isophthalic acid or the readily synthesised 5-alkoxy16 or aminoalkyl17 isophthalate ligands have been reported that do not contain additional pyridine- or imidazole-based bridging co-ligands. Herein we reported the synthesis and characterisation of 5 novel coordination polymers derived from the 5-methyl (H2mip, I), methoxy (H2MeOip, II) and tert-butyl (H2tbip, III) isophthalic acids, namely Ni2(mip)2(H2O)8·2H2O (1), Zn6(mip)5(OH)2(H2O)4·7.4H2O (2), Zn6(mip)5(OH)2(H2O)2·4H2O (3), Mn(HMeOip)2 (4), and Mn3(tbip)2(Htbip)2(EtOH)2 (5). Preliminary investigations into the nitric oxide release properties of compounds 2 and 3 are also described.
Experimental
All reagents were obtained from commercial sources and were used without further purification.Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 571424, 1378, 1363, 1337, 1297, 1271, 1261, 1185, 1136, 1100, 1061, 928, 905, 892, 873, 798, 765, 751, 702, 684, 666, 630, 575, 546, 524 cm−1.
571424, 1378, 1363, 1337, 1297, 1271, 1261, 1185, 1136, 1100, 1061, 928, 905, 892, 873, 798, 765, 751, 702, 684, 666, 630, 575, 546, 524 cm−1.
Crystallography
Crystals were coated in protective oil, prior to being mounted on a MiTeGen fibre tip (compounds 1, 2 and 5) or a Molecular Dimensions mounted 0.1 mm litholoop (compounds 3 and 4). Single crystal data were collected on a Rigaku Mercury2 SCXMini diffractor (λ = 0.71075 Å, compounds 3 and 4), or on a Bruker APEXII diffractometer (λ = 0.77490 Å, compounds 1, 2 and 5) on station 11.3.1 at the Advanced Light Source. Powder X-ray diffraction patterns were collected using Cu Kα1 radiation on a PANalytical Empyrean diffractometer. For data collected at beamline 11.3.1, appropriate scattering factors were applied using XDISP.19 Absorption corrections were applied using multi-scan methods.20 Structure solutions were obtained using either SHELXS-97 or SHELXT (beta) and refined by full matrix on F2 using SHELXL-97 and SHELX-201421 within the WinGX suite.22 All full occupancy non-hydrogen atoms were refined with anisotropic thermal displacement parameters. Aromatic and aliphatic hydrogen atoms were included at their geometrically estimated positions, as were hydrogen atoms bound to μ3-OH− anions and non-coordinated carboxylate oxygen atoms, provided they could be located. Hydrogen atoms belonging to free and coordinated water molecules were fixed at a distance of 0.90 Å from the oxygen atom and 1.47 Å from the other hydrogen bound to the same oxygen atom, and their thermal displacement parameters linked to that of the oxygen to which they are bound.Nitric oxide loading and release
Powders were weighed into individual ampules, which were then placed in a Schlenk flask. The flask was heated to 200 °C and held at this temperature under dynamic vacuum for approximately 18 hours. After this time, the flask was cooled to room temperature and filled with NO (∼1 atm static pressure) for 1 hour. Excess NO was then removed by evacuating the flask and flushing with Ar three times. The ampules were then individually flame-sealed under an argon atmosphere. NO release measurements were performed using a Sievers NOA 280i chemiluminescence Nitric Oxide Analyser. The instrument was calibrated using air passed through a zero filter (<1 ppb NO) and 87.6 ppm NO gas in N2 (Air Products). The flow rate through the instrument was set to 200 mL min−1 with a cell pressure of approximately 5.2 Torr and an oxygen pressure of 6.0 psig. Nitrogen gas of known humidity was achieved by passing dry N2 over a saturated aqueous solution of LiCl to give 11% relative humidity. Humid N2 was passed over the powders at room temperature, and the resulting NO/N2 mixture was then passed through the analyser and the NO concentration recorded at 1 second intervals. Data collection was stopped after the concentration of NO in the carrier gas dropped below 20 ppb.Results and discussion
All five compounds were prepared by solvothermal reaction between metal acetate and 5-substituted isophthalic acid in either water or aqueous alcohol. A summary of the crystallographic details is presented in Table 1. Compounds 1 and 2 crystallise in polar space groups and their Flack parameters indicate that these crystals are twinned by inversion.| Compound | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Empirical formula | C18H32Ni2O18 | C45H54.75O33.5Zn6 | C45H44O28Zn6 | C18H14MnO10 | C52H62Mn3O18 |
