
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon-rich “Click” 1,2,3-triazoles: hexaphenylbenzene and hexa-peri-hexabenzocoronene-based ligands for Suzuki–Miyaura catalysts†
James R.
Wright
ab,
James D.
Crowley
 *a and
Nigel T.
Lucas
*ab
*a and
Nigel T.
Lucas
*ab
aDepartment of Chemistry, University of Otago, P. O. Box 56, Dunedin, New Zealand. E-mail: nlucas@chemistry.otago.ac.nz; Fax: +64 3 479 7906; Tel: +64 3 479 5377
bMacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
First published on 10th October 2016
Abstract
Hexaphenylbenzene (HPB) and hexa-peri-hexabenzocoronene-(HBC) functionalised 1,2,3-triazoles have been synthesised using an optimised copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) reaction. The coordination chemistry of these ligands was explored through the synthesis of the respective palladium(II) complexes and their activity as catalysts in the Suzuki–Miyaura reaction assessed.
Polycyclic aromatic hydrocarbons (PAHs) are one of the most widely studied families of organic compounds, and present many interesting synthetic challenges. Breakthroughs in the area of PAH synthesis have led to a plethora of new materials with unique photophysical and electronic properties, and some of these materials have been incorporated into electronic devices.1
Hexa-peri-hexabenzocoronene (HBC) is a graphitic, all benzenoid PAH that has remarkable thermal and chemical stability.2 Traditionally the synthesis of HBC has been hampered by harsh conditions and as such could only be isolated in trace quantities.2,3 However, developments by the Müllen group have made HBCs more readily accessible, through the intramolecular oxidative aromatic coupling of hexaphenylbenzenes (HPBs).4 These methods now allow for the synthesis of HPBs and HBCs with a range of different substitution patterns and functionalities,5 allowing these units to be investigated in a variety of applications.6 Additionally, it has been shown that HBCs undergo facile self-assembly due to robust π⋯π interactions.7 Hexa(alkyl)-HBCs, in particular, self-assemble into highly ordered, columnar mesophases that have high charge-carrier mobility and as such act as molecular semiconductors.8 HPBs have also been shown to self-assemble9 and exploited for the fabrication of OLEDs.10 In addition to organic optoelectronic materials based on HBC and HPB, there have been a few recent reports on the use of HBC and HPB ligands for the development of hybrid metal–organic materials with new optical properties.11–13 The photophysics of HBC-acetylide Pt(II) complexes and systems that incorporate nitrogen into the HBC framework have been examined by Draper and co-workers.14 We have recently reported an HBC(tBu)5-bpy (bpy = 2,2′-bipyridine) ligand and its corresponding Re(I) chlorotricarbonyl complex; this complex exhibits a long-lived charge transfer excited state.11 Likewise, HPB ligands have been used to develop photophysically active metal complexes.15
Despite the upsurge in interest in the development of HBC ligand systems, there are few general methods that enable the generation of soluble functionalised HBC architectures. One potential solution to the solubility and reactivity problems is through the use of the copper(I) catalysed azide–alkyne cycloaddition (CuAAC) “click” reaction.16 This is an efficient and functional group tolerant way of generating 1,4-disubstituted-1H-1,2,3-triazoles (tzls), from alkynes and azides, and the triazole units have been shown to be effective N-donor ligands.17 Additionally, the solubility of HPBs and HBCs can be tuned at a late stage of synthesis from alkyne-functionalised precursors, which in turn may give rise to novel liquid-crystalline materials. Recently we have demonstrated that triazoles act as electronic insulators18 and therefore could be used in the modulation of electronics in donor–acceptor systems. Herein we report the synthesis of directly appended HPB and HBC triazoles for the first time. The triazoles are employed as ligands to form [PdCl2L2] complexes which were subsequently investigated as precatalysts in the Suzuki–Miyaura reaction.
