
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ‘photorelease, catch and photorelease’ strategy for bioconjugation utilizing a p-hydroxyphenacyl group†
D.
Madea
,
T.
Slanina
* and
P.
Klán
 *
*
Department of Chemistry and RECETOX, Faculty of Science, Masaryk University, Kamenice 5, 625 00, Brno, Czech Republic. E-mail: slanina.tomas@seznam.cz; klan@sci.muni.cz
First published on 14th October 2016
Abstract
A bioorthogonal ‘catch and photorelease’ strategy, which combines alkyne–azide cycloaddition between p-hydroxyphenacyl azide and alkyne derivatives to form a 1,2,3-triazole adduct and subsequent photochemical release of the triazole moiety via a photo-Favorskii rearrangement, is introduced. The first step can also involve photorelease of a strained alkyne and its Cu-free click reaction with azide.
Bioconjugation is a strategy to form covalent links between two molecules of biological interest,1 which is commonly used in nucleic acid research,2 pharmaceutical chemistry3 or microbiology.4 While the first procedures used non-specific approaches, such as ionic interactions with negatively charged DNA5 or reactions of nucleophilic amines,6 carboxylates7 and thiols8 with electrophiles, modern methods rely on targeted functionalization, such as biotin–streptavidin conjugates,9 Staudinger ligation10 and click reactions.4
Many bioconjugation strategies are based on bioorthogonal reactions which allow for chemical transformations of molecules without the need to protect reactive functional groups. They are used for fluorescent labeling,11,12 cell detection13 and many other applications.14 The most common types of bioorthogonal click reactions are cycloadditions, such as Cu(I)-catalyzed alkyne–azide 1,3-dipolar cycloadditions (CuAAC) or Cu-free click reactions of azides or nitrones with strained alkynes, and inverse electron-demand Diels–Alder reaction of 1,2,4,5-tetrazines with strained alkynes or alkenes.15–17 Photochemically triggered bioorthogonal reactions, which enable spatial and temporal control over the bioconjugation and can be utilized in site-specific functionalizations, are based on photolabile diazirines,18 tetrazoles,19–21 and cyclopropenones.22
The concept of ‘catch and release’, involving selective binding of a species from the reaction media, followed by its controlled liberation, has been used, for example, in combinatorial and solid-phase synthesis of complex natural products23 and biologically active molecules,24 DNA nanomachine25 applications, supramolecular host–guest chemistry26 or resin purification methods.27 The majority of these methods are based on non-photochemical processes. Nevertheless, involvement of a photochemical step (release) in this strategy has been demonstrated on photocleavable polymers,28 dendrimers,29 and liposomes.30
A reversible version of click reactions would be highly demanded in the regulation and reversible on–off switching of biological processes, advanced surface modifications, or solid-phase synthesis. However, the chemical stability of the products of click reactions makes the reverse process very difficult. Several attempts for reversible bioorthogonal reactions have been reported. Aside from the retracted scientific report,31 only a few ‘catch and release’ strategies suitable for biological applications have been reported. Catch and release DNA decoys are rare examples of non-natural DNA probes that capture and dissociate from DNA-binding proteins upon irradiation of the 4-nitroindole linker.32 Such a strategy has been demonstrated earlier by Porter and coworkers who developed a reagent containing azido and biotin groups separated by a photocleavable linker based on a benzoin photoremovable protecting group for isolation of protein adducts of lipid-derived electrophiles using streptavidin beads.33 In addition, Popik and coworkers have recently developed a reversible derivatization technique for peptides and proteins based on the reaction of photochemically generated 2-napthoquinone-3-methides.34
Photoremovable protecting groups (PPGs) allow for the precise spatial and temporal release of biologically active molecules.35 The 4-hydroxyphenacyl photoremovable (pHP) protecting group has been shown to liberate rapidly (kobs = (7–100) × 108 s−1) and efficiently (Φ = 0.1–1.0) a wide range of leaving groups.36–40
In this work, we present a ‘catch and photorelease’ strategy which combines alkyne–azide 1,3-dipolar cycloaddition between p-hydroxyphenacyl azide 1 and alkyne 2 derivatives to form a 1,2,3-triazole linkage in adduct 3 in the first (‘click’ or ‘catch’) step, and subsequent selective irreversible photochemical release of 1-H-1,2,3-triazole 4 and 4-hydroxyphenylacetic acid 5 moieties via a photo-Favorskii rearrangement41,42 (Scheme 1). In addition, the first step can involve either a Cu(I)-catalyzed or Cu-free alkyne–azide process; a strained alkyne for the latter approach can be generated from the corresponding cyclopropenone via photochemical decarbonylation.22 We show that these strategies can facilitate efficient bioorthogonal sequential connection and splitting of two (bio)molecules.
