
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic growth factor microenvironments
Manuel
Salmerón-Sánchez
 *a and
Matthew J.
Dalby
b
*a and
Matthew J.
Dalby
b
aDivision of Biomedical Engineering, School of Engineering, University of Glasgow, Rankine Building, Oakfield Avenue, Glasgow G12 8LT, UK. E-mail: Manuel.Salmeron-Sanchez@glasgow.ac.uk
bCenter for Cell Engineering, Institute of Molecular Cell and Systems Biology, University of Glasgow, Joseph Black Building, Glasgow G12 8QQ, UK
First published on 29th September 2016
Abstract
Growth factors (GF) are remarkably powerful signalling molecules that orchestrate developmental biology. GFs are currently used in medical applications with limited success but it is clear that if their potential can be harnessed for biomedicine then they could underpin the discipline of regenerative medicine. However, while we understand that biology uses cell-secreted growth factors tethered to the ECM, biologists typically employ GFs in soluble format at high concentrations. When used in vivo, this causes off-target, unwanted effects, which severely limits their use. There is a vast amount of literature dealing with material systems that control the delivery of GFs. However, it was soon observed that GFs could be more effectively presented bound to surfaces from a solid-phase state rather than in soluble form, recapitulating the way the extracellular matrix (ECM) binds GFs. In parallel, evidence was found that within the ECM, GFs can actually work in cooperation with integrins and that this produced enhanced GF signalling due to the crosstalk between both receptors. Recently this knowledge was used to engineer microenvironments that target simultaneous integrin and GF receptor engagement seeking to maximise GF effects in vitro (e.g. in terms of stem cell differentiation) but also tissue repair in vivo (e.g. bone regeneration and wound healing). This feature article introduces the concept of synergistic GF/integrin signalling and then introduces GF delivery systems that were key in the development of more advanced synergistic growth factor microenvironments.
 Manuel Salmerón-Sánchez | Manuel Salmeron-Sanchez is Professor of Biomedical Engineering at the University of Glasgow. He works on material-based strategies to engineer tissue regeneration, and to understand pathological conditions such as cancer. His group has developed novel strategies to use growth factors very efficiently, in very low doses. Manuel did a PhD in Valencia where he was full Professor before taking a Chair in Glasgow in 2013. |
 Matthew J. Dalby | Matthew J. Dalby is Professor of Cell Engineering at the University of Glasgow. He works on stem cell technologies at the interface with material systems. He did a PhD in Queen Mary University of London. In 2003 he was a BBSRC David Philips Fellow to explore mesenchymal stem cell (MSC) mechanotransduction on materials and has been an academic in Glasgow since then. He was awarded his Chair in 2014. |
1. Introduction
It is now well accepted that material-based approaches to control stem cells hold great potential. Materials can be engineered to deliver stimuli that regulate stem cell adhesion, signalling and differentiation towards defined, desired, lineages. Stiffness, nanotopography and chemistry are examples of material properties that have been used to modulate the cellular microenvironment and dictate stem cell fate.1–3 Key to achieving a desired therapeutic outcome is the need to control the stem cell local environment, or niche. However, our ability to engineer the complexity of this niche in vitro has remained, until now, a major challenge.4,5Growth factors (GF) are biomolecules that have strong influence on cells, including stimulation of proliferation, migration and differentiation. Not only they are critical in orchestrating embryonic development but also they are involved in a range of physiological and pathological process, including tissue homeostasis as well as repair and maintenance (e.g. wound healing).6,7
In addition, GFs provide potent signals that have essential influence on stem cell phenotype. For example, bone morphogenetic protein 2 (BMP-2) promotes osteogenesis; vascular endothelial growth factor (VEGF) vascularisation and transforming growth factor (TGF-β) chondrogenesis – GFs are important biochemical cues that control stem cells.8
Because of this potential to regulate regenerative biology, cocktails of GFs are traditionally used by biologists to control stem cell differentiation in vitro.9 In addition, GFs are widely used in clinical practice, with limited, and even controversial, success which involved unwanted effects including neurological problems, ectopic bone formation and risk of cancer.10 This is mainly because of the high, supraphysiological doses used in order to achieve effective local concentrations. However, if we look to developmental biology, while the body indeed presents GFs to natural stem cell niches in soluble forms secreted by cells, it mainly, however, uses tethered signals coupled to the extracellular matrix (ECM), allowing topical, low dose, administration.11,12
The controlled delivery of GFs has been used in combination with biomaterial carriers with limited success and increasing complexity of design has only produced small gains. The field has evolved from sustained released of growth factors (e.g. from hydrogels) to controlled GF binding to designed chemistries using different strategies, including covalent binding and protein engineering.13,14 Significant differences in GF activity and stability have been observed in dependence of soluble versus bound presentation, which has led to the development of surfaces that present physical and chemical cues that regulate GF activity.15,16
Significant work has been developed to engineer novel delivery systems inspired by the GF regulatory function of the ECM, i.e. bioinspired systems that recapitulate the interactions of GFs with components of the ECM. This includes heparan sulphate proteoglycans, and proteins such as fibronectin, vitronectin, tenascin C and fibrinogen. Seminal reviews addressing ECM inspired materials to control GFs with applications in bone regeneration, wound healing and angiogenesis have been recently published.17–19 The interaction of GFs with the ECM is so important that there has even been a new family of GFs engineered to have super-affinity with the natural ECM.20
This feature article highlights recent development of materials that present GFs in such a way that cell signalling is synergistically enhanced by the formation of clusters between integrins and GF receptors. Crosstalking between integrins and GF receptors is known to maximise GF effects in physiological and pathological conditions.21 Engineering this dialogue between integrins and GF receptors – synergistic growth factor microenvironments – will have important translational consequences as it has the potential to contribute towards significant reduction in GF doses, maximising effects and safety in clinical scenarios.
2. Growth factors and integrins
The way cells interact with their environment and perceive physico-chemical changes comes via cell surface receptors. Integrins are transmembrane receptors involved in cellular adhesion, connecting the extracellular environment to the cytoskeleton. Integrins are heterodimers containing two subunits (α and β) that bind specifically a number of ligand peptides in the ECM. For example, αvβ3 binds the well known arginine-glycine-aspartic acid (RGD) sequence and α2β1 is one of the collagen receptors.22 After integrin binding to the ECM, there is a process of integrin clustering into supramolecular complexes called focal adhesions which contain a number of structural and signalling molecules that link the ECM to the actin cytoskeleton.23 Super-resolution microscopy has shown that integrins and actin are vertically separated ∼40 nm by a layer of focal adhesion proteins22 Within these structures, talin regulates the nanoscale architecture of focal adhesions and integrin–talin–actin complexes works as mechanical linkages (Fig. 1).24,25 Through focal adhesions cells are able to feel the physical properties of the ECM, such as stiffness; they are mechanoreceptors which transduce physical signals, force in particular, into biochemical and signalling cascades.26,27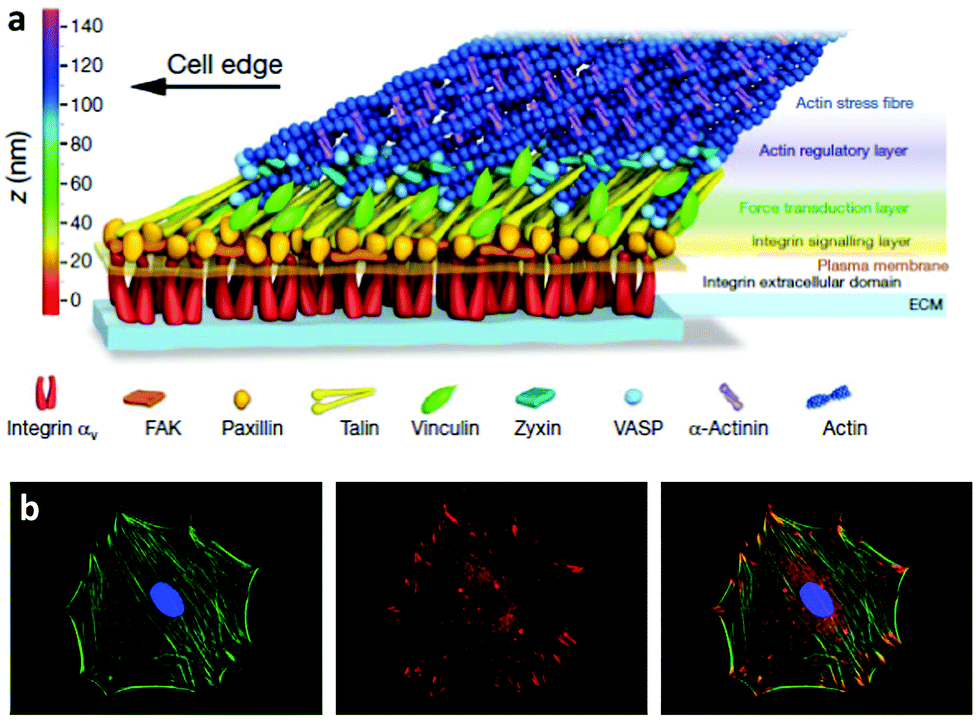 | ||
| Fig. 1 Integrins and focal adhesion. (a) The nanoscale structure of focal adhesions including integrins bound to the ECM and focal adhesion proteins that link the ECM to the actin cytoskeleton. (b) Staining of the actin cytoskeleton and focal adhesion protein vinculin. The superposition shows actin tethered to focal adhesion plaques. Reproduced with permission from ref. 24 and 28. | ||
GFs are proteins that modulate cell behaviour by regulating signalling cascades to drive cell proliferation, morphogenesis and differentiation. Cells interact with GFs via specific receptors in their membrane with complex structure and dynamics. For example, BMP-2 induces a complex at the plasma membrane of type I and type II BMP receptors, allowing the receptors to become phosphorylated and drive the down-stream signalling cascades.29 Moreover, GF receptors are laterally mobile at the plasma membrane and able to associate into microdomains to enhance signalling.30 GF receptors are able to signal from the membrane but are also able to be internalised in dependence of the localisation of the GF within the membrane and the way the presentation of the ligand occur, i.e. binding of a soluble GF might lead to endocytosis and signal modulation. Internalisation of the receptor–ligand complex is more difficult when GFs are presented bound to a substrate (e.g. the natural ECM or engineered material surfaces).31 Assembly of the ligand receptor complex initiates phosphorylation of intracellular domains activating kinases and triggering downstream signalling. For example, BMP-2 binding and receptor phosphorylation leads to the phosphorylation of intracellular Smad proteins which subsequently hetero-oligomerize with Smad4 and translocate into the nucleus, where they act as co-transcription factors with, e.g. runt related transcription factor 2 (RUNX2 – the master bone transcriptional regulator) to drive gene transcription that results in bone formation (Fig. 2).29,32 This is known as canonical BMP signalling.