| MW | 653.85 | 1523.85 | 1425.02 | 445.23 | 1139.83 |
| λ (Å) | 0.77490 | 0.77490 | 0.71075 | 0.71075 | 0.77490 |
| T (K) | 150(2) | 150(2) | 173(2) | 173(2) | 150(2) |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic | Monoclinic | Triclinic |
| Space group | Pna21 | Pnn2 | P21212 | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 15.4414(15) | 20.775(3) | 17.382(4) | 4.5131(8) | 9.1496(12) |
| b (Å) | 10.9156(11) | 27.566(3) | 10.4516(15) | 14.483(3) | 9.7908(12) |
| c (Å) | 14.5960(14) | 10.8460(13) | 15.970(3) | 12.622(3) | 16.078(2) |
| α (°) | 90 | 90 | 90 | 90 | 89.207(2) |
| β (°) | 90 | 90 | 90 | 97.792(9) | 76.087(2) |
| γ (°) | 90 | 90 | 90 | 90 | 74.643(2) |
| V (Å3) | 2460.2(4) | 6211.4(13) | 2901.3(10) | 817.4(3) | 1346.2(3) |
| Z | 4 | 4 | 2 | 2 | 1 |
| Flack parameter | 0.481(14) | 0.491(4) | 0.01(3) | — | — |
| ρ calcd (g cm−1) | 1.765 | 1.630 | 1.631 | 1.809 | 1.406 |
| μ (mm−1) | 2.040 | 2.992 | 2.524 | 0.871 | 0.960 |
| F(000) | 1360 | 3091 | 1436 | 454 | 593 |
| GoF | 1.085 | 1.042 | 1.073 | 1.032 | 1.041 |
| Reflections collected/unique | 34 124/7435 | 90 099/18 933 | 30 286/6666 | 8512/1877 | 20 014/8056 |
| No. of parameters | 397 | 767 | 358 | 135 | 343 |
| R int | 0.0613 | 0.0476 | 0.1248 | 0.0563 | 0.0538 |
| Final R indices I > 2σ(I) | R 1 = 0.0377 wR2 = 0.1051 | R 1 = 0.0533 wR2 = 0.1378 | R 1 = 0.0575 wR2 = 0.1285 | R 1 = 0.0348 wR2 = 0.0737 | R 1 = 0.0453 wR2 = 0.1204 |
| R indices (all data) | R 1 = 0.0399 wR2 = 0.1068 | R 1 = 0.0618 wR2 = 0.1432 | R 1 = 0.1102 wR2 = 0.1517 | R 1 = 0.0593 wR2 = 0.0829 | R 1 = 0.0538 wR2 = 0.1260 |
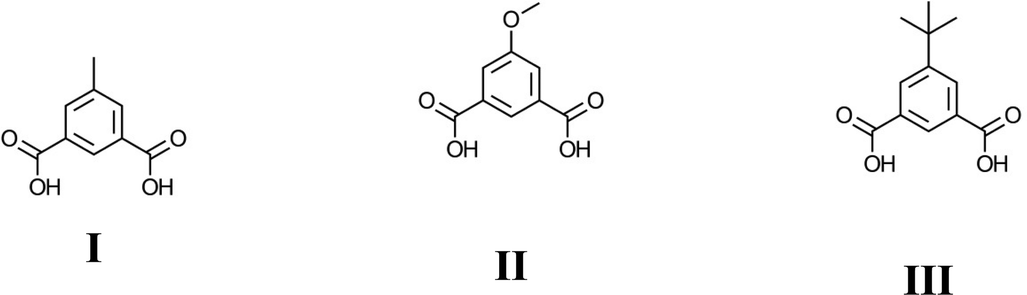
Compound 1, Ni2(mip)2(H2O)8·2H2O, crystallises in the polar space group Pna21, and the asymmetric unit contains two crystallographically distinct nickel centres, two 5-methyl isophthalate (mip) ligands and ten water molecules. Both nickel centres are bound to two trans symmetry-related mip ligands (Fig. 1) and four water molecules, leading to two distinct chains (Types 1 and 2) that extend parallel to the a-axis. The methyl isophthalate ligands do not lie in the average plane of the chain, but rather are angled such that all methyl groups on a given chain lie on one face of the chain, giving each chain a very slightly U-shaped cross section. Symmetry-related chains are arranged into layers that lie in the ab plane. Within each layer, the U-shaped chains are arranged such that all of the methyl groups lie above the same face of the layer. As seen in Fig. 1c, the methyl groups of Type 1 chains all lie on the upper face of the layer whereas the methyl groups of Type 2 layers lie on the lower face of the layer. Pairs of layers form, consisting of one Type 1 and one Type 2, in which the methyl groups of both layers are directed towards the centre of the pair. These pairs of layers then stack parallel to the c-axis in an ABAB fashion. Extensive hydrogen bonds between the coordinated water molecules and carboxylate oxygen atoms of chains belonging to different layers connect the chains into a complex three-dimensional hydrogen-bonded framework. Water molecules of crystallisation occupy the spaces between adjacent pairs of layers and are held in place by hydrogen bonds to the chains.
 | ||
| Fig. 1 The crystal structure of Ni2(mip)2(H2O)8·2H2O (compound 1). One layer of Type 1 Ni(mip)(H2O)4 chains as seen a) perpendicular and b) parallel to the plane of the layer. One layer of Type 2 Ni(mip)(H2O)4 chains as seen c) perpendicular and d) parallel to the plane of the layer. e) The stacking of pairs of layers along the c-axis as viewed along the a-axis. Non-coordinated water molecules are shown as hatched red spheres. f) Stacking of the layers as seen parallel to the b-axis. Hydrogen atoms have been omitted for clarity. | ||
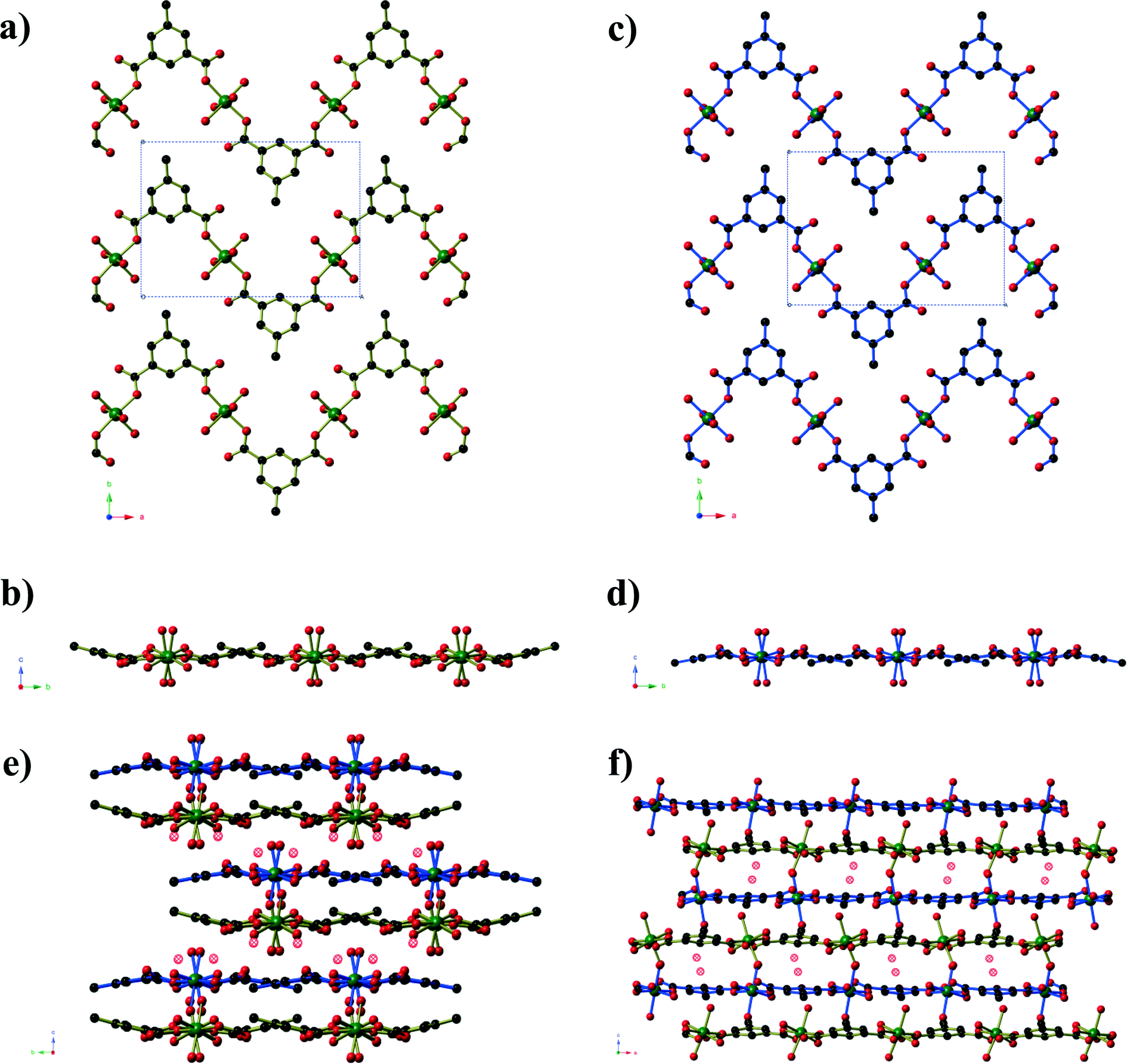