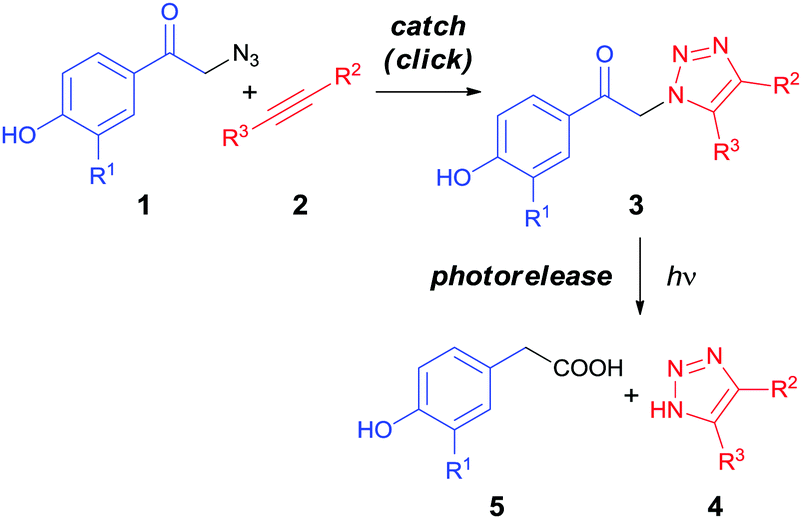 | ||
| Scheme 1 A “catch and photorelease” strategy. | ||
Two pHP triazole derivatives 3a and b were synthesized by a CuAAC click (‘catch’) approach from pHP azide (1a and b) and acetylene (2a and b) derivatives using CuSO4 and sodium ascorbate in water in 52% and 38% isolated chemical yields, respectively (Scheme 2 and ESI†).43 The O-methylglycine group in the 3-position of 1b represents a stable amide linkage between the photoactive pHP group and a peptide chain.
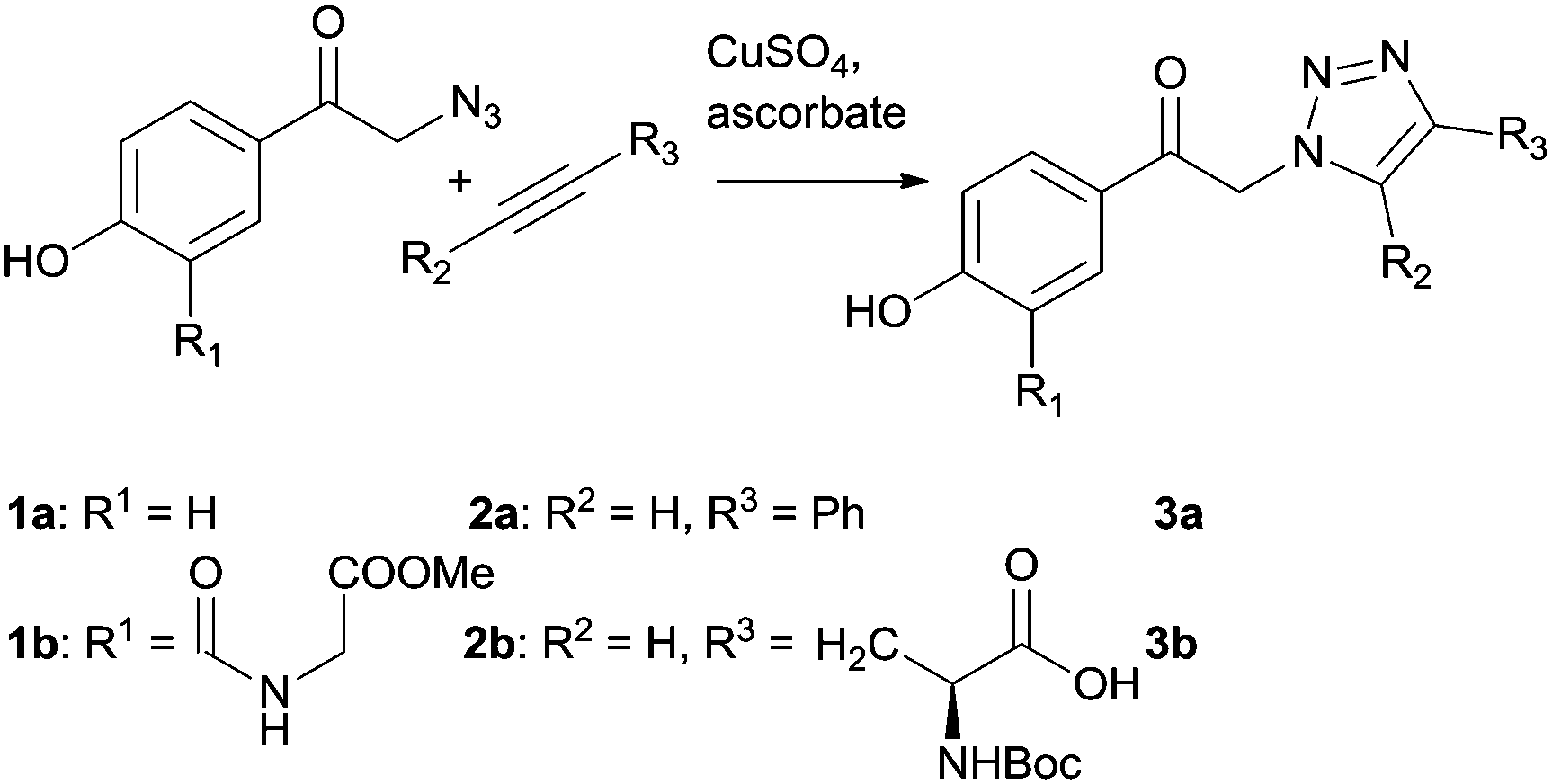 | ||
| Scheme 2 Formation of triazole derivatives in the ‘catch’ step. | ||
The second step of this strategy (Scheme 1) involves photochemical release of a triazole moiety from the pHP cage. Irradiation of 3a in CD3CN/D2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at λ = 313 nm to >99% conversion gave 4-phenyl-1-H-1,2,3-triazole (4a) as the major photoproduct in quantitative yield and 4-hydroxyphenylacetic acid (5a, 80%) and 4-hydroxybenzyl alcohol (6a, 20%) as side-products, identified by comparison of their NMR spectra with those reported before44,45 and using authentic compounds (Scheme 3; Fig. S1, ESI†). Acid 5a is a characteristic product of the photo-Favorskii rearrangement;46,47 but benzyl alcohol (such as 6a) also often accompanies the photolysis of pHP derivatives.41 The major side-photoproducts 5a and 6a are water-soluble and have negligible molar absorption coefficients at 313 nm; thus they do not act as internal optical filters.
:1, v/v) at λ = 313 nm to >99% conversion gave 4-phenyl-1-H-1,2,3-triazole (4a) as the major photoproduct in quantitative yield and 4-hydroxyphenylacetic acid (5a, 80%) and 4-hydroxybenzyl alcohol (6a, 20%) as side-products, identified by comparison of their NMR spectra with those reported before44,45 and using authentic compounds (Scheme 3; Fig. S1, ESI†). Acid 5a is a characteristic product of the photo-Favorskii rearrangement;46,47 but benzyl alcohol (such as 6a) also often accompanies the photolysis of pHP derivatives.41 The major side-photoproducts 5a and 6a are water-soluble and have negligible molar absorption coefficients at 313 nm; thus they do not act as internal optical filters.
