
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite specific protein O-glucosylation with bacterial toxins†
Y.
Sun
,
L. M.
Willis
,
H. R.
Batchelder
and
M.
Nitz
*
Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
First published on 17th October 2016
Abstract
Using a MALDI-MS based assay, the kinetic parameters for peptide glucosylation using the C. difficile toxin B glycosyltransferase domain were determined. The minimum consensus sequence for glucosylation was YXXTXFXXY and the optimal peptide found was YAPTVFDAY. Using this sequence, homogenous glucosylated proteins could be readily produced.
Glycosylation is an important and prevalent protein post-translational modification.1 In recent decades, the chemical synthesis of glycoproteins has allowed the elucidation of the roles of glycoproteins and has also inspired numerous biotechnology applications.2–4
Impressive methods are now available for the chemical and chemoenzymatic synthesis of glycoproteins.2,5,6 However, the efficiency of these methods is low when compared to the recently, developed approaches using bacterial N-linked and O-linked glycosylation machinery to produce glycoproteins directly in bacteria.7–10 The enzymes involved transfer lipid-linked glycans, generated en masse in the cytoplasm, to protein targets in the periplasm. When this approach is combined with the intermediates of bacterial O-antigen biosynthesis the resulting conjugates are valuable as potential anti-bacterial vaccine components. However, the requirement to generate lipid-linked glycosyl donors and the localization of protein glycosylation in the periplasm makes it challenging to integrate these approaches with the impressive array of oligosaccharides that have been produced in the cytosol of engineered bacteria.11 We have devised an alternative strategy for glycoprotein production that uses the toxin B glucosyltransferase from Clostridium difficile to produce site-specific homogeneous glucosylated proteins. This approach will enable glycoproteins to be produced in the bacterial cytoplasm with endogenous nucleotide sugar donors. Once this glucosyl stub is in place on the protein, further elaboration with known glycosyltransferases would be possible. Although these products will contain an unnatural glycan–protein linkage, the properties of the glycan that can influence protein trafficking, in vivo stability or immunogenicity could be exploited.
Toxin B realizes its cytotoxic effects by inactivating small GTPases through α-glucosylation of conserved active-site threonine residues.12,13 The N-terminal glucosyltransferase domain of toxin B is a family 44 glycosyltransferase (http://www.cazy.org) which uses UDP-glucose as the donor sugar and can be overexpressed as active soluble enzyme in E. coli. In vitro analysis shows similar protein glucosylation activity in the isolated domain as in the full length toxin.14 Here, we report the discovery and characterization of a nine amino acid peptide substrate for the toxin B glucosyltransferase domain (GTD) and its applications as a protein tag for site-specific and homogenous glycoprotein engineering (Fig. 1).
 | ||
| Fig. 1 Glucosylation of peptide consensus on a protein of interest (POI) with C. difficile toxin B. | ||
A thorough proteomic study of toxin A/B substrates revealed that the amino acid sequences flanking the glycosylation site are similar among GTPase substrates.15 Inspired by this finding, we speculated that a short peptide containing the consensus sequence may be an acceptable substrate for the toxin B GTD. Two 13 amino acid peptides based on the Ras (FVDEYDPTIEDSY, 1) and Rac (FPGEYIPTVFDNY, 2) families of GTPases were selected as a candidate substrates (Table 1). The glucosylation reaction was evaluated by incubation of the peptides with toxin B GTD and excess UDP-glucose (Fig. 2). An excess UDP-glucose was necessary due to the high background rate of hydrolysis catalysed by the toxin B GTD.16 MALDI-MS analysis of the de-salted peptide 2 clearly showed a mass shift consistent with glucosylation (Fig. 2a). However no glucosylation of the Ras peptide 1 was observed in the MALDI-MS analysis. 4-Fluoro-phenylalanine (F*) was incorporated into 2 as it was anticipated that the changes in 19F nuclei NMR could be used as a reporter of toxin B activity.17 Analysis of 2 by 19F NMR showed a new 19F absorbance consistent with glucosylation at a single site within the product peptide (Fig. S1, ESI†).