Penta(tert-butyl)-substituted hexaphenylbenzene (HPB)- and hexa-peri-hexabenzocoronene (HBC)-acetylenes were synthesised using established coupling conditions (ESI†).11,13 Due to the limited solubility of these species in conventional polar solvents, an optimised synthesis of the triazoles was needed (ESI†). The triazoles 1a and 1b were optimally synthesised in dichloromethane using benzyl azide, [Cu(CH3CN)4]BF4/tris(benzyltriazolylmethyl)amine (TBTA), and triethylamine at room temperature in good yields (1a: 87%, 1b: 69%; Fig. 1). 1H and 13C NMR spectroscopy supported the formation of 1a and 1b, showing signals consistent with the presence of the triazole ring, benzylic methylenes, as well as the absence of the alkynyl proton signals of the precursors. Additionally, significant downfield shifts are observed within the polyaromatic cores for the nuclei proximal to the electron-poor triazole ring (ESI†). MALDI-TOF-MS data show signals corresponding to the molecular ion as well as the loss of dinitrogen ([M–N2]+). Triazole 1b was also synthesised from 1a by oxidative aromatic cyclodehydrogenation in quantitative yield; the 1H NMR spectrum and MALDI-TOF-MS data are identical to those of 1b formed via the CuAAC method (ESI†).
 | ||
| Fig. 1 (a) Synthesis of HPB and HBC triazoles and [PdCl2L2] complexes. Conditions: (i) R-CCH (1 equiv.), benzyl azide (1.2 equiv.), NEt3 (1.2 equiv.), [Cu(MeCN)4]BF4 (0.10 equiv.), TBTA (0.10 equiv.), CH2Cl2, rt, 18 h; (ii) [PdCl2(MeCN)2] (0.5 equiv.), acetone (2a) or CHCl3 (2b), rt, 1 h; (b) synthesis of model complex. Conditions: (iii) [PdCl2(MeCN)2] (0.5 equiv.), acetone, rt, 1 h. | ||
With 1a and 1b in hand, we examined the ability of the new triazoles to act as ligands. The [PdCl2(1a–b)2] (2a–b) complexes were formed by reaction of the ligands with [PdCl2(MeCN)2] (0.5 equiv.) in acetone (2a) or chloroform (2b) solution and were isolated as yellow solids (2a: 63%, 2b: 86%; Fig. 1). Elemental analyses were consistent with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand/metal ratio. 1H and 13C NMR spectra of 2a and 2b show similar profiles to their ligand precursors. The triazole ring protons of each complex shift upfield upon complexation, presumably due to increased shielding by the benzyl ring. Conversely, the benzylic methylene protons shift downfield because of metal-induced deshielding. With respect to the HBC proton signals of 2b, the protons proximal to the triazole shift downfield upon complexation and broaden significantly due to slow rotation in solution; the remaining proton signals shift upfield consistent with the mutual face-to-face overlap of the two HBC units. MALDI-TOF-MS spectra of the complexes show weak signals associated with the complex (2a: [M–2Cl–H]+; 2b: [M]+) and stronger signals associated with the fragmentation of the respective ligands.
:1 ligand/metal ratio. 1H and 13C NMR spectra of 2a and 2b show similar profiles to their ligand precursors. The triazole ring protons of each complex shift upfield upon complexation, presumably due to increased shielding by the benzyl ring. Conversely, the benzylic methylene protons shift downfield because of metal-induced deshielding. With respect to the HBC proton signals of 2b, the protons proximal to the triazole shift downfield upon complexation and broaden significantly due to slow rotation in solution; the remaining proton signals shift upfield consistent with the mutual face-to-face overlap of the two HBC units. MALDI-TOF-MS spectra of the complexes show weak signals associated with the complex (2a: [M–2Cl–H]+; 2b: [M]+) and stronger signals associated with the fragmentation of the respective ligands.