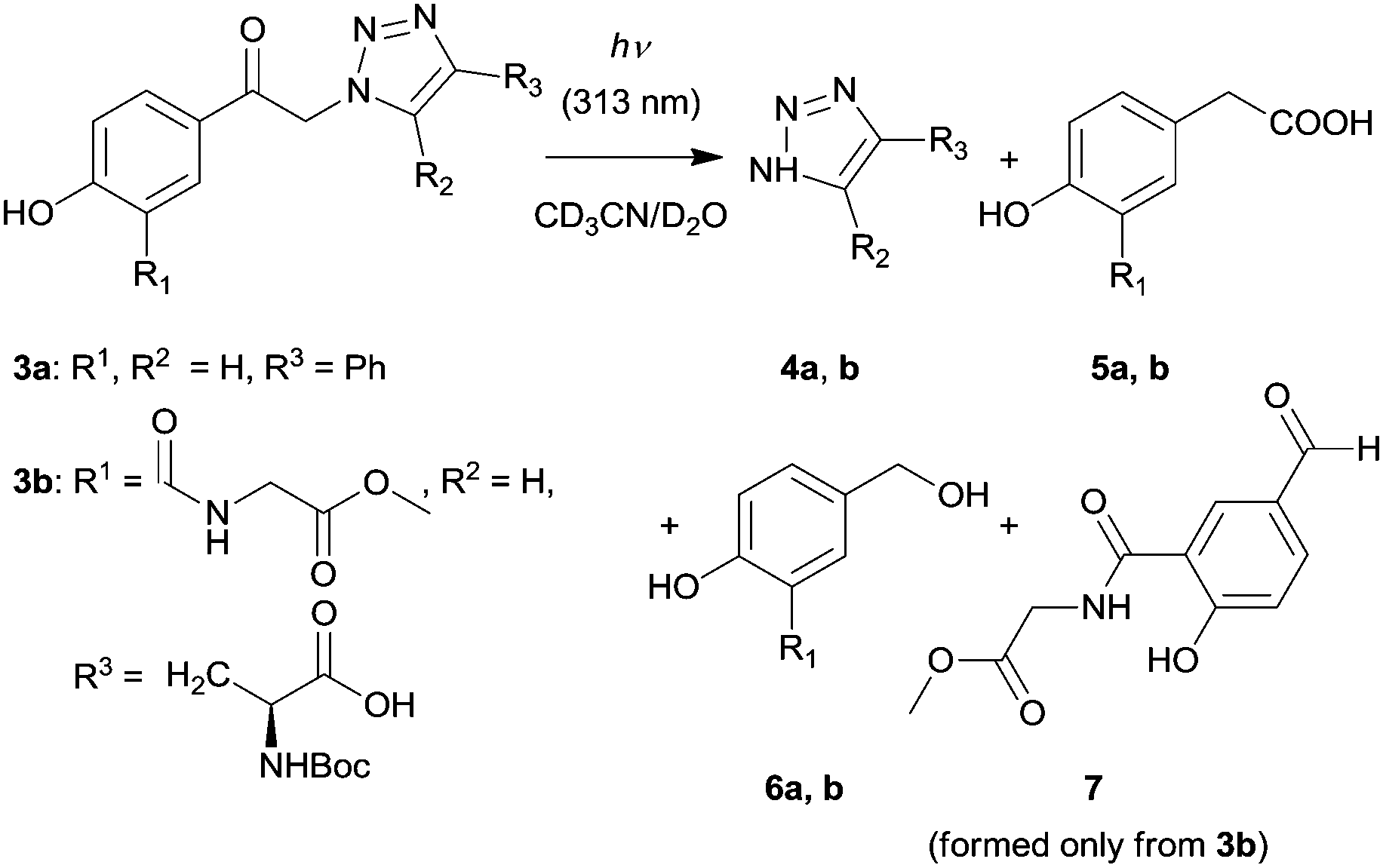 | ||
| Scheme 3 Release of 1,2,3-triazoles from the pHP PPG 3. | ||
The necessary role of water in the pHP photodegradation mechanism36,41,48 predetermines this transformation for biocompatible aqueous solutions. Acetonitrile in CD3CN/D2O (1:1, v/v) mixtures was used to increase the solubility of the systems, and it resulted in the formation of considerable amounts of the second byproduct 6, which would not be formed in pure water.41
Compound 3b represents a moiety that can interlink the photoactive pHP group with another molecule via a peptide chain. Upon irradiation, this peptide tether, which could bear another molecule or a functional group of interest, remains on the photoproduct(s) formed from the pHP chromophore. The m-substitution of the pHP group by an amido group was chosen because, unlike substituents in the p- and o-positions,47m-substituents do not significantly affect the photophysical properties of pHP.49 Exhaustive irradiation of 3b in CD3CN/D2O (1:1, v/v) at λ = 313 nm led to the formation of free triazole 4b in quantitative yield and compounds 5b (≈25%) and 6b (≈20%) as the major photo-Favorskii rearrangement products (Fig. S2, ESI†). In addition, p-hydroxybenzaldehyde derivative 7 was identified as another side product (Fig. S41, ESI†). Because this compound appeared only after prolonged irradiation, we assume that it is a secondary product formed by 6b oxidation in the presence of oxygen in air.