| Peptide | Sequence | k cat (min−1) | K M (μM) | k cat/KM (min−1 μM−1) |
|---|---|---|---|---|
| F* – 4-fluorophenylalanine. Conditions as per Fig. 2 legend. NA – no activity observed. | ||||
| 1 | FVDEYDPTIEDSY | NA | NA | — |
| 2 | FPGEYIPTVF*DNY | 0.21 ± 0.01 | 103 ± 20 | 2.0 ± 0.4 |
| 3 | PGEYIPTVF*DNY | 0.27 ± 0.02 | 170 ± 40 | 1.6 ± 0.1 |
| 4 | PTVF*D | NA | NA | — |
| 5 | IPTVF*D | NA | NA | — |
| 6 | YIPTVF*D | 0.11 ± 0.01 | 380 ± 80 | 0.29 ± 0.07 |
| 7 | EYIPTVF*D | 0.19 ± 0.01 | 770 ± 60 | 0.25 ± 0.02 |
| 8 | YIPTVF*DNY | 0.63 ± 0.03 | 340 ± 40 | 1.9 ± 0.2 |
| 9 | YIPTVF*DN | 0.31 ± 0.01 | 1100 ± 70 | 0.28 ± 0.02 |
 | ||
| Fig. 2 MALDI as the method for kinetic studies. (a) Time course of reaction by MALDI-MS. 2 (100 μM) was incubated with UDP-Glc (10 mM) and toxin B (10 μM) in reaction buffer (50 mM HEPES, 100 mM KCl, 2 mM MgCl2, 1 mM MnCl2, pH 7.5) at 37 °C. ●: native 2; ○: glucosylated 2; (b) reaction time course as monitored by MALDI; (c) estimation of initial rate (d) Michaelis–Menten curve of 2. Error bars were obtained from three independent experiments. | ||
Initially we planned to follow the kinetics of glucosylation by 19F NMR. Due to the high background rates of UDP-sugar hydrolysis, we wanted a method that could directly quantify the glucosylated peptide rather than the generated UDP. However, signal broadening, potential inequivalent relaxation due to the enzyme requirement for Mn2+, and the low concentrations of substrates in the solution, made this analysis challenging. To avoid tedious HPLC analysis, we explored monitoring the reaction with MALDI-MS. MALDI-MS has proven to be superior to HPLC and NMR analysis of low molecular weight substrates, due to its speed, low sample consumption, and high sensitivity.18 As illustrated in Fig. 2, at a known concentration of peptide, the ratio of glucosylated and non-glucosylated product can be determined, giving a percent product formation. As the peptides were often observed as their [+H]+, [+Na]+, and [+K]+ adducts, the ion counts from all three species were summed.
Initial velocity was obtained from the linear fitting of concentration glycosylated 2vs. time (Fig. 2c). Repeating this process with various peptide concentrations provided the desired Michaelis–Menten plot and the corresponding kinetic parameters (Fig. 2d). The entire experiment is material and time efficient requiring only 8–10 μL of the reaction mixture. To obtain good average signals for each sample, 60 to 100 laser shots were measured and the final accumulated spectrum was used. This analysis is based on the assumption of similar ionization efficiencies for 2 and glucosylated 2. To further validate the method, we quantified the glycosylation rate of 2 at two concentrations by HPLC in parallel. The observed rates from the HPLC analysis was within 10% of the MALDI-MS analysis in both cases (Fig. S3, ESI†).
Given the success of 2 as a peptide substrate for toxin B GTD, we explored C- and N-terminal truncations to determine the minimal substrate length. Using the MALDI-MS assay, a library of seven truncated peptides was evaluated (Table 1). The longer peptides generally gave better kcat/KM values, however, 8, a nine amino acid long peptide had similar kcat/KM to 2.
Next, the key residues within peptide 10 were evaluated using alanine scanning (Table 2). Mutation of any of the three aromatic amino acids in 8 dramatically reduced or eliminated glucosylation, suggesting these residues are key to recognition of the peptide by toxin B GTD. In contrast, the N8A (12) and I2A (17) mutants displayed increased glucosylation efficiency (kcat/KM) by a factor of two. These results suggest there is some plasticity in recognition of these positions in catalysis. However, 12 and 17 improved the efficiency of glucosylation differently: 12 had substantially higher substrate turnover (kcat) and 17 had improved substrate recognition (KM). The remaining amino acid positions 3, 5 and 7 had minimal impacts on catalysis. Based on these preliminary studies a YXXTXFXXY consensus sequence is proposed for glucosylation. To examine the effect of combining both beneficial mutations in 12 and 17, peptide 19 was synthesized. The kcat/KM value of 19 is better than either I2A or N8A mutants alone with the increased efficiency mainly achieved by improving substrate recognition (KM).
