
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate scope for trimethyllysine hydroxylase catalysis†
Abbas H. K.
Al Temimi‡
,
Bas J. G. E.
Pieters‡
,
Y. Vijayendar
Reddy
,
Paul B.
White
and
Jasmin
Mecinović
*
Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: j.mecinovic@science.ru.nl; Fax: +31 (0)24 3653391; Tel: +31 (0)24 3652381
First published on 5th October 2016
Abstract
Trimethyllysine hydroxylase (TMLH) is a non-haem Fe(II) and 2-oxoglutarate dependent oxygenase that catalyses the C-3 hydroxylation of an unactivated C–H bond in L-trimethyllysine in the first step of carnitine biosynthesis. The examination of trimethyllysine analogues as substrates for human TMLH reveals that the enzyme does hydroxylate substrates other than natural L-trimethyllysine.
Carnitine (L-3-hydroxy-4-N,N,N-trimethylaminobutyrate) is an important metabolite that plays the central role in the transport of long chain fatty acids from cytosol to the mitochondrial matrix in most eukaryotes and several prokaryotes.1–3 The biosynthetic pathway leading to carnitine involves four enzymatic steps starting from L-trimethyllysine (TML, 1), which is derived from the hydrolytic degradation of posttranslationally modified trimethyllysine-containing proteins (Fig. 1).3,4 The first and the last steps in carnitine biosynthesis are catalysed by Fe(II) and 2-oxoglutarate (2OG) dependent oxygenases, trimethyllysine hydroxylase (TMLH) and γ-butyrobetaine hydroxylase (BBOX), respectively (Fig. 1). Human TMLH and BBOX exhibit a relatively high degree of homology, with several key residues conserved, including: (i) the iron chelating triad His-Asp-His, (ii) the active site Arg that forms a salt-bridge with the C-5 carboxylate of the 2OG co-substrate, and (iii) the aromatic cage that consists of Tyr and Trp, and associates with the positively charged trimethylammonium group of trimethyllysine (for TMLH) or γ-butyrobetaine (for BBOX).5 A notable difference between these two oxygenases includes the site that is presumably responsible for specific binding of the α-ammonium group of trimethyllysine (Asp131 in TMLH, Asn191 in BBOX).5
 | ||
| Fig. 1 The carnitine biosynthesis. | ||
In contrast to recent structural, mechanistic and inhibition studies on BBOX,5–13 little is known about the mechanism and substrate requirements for TMLH catalysis. Early work on cellular TMLH, based on column chromatography, radioactive labelling, oxidative cleavage reactions and limited NMR analyses, provided basic evidence that the enzyme catalyses the C-3 hydroxylation of trimethyllysine and highlighted that trimethyllysine is a natural substrate.14–17 Previous biochemical studies, however, did not provide any insight into whether TMLH also has a potential to catalyse the hydroxylation of other natural and unnatural trimethyllysine analogues. Herein, we report the examination of trimethyllysine analogues as substrates for TMLH, employing the MS and NMR-based assays.