It has been shown that poor leaving groups such as alcohols or amines possessing pKa values above ≈11 cannot be efficiently released from the pHP group.35,38 For example, phenolate (pKa ≈ 10) or p-cyanophenolate (pKa ≈ 7.2) is released with a quantum yield of 0.04 and 0.11, respectively.38 Therefore, because the pKa value of 1H-1,2,3-triazole is 9.4,50 a rather low release quantum yield from its pHP derivative 3a, Φdis = 9 × 10−4 (Table 1), was anticipated.
| pHP derivative | Φ dis/% |
|---|---|
| a Irradiated at λ = (313 ± 1.5) nm (3a, 3d) or (320 ± 10) nm (3c, 3e); 4-hydroxyphenacyl fluoride39 was used as an actinometer. b In a mixture of acetonitrile (10%) and acetate buffer (pH = 5.0). c In a mixture of acetonitrile (20%) and acetate buffer (pH = 5.0). | |
| 3a | 0.09 ± 0.02 |
| 3c | 2.9 ± 0.4 |
| 3d | 83 ± 4 |
| 3e | 0.39 ± 0.05 |
To increase the quantum yield of photorelease, two modified click adducts 3c and 3d were synthesized using a SPAAC (strain-promoted azide–alkyne cycloaddition) and a CuAAC procedure, respectively (Scheme 4 and ESI†). Compound 3c possesses two electron withdrawing fluorine atoms in the near proximity of the triazole core, which lower its pKa value and make it a better leaving group. Indeed, the Φdis value of 3c was enhanced by a factor of ≈30 compared to that of 3a (Table 1). Because the triazoles 3a, 3b and 3d were synthesized by CuAAC in the presence of CuI ions, which are toxic due to the generation of reactive oxygen species,51 the non-catalyzed click (SPAAC) synthesis of the DIFO-derivative523c represents a more biocompatible method.
 | ||
| Scheme 4 Photochemistry of 3c and d. | ||
Because controlling the pKa values of triazoles is structurally limited, we adopted a different approach. The poor leaving ability of alcohols, phenols or amines can be enhanced by connecting them to the chromophore through a carbonate53,54 or carbamate55 linker. Here we modified the triazole moiety through a methylcarbonyloxy linker in 3d, where the leaving group is a carboxylate; thus the pKa value of its conjugate acid is approximately 4. The Φdis value of 0.83, which is 3 orders of magnitude higher than that of 3a, is within the values found for other pHP esters (Φdis = 0.4–0.94)38 and represents a significant improvement for any future applications. A measure of the photorelease (the uncaging cross section), the product of the quantum yield and the decadic molar absorption coefficient, Φε(λirr),35 for 3d at 313 nm (ε313 = 2.73 × 103 M−1 cm−1) is remarkably large (2.27 × 103 M−1 cm−1).
We further modified the first step of the ‘catch and photorelease’ strategy by introducing an angle-strained cycloalkyne which allows for efficient Cu-free click chemistry via facile 1,3-dipolar cycloaddition (SPAAC).17,56,57 Such a reactive alkyne can be efficiently photochemically produced in situ from its cyclopropenone precursor, which is an elegant solution to spatial and temporal control of the release.22,58 In this work, cyclopropenone 8 was synthesized by a 2-step procedure in 27% overall yield (Scheme 5 and ESI†) using a LiNK metalation-coupling procedure.59 This is a significant improvement when compared to a previously reported 4-step synthesis60 of cyclopropenone 8. It has been shown that this compound photodecarbonylates with a quantum yield of 0.33 to give a strained alkyne 9, which can undergo non-photochemical SPAAC reaction with azides.22 Thus, a solution of 8 and pHP azide (1a; 1 equiv., c ≈ 5 mg mL−1) in acetonitrile was irradiated at a wavelength of 350 nm, at which only 8 absorbs (Fig. S6, ESI†). The generated alkyne 9 underwent in situ non-photochemical SPAAC reaction with 1a to form 3e (quantitative yield determined by 1H NMR; 76% isolated yield). Upon irradiation at 313 nm, this pHP derivative was converted quantitatively into triazole 4e and p-hydroxybenzyl derivatives 5a and 6a, which could bear a peptide tether to connect the released moiety with a biomolecule, via the photo-Favorskii rearrangement. The disappearance quantum yield of 3e was higher than that of 3a because 4e is a better leaving group compared to 4a.