Compounds 2 and 3 are formed by reaction of zinc acetate and 5-methyl isophthalic acid, differing only in which organic solvent is used in conjunction with water. The two frameworks obtained, however, are vastly different to one another and are distinct from the five literature coordination frameworks23 and metal–organic polyhedra24 that are also composed of zinc(II) and 5-methyl isophthalate in the absence of additional metal ions or bridging ligands. Compound 2, produced from aqueous methanol, has composition Zn6(mip)5(OH)2(H2O)4·xH2O (x ≈ 7. 4) forms in orthorhombic space group Pnn2. The asymmetric unit contains six crystallographically distinct ZnII centres, 5 distinct mip ligands, 2 μ3-OH− anions, three coordinated water molecules and several disordered guest water molecules. There is some rotational disorder of the mip ligands, hinged about the two carboxylate groups, and the coordinated water molecules. The ZnII centres are arranged to form clusters of composition Zn6(OH)2(CO2)10(H2O)3 (Fig. 2). The clusters act as a 6-connecting node, binding to four symmetry-related clusters in the bc-plane via pairs of bridging mip ligands and to two symmetry-related clusters above and below this plane via a single mip ligand. The resulting three-dimensional coordination framework has the α-polonium topology. Narrow channels (approx. 4 × 6 Å) extend parallel to the c-axis that are bordered on one ‘end’ by the methyl groups of the mip ligand and on the other by carboxylate groups. The hydrophobic section of the channels is completely blocked by the ligands; however, disordered water molecules occupy the hydrophilic section. Disordered water molecules also occupy the smaller channels (3 × 4.5 Å) that lie between the clusters.
 | ||
| Fig. 2 a) The Zn6(OH)2(CO2)10(H2O)3 cluster found in Zn6(mip)5(OH)2(H2O)4·xH2O (x ≈ 7.4, compound 2). The two μ3-OH anions are shown in blue, and major sites of the disordered coordinated water molecules are shown as hatched red circles. b) A view along the channels of the α-Po net of Zn6(mip)5(OH)2(H2O)4·xH2O (x ≈ 7.4). Partial occupancy water molecules are shown as hatched blue spheres. Hydrogen atoms have been omitted for clarity. c) The connectivity of the Zn6(OH)2(CO2)10(H2O)3 clusters which act as six-connecting nodes in the α-Po net. The Zn centres belonging to the central and peripheral clusters are shown in purple and green, respectively. Terminal methyl groups, coordinated water molecules and monodentate carboxylate groups have been omitted for clarity. | ||
Compound 3, formed from aqueous ethanol, also has composition Zn6(mip)5(OH)2(H2O)2·xH2O (x = 4) and crystallises in orthorhombic space group P21212. The asymmetric unit contains three crystallographically distinct ZnII centres, two and a half mip ligands, one μ3-OH, one coordinated water molecule and two full occupancy guest water molecules. Unlike in compound 2, the Zn centres in compound 3 are arranged into Zn3(OH) units (Fig. 3) that are connected into ‘columns’ by μ2 carboxylate oxygen atoms and Type 3 mip ligands. The remaining two mip ligands connect these ‘columns’ into corrugated sheets that extend parallel to the ab-plane, in which the Types 1 and 2 mip ligands form the peaks and troughs of the sheet and the methyl groups of Type 3 mip ligands project outward from the plane of the sheet on either side of the peaks and troughs. The Type 1 and 2 mip ligands alternate parallel to the b-axis, where each Type 1 ligand is involved in face-to-face aromatic interactions with two Type 2 ligands, and vice versa, with close contact C⋯C distances of 3.4 to 3.8 Å. Sheets stack along the c-axis, with the methyl groups of Type 3 mip ligands interdigitating with those belonging to adjacent sheets. Water molecules occupy the ‘channels’ between the corrugated sheets.
 | ||
| Fig. 3 a) A view of the columns of Zn3(OH) clusters in Zn6(mip)5(OH)2(H2O)2·xH2O (x = 4, compound 3). b) One sheet of Zn6(mip)5(OH)2(H2O)2·xH2O (x = 4) as seen from along the plane of the sheet. The columns from Fig. 3a are highlighted using pink Zn–O bonds and extend into the page. c) The packing of the sheets of Zn6(mip)5(OH)2(H2O)2·xH2O (x = 4). The μ3-OH anions are shown as blue spheres, whilst guest water molecules are shown as hatched blue spheres. | ||
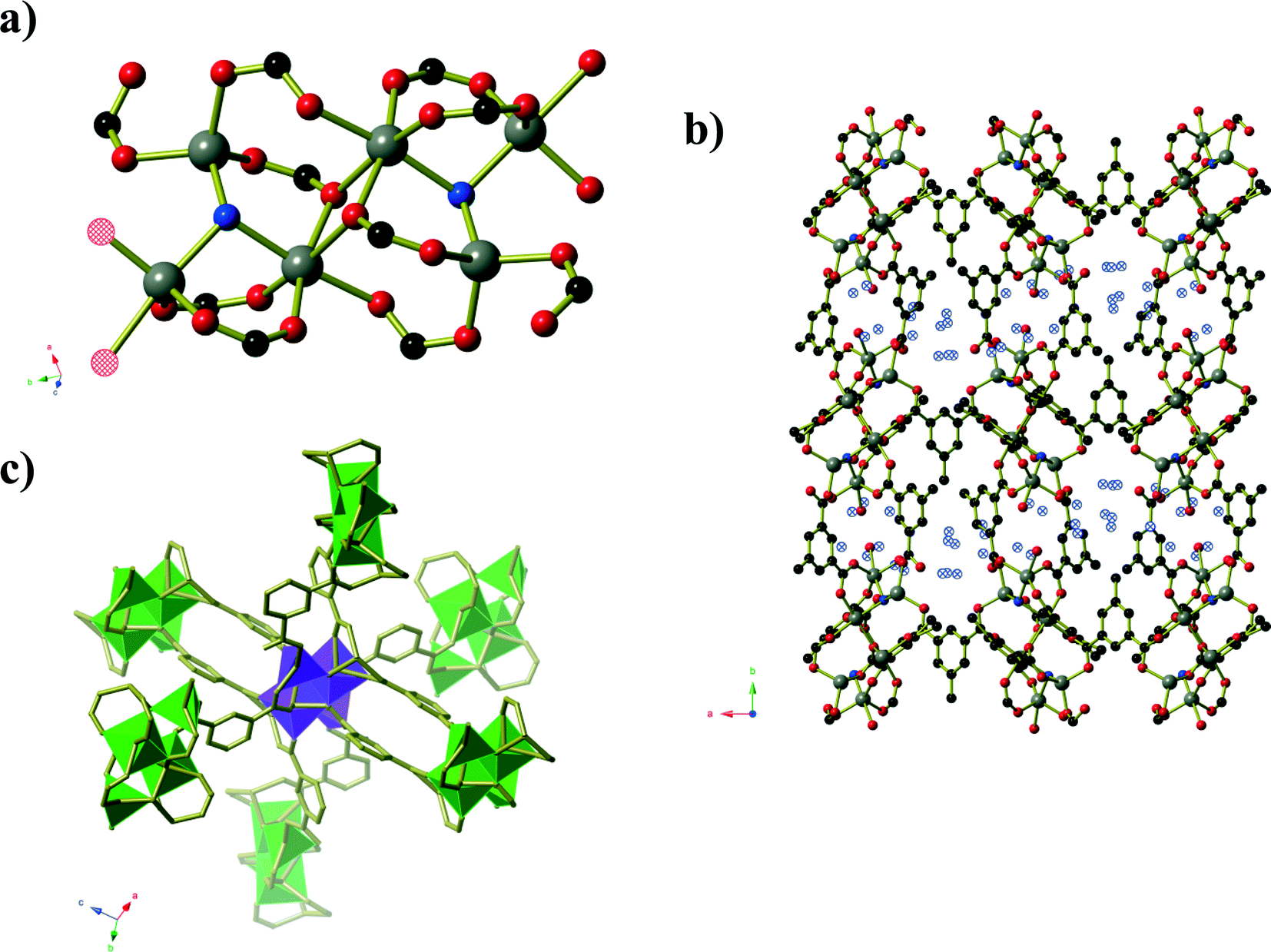