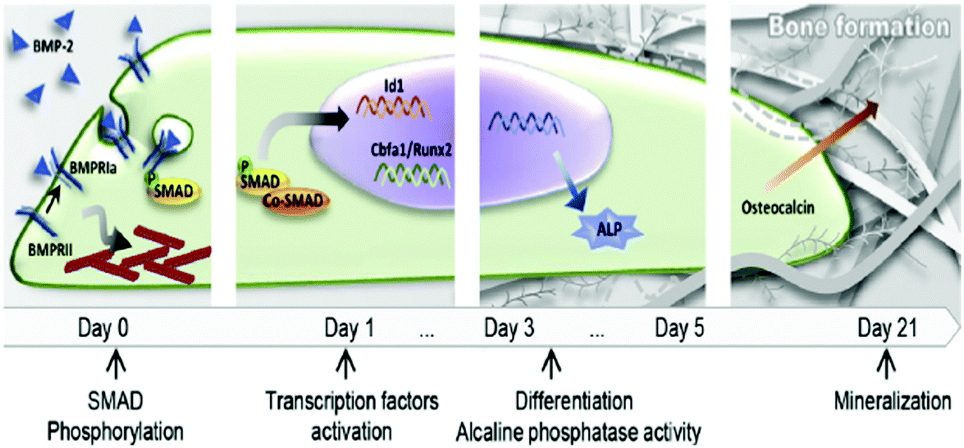 | ||
| Fig. 2 Schematic representation of the classical action of soluble GFs (e.g. BMP-2). BMP-2/receptor interaction promotes Smad phosphorylation and translocation of transcription factors into the nucleus that lead to differentiation and production of mature ECM. Reproduced with permission from ref. 15. | ||
3. Crosstalk between integrins and growth factor receptors
There is still a traditional view of the ECM as a reservoir of GFs that can be released from the mesh of protein fibrils to function as soluble ligands. However, there are several examples of GFs that actually bind to their cell-based receptors as ‘solid phase’ ligands, bound to the ECM. There are also increasing number of examples of GFs binding to ECM proteins themselves, supporting the idea that the presentation of GFs is an important function of the ECM.11One of the primary functions of the ECM is to provide cells with a network of proteins for integrin-mediated adhesion.12 The crosstalk between integrins and GFs was revealed in biological sciences more than a decade ago: cell adhesion is necessary to implement activation of GF receptors, and then GFs are necessary to stimulate cell adhesion, migration and integrin-dependent signals.21 It is still an open question whether this crosstalk between integrins and GF receptors involves co-localisation of both proteins within the same region within the cell membrane or only cooperation in the downstream signal pathway (Fig. 3).11,33
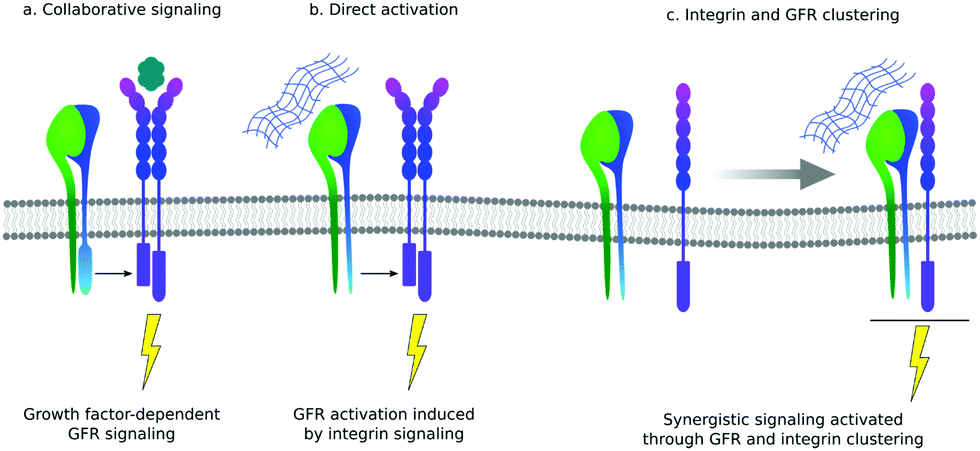 | ||
| Fig. 3 Different mechanisms of interaction between integrins and GF receptors (GFR). (a) Collaborative signalling means that often integrins are required for GFR activation even after GF binding. (b) Integrins have been reported to activate GFRs even in the absence of GFs (direct activation). (c) Binding of integrin to the ECM in molecular proximity to GFRs bound to GFs can induce the formation of stable integrin–GFRs complexes that result in receptor crosstalk and synergistic signalling. | ||
GF receptors can be activated through integrin association leading to the formation of integrin–GF receptor clusters that can trigger GF signalling even in the absence of GFs. For example, mesenchymal stem cell (MSC) adhesion to fibronectin induced α5β1 integrin dependent phosphorylation of PDGF receptor-β in the absence of GFs. This synergistic relationship between integrins and PDGF receptors was essential for MSC migration, which is a physiological mechanism to recruit mesenchymal cells to sites of vascular remodelling.35
Here, however, we are more interested in the importance of direct physical interaction, i.e. co-localisation between integrins and GF receptors, as this article is focused on how to engineer microenvironments that recruit, in the same nanoscale cluster, both receptors. For example, α5β1 forms a stable interaction with Tie2, which is a receptor for Ang-1 (angiopoeitin-1) and the co-receptor cluster is required for Ang-1 mediated blood vessel formation in vivo and enhancement of endothelial cell migration in vitro (Fig. 3).36 It is interesting that this synergistic mechanism in vitro was only enhanced when cells were plated on fibronectin and that this is related to the ability of fibronectin to bind GFs next to the integrin binding regions in a way that was described five years later, and as will be discussed later.37 Integrin α5β1 bound to fibronectin has been described to co-localise with other GF receptors, such as receptors for bFGF, insulin and EGF to activate Rac and promote endothelial cell-cycle progression. Using the same GFs with cells attached on laminin (using a α2β1) the effect of the GF was not observed which led to the conclusion that physical interactions and integration of both integrin and GF receptor signalling localised to the same focal adhesions was needed.38 Notwithstanding their role in biology to enhance signalling and control physiological and pathological processes, precise molecular details of the interaction between integrins and GF receptors clusters still require elucidation.33,34,39
4. Presentation of growth factors – looks matters
4.1. From surface bound GFs to engineered integrin/GF microenvironments
As we have mentioned, in in vitro and in vivo experiments it is still common to use GFs in solution either directly added to the culture media or released from a biomaterial carrier.13,40 This thus ignores that GFs in vivo are bound to the ECM, both to proteins and glycosaminoglcans.11 Solid-phase presentation of GFs bound to a surface is more efficient compared to soluble administration, resulting in enhanced biological function with lower doses used.8,17,18Fig. 4 shows a sketch of classical (soluble) vs. surface-bound presentation of GFs.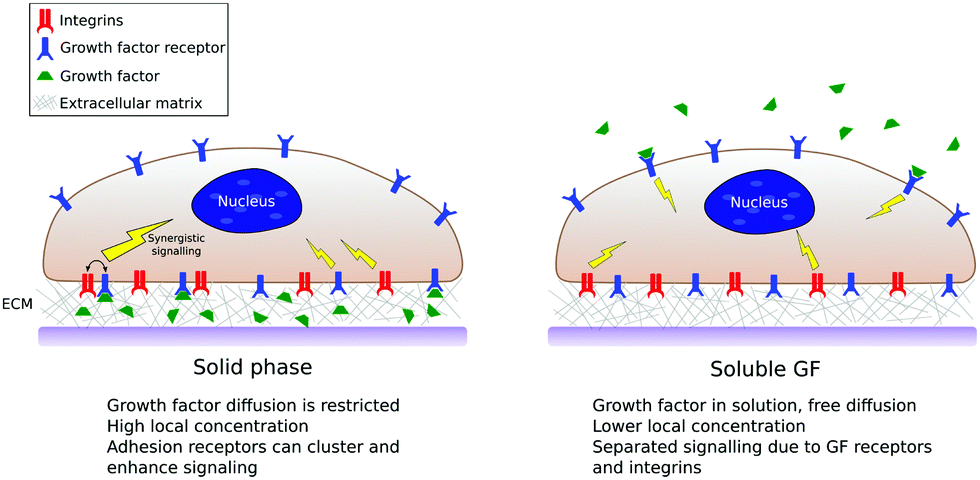 | ||
| Fig. 4 The scheme summarises the main differences between soluble vs. solid-phase presentation of GFs. GFs presented from surfaces are spatially confined and have limited diffusion. Even if the surface concentration of GFs is low, they are presented in highly local concentrations and have the potential to establish crosstalk with adhesion receptors. | ||
The pioneering work of Griffith et al. revealed that Epidermal GF (EGF) could be tethered to surfaces retaining its biological activity and with superior control of temporal and spatial availability in the extracellular matrix.16 They used star PEG on glass on which EGF was conjugated and the superior effect on hepatocytes assessed. However, besides this remarkable success that suggested an imminent change of paradigm in the way GFs could be used – from soluble delivery to matrix-bound presentation – it took more than a decade to investigate the presentation of other GFs from surfaces. Biotynilated IGF-1 was tethered on self-assembled peptides which activated Akt, decreased the activation of caspase-3 and increased expression of troponin-I in cardiomyocytes in vitro, as well as improved systolic function after infarction in vivo.41 VEGF was shown to be retained within collagen gels (through an ECM binding domain). This matrix bound VEGF elicited prolonged activation of VEGF receptor VEGFR2 and reciprocal responses on β1 integrin, a response which was absent upon exposure to soluble VEGF.42 One of the first works which showed the differential effect of surface-bound versus soluble presentation of GFs on stem cells using EGF on glass revealed that surface-tethered EGF promoted both cell spreading and survival more strongly than saturating concentrations of soluble EGF.43