A recently developed protocol for the efficient bacterial production of stable and active human TMLH-a isoform as the MBP-TMLH fusion protein prompted us to explore its substrate scope.18 We first tested L-trimethyllysine as a substrate for the recombinantly expressed MBP-TMLH-a (hereafter referred to TMLH) using the highly sensitive MS and information rich NMR assays. The incubation of trimethyllysine (500 μM) in the presence of TMLH (3 μM), FeSO4, 2OG and ascorbate at 37 °C for 30 minutes resulted in almost quantitative production of 3-hydroxy-L-trimethyllysine (90% by MS and 81% by 1H NMR) as indicated by the mass shift of +16 Da by MS (Fig. S1, ESI†), and the integration of NMe3+ peaks of the trimethyllysine starting material and the 3-hydroxy-L-trimethyllysine product by 1H NMR (Fig. 2A). 1H NMR data and more detailed 2D NMR analyses provided unambiguous evidence that the enzymatic hydroxylation occurs at the C-3 site (Fig. 2 and Fig. S2, S3, ESI†). The 1H–1H COSY spectrum revealed that the new downfield resonances at 4.07 and 3.78 ppm are coupled to each other (Fig. 2B), and subsequent analysis of the TOCSY spectrum revealed a coupling network consistent with a trimethyllysine derivative (Fig. S2 and S3, ESI†). The multiplicity-edited 1H–13C HSQC spectrum confirmed that these resonances were methines (as opposed to methylenes) with 13C chemical shifts consistent with a Cα (59.3 ppm) carbon and a secondary alcohol (Cβ, 68.8 ppm) carbon (Fig. 2C). Our advanced NMR analyses, however, were not able to conclusively assign the stereochemistry at the C-3, presumably due to freely rotatable Cα–Cβ bond. Based on the results from previous studies, the absolute configuration at the C-3 has not been unquestionably established, although it has been suggested that 3-hydroxy-L-trimethyllysine possesses the 2S, 3S stereochemistry (i.e. the erythro form).15 It is envisioned that the further work using the synthetic enantiopure (3S)- and (3R)-3-hydroxy-L-trimethyllysine, will provide the unambiguous confirmation of the exact stereochemistry of the TMLH-catalysed formation of the 3-hydroxy-L-trimethyllysine product. Our NMR analyses, however, revealed that the TMLH-catalysed C-3 hydroxylation of trimethyllysine is tightly coupled to the conversion of 2OG to succinate (the ratio succinate formation![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hydroxylation was observed to be 1.1). This result is in agreement with mechanistic studies on other non-haem Fe(II) and 2OG oxygenases, including BBOX.5,7,9
:hydroxylation was observed to be 1.1). This result is in agreement with mechanistic studies on other non-haem Fe(II) and 2OG oxygenases, including BBOX.5,7,9
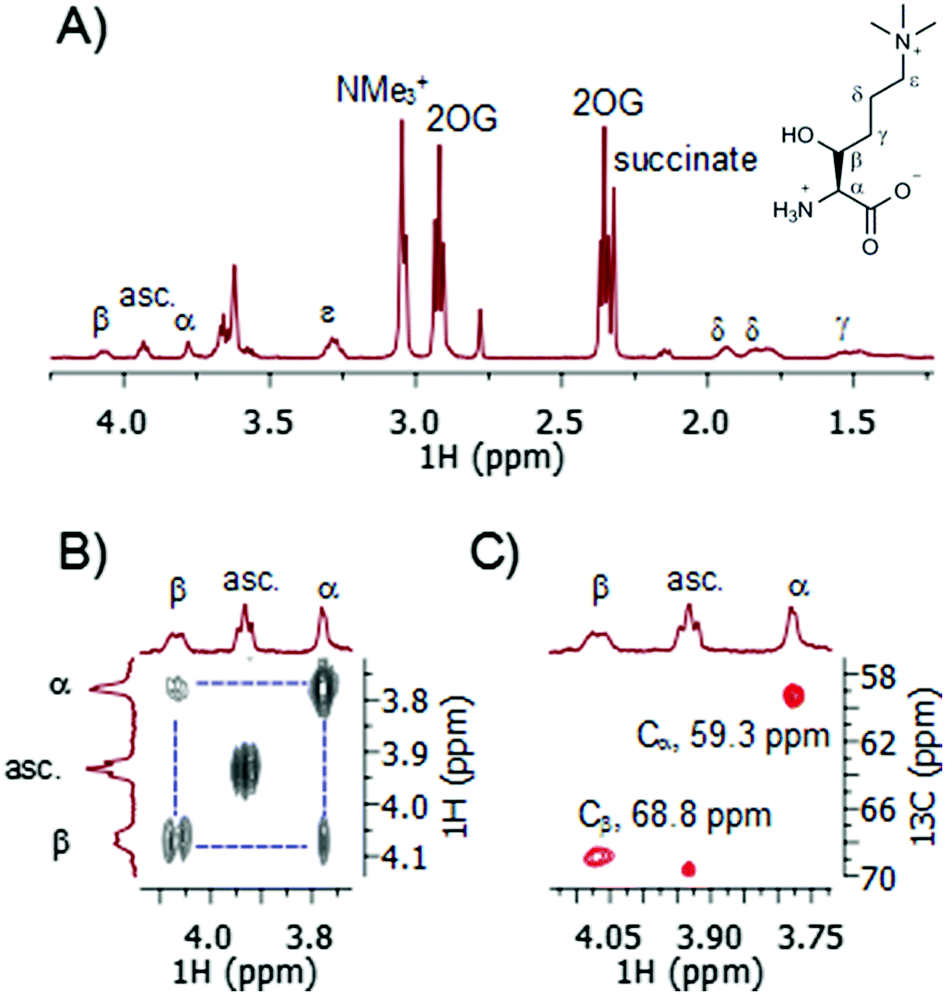 | ||