X-ray quality crystals of both complexes were obtained by either a layered diffusion of methanol into a solution (2a) or the slow evaporation of a CH2Cl2 solution (2b). Both complexes show the expected connectivity of trans-N-donor complexes and a geometry about the palladium(II) centres typical of palladium-triazole complexes.19 The Pd–N(tzl) distances range from 1.993(4)–2.029(5) Å and sit within the lower range of reported Pd–N(tzl) distances (1.983–2.166 Å). Although the N–Pd–N bond angle of 2b at 177.9(2)° is close to the mean of reported complexes trans-bis(tzl) palladium complexes (176.4°), that of 2a (171.3(2)°) is rather acute (Fig. 2), presumably because of increased ligand size and resultant intramolecular interactions. The complexes are isostructural in the sense that the polyaromatic cores of each ligand in the respective complexes are intramolecularly stacked and eclipsed. The stacking in 2a appears to be stabilised by multiple edge-to-face C–H⋯π interactions. Each molecule of 2a co-crystallised with seven CH2Cl2 solvate molecules, five of which are situated within clefts of HPB moieties and stabilised by two-fold C–H⋯π interactions (ESI†). For 2b, however, the HBC moieties are stabilised through offset face-to-face π-interactions facilitated by the π-electron-rich nature of the HBC unit (central ring inter-centroid distance = 3.486 Å), thus supporting the NMR evidence of stacking in solution. The chloro ligands of 2b are positioned within bay positions of the HBC units and are stabilised by C–H⋯Cl interactions (ESI†).
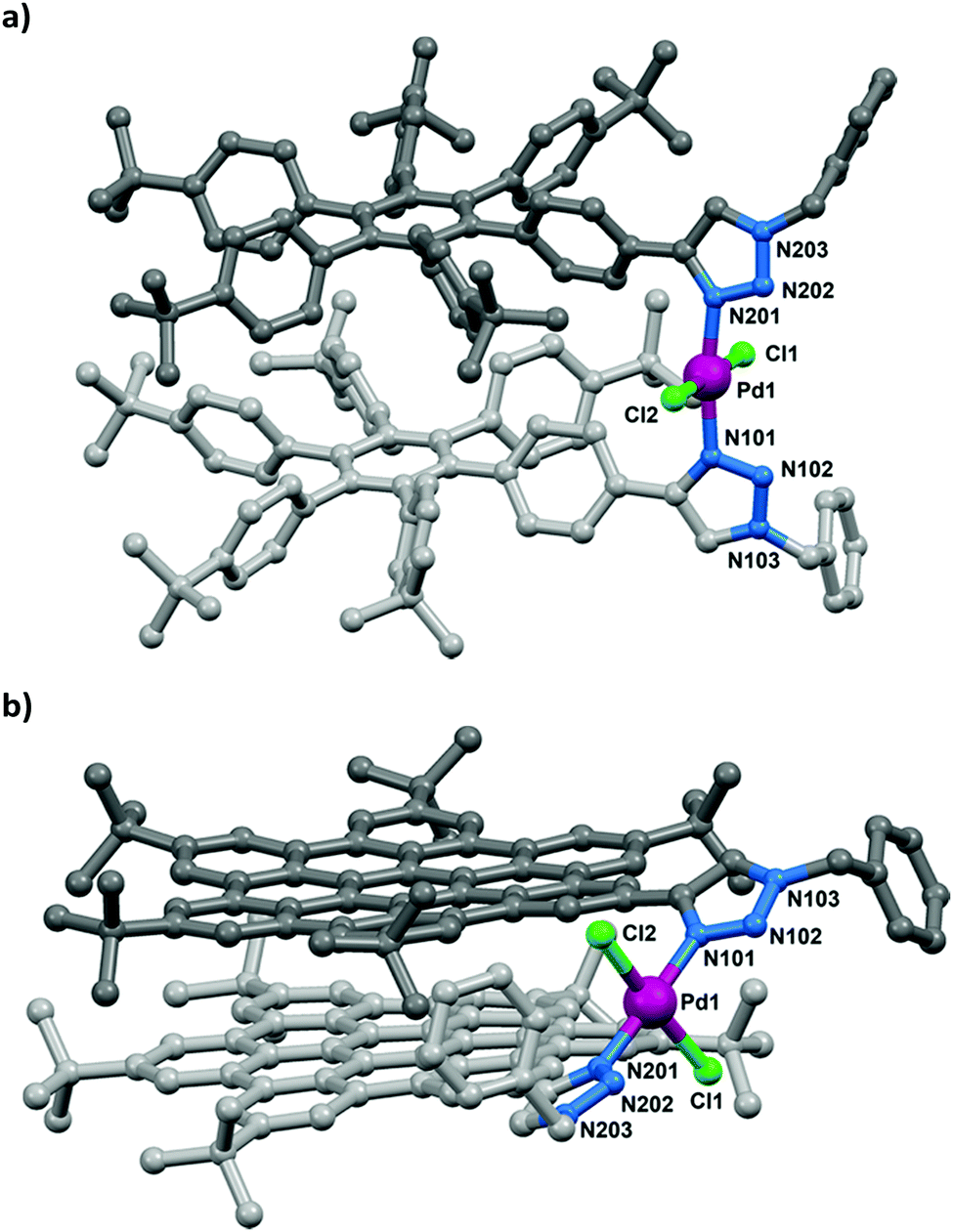 | ||