Cavalcanti-Adam showed that matrix immobilised BMP-2 promoted cell migration and signalling compared to the soluble delivery of BMP-2.45 To investigate the role of GF density, they used block copolymer micelle nanolithography to fabricate substrates with precise nanoscale distribution of gold nanoparticles functionalised with BMP-2.46 They showed that surface presentation of BMP was highly efficient at triggering the Smad-transcriptional pathway, when compared to soluble administration of the GF. More so that surface-immobilized BMP-2 at a concentration as low as 0.2 ng cm−2 was still able to induce Smad-dependent signalling due to local and sustained presentation that did not perturb GF bioactivity. Segura et al. showed that not only was important to have GFs presented from surfaces but also that the density and organisation of GFs should be considered to determine GF activity and efficiency bound to surfaces.47 They immobilised VEGF to nanoparticles that were loaded in a fibrin matrix together with endothelial cells (HUVECs). VEGF was bound in high and low densities to study the influence of presentation in heterogeneous nanodomains with homogeneous distribution and they found the vascularisation potential to increase in the high binding density format.47 This result was related to the creation of GF reservoirs, mimicking the physiological environment.11
Some of the pioneering works on ‘solid-phase’ presentation of GFs used covalent tethering of GFs to surfaces.16 However, exploiting the natural affinity of ECM components (GAGs and structural proteins) towards GFs has resulted in the development of affinity-based release systems, to control release of therapeutics locally.48,49 In addition, affinity-based systems allow engineering ‘solid-phase’ presentation of GFs that can be further internalised by cells after binding, as this is key to activate certain signalling pathways.31 Seeking to engineer localised GFs reservoirs, Picart et al. showed that the well-known layer-by-layer (LbL) technique using polyelectrolytes was a powerful and versatile strategy to work as a delivery reservoir for BMP-2. LbL involves the use alternate positive and negative charged molecules that are assembled into layers.50 Picart et al. used poly(L-lysine)/hyaluronan (PLL/HA) polyelectrolyte multilayer films to show that the amount of BMP-2 loaded in LbL films could be tailored by varying the film thickness, i.e. the number of layers fabricated into the system, as well as the concentration of the GF in the initial solution.51 They showed that BMP-2 trapped in this LbL system was released to the media in very low amounts and moreover it remained bioactive within the film for more than 10 days to promote osteogenic differentiation of C2C12 myoblasts.44 Interestingly, they showed that the activity of the GF was related to the maintenance of its secondary structure within the LbL film.52 This work fully supported the idea that GF presentation rather than controlled delivery was key to enhance the bioactivity of GFs. In addition to this, they showed, for the first time, that presentation of GFs bound to biomaterial systems did interfere with other physical properties of the ECM such as mechanical stiffness.
Cells are known to respond to the stiffness of the environment. It has been shown that cells differentiate into osteoblast on stiff substrates (>25 kPa) but not on soft ones (<10 kPa).53 Even when osteogenic media was used, cells were unable to differentiate into osteoblast if the stiffness of the substrate was too low.53 This reveals the importance of the physical environment in cell behaviour. However, in a very elegant work, Picart et al. showed that whereas on stiff membranes cell respond similarly to BMP-2, either bound to the surface or soluble in the culture media, on soft films cells were indeed responsive to matrix bound BMP-2 (but not to soluble BMP-2).44 These experiments were key to reveal not only that the effect of GFs was more effective if presented from surfaces, but also that the interplay between different receptors (e.g. integrins – mechanosensors able to discriminate matrix stiffness54 – and GF receptors) was key in the design of biomaterial systems.
The key role of the interplay between GFs presentation and the physical properties of the synthetic matrix used as a vehicle for GF presentation has been further demonstrated using self-assembling peptide amphiphiles (PA). PA molecules can self-assemble into supramolecular nanofibers that can be designed to bind specific GFs.55 Recently, Stupp et al. modulated intermolecular interactions within supramolecular assemblies and their role in GF signalling. To do so, they designed two systems that differed in the degree of intermolecular hydrogen bonding (choosing primary amino acid sequences with varied β-sheet propensity) and found that the structures with weaker internal cohesion significantly enhanced GF signalling and osteogenic commitment using BMP-2. This was related to the ability of these structures to promote enhanced raft mobility within the cell membrane which in turn enhanced cell signalling. This work identified a new mechanism – membrane mobility – as a way to enhance the effect of GFs presented from biomaterial nanofibers.56 It is interesting to note in a parallel work the mobility of fibronectin adsorbed on polymers with different chain dynamics has been correlated with cell adhesion and cell differentiation.57
In relation to the interplay between GF delivery systems and cell adhesion, it has been shown that the soluble administration of GFs was more effective if cells were previously targeted for integrins to bind specifically to the GF delivery system. These works further revealed the importance of both receptors in maximising GF effects. For example, Garcia et al. showed that protease-degradable hydrogels released controlled amounts of VEGF that were more effective in promoting vascularisation in a rat subcutaneous implant when the hydrogel had been functionalised with RGD molecules.58 Integrin-modulation of GFs effects were also revealed in protease-degradable hydrogels functionalised with a triple-helical, α2β1 integrin-specific peptide (GFOGER) as a BMP-2 delivery system. These engineered hydrogels increased osteoprogenitor localization in the defect site, enhanced bone formation and induced defect bridging and mechanically robust healing at low BMP-2 doses.59 Importantly, similar doses led to no bone regeneration when delivered from simple collagen sponges.