| Fig. 2 NMR analyses of TMLH-catalysed C-3 hydroxylation of trimethyllysine. (A) 1H NMR data and the assignment of 3-hydroxytrimethyllysine. (B) COSY data showing the Hα–Hβ coupling. (C) HSQC data showing the Cα–Hα and Cβ–Hβ couplings. (asc. = ascorbate). | ||
Control experiments under standard conditions, but in the absence of TMLH, showed no formation of 3-hydroxytrimethyllysine, indicating that the hydroxylation is TMLH-catalysed (Fig. S4, ESI†). MS and NMR analyses, furthermore, illustrated that the presence of Fe(II), 2OG and ascorbate, respectively, is required for the efficient enzymatic hydroxylation of trimethyllysine (Fig. S5–S7, ESI†). These results are in line with previous biochemical studies on mitochondrial TMLH extracts.14,16
We also carried out the enzymatic assay in H218O (90% 18O, phosphate buffer, pH 7.0) under standard conditions. MS analyses showed that 18O was not incorporated in 3-hydroxytrimethyllysine product, implying that the oxygen atom in 3-hydroxytrimethyllysine derives from molecular oxygen, and not from water (Fig. S8, ESI†). This result is consistent with the catalytic cycle and the oxygen labelling studies on several other members of Fe(II)/2OG oxygenases.7,19
Although there is some evidence that trimethyllysine that is required for carnitine biosynthesis in mammals can be obtained via food (e.g. vegetables),20 the most likely source of trimethyllysine in cells are endogeneous trimethyllysine-containing proteins, most commonly histones, calmodulin, cytochrome C and myosine.21 We have tested physiologically important and abundant histones H3K4me3 and H3K9me3 as potential substrates for TMLH. Our MALDI-TOF MS analyses showed that H3K4me3 and H3K9me3 histone peptides are not hydroxylated in the presence of TMLH under standard conditions (Fig. S9, ESI†). These results on representative trimethyllysine-containing proteins/peptides suggest that it is unlikely that 3-hydroxytrimethyllysine could also be obtained via TMLH-catalysed C-3 hydroxylation of trimethyllysine that is present in various proteins, followed by the proteolytic degradation of such hydroxylated proteins.
After verifying that trimethyllysine, in its free form, is an excellent substrate for TMLH, we examined other naturally occurring lysine derivatives (Fig. 3). Dimethyllysine (2) was observed to be a poor substrate for TMLH, yielding only a small amount (25% conversion) of hydroxylated product by MS assay, and only traces of the product were detected by NMR (Fig. S10, ESI†). Methyllysine (3) and lysine (4) were not hydroxylated in the presence of TMLH under standard conditions (Fig. S11 and S12, ESI†), even after prolonged incubation (2 hours), highlighting that the presence of methyl groups on the ε-N site of lysine has a profound effect on substrate efficiency, presumably via the contribution of the energetically favourable cation–π interactions.