| Fig. 2 Ball-and-stick representations of the X-ray crystal structures of (a) 2a·7CH2Cl2 and (b) 2b. Hydrogen atoms and solvate molecules have been omitted for clarity. Key bond lengths (Å) and angles (°) for 2a: Pd1–N101 = 2.029(5) Å, Pd1–N201 = 1.994(5) Å, Pd1–Cl1 = 2.297(2) Å, Pd1–Cl2 2.279(2) Å, N101–Pd1–N201 = 171.3(4)°, Cl1–Pd1–Cl2 = 178.95(7)°. 2b: Pd1–N101 = 1.993(4) Å, Pd1–N201 = 1.996(4) Å, Pd1–Cl1 = 2.320(2) Å, Pd1–Cl2 = 2.316(2) Å, N101–Pd1–N201 = 177.9(2)°, Cl1–Pd1–Cl2 = 174.18(5)°. | ||
As palladium complexes are used as catalysts in a wide range of reactions,20 we investigated the catalytic potential of 2a and 2b. Triazoles feature prominently as supporting ligands for a variety of metal-based catalysts.21,22 Although triazoles often comprise part of bidentate ligands, few catalytically-active complexes employ triazoles as monodentate ligands.22 Palladium(II) triazole dendrimers have shown good catalytic activity and high turnover in the Suzuki–Miyaura reaction;23 accordingly we examined 2a and 2b, along with phenyl-substituted model 2c19 (Fig. 1b) and commercially available [PdCl2(PPh3)2] (3), as catalysts in the Suzuki–Miyaura cross-coupling reaction (Table 1 and ESI†). Upon addition of model 2c (5 mol%) to phenylboronic acid and 4-bromoacetophenone at 80 °C for 4 hours, an 84% yield of 4-acetylbiphenyl was obtained confirming this bis(monodentate)triazole complex is catalytically active. Repeating the reaction under identical conditions with 2a and 2b resulted in slightly improved yields of 94% and 92%, respectively. Commercially available [PdCl2(PPh3)2] (3) afforded the product in 90% yield.