Sobel et al. used recombinant fibronectin fragments to identify the heparin II binding region of fibronectin (FNIII12–14) as a VEGF binding site and reported that only bivalent fibronectin constructs encompassing the integrin binding site and VEGF binding domains significantly promoted endothelial migration, proliferation and signalling.60 Building on this result, Martino et al. showed that FNIII12–14 was actually a highly promiscuous GF binding region able to sequester not only VEGF but also a large number of GFs from different families (Fig. 5).37 They used this information to produce a major breakthrough in the design of material systems to present GFs; the concept of utilizing crosstalk between integrins and GF receptors in a synthetic system. To do so, they started with a fibrin matrix that was functionalised with two recombinant fragments of fibronectin pieced together. FNIII9–10,61 which contains the well know RGD site to promote cell adhesion, and FNIII12–14, the classical heparin II binding site that promiscuously binds GFs37 (Fig. 5). They showed that the resulting FNIII9–10/12–14 fragment fused to the fibrin matrix induced in vitro tube-like structure formation in endothelial cells using VEGF-A, smooth muscle cell sprouting in response to PDGF-BB and mesenchymal stem cell (MSC) differentiation towards osteogenic lineages using BMP-2. Importantly, they also showed that the system worked in vivo by promoting (a) skin healing (wound healing) in a diabetic mice model using a combination of VEGF-A165 and PDGF-BB and (b) bone regeneration in a critical size calvarial defect in a skeletally mature rat using a combination of BMP-2 and PDGF-BB.62 This work showed that the biological concept of crosstalk between integrins and GF receptors used by ‘biology’ could actually be engineered into a material-based system that could stimulate cell behaviour in vitro and promote tissue repair in vivo. From a clinical perspective, it illustrated that significantly lower doses of GFs could be effectively used to promote tissue healing following tissue engineering principles. This material system incorporated ∼100 ng of GFs which was significantly lower than the >10 μg previously used to promote regeneration in the same model.62 This work represented a major landmark in the way GFs had been used and put synergistic signalling in the map as a way to use GFs efficiently and safely.
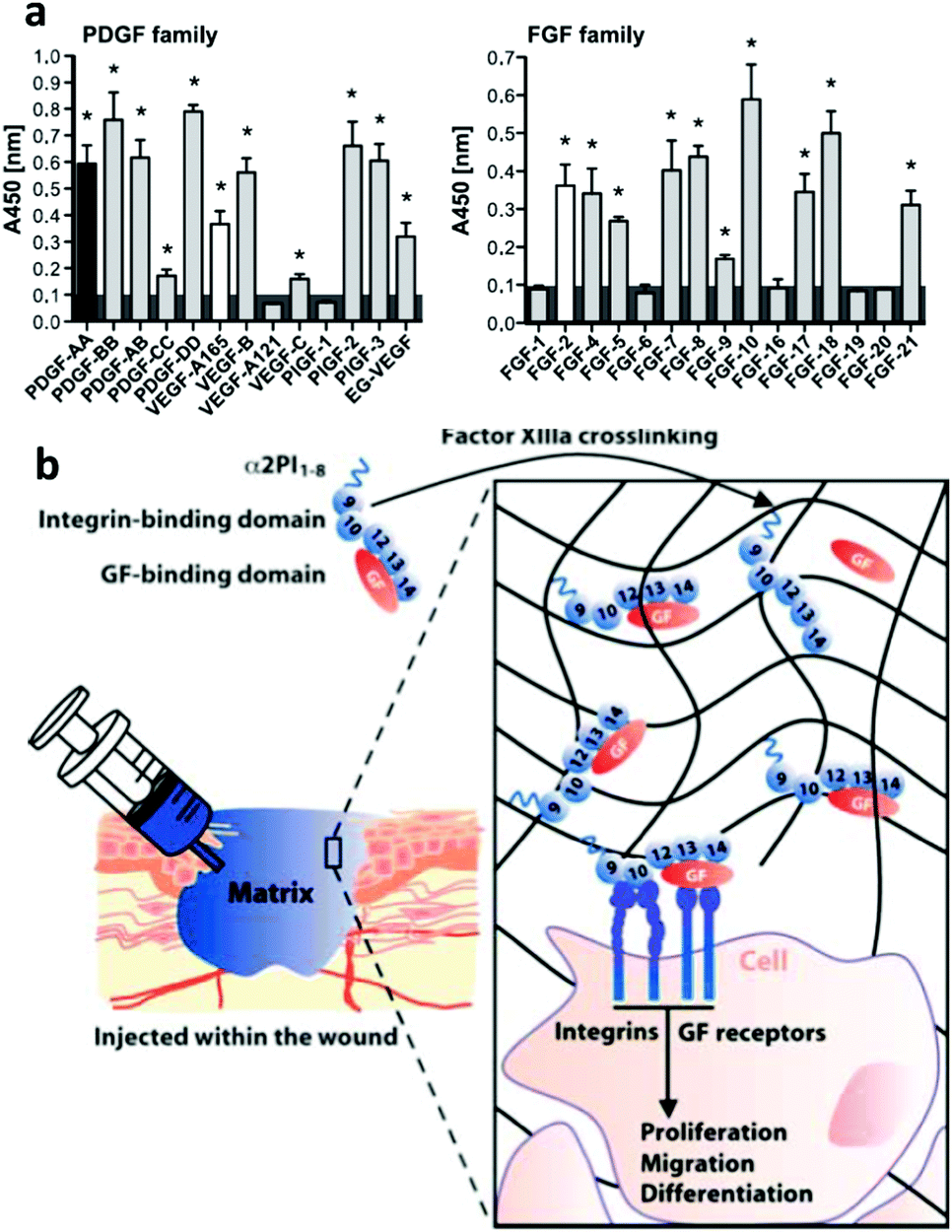 | ||
| Fig. 5 Engineered GF microenvironments based on FNIII12–14 incorporated in fibrin matrices. (a) Examples of GFs with affinity for FNIII12–14 measured by ELISA. Binding was promiscuous, known interactions shown in black or white. Overall, more than 25 new interactions were described. (b) The sketch shows a recombinant fibronectin fragment that consist of FNIII9–10 (the integrin binding site) linked to FNIII12–14 (the GF binding site) and a substrate sequence to be covalently bound into a fibrin matrix. This engineered microenvironment promotes GF binding in synergy with integrins and enhanced cellular processes. Reproduced with permission of ref. 37 and 62. | ||
4.2. Integrins in engineered synergistic microenvironments
There is debate over which integrins are involved in the adhesion/GF synergistic process and whether this depends on the type of GF and/or the biomaterial system itself. Originally, fibrin matrices modified with FNIII9–10/12–14 showed that the synergy with GFs was principally mediated through α5β1 (versus αvβ3, another important RGD receptor) as revealed by blocking experiments with antagonistic antibodies (Fig. 6).62 However, previous work had described a critical role of integrin αvβ3 in physiological angiogenesis and the synergistic role with VEGF receptor-2.63 Following from this earlier work suggesting a major role for αvβ3, Traub et al. engineered FNIII10 (RGD sequence without the PHSRN and so only ligates to αvβ3 and not α5β1)64 fused to VEGF-A165 in a fibrin matrix. This system presents the GF in close apposition to the αvβ3 binding site of fibronectin.65 They found that while HUVECs were able to bind VEGF within the system and activate both VEGFR-2 and αvβ3, the angiogenic response was reduced in comparison to the pro-angiogenic effect of fibrin-immobilised VEGF only; inferring a more important role for α5β1.65 | ||
| Fig. 6 Controversies in integrin activation in synergy with BMP-2. Both α5β1 and αvβ3 have been reported to be involved in synergistic integrin/BMP-2 receptor signalling. (a) MSC osteogenic differentiation with fibrin matrix functionalised with fibronectin recombinant proteins (FNIII9–10 for integrin binding, FNIII12–14 for GF binding and FNIII9–10/12–14 for synergistic activation of receptors). Expression of osteoblastic genes by qPCR. Note that osteogenic differentiation was prevented by blocking α5β1 but not affected by αvβ3. (b) Cells (C2C12) seeded on LbL soft films with BMP-2 bound to the surface and stained for integrins β1, β3, α5, αv. Inserts show detail of focal adhesion structures containing these integrin subunits. (c) Incubation with anti-β3 antibodies decreased drastically the number of cells and cell size; no effect was observed by incubating with anti-β1 antibodies whereas the number of cells was drastically reduces using anti-β3 Reproduced with permission from ref. 62 and 68. | ||
Similar behaviour was found using the heparin-binding domain of fibrinogen incorporated into synthetic PEG matrices as described next. Martino et al. showed that the fibrinogen domain (Fg b15–66(2)) binds several GFs from the PDGF/VEGF and FGF families and that incorporated together with RGD (no synergy sequence and so αvβ3 binding only) into a PEG hydrogel crosslinked with a MMP-degradable sequence, the system was able to promote wound healing in a diabetic mouse.66 Rather unexpectedly, but in agreement with Traub et al.,65 they obtained better wound healing when the RGD sequence was removed from the matrix.66 This finding per se does not mean that integrin binding is not involved in enhancing the role of GFs to promote tissue healing but most likely that α5β1, being unable to bind to RGD (as the synergy sequence was not included),67 was not allowed to play its critical role in VEGF mediated wound healing.62 Further experiments with this system including integrin-specific fibronectin fragments are deemed necessary to assess this hypothesis and fully establish the role of α5β1 in cooperation with GF receptors.66
In an effort to understand signalling mechanisms, including integrin and GF receptors signals, as well as their interplay with the physical properties of the matrix (stiffness), Albiges-Rizo et al. investigated how matrix-bound BMP-2 activated the Smad cascade in relation to co-ordinated integrin and BMP-2 receptor signalling.68 They used LBL films made of poly(L-lysine) and hyaluronan with modulated and controlled stiffness. Interestingly, they found that BMP-2 receptors and β3 integrins (but not β1) co-ordinately controlled cell spreading and Smad signalling, which complicates the above described roles of β3 and β1 (Table 1).
| Integrin | GF | Synergistic effect |
|---|---|---|
| α5β1 | BMP-2/PDGF | Osteogenesis62 |
| α5β1 | PDGF | Stem cell recruitment62 |
| α5β1 | VEGF | Organisation of endothelial cells and wound healing62 |
| α5β1 | BMP-2 | Osteogenesis via Smad signalling69 |
| αvβ3 | VEGF | Limited angiogenic response65 |
| αvβ3 | PDGF/FGF | Wound healing66 |
| αvβ3 | BMP-2 | Osteogenesis via Smad cascades68 |
Soluble GFs are non-effective when cells are cultured on soft matrixes, for example, BMP-2 cannot drive bone formation. However, solid-phase GFs are effective. BMP-2 presented in surface bound format overrode the stiffness response of cells to soft matrixes and permitted osteogenic differentiation. It is proposed this occurs through actin and adhesion dynamics via cooperation between both integrin and GF receptors. The authors concluded that the presentation of matrix bound BMP-2 to receptors activated the αvβ3 integrin hence mediating cell spreading and cell migration thanks to interactions that involved BMP-2 and fibronectin.