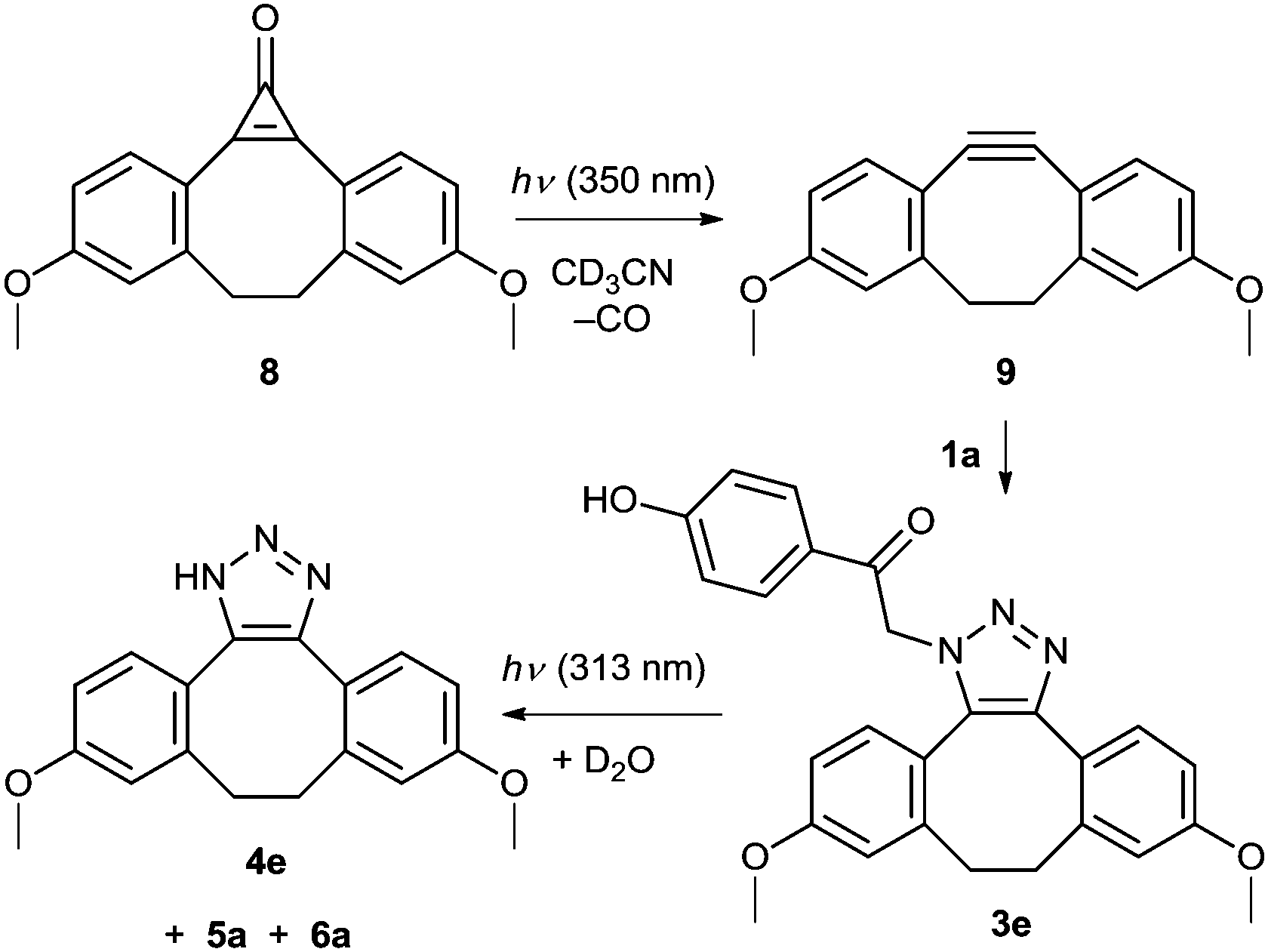 | ||
| Scheme 5 ‘Release, catch and release’ strategy. | ||
Our ‘photorelease, catch and photorelease’ strategy is a one-pot multistep procedure, which is solely controlled by light, and does not require any other reagents besides water. In addition, both photochemical steps are orthogonal. The pHP group does not absorb at a wavelength of 350 nm (Fig. S39, ESI†) used for generation of strained alkyne 9; thus cycloadduct 3e remains unreacted in the solution during the first irradiation step. The pHP chromophore transformations rely on irradiation at wavelengths in the UV region (<≈320 nm), which may limit their bioapplications. However, 2-photon excitation could overcome this issue, as was recently successfully demonstrated by Givens and coworkers on pHP-caged diethyl phosphate and ATP using a 550 nm laser.40
In conclusion, we synthesized a series of substituted pHP azides and alkynes which can be interlinked using an alkyne–azide 1,3-dipolar cycloaddition protocol and subsequently be released upon irradiation with UV-light via a photo-Favorskii rearrangement. We show that the reactions can be accomplished in aqueous media favorable for biological applications and that the linker can be used to connect/disconnect two (bio)molecules. A rational design of the leaving group (triazole) structure led to the development of a linker that cleaves upon irradiation with near-unity quantum yield. The second generation of this strategy encompasses chromatically orthogonal photoproduction of a strained alkyne which allows for Cu-free click chemistry. We propose that the systems could be used in bioconjugation or drug-delivery applications.