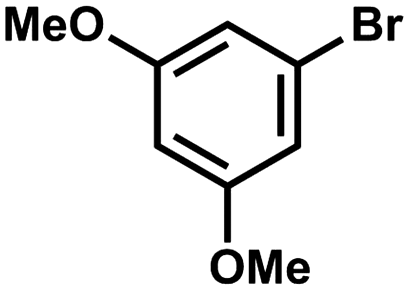

To further probe whether the incorporation of polyaromatic groups in ligands 1a and 1b has an effect on the catalytic activity, the reaction was repeated with the less activated 3,5-dimethoxybromobenzene substrate. Utilising the same reaction conditions (but a longer reaction time) complexes 2a and 2b, along with model 2c and 3, were all found to be active. Surprisingly, employing 2a and 2c resulted in a significantly lower yield of the biphenyl product (30–39%), while HBC-containing 2b and 3 produced good yields of 69% and 76%, respectively. The reactions with 2b and 3 were also monitored via1H NMR spectroscopy and revealed a small difference in activity. It is noteworthy that for this system, conversion ceased after ca. 5 hours, indicative of catalyst decomposition for both species (ESI†). This outcome was supported by an increase in the intensity of free ligand signals in the 1H NMR spectra. To test the limits of the triazole catalysts 2a–c, in comparison with 3, the haloarene substrate 4′-chloroacetophenone was reacted under similar conditions. Conversions dropped markedly for all catalysts with low yields of 4-acetylbiphenyl (0–14%). The HBC-based complex 2b was catalytically active while the HPB analogue 2a was inactive; remarkably, the Ph-bearing model 2c showed comparable activity to 2b, albeit low in both cases (13–14%). Subsequent monitoring by 1H NMR spectroscopy indicated rapid catalyst decomposition (ca. 2 hours) for both 2b and 3 (ESI†), rendering the longer reaction times unnecessary.
The activity of Werner complexes 2a–c (cf. phosphine complex 3) is intriguing because it is expected that N-donor ligands have poor donor properties. To investigate this, the donor strengths of all ligands were assessed using a 13C NMR technique developed by Huynh and coworkers which involved the synthesis of [PdBr2(iPr2-bimy)(L)] complexes (4a–c,5; ESI†).24 The data revealed the following: The phosphine ligand has a greater donor strength than all triazole-based ligands, and the triazole ligand with the more electron-rich HBC core (2b) has a greater donor strength than the phenyl-appended model analogue 2c. Thus, the catalytic activity cannot be explained by electronic arguments alone.
There are several postulates to explain the activity of 2b, each involving the influence of the HBC core; the interaction of the two HBC units within the complex transforms the triazoles into one pseudo-bidentate ligand and/or that the HBC cores help to stabilise catalytically-active palladium nanoparticles that form during the reaction. In the former, intramolecular π-stacking may confer greater stability, however, molecular modelling (ESI†) suggests that while the cis-arrangement required for reductive elimination is accessible, it is higher in energy than the trans-isomer observed crystallographically. More likely here, is that nanoparticles are playing a significant role in the catalysis. A repeat catalytic run under the conditions of entry 6 (Table 1), but in the presence of a mercury drop, led to a fall in yield from 69% to 22%. It is generally thought that N-donor ligands do not effectively stabilise monoatomic Pd(0) resulting in Pd nanoparticle formation,25 and polyaromatic groups can help slow aggregation of the particles to Pd black precipitates.26 Further work will concentrate on determining the exact nature of the catalytically-active species and the strength of the HBC–HBC interaction in solution.
1,4-Disubstituted-1,2,3-triazoles functionalised with carbon-rich hexaphenylbenzen and hexa-peri-hexabenzocoronene substituents have been synthesised, using the CuAAC methodology. Furthermore, the triazole group was found to be tolerant of the oxidative dehydrogenation conditions used for converting HPBs to HBCs. These HPB and HBC compounds have been used as ligands to generate trans-[PdCl2L2] complexes which show π-interactions between each ligand in the solid state, and in solution for complex 2b. The triazole-palladium complexes are catalytically active in the Suzuki–Miyaura cross-coupling reaction, with the HBC-bearing 2b appearing to be the best across the substrates surveyed. Both catalysts have limited lifetimes (2–5 hours) as made evident through 1H NMR studies.
The versatile and functional group tolerant CuAAC “click” approach developed herein should enable the synthesis of a wide range of HBC and HPB 1,2,3-triazole ligands which could be exploited to generate new catalysts and hybrid metal–organic materials with tunable optical properties.
We thank the University of Otago for support including a PhD scholarship for J. R. W. and a Near Miss Research Grant.