As there was no exogenous fibronectin in their system, for this mechanism to occur, the authors hypothesised that fibronectin secreted by cells was actually the ligand for αvβ3. They suggested that αvβ3 integrin bound to (cell-secreted) fibronectin distributed around cells. The co-localisation of BMP-2 to the integrin binding sequence was proposed to help elongate adhesion and drive traction-mediated cell spreading processes.37 These last two findings we have described rather contradict previous results by Martino et al. where the role of α5β1 was predominant over αvβ3.62 Together this shows that this is likely a complex story with much further work needed.
One should note, however, that there are fundamental conceptual differences in material systems used to present GFs in ref. 68 and 62: while Martino et al. engineered a system which contained integrin binding (FNIII9–10) as well as GF binding regions (FNIII12–14) to target α5β1 and not αvβ3; the layer-by-layer material system used by Albiges-Rizo et al. was designed to present surface-bound BMP-2.68 The question arises whether the role of vitronectin should have been considered in the attempt to understand this system, as it is known that vitronectin is present in high amount in the serum included in the culture medium and also secreted by cells.70
That the presentation of GFs bound to the ECM rather than in soluble format is essential to enhance the efficiency of GFs was demonstrated by engineering GFs with super-affinity to the ECM. Martino et al. discovered a domain in placenta growth factor-2 (PlGF2123–144) that binds strongly to a large number of ECM proteins, including fibronectin, vitronectin, fibrinogen and collagen I. Then, PDGF-BB was modified to have PlGF2123–144 fused to GFs and topically applied in a very low concentration in vivo (∼200 ng). This led to a significant faster wound healing closure and granulation tissue. Similarly, in the context of bone repair, PlGF2123–144 fused to BMP-2 and PDGF-BB led to full regeneration in a critical-size calvarial defect using low concentrations of GFs (∼200 ng).20 This work shows that in the clinical context, the sole delivery of GFs without a biomaterial carrier might work well as long as the GF has been engineer to bind the host ECM and, paradoxically, reveals the importance of GF presentation to maximise efficiency, even if in this case GFs were delivered topically and in solution, without carriers.20
5. Material-driven assembly of proteins and synergistic growth factor microenvironments
Some ECM proteins have domains to bind GFs that are hidden in the globular conformation of the protein but become available once these proteins are incorporated into a fibrillar ECM. This happens for example for fibronectin.71 As discussed, FNIII12–14 is a promiscuous region to bind GFs.37 However, fibronectin needs to be cell-assembled into a fibrillar protein network for these GF-binding domains to be available – this is the natural process of cell-mediated fibronectin assembly.71 There has been significant efforts to engineer material that assemble proteins seeking to recapitulate the ECM. Here we show that this material-driven assembly of proteins results in systems that promote high efficiency presentation of GFs.5.1. Engineering protein assembly
It is known that certain materials have the ability to induce the organisation of ECM proteins upon adsorption from solutions.72 In particular, there has been a significant amount of work seeking to engineer fibronectin matrices that can recapitulate biological features of the natural protein within the ECM.73 Designing engineered systems which are biomimetic of the natural ECM has been a challenge in regenerative medicine as the ECM promotes cell adhesion, migration, signalling and controls the availability of bioactive molecules, in particular GFs, as has been discussed.11Fibronectin is a ubiquitous protein with globular conformation in solution but that is organised into fibrillar structures through cell-mediated re-arrangement. This is a biologically regulated mechanism which involves integrin binding, reorganisation of the actin cytoskeleton and then, through cytoskeletal contraction, exertion of forces upon fibronectin to open up the molecule and present key sites for fibronectin–fibronectin interactions. There is a large body of research and excellent reviews about this physiological process of cell-mediated fibronectin fibrillogenesis.71,74
Cell-free routes to induce fibronectin fibrillogenesis have been proposed and strategies involve first unfolding of fibronectin dimers from their globular conformation, leading to fibronectin–fibronectin interactions and then fibril formation in absence of cells. Routes that have been proposed in the literature include the use of chemical agents (addition of reducing or oxidising agents to the protein solution; use of denaturing anionic compounds), use of peptidic fibronectin fragments; force-based assembly, via application of mechanical tension or shear forces; and surface-initiated assembly (see ref. 73 for a complete classification and discussion of published methods).