| Peptide | Sequence | k cat (min−1) | K M (μM) | k cat/KM (min−1 μM−1) |
|---|---|---|---|---|
| 10 | Y1I2P3T4V5F6D7N8Y9 | 10 ± 3 | 2200 ± 1000 | 4.5 ± 2 |
| 11 | YIPTVFDNA | 0.95 ± 0.1 | 3000 ± 500 | 0.32 ± 0.06 |
| 12 | YIPTVFDAY | 17 ± 3 | 1900 ± 400 | 8.9 ± 2 |
| 13 | YIPTVFANY | 3.6 ± 0.7 | 840 ± 300 | 4.3 ± 1 |
| 14 | YIPTVADNY | NA | NA | — |
| 15 | YIPTAFDNY | 9.5 ± 2 | 2500 ± 600 | 3.8 ± 1 |
| 16 | YIATVFDNY | 1.0 ± 0.1 | 300 ± 80 | 3.3 ± 0.9 |
| 17 | YAPTVFDNY | 4.3 ± 0.5 | 480 ± 100 | 9.0 ± 2 |
| 18 | AIPTVFDNY | NA | NA | — |
| 19 | YAPTVFDAY | 1.9 ± 0.1 | 140 ± 20 | 13 ± 2 |
After developing promising peptide substrates for the toxin B GTD, we investigated the use of these peptides as protein tags for generating site-specific and homogeneous glycoproteins. Based on our kinetic results, we chose peptides 12, 17, and 19 as potential tag candidates. All three peptides were genetically fused to the C-terminus of maltose binding protein (MBP) after a thrombin-recognition site (Fig. S5, ESI†).
Purified tagged proteins, when treated with the toxin B GTD and UDP-Glc, were readily glucosylated as indicated by electrospray ionization (ESI) mass spectroscopy (Fig. S5, ESI†). The conversion of all three constructs exceeded 60%, with the most promising sequence being MBP-12 (Fig. S5, ESI†) with a glycosylation yield of approximately 80% based on ESI-MS (80%, Fig. S5, ESI†). Driven by these preliminary results, we optimized the glucosylation conditions on MBP-12. High glycosylation yield is required for the synthesis of homogenous glycoproteins, as purifying glycosylated proteins from non-glycosylated ones is challenging. Satisfyingly it was found by increasing the concentration of UDP-Glc (20 mM) and adding alkaline phosphatase led to glucosylated products of over 96% yield, as determined by ESI-MS (Fig. 3a and Fig. S9, ESI†). As incorporation of a peptide tag to the C-terminus is not always feasible, and to demonstrate the generality of our method, we prepared construct 12-MBP (Fig. 3b and Fig. S9, ESI†), which has peptide 12 fused directly to the N-terminus of MBP with no added linker. As demonstrated in Fig. 3b, all of the 12-MBP was converted to the mono-glucosylated form in the presence of the toxin B GTD. Under the same reaction conditions, no glycosylation of native MBP was observed, indicating that the site-specific glycosylation reaction was driven by peptide-based toxin B substrates (Fig. S5, ESI†).