Compound 4 forms as a three-dimensional coordination polymer of composition Mn(HMeOip)2 in space group P21/c. One MnII centre and one HMeOip− monoanion make up the asymmetric unit. The HMeOip− ligand acts as a 3-connecting node, binding to one metal centre through the carbonyl oxygen of the carboxylic acid, and the carboxylate group binds to a further two, one per oxygen atom (Fig. 4). The MnII centre adopts an octahedral coordination environment in which four bridging carboxylate groups are arranged in a square plane whilst the two monodentate carboxylic acid groups bind in a trans arrangement. Bridging carboxylate groups connect the Mn centres into chains of Mn(CO2)2Mn that extend parallel to the a-axis. These chains are then cross-linked by the coordinated carboxylic acid to afford a three-dimensional network with rutile topology. Intra-chain hydrogen bonds (O⋯O separation of 2.657(3) Å) occur between the carboxylic OH group and one oxygen atoms of the bridging carboxylate group.
 | ||
| Fig. 4 a) Chains of carboxylate bridged MnII centres in Mn(HMeOip)2 (compound 4). b) The full Mn(HMeOip)2 framework. c) A schematic representation of the rutile topology, in which the three-connecting HMeOip− ligands are represented by purple spheres. Hydrogen atoms have been omitted for clarity. | ||
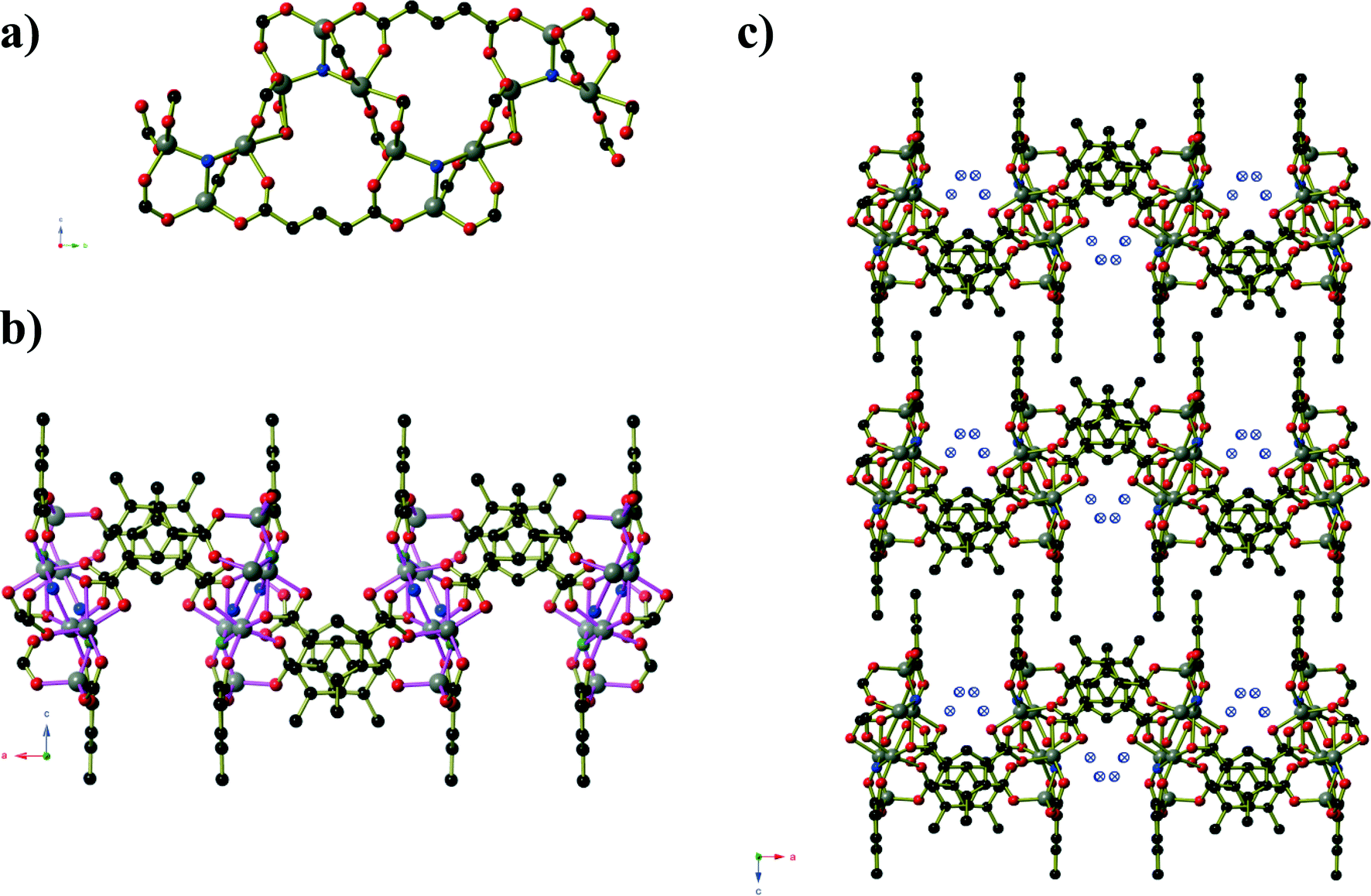
Compound 5 is analogous to the literature compound Mn3(tbip)2(Htbip)2(iPrOH)2 with ethanol replacing the coordinated isopropanol molecules.25 The compound forms in space group P and the asymmetric unit contains two Mn(II) centres, a coordinated ethanol molecule, one tbip2− ligand and one Htbip− ligand. The fully deprotonated tbip2− units connect the Mn centres into a two dimensional sheet (Fig. 5) that extends parallel to the ab-plane. Within this sheet, the Mn centres are grouped into trios containing one central octahedral Mn centre and two peripheral trigonal bipyramidal Mn centres that are connected via μ2 carboxylate oxygen atoms and bridging carboxylate groups that lie in the basal planes of the three Mn centres. These trios are connected by bridging carboxylate groups (also binding in the basal planes of the Mn centres) into chains running parallel to the [1 0 0] direction. Pairs of tbip2− ligands connect these chains together, oriented at an angle of approximately 34° to and sitting slightly above and below the plane of the sheet. These pairs of ligands are arranged such that they form a Piedfort unit with close contact C⋯C separations of 3.67 to 3.80 Å. The axial coordination sites on the octahedral Mn centres are occupied by the deprotonated carboxylate group of the Htbip− ligands, the second oxygen atoms of which binds in one axial site of an adjacent trigonal bipyramidal Mn centre. A coordinated ethanol molecule occupies the remaining coordination site on the trigonal bipyramidal Mn centres and forms an intramolecular hydrogen bond to the coordinated carboxylate oxygen atom on the adjacent octahedral Mn centre. The carboxylic acid groups of the Htbip− ligands project outwards from the plane of the sheet, where they form complementary pairs of hydrogen bonds with symmetry-related ligands on adjacent sheets. These hydrogen bonds connect the 2D framework of Mn3(tbip)2(Htbip)2(EtOH)2 into a three-dimensional hydrogen-bonded network.
 | ||
| Fig. 5 a) One sheet of Mn3(tbip)2(Htbip)2(EtOH)2 (compound 5) as viewed perpendicular to the plane of the sheet. The atoms of the coordinated ethanol molecules are shown using hatched spheres. The Htbip− ligands have been omitted for clarity. b) View along the edge of a complete sheet. c) The hydrogen-bonding interactions between adjacent sheets. Hydrogen bonds are shown as black and white striped bonds. d) The packing of the sheets along the c-axis. | ||
Nitric oxide release