In the case of surface initiated fibronectin assembly, hydrophilic and negatively charged surfaces were shown to promote the extension of fibronectin upon adsorption, whereas hydrophobic surfaces provoked the disruption of the secondary structures of the protein.64,75 Pernodet et al. showed that interactions of sulphonated polystyrene with the III12–14 modules of fibronectin unfold the molecule and trigger the process of fibronectin assembly.76 Feinberg and Parker demonstrated that surface initiated assembly could be used to engineer multiscale, free-standing nanofabrics using a variety of ECM proteins (fibronectin, laminin, fibrinogen, collagens).77 Using much simple mechanisms, we showed that certain surface chemistries – in particular poly(alkyl acrylates) with length of side chain above two (e.g. ethyl, butyl, hexyl) have the ability to promote first the unfolding of fibronectin upon adsorption on the polymer surface and the fibronectin–fibronectin interactions leading to the organisation of fibronectin nanonetworks on the polymer surface (Fig. 7).78–80 We have shown that this assembly of fibronectin at the material interface depends on the underlying chemistry. E.g. spontaneous organisation does occur on PEA but does not on poly(methyl acrylate) PMA, and the degree of fibronectin assembly and nanonetwork interconnection can be controlled by copolymerising EA and MA (Fig. 7).81
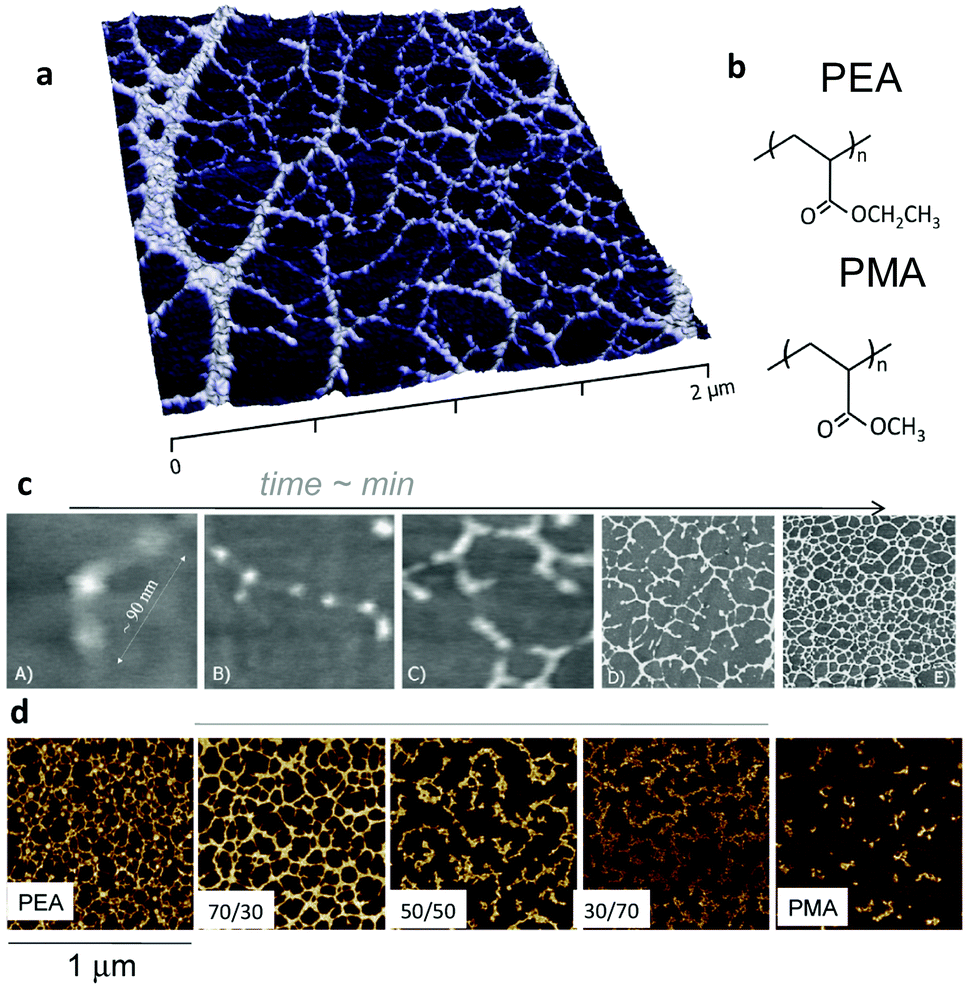 | ||
| Fig. 7 Material-driven fibronectin assembly on PEA. (a) AFM picture of fibronectin organised into nanonetworks on PEA after adsorption from a protein solution. (b) Chemical structure of PEA and PMA. Fibronectin assembly occurs on PEA but not on PMA notwithstanding chemical similarities. (c) Dynamics of fibronectin assembly on PEA followed by AFM. Individual fibronectin molecules are unfolded upon contact with the material surface (A) and fibronectin–fibronectin interactions occur (B and C) leading to the self-organisation into nanofibrils (D and E). (d) Co-polymerisation of ethyl acrylate and methyl acrylate in different rations result in controlled degree of assembly of fibronectin. Reproduced with permission from ref. 78 and 81. | ||
5.2. Protein assembly and GFs
It can be asked why the assembly of fibronectin fibrils is dependent upon the engineering of GF microenvironments? Fibronectin contains three kinds of domains which mediate interactions with other fibronectin molecules, other ECM molecules and cells (Fig. 8) as we have discussed.61 Particularly, we note the FNIII12–14 (heparin II binding region) is a highly promiscuous ligand for GFs.37 This region is located next to the integrin binding region (FNIII9–10) with consequence for synergistic integrin/GF signalling. To be exploited in cell engineering, however, the fibronectin peptide must be open, unfolded, involving the relevant regions (Fig. 8).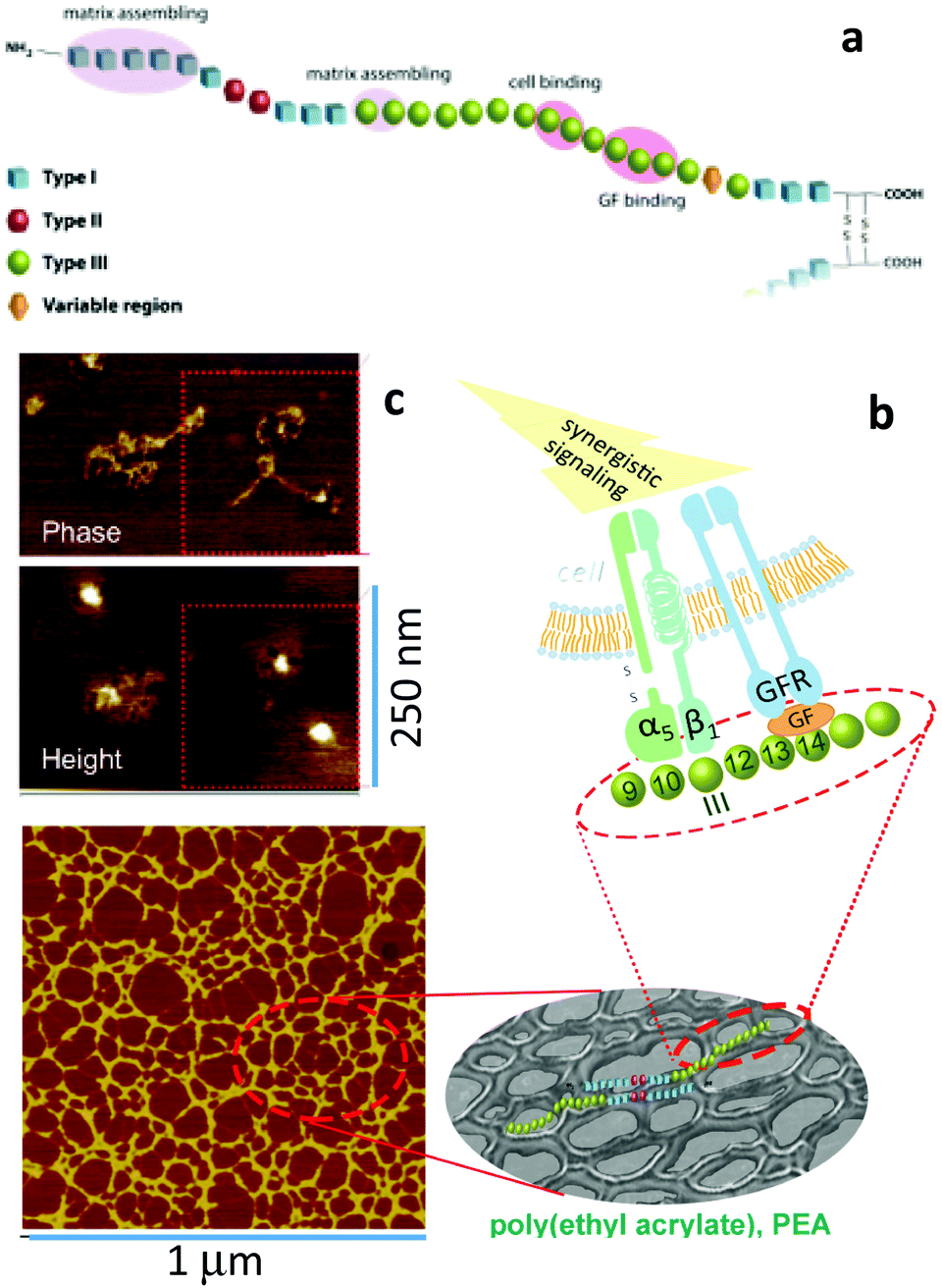 | ||
| Fig. 8 Material-driven fibronectin assembly. (a) Sketch of fibronectin which shows the three different kind of domains with particular emphasis on III9–10 (which contains RGD and site PHSRN to promote a5b1 integrin binding) and III12–15 (the GF binding site). (b) fibronectin assembly on the surface of PEA promote GF sequestration and then simultaneous integrin and GF receptor activation leading to synergistic signalling. (c) AFM images of individual fibronectin molecules unfolded on PEA with two GF molecules on top of them. An anti-GF antibody labelled with a gold nanoparticle was used to univocally identified BMP-2 (height image). Reproduced with permission from ref. 69. | ||
We showed that poly(ethyl acrylate) induces the spontaneous organisation of fibronectin molecules into nanonetworks79,80 and that this assembled fibronectin structure has the ability to present BMP-2 in synergy with α5β1 integrins, i.e., the molecule is open and the FNIII9–10 and III12–14 regions available. Indeed, we used AFM to show that BMP-2 directly bound fibronectin networks on PEA and then co-localisation of integrins and GF receptors leading to enhanced canonical Smad signalling (Fig. 8).69
Phenomenologically, the system used very low doses of BMP-2 (25 ng ml−1) to promote mesenchymal stem cell differentiation in vitro towards osteogenic lineages as well as bone regeneration in a critical size defect in the mouse radius (Fig. 9).69
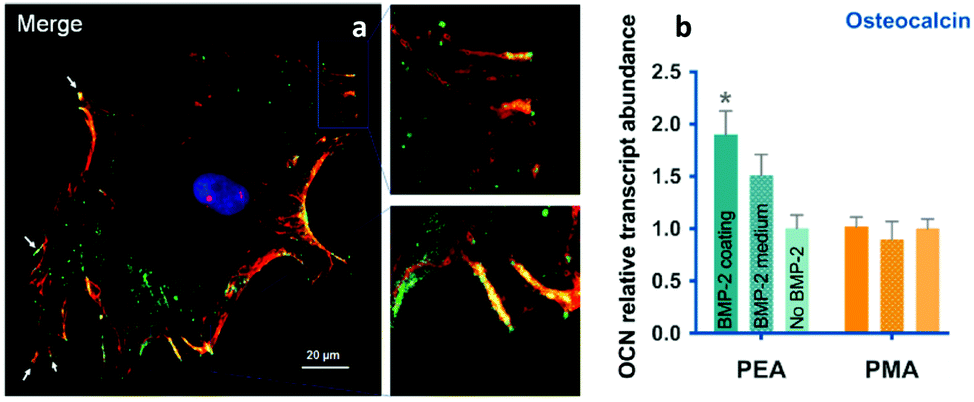 | ||
| Fig. 9 BMP-2 presentation from material-driven fibronectin matrices drives co-localisation of integrins (red) and GF receptors (green) in (a) and promotes MSC differentiation more efficiently than soluble delivery of BMP-2. Reproduced with permission from ref. 69. | ||
The ability of the PEA-controlled fibronectin presentation to drive paired integrin and BMP receptor localisation required to drive synergistic signalling was shown by co-immunoprecipitation and co-localisation of integrin β1, and the BMP-2 receptor, BMPR1a (Fig. 9). This synergistic GF effect was mediated by β1 integrins, in line with previous results using recombinant fibronectin fragments in fibrin gels62 and contrarily to results with BMP-2 in LbL films where αv was suggested to be the predominant protein – but please note that this is an area of debate.15 β1 and BMPR1a co-localisation had clear effects on subsequent cell signalling. It was shown that Smads were phosphorylated by BMPR1a and then translocate into the nucleus to activate RUNX2 and supported the conclusion that enhanced canonical BMP-2 signalling was a consequence of the simultaneous occupancy of integrins and BMP-2 receptors.