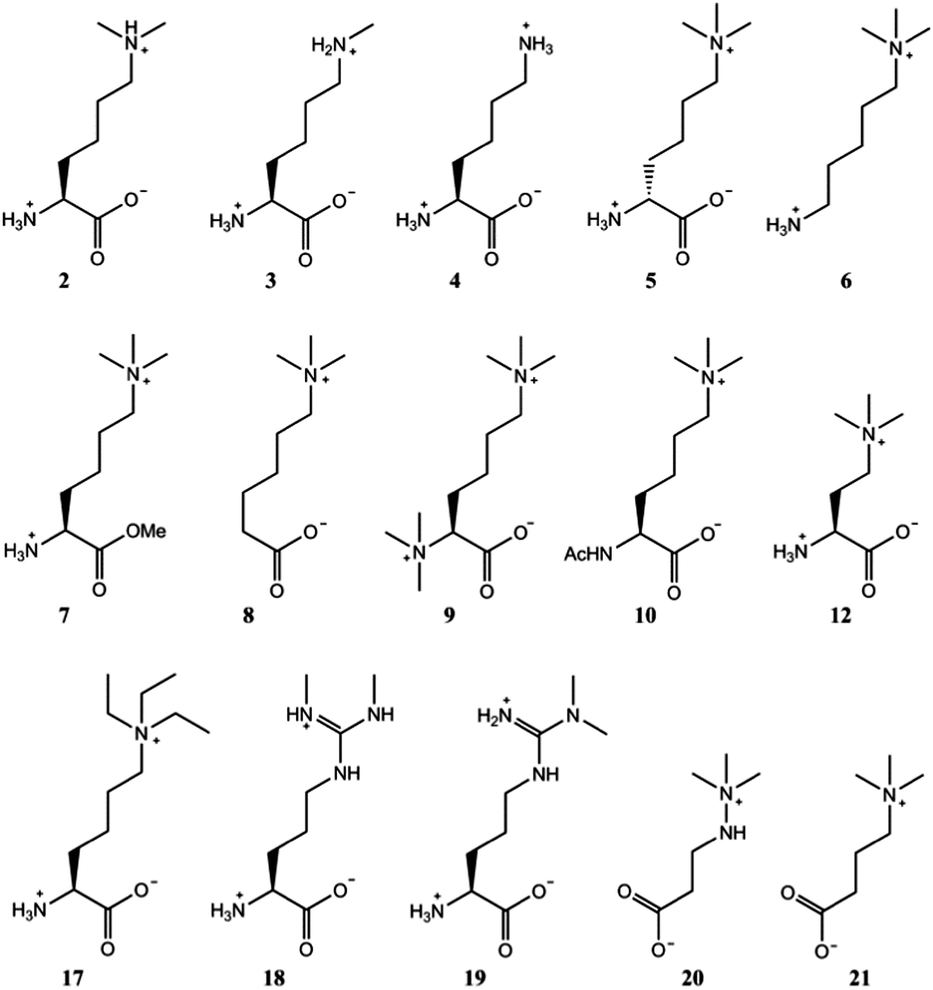 | ||
| Fig. 3 Trimethyllysine analogues that are not substrates for TMLH. 2 was observed to be a poor TMLH substrate by MS and NMR analyses. | ||
In contrast to L-trimethyllysine, its enantiomer D-trimethyllysine (5) did not undergo TMLH-catalysed hydroxylation reaction, as indicated by MS and NMR analyses (Fig. 3 and Fig. S13, ESI†). We did not observe any 3-hydroxy-D-trimethyllysine, nor the TMLH-mediated conversion of 2OG to succinate. It is indeed interesting that the stereochemistry on C-2 (i.e. the α carbon) fully determines the substrate efficiency.
We then carried out structure–activity relationship studies with the aim of determining which other features of L-trimethyllysine are responsible for its ability to act as a specific substrate for TMLH. 5-Trimethylamino-1-aminopentane (6), which does not possess the C-1 carboxylate functionality, was not hydroxylated in the presence of TMLH (Fig. 3 and Fig. S14, ESI†). Similarly, the methyl ester of trimethyllysine (7) was found not to be a TMLH substrate (Fig. 3 and Fig. S15, ESI†). These two analogues showcase that the negatively charged C-1 carboxylate moiety of trimethyllysine is required for productive enzymatic hydroxylation. We also investigated whether the α-ammonium group of trimethyllysine contributes to substrate recognition. The absence of the α-ammonium group (as in 6-trimethylaminohexanoic acid, 8) resulted in an inactive substrate (Fig. 3 and Fig. S16, ESI†). In addition, the positively charged and bulkier hexamethyllysine (9) and the neutral N-acetyltrimethyllysine (10) were observed not to be substrates for TMLH in our assays (Fig. 3 and Fig. S17, S18, ESI†). These results illustrate the essential role of the free α-ammonium group, possibly via the formation of the energetically favourable salt bridge with Asp131 of TMLH. Our NMR work also showed that none of trimethyllysine analogues that have altered α-ammonium or C-1 carboxylate functionalities stimulated TMLH-mediated uncoupled turnover of 2OG to succinate.