Preliminary screening was conducted on compounds 2 and 3 for their nitric oxide release capacity. These compounds were selected because they contained coordinated solvent molecules that may be removed to generate a vacant coordination site whilst potentially leaving the coordination framework intact. compound 5 was not considered as removal of the coordinated ethanol molecules would most likely disrupt the inter-sheet hydrogen bonds. Thermogravimetric analyses were conducted on all 5 compounds, and are presented in ESI.† Samples were dehydrated at 200 °C under dynamic vacuum for approximately 18 hours, before being exposed to a nitric oxide atmosphere for approximately 1 hour. The samples were flame-sealed in an argon atmosphere. Nitric oxide release was triggered by exposure to humid N2 (∼11% RH). Initial measurements showed that the NO release from these compounds was very time sensitive, so repeat measurements were conducted in two phases – as soon as possible after NO loading, and then after 1 week (Tables 2 and 3). The NO-release from compound 3 proved to be erratic. Both samples showed a small initial release on the day of loading, in the range of 0.16–0.21 μmol g−1 for compound 2 and 9.5–17.5 μmol g−1 for compound 3. These values are small but occur over a period of minutes for compound 2 and hours for compound 3, which is consistent with the loss of NO that has been included into the pores of the material rather than simply being physisorbed onto the surface of the powder. The difference in the amount of NO released from compounds 2 and 3 we attribute to the larger cross-sectional area of the channels in compound 3 compared to compound 2, as may be seen by comparison of Fig. 2b and 3c. The channels in compound 2 have a corrugated surface and are approximately 6 Å high and 2 Å wide, whereas those in compound 3 are approximately 8.5 Å high and between 2.7 and 5.5 Å wide. The larger the cross-sectional area of the channel opening allows for easier ingress and egress of the NO and water molecules. The amount of NO released less than 1 day after loading is comparable to the release from other well-known materials such as HKUST-1 (NO release 2 μmol g−1) and this amount of NO has been shown to have biological effects (e.g. anti-platelet activation).26 After standing for one week, however, the amount of released NO drops by a factor of 40 for compound 2 and by two orders of magnitude for compound 3. The release time was also significantly lower after this short storage time, signifying that the NO-loaded compounds have a very short shelf-life and these compounds would not be suitable for use as biomedical NO delivery systems.| Zn6(mip)5(OH)2(H2O)4·xH2O (x ≈ 7.4) | Pre-loaded weight (mg) | Loaded weight | Total NO released (μmol g−1) | Release time (min) | Peak NO release |
|---|---|---|---|---|---|
| 1 | 15.5 | 13.1 | 0.21 | 4.5 | 2.4 ppm |
| 2 | 15.2 | 13.0 | 0.18 | 4.5 | 2.0 ppm |
| 3 | 15.5 | 13.1 | 0.18 | 4.5 | 1.8 ppm |
| 4 | 15.1 | 12.8 | 0.16 | 4.5 | 1.5 ppm |
| 5 (after 1 week) | 15.3 | 13.5 | 0.0059 | <1 | 81.8 ppb |
| 6 (after 1 week) | 15.4 | 13.3 | 0.0036 | <1 | 62.8 ppb |
| 7 (after 1 week) | 15.4 | 12.9 | 0.0027 | <1 | 43.3 ppb |
| 8 (after 1 week) | 15.5 | 13.1 | 0.0051 | <1 | 93.2 ppb |
| Zn6(mip)5(OH)2(H2O)4·xH2O (x ≈ 4) | Pre-loaded weight (mg) | Loaded weight | Total NO released (μmol g−1) | Release time (min) | Peak NO release |
|---|---|---|---|---|---|
| 1 | 15.1 | 14.2 | 17.10 | 93 | 12.9 ppm |
| 2 (after 1 day) | 15.1 | 14.6 | 9.87 | 60 | 2.8 ppm |
| 3 (after 1 day) | 15.3 | 13.9 | 9.72 | 118 | 3.2 ppm |
| 4 (after 1 week) | 15.3 | 13.8 | 0.46 | 10 | 1.2 ppm |
| 5 (after 1 week) | 15.2 | 13.8 | 0.98 | 19 | 1.3 ppm |
| 6 (after 1 week) | 15.0 | 13.6 | 2.75 | 59 | 1.2 ppm |
Powder diffraction patterns were collected on both compounds before and after NO-loading and release experiments. This showed that the PXRD pattern for compound 3 does not change as a result of the NO-loading and release, indicating that the framework remains intact throughout the dehydration–NO-loading–rehydration process. The PXRD pattern for compound 2 after NO release contains one extra peak at 2θ = 7°, which may be indicative of a contaminent being formed from the partial decomposition of compound 2. Variable temperature PXRD studies showed that compound 2 remains largely unchanged when heated to 150 °C, after this temperature some degradation of the sample occurs (Fig. 6). Upon heating to 250 °C, the sample is completely destroyed and further powder diffraction studies showed that the residue contains a peak centred at 2θ = 7°.