Notes and references
- D. Braga and G. Horowitz, Adv. Mater., 2009, 21, 1473 CrossRef CAS; H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158 CrossRef PubMed; Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372 CrossRef PubMed; W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC; S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863 CrossRef PubMed; K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, Macromolecules, 2013, 46, 8059 CrossRef; X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef; A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef PubMed.
- E. Clar, C. T. Ironside and M. Zander, J. Chem. Soc., 1959, 142 RSC; A. Halleux, R. H. Martin and G. S. D. King, Helv. Chim. Acta, 1958, 41, 1177 CrossRef CAS.
- W. Hendel, Z. H. Khan and W. Schmidt, Tetrahedron, 1986, 42, 1127 CrossRef CAS.
- A. Stabel, P. Herwig, K. Müllen and J. P. Rabe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1609 CrossRef CAS; V. S. Iyer, M. Wehmeier, J. D. Brand, M. A. Keegstra and K. Müllen, Angew. Chem., Int. Ed. Engl., 1997, 36, 1604 CrossRef.
- R. Yamaguchi, S. Ito, B. S. Lee, S. Hiroto, D. Kim and H. Shinokubo, Chem. – Asian J., 2013, 8, 178 CrossRef CAS PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481 CrossRef CAS PubMed; Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051 CrossRef PubMed.
- W. Pisula, X. Feng and K. Müllen, Chem. Mater., 2011, 23, 554 CrossRef CAS.
- S. Hiraoka, K. Harano, T. Nakamura, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7006 CrossRef CAS PubMed; S. Hiraoka, K. Harano, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 14368 CrossRef PubMed.
- J.-s. Y. T Ma, S.-l. Lou, X.-q. Tang and Y.-d. Jiang, Chem. Res. Chin. Univ., 2009, 25, 590 Search PubMed; K. R. J. Thomas, M. Velusamy, J. T. Lin, C. H. Chuen and Y.-T. Tao, J. Mater. Chem., 2005, 15, 4453 RSC.
- A. B. S. Elliott, R. Horvath, X.-Z. Sun, M. G. Gardiner, K. Müllen, N. T. Lucas, M. W. George and K. C. Gordon, Inorg. Chem., 2016, 55, 4710 CrossRef CAS PubMed.
- D. Nolan, B. Gil, L. Wang, J. Zhao and S. M. Draper, Chem. – Eur. J., 2013, 19, 15615 CrossRef CAS PubMed; F. A. Murphy and S. M. Draper, J. Org. Chem., 2010, 75, 1862 CrossRef PubMed; N. T. Lucas, H. M. Zareie and A. M. McDonagh, ACS Nano, 2007, 1, 348 CrossRef PubMed; Y. Fogel, M. Kastler, Z. Wang, D. Andrienko, G. J. Bodwell and K. Müllen, J. Am. Chem. Soc., 2007, 129, 11743 CrossRef PubMed; B. E. Hamaoui, L. Zhi, J. Wu, J. Li, N. T. Lucas, Ž. Tomović, U. Kolb and K. Müllen, Adv. Funct. Mater., 2007, 17, 1179 CrossRef; K.-Y. Kim, S. Liu, M. E. Köse and K. S. Schanze, Inorg. Chem., 2006, 45, 2509 CrossRef PubMed; B. E. Hamaoui, F. Laquai, S. Baluschev, J. Wu and K. Müllen, Synth. Met., 2006, 156, 1182 CrossRef; P. T. Herwig, V. Enkelmann, O. Schmelz and K. Müllen, Chem. – Eur. J., 2000, 6, 1834 CrossRef.
- D. Nolan, B. Gil, F. A. Murphy and S. M. Draper, Eur. J. Inorg. Chem., 2011, 3248 CrossRef CAS.