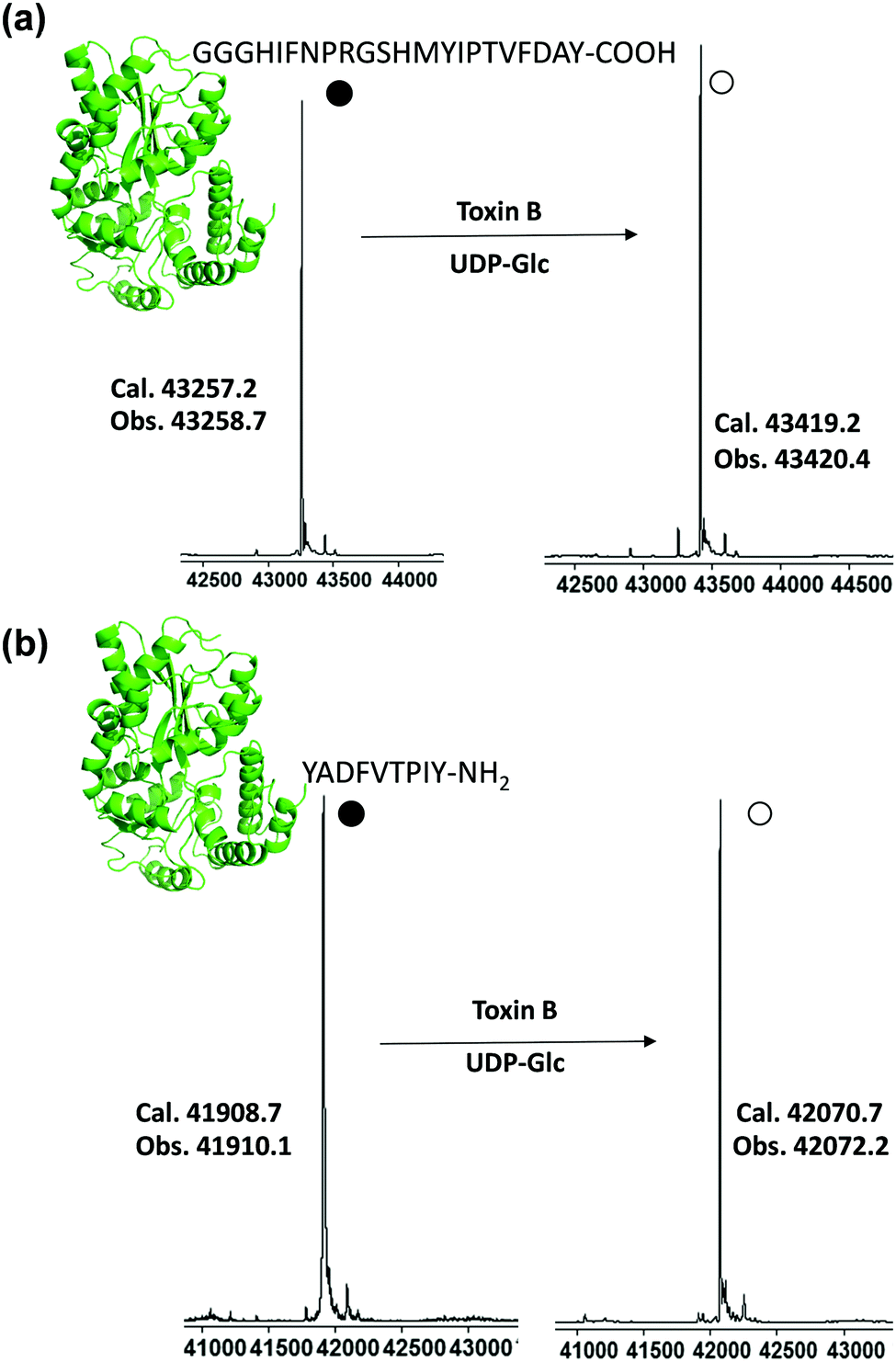 | ||
| Fig. 3 Glucosylation of MBP-tagged proteins. Protein constructs MBP-12 and 12-MBP (100 μM) were incubated with UDP-Glc (20 mM), toxin B GTD (10 μM), and alkaline phosphatase (5 units per mL) in buffer (50 mM HEPES, 100 mM KCl, 2 mM MgCl2, 1 mM MnCl2, pH 7.5) at 37 °C overnight. Glucosylation was evaluated by ESI-MS (a) glucosylation of C-terminal tag MBP-12; (b) glucosylation of N-terminal tag construct 12-MBP. | ||
Here we demonstrate bacterial toxins may be useful reagents for generating novel glycoproteins. Kinetic analysis, using MALDI-MS, has revealed the key residues in a nine amino acid consensus sequence. These peptides can be used as fusion tags to generate defined glycoproteins. Although this is an unnatural glycosylation it provides a method to generate the important glycan–protein linkage, for which there are few methods. We envisage elaborating the glucoside with known glycosyltransferases such as the Neisseria meningitidis β1–4GalT (NmLgtB) which has been shown to have a broad substrate tolerance for both α and β glucoside configured acceptors.19 This approach may allow for the oligosaccharides commonly found on glycolipids to be produced. Furthermore, it may be possible to alter the substrate specificity of the glucosyltransferase as mutants of toxin B which transfer GlcNAc have been reported.16
Special thanks to Roman Melnyk for supplying the TcdB GTD expression plasmid.20 This work was funded by the Natural Sciences and Engineering Research Council (NSERC).