PEA provides a highly facile coating can be applied through several technologies to provide bioactive coatings of biomedical devices, including complex 3D geometries by spin coating, solvent casting and plasma polymerisation.82 This flexibility allows us to envisage using the technology to engineer synergistic GF microenvironments in a broad range of devices and implants.
6. Conclusions and outlook
While widely employed in clinic for e.g. bone repair, the use of GFs has been only partially successful and even controversial. Engineering synergistic integrin GF receptor systems has the potential to drastically reduce GF dose and topically deliver the GFs to the site of regenerative demand, maximising effects by targeting integrins and GF receptors in synergy. Furthermore, the GFs remain bound and localized to the material and so off target effects should be reduced.The use of GFs in biology and medicine has evolved from uncontrolled delivery to systems that release GFs upon demand (e.g. systems based in protein engineering that cleave GFs in dependence of the concentration of cell-secreted proteases). It is now accepted that presentation of GFs from ‘solid-phase’ is more efficient and somehow recapitulates the way GFs are used by nature in the ECM. A new paradigm has emerged in which the presentation of GFs can be engineered to promote crosstalk with integrins, driving integrin/GF receptor synergistic singling and maximising the effect of GFs at minimal GF concentrations. We envisage that the translation of these systems in clinical applications will require simple engineering to go through regulatory hurdles. We believe the use of simple polymers in combination with recombinant proteins will unlock translation in coming years.
Acknowledgements
Financial support from the European Research Council (HealInSynergy 306990) and the UK Medical Research Council (MR/L022710/1) are acknowledged. We thank Dr Aleixandre Rodrigo-Navarro for preparation of Fig. 3 and 4.Notes and references
- S. W. Crowder, V. Leonardo, T. Whittaker, P. Papathanasiou and M. M. Stevens, Cell Stem Cell, 2016, 18, 39–52 CrossRef CAS PubMed.
- M. J. Dalby, N. Gadegaard and R. O. Oreffo, Nat. Mater., 2014, 13, 558–569 CrossRef CAS PubMed.
- W. L. Murphy, T. C. McDevitt and A. J. Engler, Nat. Mater., 2014, 13, 547–557 CrossRef CAS PubMed.
- M. P. Lutolf and H. M. Blau, Adv. Mater., 2009, 21, 3255–3268 CrossRef CAS PubMed.
- S. Allazetta and M. P. Lutolf, Curr. Opin. Biotechnol., 2015, 35, 86–93 CrossRef CAS PubMed.
- G. S. Schultz and A. Wysocki, Wound Repair Regen., 2009, 17, 153–162 CrossRef PubMed.
- H. K. Kleinman, D. Philp and M. P. Hoffman, Curr. Opin. Biotechnol., 2003, 14, 526–532 CrossRef CAS PubMed.
- A. C. Mitchell, P. S. Briquez, J. A. Hubbell and J. R. Cochran, Acta Biomater., 2016, 30, 1–12 CrossRef CAS PubMed.
- K. Takahashi and S. Yamanaka, Cell, 2006, 126, 663–676 CrossRef CAS PubMed.
- E. J. Carragee, E. L. Hurwitz and B. K. Weiner, Spine J., 2011, 11, 471–491 CrossRef PubMed.
- R. O. Hynes, Science, 2009, 326, 1216–1219 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- J. J. Rice, M. M. Martino, L. De Laporte, F. Tortelli, P. S. Briquez and J. A. Hubbell, Adv. Healthcare Mater., 2013, 2, 57–71 CrossRef CAS PubMed.
- K. Lee, E. A. Silva and D. J. Mooney, J. R. Soc., Interface, 2011, 8, 153–170 CrossRef CAS PubMed.
- E. Migliorini, A. Valat, C. Picart and E. A. Cavalcanti-Adam, Cytokine Growth Factor Rev., 2016, 27, 43–54 CrossRef CAS PubMed.
- P. R. Kuhl and L. G. Griffith-Cima, Nat. Med., 1996, 2, 1022–1027 CrossRef CAS PubMed.
- M. M. Martino, P. S. Briquez, K. Maruyama and J. A. Hubbell, Adv. Drug Delivery Rev., 2015, 94, 41–52 CrossRef CAS PubMed.
- P. S. Briquez, J. A. Hubbell and M. M. Martino, Adv. Wound Care, 2015, 4, 479–489 CrossRef PubMed.
- P. S. Briquez, L. E. Clegg, M. M. Martino, F. Mac Gabham and J. A. Hubbell, Nat. Rev. Mater., 2016, 1, 15006 CrossRef.
- M. M. Martino, P. S. Briquez, E. Guc, F. Tortelli, W. W. Kilarski, S. Metzger, J. J. Rice, G. A. Kuhn, R. Muller, M. A. Swartz and J. A. Hubbell, Science, 2014, 343, 885–888 CrossRef CAS PubMed.
- P. M. Comoglio, C. Boccaccio and L. Trusolino, Curr. Opin. Cell Biol., 2003, 15, 565–571 CrossRef CAS PubMed.
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS PubMed.
- B. Geiger, A. Bershadsky, R. Pankov and K. M. Yamada, Nat. Rev. Mol. Cell Biol., 2001, 2, 793–805 CrossRef CAS PubMed.
- P. Kanchanawong, G. Shtengel, A. M. Pasapera, E. B. Ramko, M. W. Davidson, H. F. Hess and C. M. Waterman, Nature, 2010, 468, 580–584 CrossRef CAS PubMed.
- J. Liu, Y. Wang, W. I. Goh, H. Goh, M. A. Baird, S. Ruehland, S. Teo, N. Bate, D. R. Critchley, M. W. Davidson and P. Kanchanawong, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4864–E4873 CrossRef CAS PubMed.
- C. Grashoff, B. D. Hoffman, M. D. Brenner, R. Zhou, M. Parsons, M. T. Yang, M. A. McLean, S. G. Sligar, C. S. Chen, T. Ha and M. A. Schwartz, Nature, 2010, 466, 263–266 CrossRef CAS PubMed.
- D. W. Dumbauld, T. T. Lee, A. Singh, J. Scrimgeour, C. A. Gersbach, E. A. Zamir, J. Fu, C. S. Chen, J. E. Curtis, S. W. Craig and A. J. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9788–9793 CrossRef CAS PubMed.
- C. Gonzalez-Garcia, S. R. Sousa, D. Moratal, P. Rico and M. Salmeron-Sanchez, Colloids Surf., B, 2010, 77, 181–190 CrossRef CAS PubMed.
- D. Yadin, P. Knaus and T. D. Mueller, Cytokine Growth Factor Rev., 2016, 27, 13–34 CrossRef CAS PubMed.
- J. Nickel, W. Sebald, J. C. Groppe and T. D. Mueller, Cytokine Growth Factor Rev., 2009, 20, 367–377 CrossRef CAS PubMed.
- M. Ehrlich, Cytokine Growth Factor Rev., 2016, 27, 35–42 CrossRef CAS PubMed.
- J. Massague, Annu. Rev. Biochem., 1998, 67, 753–791 CrossRef CAS PubMed.
- C. H. Streuli and N. Akhtar, Biochem. J., 2009, 418, 491–506 CrossRef CAS PubMed.
- J. Ivaska and J. Heino, Annu. Rev. Cell Dev. Biol., 2011, 27, 291–320 CrossRef CAS PubMed.
- J. Veevers-Lowe, S. G. Ball, A. Shuttleworth and C. M. Kielty, J. Cell Sci., 2011, 124, 1288–1300 CrossRef CAS PubMed.
- I. Cascone, L. Napione, F. Maniero, G. Serini and F. Bussolino, J. Cell Biol., 2005, 170, 993–1004 CrossRef CAS PubMed.
- M. M. Martino and J. A. Hubbell, FASEB J., 2010, 24, 4711–4721 CrossRef CAS PubMed.
- A. Mettouchi, S. Klein, W. Guo, M. Lopez-Lago, E. Lemichez, J. K. Westwick and F. G. Giancotti, Mol. Cell, 2001, 8, 115–127 CrossRef CAS PubMed.
- M. F. Brizzi, G. Tarone and P. Defilippi, Curr. Opin. Cell Biol., 2012, 24, 645–651 CrossRef CAS PubMed.