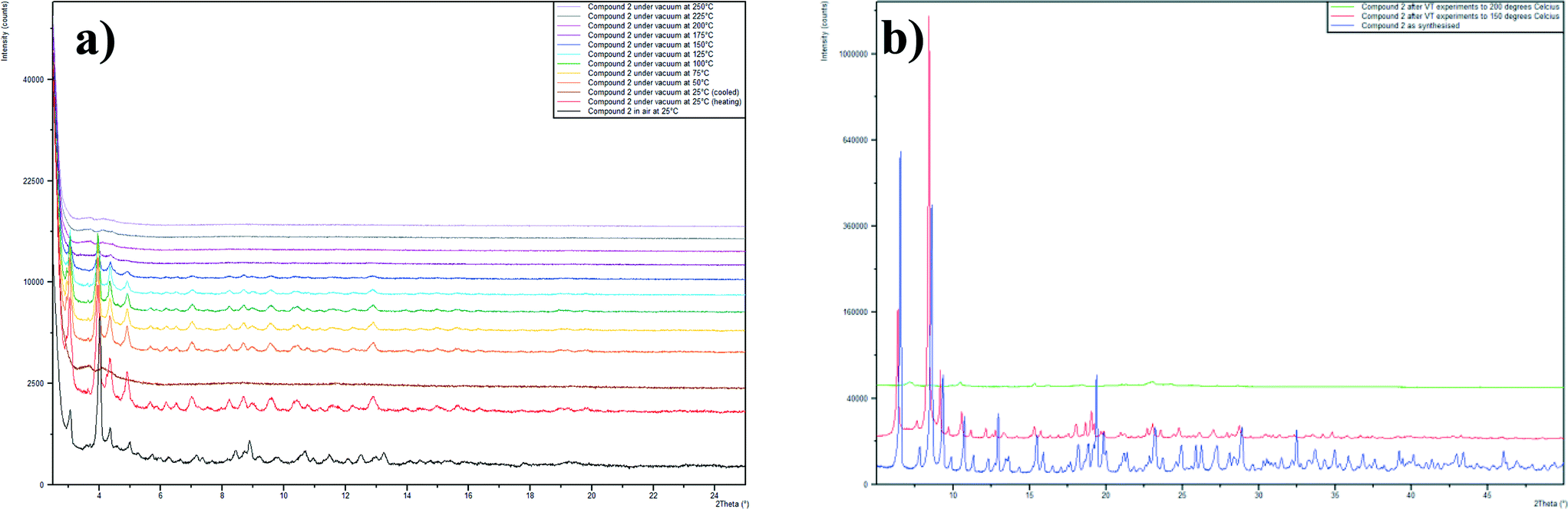 | ||
| Fig. 6 a) Variable temperature PXRD traces for compound 2. b) PXRD patterns on compound 2 on samples that are as synthesised (blue), after VT studies to 150 °C (red) and after VT studies to 250 °C (green). | ||
Conclusions
One derivative and four novel coordination polymers have been prepared by solvothermal reaction of metal acetate and 5-substituted isophthalic acid in either water or aqueous short-chain alcohols. In the case of zinc and 5-methyl isophthalate, varying the length of the carbon chain in the organic solvent can influence the connectivity and dimensionality of the resulting coordination framework. Preliminary NO release screening showed that compounds 2 and 3 are able to deliver small amounts of NO, however the shelf life of the NO-loaded compounds is limited.Acknowledgements
This work was funded by the British Heart Foundation (NH/11/8/29253) and the EPSRC (EP/K005499/1). Crystallographic data for compound 1, 2 and 5 were collected at station 11.3.1 at the Advanced Light Source, Berkeley, CA, USA. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.References
- F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tome, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088 RSC; R. Matsuda, Bull. Chem. Soc. Jpn., 2013, 86, 1117 CrossRef CAS.
- Y. Yan, S. Yang, A. J. Blake and M. Schröder, Acc. Chem. Res., 2014, 47, 296 CrossRef CAS PubMed; M. Tagliabue, C. Rizzo, R. Millini, P. D. C. Dietzel, R. Blom and S. Zanardi, J. Porous Mater., 2010, 18, 9378 Search PubMed.
- A. C. McKinlay, P. K. Allan, C. L. Renouf, M. J. Duncan, P. S. Wheatley, S. J. Warrender, D. Dawson, S. E. Ashbrook, B. Gil, B. Marszalek, T. Düren, J. J. Williams, C. Charrier, D. K. Mercer, S. J. Teat and R. E. Morris, APL Mater., 2014, 2, 124108 CrossRef CAS; P. K. Allan, P. S. Wheatley, D. Aldous, M. I. Mohideen, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. de Weireld, S. Vaesen and R. E. Morris, Dalton Trans., 2012, 41, 4060 RSC.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef CAS PubMed.
- H. F. Zhu, B. Ka and F. Murad, World J. Surg., 2007, 31, 624 CrossRef CAS PubMed; M. R. Miller and I. L. Megson, Br. J. Pharmacol., 2007, 151, 305 CrossRef PubMed.