Support for this work was provided by the Czech Science Foundation (GA13-25775S) and by the Czech Ministry of Education, Youth and Sports (LO1214 and LM2015051).
References
- J. Kalia and R. T. Raines, Curr. Org. Chem., 2010, 14, 138–147 CrossRef CAS PubMed.
- R. Sunasee and R. Narain, Chemistry of Bioconjugates, John Wiley & Sons, Inc., 2014, pp. 1–75 Search PubMed.
- F. M. Veronese and M. Morpurgo, Farmaco, 1999, 54, 497–516 CrossRef CAS PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- B. S. Gaylord, A. J. Heeger and G. C. Bazan, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10954–10957 CrossRef CAS PubMed.
- A. Varadarajan, R. M. Sharkey, D. M. Goldenberg and M. F. Hawthorne, Bioconjugate Chem., 1991, 2, 102–110 CrossRef CAS PubMed.
- K. A. Totaro, X. Liao, K. Bhattacharya, J. I. Finneman, J. B. Sperry, M. A. Massa, J. Thorn, S. V. Ho and B. L. Pentelute, Bioconjugate Chem., 2016, 27, 994–1004 CrossRef CAS PubMed.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed.
- M. Howarth, D. J. F. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS PubMed.
- J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873–877 CrossRef CAS PubMed.
- J. R. G. Navarro, G. Conzatti, Y. Yu, A. B. Fall, R. Mathew, M. Edén and L. Bergström, Biomacromolecules, 2015, 16, 1293–1300 CrossRef CAS PubMed.
- J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CAS.
- J. B. Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee and R. Weissleder, Nat. Nanotechnol., 2010, 5, 660–665 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- A. L. MacKinnon and J. Taunton, Current Protocols in Chemical Biology, John Wiley & Sons, Inc., 2009 Search PubMed.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., Int. Ed., 2008, 47, 2832–2835 CrossRef CAS PubMed.
- W. Song, Y. Wang, Z. Yu, C. I. R. Vera, J. Qu and Q. Lin, ACS Chem. Biol., 2010, 5, 875–885 CrossRef CAS PubMed.
- P. An, Z. Yu and Q. Lin, Chem. Commun., 2013, 49, 9920 RSC.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 2566 RSC.
- M. C. Pirrung, L. N. Tumey, A. L. McClerren and C. R. H. Raetz, J. Am. Chem. Soc., 2003, 125, 1575–1586 CrossRef CAS PubMed.
- E. M. McConnell, R. Bolzon, P. Mezin, G. Frahm, M. Johnston and M. C. DeRosa, Bioconjugate Chem., 2016, 27, 1493–1499 CrossRef CAS PubMed.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H.-F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976–12979 CrossRef CAS PubMed.
- T. Fujii, N. Kato, I. Iwakura, Y. Manabe and M. Ueda, Chem. Lett., 2008, 37, 52–53 CrossRef CAS.
- M. D. Green, A. A. Foster, C. T. Greco, R. Roy, R. M. Lehr, I. I. I. T. H. Epps and M. O. Sullivan, Polym. Chem., 2014, 5, 5535–5541 RSC.
- A. Sharma, B. König and N. Jayaraman, New J. Chem., 2014, 38, 3358 RSC.
- S. J. Leung and M. Romanowski, Adv. Mater., 2012, 24, 6380–6383 CrossRef CAS PubMed.
- The report ‘Unclicking the click: mechanically facilitated 1,3-dipolar cycloreversions’ by Bielawski and coworkers (Science, 2011, 333, 1606–1609) has been retracted by Science.
- N. B. Struntz and D. A. Harki, ACS Chem. Biol., 2016, 11, 1631–1638 CrossRef CAS PubMed.
- H. Y. H. Kim, K. A. Tallman, D. C. Liebler and N. A. Porter, Mol. Cell. Proteomics, 2009, 8, 2080–2089 CAS.