- C. Delaney, G. M. Ó. Máille, B. Twamley and S. M. Draper, Org. Lett., 2016, 18, 88 CrossRef CAS PubMed; L. P. Wijesinghe, B. S. Lankage, G. M. O. Maille, S. D. Perera, D. Nolan, L. Wang and S. M. Draper, Chem. Commun., 2014, 50, 10637 RSC; D. J. Gregg, C. M. A. Ollagnier, C. M. Fitchett and S. M. Draper, Chem. – Eur. J., 2006, 12, 3043 CrossRef PubMed; D. J. Gregg, C. M. Fitchett and S. M. Draper, Chem. Commun., 2006, 3090 RSC; D. J. Gregg, E. Bothe, P. Höfer, P. Passaniti and S. M. Draper, Inorg. Chem., 2005, 44, 5654 CrossRef PubMed; S. M. Draper, D. J. Gregg, E. R. Schofield, W. R. Browne, M. Duati, J. G. Vos and P. Passaniti, J. Am. Chem. Soc., 2004, 126, 8694 CrossRef PubMed; S. M. Draper, D. J. Gregg and R. Madathil, J. Am. Chem. Soc., 2002, 124, 3486 CrossRef PubMed.
- S. Hiraoka, Y. Yamauchi, R. Arakane and M. Shionoya, J. Am. Chem. Soc., 2009, 131, 11646 CrossRef CAS PubMed; K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300 CrossRef PubMed; S. Hiraoka, K. Harano, M. Shiro, Y. Ozawa, N. Yasuda, K. Toriumi and M. Shionoya, Angew. Chem., Int. Ed., 2006, 45, 6488 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC; J. D. Crowley and D. A. McMorran, in Click Triazoles, ed. J. Košmrlj, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, p. 31 RSC; H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675 RSC; D. Schweinfurth, N. Deibel, F. Weisser and B. Sarkar, Nachr. Chem., 2011, 59, 937 CrossRef CAS.
- T. Y. Kim, A. B. S. Elliott, K. J. Shaffer, C. J. McAdam, K. C. Gordon and J. D. Crowley, Polyhedron, 2013, 52, 1391 CrossRef CAS.
- J. D. Crowley and E. L. Gavey, Dalton Trans., 2010, 39, 4035 RSC.
- Palladium in Organic Synthesis, ed. J. Tsuji, Springer Berlin Heidelberg, 2005 Search PubMed.
- D. Huang, P. Zhao and D. Astruc, Coord. Chem. Rev., 2014, 272, 145 CrossRef CAS.
- D. Wang, L. N. S. Gautam, C. Bollinger, A. Harris, M. Li and X. Shi, Org. Lett., 2011, 13, 2618 CrossRef CAS PubMed; W. Yan, X. Ye, N. G. Akhmedov, J. L. Petersen and X. Shi, Org. Lett., 2012, 14, 2358 CrossRef PubMed.
- L. Wang, D. J. Kiemle, C. J. Boyle, E. L. Connors and I. Gitsov, Macromolecules, 2014, 47, 2199 CrossRef CAS.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395 CrossRef CAS.
- T. Iwasawa, M. Tokunaga, Y. Obora and T. Tsuji, J. Am. Chem. Soc., 2004, 126, 6554 CrossRef CAS PubMed; J. Cookson, Platinum Met. Rev., 2012, 56, 83 CrossRef; L. Li, T. Wu, J. Wang and R. Wang, ChemPlusChem, 2014, 79, 257 CrossRef.
- C. Deraedt, L. Salmon and D. Astruc, Adv. Synth. Catal., 2014, 356, 2525 CrossRef CAS; M. Gómez-Martínez, E. Buxaderas, I. M. Pastor and D. A. Alonso, J. Mol. Catal. A: Chem., 2015, 404–405, 1 CrossRef; S. S. Kotha, N. Sharma and G. Sekar, Tetrahedron Lett., 2016, 57, 1410 CrossRef; B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54 Search PubMed; M. Pérez-Lorenzo, J. Phys. Chem. Lett., 2012, 3, 167 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C NMR, MALDI-TOF-MS, crystallographic data, ORTEP and packing diagrams, molecular modelling. CCDC 1502527–1502529. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc07413h |
| This journal is © The Royal Society of Chemistry 2016 |