Notes and references
- G. W. Hart and R. J. Copeland, Cell, 2010, 143, 672–676 CrossRef CAS PubMed.
- L. X. Wang and M. N. Amin, Chem. Biol., 2014, 21, 51–66 CrossRef CAS PubMed.
- R. J. Sola and K. Griebenow, BioDrugs, 2010, 24, 9–21 CrossRef CAS PubMed.
- Y. X. Zhu, X. M. Li, J. Kyazike, Q. Zhou, B. L. Thurberg, N. Raben, R. J. Mattaliano and S. H. Cheng, J. Biol. Chem., 2004, 279, 50336–50341 CrossRef CAS PubMed.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS PubMed.
- R. Okamoto, M. Izumi and Y. Kajihara, Curr. Opin. Chem. Biol., 2014, 22, 92–99 CrossRef CAS PubMed.
- M. F. Feldman, M. Wacker, M. Hernandez, P. G. Hitchen, C. L. Marolda, M. Kowarik, H. R. Morris, A. Dell, M. A. Valvano and M. Aebi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3016–3021 CrossRef CAS PubMed.
- M. Wacker, D. Linton, P. G. Hitchen, M. Nita-Lazar, S. M. Haslam, S. J. North, M. Panico, H. R. Morris, A. Dell, B. W. Wren and M. Aebi, Science, 2002, 298, 1790–1793 CrossRef CAS PubMed.
- C. Pan, P. Sun, B. Liu, H. Liang, Z. Peng, Y. Dong, D. Wang, X. Liu, B. Wang, M. Zeng, J. Wu, L. Zhu and H. Wang, mBio, 2016, 7, e00443 CrossRef PubMed.
- J. D. Valderrama-Rincon, A. C. Fisher, J. H. Merritt, Y. Y. Fan, C. A. Reading, K. Chhiba, C. Heiss, P. Azadi, M. Aebi and M. P. DeLisa, Nat. Chem. Biol., 2012, 8, 434–436 CrossRef CAS PubMed.
- J. Harle and S. Panke, Curr. Org. Chem., 2014, 18, 987–1004 CrossRef CAS.
- I. R. Vetter, F. Hofmann, S. Wohlgemuth, C. Herrmann and I. Just, J. Mol. Biol., 2000, 301, 1091–1095 CrossRef CAS PubMed.
- T. Jank, T. Giesemann and K. Aktories, Glycobiology, 2007, 17, 15r–22r CrossRef CAS PubMed.
- F. Hofmann, C. Busch, U. Prepens, I. Just and K. Aktories, J. Biol. Chem., 1997, 272, 11074–11078 CrossRef CAS PubMed.
- J. Zeiser, R. Gerhard, I. Just and A. Pich, J. Proteome Res., 2013, 12, 1604–1618 CrossRef CAS PubMed.
- T. Jank, T. Giesemann and K. Aktories, J. Biol. Chem., 2007, 282, 35222–35231 CrossRef CAS PubMed.
- M. A. Danielson and J. J. Falke, Annu. Rev. Biophys. Biomol. Struct., 1996, 25, 163–195 CrossRef CAS PubMed.
- K. D. Greis, Mass Spectrom. Rev., 2007, 26, 324–339 CrossRef CAS PubMed.
- K. Lau, V. Thon, H. Yu, L. Ding, Y. Chen, M. M. Muthana, D. Wong, R. Huang and X. Chen, Chem. Commun., 2010, 46, 6066–6068 RSC.
- J. Tam, G. L. Beilhartz, A. Auger, P. Gupta, A. G. Therien and R. A. Melnyk, Chem. Biol., 2015, 22, 175–185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Containing full experimental details and additional figures. See DOI: 10.1039/c6cc06223g |
| This journal is © The Royal Society of Chemistry 2016 |