- S. Arumugam, J. Guo, N. E. Mbua, F. Friscourt, N. Lin, E. Nekongo, G.-J. Boons and V. V. Popik, Chem. Sci., 2014, 5, 1591 RSC.
- P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef CAS PubMed.
- R. S. Givens, A. Jung, C.-H. Park, J. Weber and W. Bartlett, J. Am. Chem. Soc., 1997, 119, 8369–8370 CrossRef CAS.
- R. S. Givens, J. F. W. Weber, P. G. Conrad, G. Orosz, S. L. Donahue and S. A. Thayer, J. Am. Chem. Soc., 2000, 122, 2687–2697 CrossRef CAS.
- R. S. Givens, M. Rubina and J. Wirz, Photochem. Photobiol. Sci., 2012, 11, 472 CAS.
- T. Slanina, P. Sebej, A. Heckel, R. S. Givens and P. Klan, Org. Lett., 2015, 17, 4814–4817 CrossRef CAS PubMed.
- A. L. Houk, R. S. Givens and C. G. Elles, J. Phys. Chem. B, 2016, 120, 3178–3186 CrossRef CAS PubMed.
- R. S. Givens, D. Heger, B. Hellrung, Y. Kamdzhilov, M. Mac, P. G. Conrad, E. Cope, J. I. Lee, J. F. Mata-Segreda, R. L. Schowen and J. Wirz, J. Am. Chem. Soc., 2008, 130, 3307–3309 CrossRef CAS PubMed.
- R. S. Givens, M. Rubina and K. F. Stensrud, J. Org. Chem., 2013, 78, 1709–1717 CrossRef CAS PubMed.
- The yields were diminished upon purification; the reaction conversion was complete and no other products were found.
- Y. Y. Kuang, K. Baakrishnan, V. Gandhi and X. H. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS PubMed.
- O. Cloarec, A. Campbell, L. H. Tseng, U. Braumann, M. Spraul, G. Scarfe, R. Weaver and J. K. Nicholson, Anal. Chem., 2007, 79, 3304–3311 CrossRef CAS PubMed.
- K. Stensrud, J. Noh, K. Kandler, J. Wirz, D. Heger and R. S. Givens, J. Org. Chem., 2009, 74, 5219–5227 CrossRef CAS PubMed.
- P. Sebej, B. H. Lim, B. S. Park, R. S. Givens and P. Klan, Org. Lett., 2011, 13, 644–647 CrossRef CAS PubMed.
- T. Solomek, D. Heger, B. P. Ngoy, R. S. Givens and P. Klan, J. Am. Chem. Soc., 2013, 135, 15209–15215 CrossRef CAS PubMed.
- R. S. Givens, K. Stensrud, P. G. Conrad, A. L. Yousef, C. Perera, S. N. Senadheera, D. Heger and J. Wirz, Can. J. Chem., 2011, 89, 364–384 CrossRef CAS PubMed.
- A. C. Tomé, in Science of Synthesis, 13: Hetarenes and Related Ring Systems, ed. R. C. Storr and T. L. Gilchrist, Georg Thieme Verlag, Stuttgart, 2004, vol. 13, pp. 415–602 Search PubMed.
- V. Hong, N. F. Steinmetz, M. Manchester and M. G. Finn, Bioconjugate Chem., 2010, 21, 1912–1916 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- A. Banerjee, K. Lee and D. E. Falvey, Tetrahedron, 1999, 55, 12699–12710 CrossRef CAS.
- J. Literak, J. Wirz and P. Klan, Photochem. Photobiol. Sci., 2005, 4, 43–46 CAS.
- L. Kammari, L. Plistil, J. Wirz and P. Klan, Photochem. Photobiol. Sci., 2007, 6, 50–56 CAS.
- M. F. Debets, J. C. M. van Hest and F. P. J. T. Rutjes, Org. Biomol. Chem., 2013, 11, 6439 CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- A. Poloukhtine and V. V. Popik, J. Phys. Chem. A, 2006, 110, 1749–1757 CrossRef CAS PubMed.
- M. Blangetti, P. Fleming and D. F. O'Shea, J. Org. Chem., 2012, 77, 2870–2877 CrossRef CAS PubMed.
- F. R. Fischer and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7257–7260 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; spectroscopy; synthesis and compound characterization; and NMR and HRMS and optical spectra. See DOI: 10.1039/c6cc07496k |
| This journal is © The Royal Society of Chemistry 2016 |