The effect of the chain length on the substrate efficiency was also investigated. Trimethylornithine (11), a one methylene shorter analogue of trimethyllysine, underwent TMLH-catalysed C-3 hydroxylation (75% conversion) under standard assay conditions (Fig. 4 and Fig. S19–S21, ESI†). The removal of two methylene groups (as in trimethyldiaminobutyric acid, 12), however, led to no observed hydroxylation by MS and NMR (Fig. 3 and Fig. S22, ESI†). Longer trimethylhomolysine (13) was found to be a good TMLH substrate (70% conversion, Fig. S23, ESI†), but comparatively worse than trimethyllysine. Out of two potential hydroxylation sites (C-3 and C-4), the hydroxylation of 13 exclusively occurs at the C-3 site; we could not assign the stereochemistry at C-3 due to rotatable Cα–Cβ single bond (Fig. 4 and Fig. S24, S25, ESI†). Collectively, these results show that TMLH has the ability to catalyse the hydroxylation of unactivated C–H bonds in slightly shorter or longer trimethyllysine analogues, suggesting that there is some structural flexibility that enables the enzymatically productive recognition processes; similar trends were observed for BBOX-catalysed hydroxylation of γ-butyrobetaine and its simplest analogues.5,9 In contrast to the natural metabolite 3-hydroxy-L-trimethyllyine, hydroxylated trimethylornithine and trimethylhomolysine products have not yet been found in natural sources. The observation that TMLH catalyses the C-3 hydroxylation of trimethylornithine might be biologically relevant, as trimethylornithine-containing lipids have been characterised in Planctomycetes.22
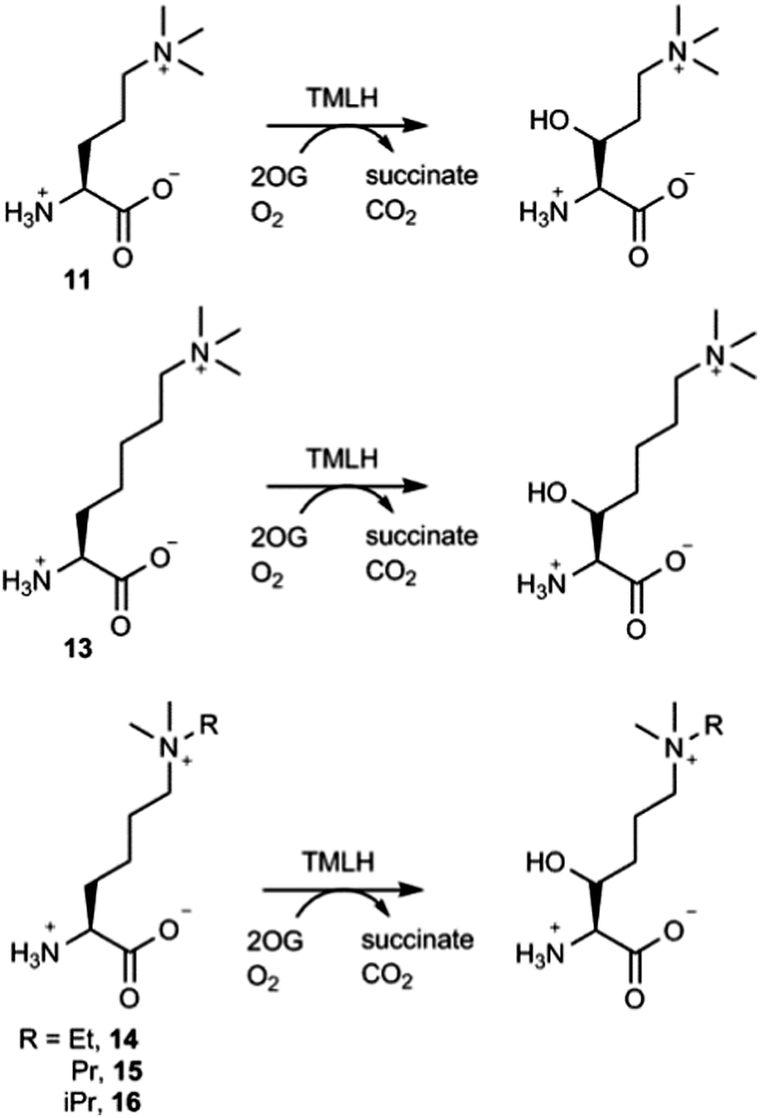 | ||
| Fig. 4 TMLH-catalysed hydroxylation of trimethyllysine analogues. | ||
Having shown that TMLH has the ability to accept one methylene shorter and longer analogues of trimethyllysine, we next investigated whether TMLH catalyses the hydroxylation of trimethyllysine analogues that bear bulkier trialkylammonium functionality. Dimethylethyllysine (14), dimethylpropyllysine (15), and dimethylisopropyllysine (16) were all efficiently hydroxylated in the presence of TMLH under standard conditions (Fig. 4 and Fig. S26–S34, ESI†). 2D NMR analyses revealed that, as in the case of trimethyllysine, these three substrates are hydroxylated at the C-3 site (none of the three hydroxylated products has been previously (bio)synthesised). NMR experiments confirmed that the enzymatic hydroxylation is well coupled to the conversion of 2OG to succinate. A sterically more demanding triethyllysine (17) was found to be a very poor substrate (traces detected) for TMLH by MS, whereas we could not detect the hydroxylated product by NMR (Fig. 3 and Fig. S35, ESI†). These results suggest that the putative aromatic cage of TMLH, comprised of Tyr217, Trp221 and Tyr234, is arranged in the way that allows binding of alkylated dimethyllysines, whereas it does not accept significantly bulkier trimethyllysine analogues. In this regard, inhibition studies on the related BBOX showed that its aromatic cage also poses constraints on substrates that bear bulkier trialkylammonium groups, but not on the alkyldimethyl derivatives of γ-butyrobetaine.12