- V. Sacchi, R. Mittermayr, J. Hartinger, M. M. Martino, K. M. Lorentz, S. Wolbank, A. Hofmann, R. A. Largo, J. S. Marschall, E. Groppa, R. Gianni-Barrera, M. Ehrbar, J. A. Hubbell, H. Redl and A. Banfi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6952–6957 CrossRef CAS PubMed.
- M. E. Davis, P. C. Hsieh, T. Takahashi, Q. Song, S. Zhang, R. D. Kamm, A. J. Grodzinsky, P. Anversa and R. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS PubMed.
- T. T. Chen, A. Luque, S. Lee, S. M. Anderson, T. Segura and M. L. Iruela-Arispe, J. Cell Biol., 2010, 188, 595–609 CAS.
- V. H. Fan, K. Tamama, A. Au, R. Littrell, L. B. Richardson, J. W. Wright, A. Wells and L. G. Griffith, Stem Cells, 2007, 25, 1241–1251 CrossRef CAS PubMed.
- T. Crouzier, L. Fourel, T. Boudou, C. Albiges-Rizo and C. Picart, Adv. Mater., 2011, 23, H111–H118 CrossRef CAS PubMed.
- K. Hauff, C. Zambarda, M. Dietrich, M. Halbig, A. L. Grab, R. Medda and E. A. Cavalcanti-Adam, Front. Bioeng. Biotechnol., 2015, 3, 62 Search PubMed.
- E. H. Schwab, T. L. Pohl, T. Haraszti, G. K. Schwaerzer, C. Hiepen, J. P. Spatz, P. Knaus and E. A. Cavalcanti-Adam, Nano Lett., 2015, 15, 1526–1534 CrossRef CAS PubMed.
- S. M. Anderson, S. N. Siegman and T. Segura, Biomaterials, 2011, 32, 7432–7443 CrossRef CAS PubMed.
- H. S. Azevedo and I. Pashkuleva, Adv. Drug Delivery Rev., 2015, 94, 63–76 CrossRef CAS PubMed.
- V. Delplace, J. Obermeyer and M. S. Shoichet, ACS Nano, 2016, 10, 6433–6436 CrossRef CAS PubMed.
- J. M. Silva, R. L. Reis and J. F. Mano, Small, 2016, 12, 4308–4342 CrossRef CAS PubMed.
- T. Crouzier, K. Ren, C. Nicolas, C. Roy and C. Picart, Small, 2009, 5, 598–608 CrossRef CAS PubMed.
- F. Gilde, O. Maniti, R. Guillot, J. F. Mano, D. Logeart-Avramoglou, F. Sailhan and C. Picart, Biomacromolecules, 2012, 13, 3620–3626 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- P. Roca-Cusachs, T. Iskratsch and M. P. Sheetz, J. Cell Sci., 2012, 125, 3025–3038 CrossRef CAS PubMed.
- S. S. Lee, E. L. Hsu, M. Mendoza, J. Ghodasra, M. S. Nickoli, A. Ashtekar, M. Polavarapu, J. Babu, R. M. Riaz, J. D. Nicolas, D. Nelson, S. Z. Hashmi, S. R. Kaltz, J. S. Earhart, B. R. Merk, J. S. McKee, S. F. Bairstow, R. N. Shah, W. K. Hsu and S. I. Stupp, Adv. Healthcare Mater., 2015, 4, 131–141 CrossRef CAS PubMed.
- C. J. Newcomb, S. Sur, S. S. Lee, J. M. Yu, Y. Zhou, M. L. Snead and S. I. Stupp, Nano Lett., 2016, 16, 3042–3050 CrossRef CAS PubMed.
- F. Bathawab, M. Bennett, M. Cantini, J. Reboud, M. J. Dalby and M. Salmeron-Sanchez, Langmuir, 2016, 32, 800–809 CrossRef CAS PubMed.
- E. A. Phelps, N. Landazuri, P. M. Thule, W. R. Taylor and A. J. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3323–3328 CrossRef CAS PubMed.
- A. Shekaran, J. R. Garcia, A. Y. Clark, T. E. Kavanaugh, A. S. Lin, R. E. Guldberg and A. J. Garcia, Biomaterials, 2014, 35, 5453–5461 CrossRef CAS PubMed.
- E. S. Wijelath, S. Rahman, M. Namekata, J. Murray, T. Nishimura, Z. Mostafavi-Pour, Y. Patel, Y. Suda, M. J. Humphries and M. Sobel, Circ. Res., 2006, 99, 853–860 CrossRef CAS PubMed.
- R. Pankov and K. M. Yamada, J. Cell Sci., 2002, 115, 3861–3863 CrossRef CAS PubMed.
- M. M. Martino, F. Tortelli, M. Mochizuki, S. Traub, D. Ben-David, G. A. Kuhn, R. Muller, E. Livne, S. A. Eming and J. A. Hubbell, Sci. Transl. Med., 2011, 3, 100ra189 Search PubMed.
- P. Carmeliet, Nat. Med., 2002, 8, 14–16 CrossRef CAS PubMed.
- A. J. Garcia, M. D. Vega and D. Boettiger, Mol. Biol. Cell, 1999, 10, 785–798 CrossRef CAS PubMed.
- S. Traub, J. Morgner, M. M. Martino, S. Honing, M. A. Swartz, S. A. Wickstrom, J. A. Hubbell and S. A. Eming, Biomaterials, 2013, 34, 5958–5968 CrossRef CAS PubMed.
- M. M. Martino, P. S. Briquez, A. Ranga, M. P. Lutolf and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4563–4568 CrossRef CAS PubMed.
- A. J. Garcia, J. E. Schwarzbauer and D. Boettiger, Biochemistry, 2002, 41, 9063–9069 CrossRef CAS PubMed.
- L. Fourel, A. Valat, E. Faurobert, R. Guillot, I. Bourrin-Reynard, K. Ren, L. Lafanechere, E. Planus, C. Picart and C. Albiges-Rizo, J. Cell Biol., 2016, 212, 693–706 CrossRef CAS PubMed.
- V. Llopis-Hernandez, M. Cantini, C. Gonzalez-Garcia, Z. A. Cheng, J. Yang, P. M. Tsimbouri, A. J. Garcia, M. J. Dalby and M. Salmeron-Sanchez, Sci. Adv., 2016, 2, e1600188 Search PubMed.
- R. Pankov, E. Cukierman, B. Z. Katz, K. Matsumoto, D. C. Lin, S. Lin, C. Hahn and K. M. Yamada, J. Cell Biol., 2000, 148, 1075–1090 CrossRef CAS PubMed.
- Y. Mao and J. E. Schwarzbauer, Matrix Biol., 2005, 24, 389–399 CrossRef CAS PubMed.
- M. Cantini, C. Gonzalez-Garcia, V. Llopis-Hernandez and M. Salmeron-Sanchez, ACS Symp. Ser., 2012, 1120, 471–496 CrossRef CAS.
- V. Llopis-Hernandez, M. Cantini, C. Gonzalez-Garcia and M. Salmeron-Sanchez, Int. Mater. Rev., 2015, 60, 245–263 CrossRef CAS.
- J. E. Schwarzbauer and D. W. DeSimone, Cold Spring Harbor Perspect. Biol., 2011, 3, a005041 CrossRef PubMed.
- B. G. Keselowsky, D. M. Collard and A. J. Garcia, J. Biomed. Mater. Res., Part A, 2003, 66, 247–259 CrossRef PubMed.
- N. Pernodet, M. Rafailovich, J. Sokolov, D. Xu, N. L. Yang and K. McLeod, J. Biomed. Mater. Res., Part A, 2003, 64, 684–692 CrossRef PubMed.
- A. W. Feinberg and K. K. Parker, Nano Lett., 2010, 10, 2184–2191 CrossRef CAS PubMed.
- D. Gugutkov, C. Gonzalez-Garcia, J. C. R. Hernandez, G. Altankov and M. Salmeron-Sanchez, Langmuir, 2009, 25, 10893–10900 CrossRef CAS PubMed.
- P. Rico, J. C. Rodriguez Hernandez, D. Moratal, G. Altankov, M. Monleon Pradas and M. Salmeron-Sanchez, Tissue Eng., Part A, 2009, 15, 3271–3281 CrossRef CAS PubMed.
- M. Salmeron-Sanchez, P. Rico, D. Moratal, T. T. Lee, J. E. Schwarzbauer and A. J. Garcia, Biomaterials, 2011, 32, 2099–2105 CrossRef CAS PubMed.
- H. Mnatsakanyan, P. Rico, E. Grigoriou, A. M. Candelas, A. Rodrigo-Navarro, M. Salmeron-Sanchez and R. Sabater i Serra, ACS Appl. Mater. Interfaces, 2015, 7, 18125–18135 CAS.
- M. Cantini, P. Rico, D. Moratal and M. Salmeron-Sanchez, Soft Matter, 2012, 8, 5575–5584 RSC.
| This journal is © The Royal Society of Chemistry 2016 |