- R.-Q. Zou, H. Sakurai, S. Han, R.-Q. Zhong and Q. Xu, J. Am. Chem. Soc., 2007, 129, 8402 CrossRef CAS PubMed.
- L.-F. Ma, L.-Y. Wang, D.-H. Lu, S. R. Batten and J.-G. Wang, Cryst. Growth Des., 2009, 9, 1741 CAS.
- H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167 CrossRef CAS PubMed; M. I. H. Mohideen, B. Xiao, P. S. Wheatley, A. C. McKinlay, Y. Li, A. M. Z. Slawin, D. W. Aldous, N. F. Cessford, T. Düren, X. Zhao, R. Gill, K. M. Thomas, J. M. Griffin, S. E. Ashbrook and R. E. Morris, Nat. Chem., 2011, 3, 304 CrossRef PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Science, 1999, 402, 276 CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- O. I. Lebedev, F. Millange, C. Serre, G. van Tendeloo and G. Ferey, Chem. Mater., 2005, 17, 6525 CrossRef CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. P. Williams, Science, 1999, 283, 1148 CrossRef CAS PubMed.
- G. Ferey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296 CrossRef CAS PubMed.
- (a) R.-Q. Zou, H. Sakurai, S. Han, R.-Q. Zhong and Q. Xu, J. Am. Chem. Soc., 2007, 129, 8402 CrossRef CAS PubMed; (b) Z.-L. Yu and J.-L. Hu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1522 CAS; (c) H. Chun and H. Jung, Inorg. Chem., 2009, 48, 417 CrossRef CAS PubMed; (d) W.-X. Chen, Y.-P. Ren, L.-S. Long, R.-B. Huang and L.-S. Zheng, CrystEngComm, 2009, 11, 1522 RSC; (e) L. Wang, J. Liu, C.-Z. Zheng and Y.-J. Wang, Huaxue Yu Shengwu Gongcheng, 2011, 28, 17 CAS; (f) Q.-B. Bo, Z.-W. Zhang, J.-L. Miao, D.-Q. Wang and G.-X. Sun, CrystEngComm, 2011, 13, 1765 RSC; (g) Q.-B. Bo, H.-Y. Wang, D.-Q. Wang, Z.-W. Zhang, J.-L. Miao and G.-X. Sun, Inorg. Chem., 2011, 50, 10163 CrossRef CAS PubMed; (h) H. Y. Zhu, J. W. Ji, Y. W. Li, C. W. Li and K. Z. Zhu, Koord. Khim., 2012, 38, 157 Search PubMed; (i) J.-W. Ji, X.-F. Wang, B.-Y. Li and Z.-B. Han, Solid State Sci., 2012, 14, 335 CrossRef CAS; (j) S. Du, C. Hu, J.-C. Xiao, H. Tan and W. Liao, Chem. Commun., 2012, 48, 9177 RSC; (k) Q.-B. Bo, H.-Y. Wang, J.-L. Miao and D.-Q. Wang, RSC Adv., 2012, 2, 11650 RSC; (l) X.-L. Deng, S.-Y. Yang, R.-F. Jin, J. Tao, C.-Q. Wu, Z.-L. Li, L.-S. Long, R.-B. Huang and L.-S. Zheng, Polyhedron, 2013, 50, 219 CrossRef CAS; (m) Q.-B. Bo, H.-Y. Wang and D.-Q. Wang, New J. Chem., 2013, 37, 380 RSC; (n) L.-P. Xue, Y.-C. Liu, Y.-H. Han, C.-B. Tain, Q.-P. Li, P. Lin and S.-W. Du, Chin. J. Struct. Chem., 2014, 33, 1191 CAS; (o) C.-B. Tian, R.-P. Chen, C. He, W.-J. Li, Q. Wei, X.-D. Zhang and S.-W. Du, Chem. Commun., 2014, 50, 1915 RSC; (p) L. Jin, L.-L. Zha, S. Gao, S.-Y. Yang and R.-B. Huang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2015, 71, m1 CAS.
- (a) L. Pan, B. Parker, X. Huang, D. H. Olson, J. Y. Lee and J. Li, J. Am. Chem. Soc., 2006, 128, 4180 CrossRef CAS PubMed; (b) S. Ma, D. Sun, X.-S. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2007, 46, 2458 CrossRef CAS PubMed; (c) X.-L. Li and M.-L. Huang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1501 CAS; (d) X.-L. Li and M.-L. Huang, Z. Kristallogr. - New Cryst. Struct., 2009, 224, 127 CAS; (e) Y.-T. Fan, B. Li, L.-F. Ma and L.-Y. Wang, Chin. J. Struct. Chem., 2009, 28, 1061 CAS; (f) Z.-X. Du, L.-Y. Wang and H.-W. Hou, Z. Anorg. Allg. Chem., 2009, 635, 1659 CrossRef CAS; (g) L.-F. Ma, L.-Y. Wang, D.-H. Lu, S. R. Batten and J.-G. Wang, Cryst. Growth Des., 2009, 9, 1741 CrossRef CAS; (h) D.-S. Zhou, F.-K. Wang, S.-Y. Yang, Z.-X. Xie and R.-B. Huang, CrystEngComm, 2009, 11, 2548 RSC; (i) J.-D. Lin, X.-F. Long, P. Lin and S.-W. Du, Cryst. Growth Des., 2010, 10, 146 CrossRef CAS; (j) J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893 CrossRef CAS PubMed; (k) J.-D. Lin, S.-T. Wu, Z.-H. Li and S.-W. Du, Dalton Trans., 2010, 39, 10719 RSC; (l) C.-B. Tian, Z.-Z. He, Z.-H. Li, P. Lin and S.-W. Du, CrystEngComm, 2011, 13, 3080 RSC; (m) B. Kilbas, S. Mirtschin, T. Riis-Johannessen, R. Scopelliti and K. Severin, Inorg. Chem., 2012, 51, 5795 CrossRef CAS PubMed; (n) L.-J. Chen, J.-B. Su, R.-B. Huang, S. Lin, M.-X. Yang and H. Huang, Dalton Trans., 2011, 40, 9731 RSC; (o) X.-F. Wang, L. Li, Y.-M. Kong and Y. Liu, Inorg. Chem. Commun., 2012, 21, 72 CrossRef CAS; (p) S.-H. Li and J.-B. Guo, Z. Kristallogr. - New Cryst. Struct., 2012, 227, 389 CrossRef CAS; (q) Y.-F. Liang, Y.-R. Zhang and Z.-B. Han, Z. Anorg. Allg. Chem., 2012, 638, 423 CrossRef CAS; (r) R.-F. Jin, S.-Y. Yang, H.-M. Li, L.-S. Long, R.-B. Huang and L.-S. Zheng, CrystEngComm, 2012, 14, 1301 RSC; (s) Q.-B. Bo, H.-Y. Wang, J.-L. Miao and D.-Q. Wang, RSC Adv., 2012, 2, 11650 RSC; (t) S.-Y. Yang, H.-B. Yuan, X.-B. Xu and R.-B. Huang, Inorg. Chim. Acta, 2013, 403, 53 CrossRef CAS; (u) Q.-B. Bo, H.-Y. Wang and D.-Q. Wang, New J. Chem., 2013, 37, 380 RSC; (v) K. Noh and J. Kim, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m579 CAS; (w) J. Wang, J.-Q. Tao, X.-J. Xu and C.-Y. Tan, Zh. Strukt. Khim., 2013, 54, 759 CAS; (x) C. Tian, Z. Lin and S. Du, Cryst. Growth Des., 2013, 13, 3746 CrossRef CAS; (y) A. K. Chaudhari, S. Mukherjee, S. S. Nagarkar, B. Joarder and S. K. Ghosh, CrystEngComm, 2013, 15, 9465 RSC.
- (a) G.-L. Wen, Z. Kristallogr. - New Cryst. Struct., 2009, 224, 497 CAS; (b) S.-M. Shen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1173 CAS; (c) F.-P. Liang, Q.-L. Meng, L.-F. Ma and L.-Y. Wang, Chin. J. Struct. Chem., 2010, 29, 157 CAS; (d) H.-J. Zhang, Z. Kristallogr. - New Cryst. Struct., 2011, 226, 629 CAS; (e) X.-H. Huang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 483 CAS; (f) C. Tian, Z. Lin and S. Du, Cryst. Growth Des., 2013, 13, 3746 CrossRef CAS; (g) A. Dorazco-Gonzalez, S. Martinez-Vargas, S. Hernandez-Ortega and J. Valdes-Martinez, CrystEngComm, 2013, 15, 5961 RSC; (h) X.-H. Chang, J.-H. Qin, M.-L. Han, L.-F. Ma and L.-Y. Wang, CrystEngComm, 2014, 16, 870 RSC; (i) H. Abourahma, G. J. Bodwell, J. Lu, B. Moulton, I. R. Pottie, R. B. Walsh and M. J. Zaworotko, Cryst. Growth Des., 2003, 3, 513 CrossRef CAS; (j) J. J. Perry, G. J. McManus and M. J. Zaworotko, Chem. Commun., 2004, 2534 RSC; (k) X. Zhang, J.-X. Yang and Y.-G. Yao, J. Inorg. Organomet. Polym. Mater., 2012, 22, 1189 CrossRef CAS; (l) X. Li, J. Li, M.-K. Li and Z. Fei, J. Mol. Struct., 2014, 1059, 294 CrossRef CAS; (m) D.-W. Zhang and G.-Y. Zhao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2015, 45, 524 CrossRef CAS.
- (a) S. Xiaofei, Y. J. X. X. Hong and L. Xiaoju, Chin. J. Struct. Chem., 2015, 34, 562 Search PubMed; (b) J. Xu, Z.-S. Bai, T. Okamura, M.-S. Chen, W.-Y. Sun and N. Ueyama, Polyhedron, 2009, 28, 2480 CrossRef CAS; (c) J. Xu, Z. Su, M.-S. Chen, S.-S. Chen and W.-Y. Sun, Inorg. Chim. Acta, 2009, 362, 4002 CrossRef CAS; (d) X.-C. Cheng, X.-H. Zhu and H.-W. Kuai, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 1007 CAS; (e) H.-W. Kuai, X.-C. Cheng, L.-D. Feng and X.-H. Zhu, Z. Anorg. Allg. Chem., 2011, 637, 156 CrossRef; (f) X.-H. Zhu and X.-C. Cheng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1862 CAS; (g) X.-J. Li, X.-H. Xu and X.-F. Guo, Chin. J. Struct. Chem., 2014, 33, 721 CAS; (h) Y. Wang, T. Wang, G.-X. Liu and Y.-C. Chen, Wuji Huaxue Xuebao, 2010, 26, 1467 CAS; (i) X. Liu, M. Oh and M. S. Lah, Cryst. Growth Des., 2011, 11, 5064 CrossRef CAS; (j) Y. Wang, Y.-J. Dong, J.-H. Zhou, S.-N. Wang, G.-X. Liu and Y.-C. Chen, Wuji Huaxue Xuebao, 2012, 28, 2407 CAS; (k) M. C. Das and P. K. Bharadwaj, J. Am. Chem. Soc., 2009, 131, 10942 CrossRef CAS PubMed; (l) M. C. Das and P. K. Bharadwaj, Chem. – Eur. J., 2010, 16, 5070 CrossRef CAS PubMed.
- (a) A. J. Guenthner, M. C. Davis, M. D. Ford, J. T. Reams, T. J. Groshens, L. C. Baldwin, L. M. Lubin and J. M. Mabry, Macromolecules, 2012, 45, 9707 CrossRef CAS; (b) L. J. McCormick, S. A. Morris, S. J. Teat, M. J. McPherson, A. M. Z. Slawin and R. E. Morris, Dalton Trans., 2015, 44, 17686 RSC.
- L. Kissel and R. H. Pratt, XDISP Program in WinGX, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 170 CrossRef.
- (a) SADABS: Area-Detector Absorption Correction, Siemens Industrial Automation, Inc., Madison, WI, 1996 Search PubMed; (b) CrystalClear 2.1, Rigaku Corporation, CrystalClear Software User's Guide, Molecular Structure Corporation, 2014; Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- (a) Q.-B. Bo, H.-Y. Wang, D.-Q. Wang, Z.-W. Zhang, J.-L. Miao and G.-X. Sun, Inorg. Chem., 2011, 50, 10163–10177 CrossRef CAS PubMed; (b) Z. Yanq, L. Wang, J. Liu and C. Zhen, Yingyong Huagong, 2011, 40, 589–591 Search PubMed; (c) H. Chun and H. Jung, Inorg. Chem., 2009, 48, 417–419 CrossRef CAS PubMed; (d) Q.-B. Bo, H.-Y. Wang and D.-Q. Wang, New J. Chem., 2013, 37, 380–390 RSC.
- H. Chun, J. Am. Chem. Soc., 2008, 130, 800–801 CrossRef CAS PubMed.
- C.-B. Tian, Z.-Z. He, Z.-H. Li, P. Lin and S.-W. Du, CrystEngComm, 2011, 13, 3080–3086 RSC.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1417516–1417520 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce02091c |
| This journal is © The Royal Society of Chemistry 2016 |