Naturally occurring symmetric and asymmetric dimethylarginines (18 and 19) are produced by proteolytic degradation of methylated proteins.23 Dimethylated arginines possess both a fixed positive charge and hydrophobic methyl groups at the end of the side chain; these features are similar to those of trimethyllysine, although the geometry and the chain length differ. Despite the chemical similarities with trimethyllysine, dimethylated arginines were found not to be substrates for TMLH in our MS and NMR assays (Fig. 3 and Fig. S36, S37, ESI†). It is possible that the planar guanidinium group of dimethylarginine exhibits a geometry and size that prevents an efficient association with the aromatic cage of TMLH.
Targeting carnitine biosynthesis by small molecules has been recognised as a promising strategy for treatment of cardiovascular diseases. Mildronate (also known as meldonium, 20) is a clinically used γ-butyrobetaine competitive and specific BBOX inhibitor that undergoes the unusual Steven's-type rearrangement.6,24 Our MS and NMR studies revealed that mildronate is neither a substrate (at 500 μM) nor inhibitor (at 1 mM) of TMLH under standard conditions (Fig. 3 and Fig. S38, S39, ESI†). These data indicate that, out of the two Fe(II)/2OG oxygenases involved in carnitine biosynthesis, mildronate only acts as an inhibitor of BBOX (Fig. 1). In line with these results, our studies showcase that γ-butyrobetaine (21), the natural substrate for BBOX, does not undergo TMLH-catalysed hydroxylation to yield L-carnitine (Fig. 1, 3 and Fig. S40, ESI†). The work on TMLH reported here, together with recent studies on BBOX,5,9 demonstrate that TMLH and BBOX are highly specific enzymes that only catalyse hydroxylation of carnitine biosynthesis intermediates L-trimethyllysine and γ-butyrobetaine, respectively.
In conclusion, we have demonstrated that TMLH has the ability to catalyse the hydroxylation of an unactivated C–H bond in several trimethyllysine analogues. Our structure–activity relationship studies highlight the essential contributions from various structural elements of trimethyllysine that are required for efficient substrate hydroxylation. This study raises the possibility that the engineered TMLHs could be used as biocatalysts for the hydroxylation of structurally diverse unnatural amino acids. This work, furthermore, spotlights the opportunities in drug discovery by targeting carnitine biosynthesis via TMLH inhibition.
We thank Dr Andris Kazaks (Latvian Biomedical Research and Study Centre) for kindly providing the MBP-TMLH plasmid.
Notes and references
- J. Kerner and C. Hoppel, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2000, 1486, 1–17 CrossRef CAS.
- A. Steiber, J. Kerner and C. L. Hoppel, Mol. Aspects Med., 2004, 25, 455–473 CrossRef CAS PubMed.
- F. M. Vaz and R. J. A. Wanders, Biochem. J., 2002, 361, 417–429 CrossRef CAS PubMed.
- K. Strijbis, F. M. Vaz and B. Distel, IUBMB Life, 2010, 62, 357–362 CAS.
- I. K. H. Leung, T. J. Krojer, G. T. Kochan, L. Henry, F. von Delft, T. D. W. Claridge, U. Oppermann, M. A. McDonough and C. J. Schofield, Chem. Biol., 2010, 17, 1316–1324 CrossRef CAS PubMed.
- L. Henry, I. K. H. Leung, T. D. W. Claridge and C. J. Schofield, Bioorg. Med. Chem. Lett., 2012, 22, 4975–4978 CrossRef CAS PubMed.
- J. J. A. G. Kamps, A. Khan, H. Choi, R. K. Lesniak, J. Brem, A. M. Rydzik, M. A. McDonough, C. J. Schofield, T. D. W. Claridge and J. Mecinović, Chem. – Eur. J., 2016, 22, 1270–1276 CrossRef CAS PubMed.
- A. M. Rydzik, R. Chowdhury, G. T. Kochan, S. T. Williams, M. A. McDonough, A. Kawamura and C. J. Schofield, Chem. Sci., 2014, 5, 1765–1771 RSC.
- A. M. Rydzik, I. K. H. Leung, G. T. Kochan, N. D. Loik, L. Henry, M. A. McDonough, T. D. W. Claridge and C. J. Schofield, Org. Biomol. Chem., 2014, 12, 6354–6358 CAS.
- A. M. Rydzik, I. K. H. Leung, G. T. Kochan, M. A. McDonough, T. D. W. Claridge and C. J. Schofield, Angew. Chem., Int. Ed., 2014, 53, 10925–10927 CrossRef CAS PubMed.
- A. M. Rydzik, I. K. H. Leung, A. Thalhammer, G. T. Kochan, T. D. W. Claridge and C. J. Schofield, Chem. Commun., 2014, 50, 1175–1177 RSC.
- K. Tars, J. Leitans, A. Kazaks, D. Zelencova, E. Liepinsh, J. Kuka, M. Makrecka, D. Lola, V. Andrianovs, D. Gustina, S. Grinberga, E. Liepinsh, I. Kalvinsh, M. Dambrova, E. Loza and O. Pugovics, J. Med. Chem., 2014, 57, 2213–2236 CrossRef CAS PubMed.
- K. Tars, J. Rumnieks, A. Zeltins, A. Kazaks, S. Kotelovica, A. Leonciks, J. Sharipo, A. Viksna, J. Kuka, E. Liepinsh and M. Dambrova, Biochem. Biophys. Res. Commun., 2010, 398, 634–639 CrossRef CAS PubMed.
- J. D. Hulse, S. R. Ellis and L. M. Henderson, J. Biol. Chem., 1978, 253, 1654–1659 CAS.
- R. F. Novak, T. J. Swift and C. L. Hoppel, Biochem. J., 1980, 188, 521–527 CrossRef CAS PubMed.
- D. S. Sachan and C. L. Hoppel, Biochem. J., 1980, 188, 529–534 CrossRef CAS PubMed.
- F. M. Vaz, R. Ofman, K. Westinga, J. W. Back and R. J. A. Wanders, J. Biol. Chem., 2001, 276, 33512–33517 CrossRef CAS PubMed.
- A. Kazaks, M. Makrecka-Kuka, J. Kuka, T. Voronkova, I. Akopjana, S. Grinberga, O. Pugovics and K. Tars, Protein Expression Purif., 2014, 104, 1–6 CrossRef CAS PubMed.
- R. W. D. Welford, J. M. Kirkpatrick, L. A. McNeill, M. Puri, N. J. Oldham and C. J. Schofield, FEBS Lett., 2005, 579, 5170–5174 CrossRef CAS PubMed.
- L. Servillo, A. Giovane, D. Cautela, D. Castaldo and M. L. Balestrieri, PLoS One, 2014, 9, e84589 Search PubMed.
- G. Huszar, J. Mol. Biol., 1975, 94, 311–326 CrossRef CAS PubMed.
- E. K. Moore, E. C. Hopmans, W. I. C. Rijpstra, L. Villanueva, S. N. Dedysh, I. S. Kulichevskaya, H. Wienk, F. Schoutsen and J. S. Sinninghe Damsté, Appl. Environ. Microbiol., 2013, 79, 6874–6884 CrossRef CAS PubMed.
- T. Teerlink, Vasc. Med., 2005, 10, S73–S81 CrossRef PubMed.
- M. Dambrova, E. Liepinsh and I. Kalvinsh, Trends Cardiovasc. Med., 2002, 12, 275–279 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, MS experiments, NMR analyses. See DOI: 10.1039/c6cc07845a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |