
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular aggregates as sensory ensembles
Qian
Wang
*,
Zhao
Li
,
Dan-Dan
Tao
,
Qian
Zhang
,
Peng
Zhang
,
Dai-Ping
Guo
and
Yun-Bao
Jiang
*
Department of Chemistry, College of Chemistry and Chemical Engineering, MOE Key Laboratory of Spectrochemical Analysis and Instrumentation, and the Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), Xiamen University, Xiamen 361005, China. E-mail: littlewint@gmail.com; ybjiang@xmu.edu.cn
First published on 31st August 2016
Abstract
As a new emerging area in chemical sensing, sensing using supramolecular aggregates exhibits unique advantages over that using conventional small-molecule chemical sensors, in terms of high sensitivity and selectivity, and the simplicity of the sensory building blocks. This Feature Article outlines the recent research progress made in sensing based on induced supramolecular aggregation–disaggregation. The reviewed sensory building blocks, in general, in the form of a small molecular sensor, yet with a much simpler structure, which form aggregates, are those of perylene derivatives, pyrene derivatives, tetraphenylethylene derivatives, metallophilic species and metal–organic frameworks.
 From left to right: Yun-Bao Jiang, Qian Wang, Dan-Dan Tao, Zhao Li, Qian Zhang, Peng Zhang and Dai-Ping Guo | Dr Yun-Bao Jiang was born in central China and studied chemistry at Xiamen University, earning a PhD degree in 1990 under the supervision of late Professor Guo-Zhen Chen. Dr Jiang is currently a professor of chemistry, with research interests in chemo-/bio-sensing using supramolecular interactions which influence excited-state electron and/or energy transfer. He was awarded the distinguished young investigator grant by the NSF of China and has led innovation research teams financially supported, respectively, by the Ministry of Education and the NSF of China. He is currently on the board or the advisory board of Photochem. Photobiol. Sci., Supramol. Chem., MAF, and ACS Sensors. Dr Qian Wang obtained her PhD in 2016 for her work on aggregation in β-amyloids under the supervision of Prof. Jiang and Prof. H. Peter Lu from Xiamen University and joined Xiamen University as an engineer for FCS and fluorescence imaging, working together with Ms Dai-Ping Guo; Dr Zhao Li obtained her PhD in 2008 from Xiamen University and is currently an associate professor; Dan-Dan Tao, Qian Zhang and Peng Zhang are PhD candidates in Prof. Jiang's lab. |
Introduction
A molecular chemosensor in general consists of a molecular recognition site and a signal reporting group, which are either directly connected or linked by a spacer.1 In order to achieve high sensing performance, for instance, high sensitivity and selectivity, the sensory molecule needs to be sophisticatedly designed. Inspired by the chelating effect observed in coordination chemistry, such as that observed with metal–EDTA complexes in which four amino acid binding sites are covalently linked within the same EDTA framework, and the specific bio-recognition which relies on multiple weak and less selective interactions operating in a cooperative manner, sensing using aggregates that hold a more simply designed chemosensor(s) by noncovalent interactions was developed.2 Here the chemosensor-like building block itself may exhibit less effective performance than the otherwise molecular chemosensor would have done, yet in its aggregated state a comparable or better performance can be achieved. Supramolecular chemistry, landmarked by the 1987 Nobel Prize of chemistry awarded to Lehn, Cram and Pedersen,3 dealing with a complex of molecules that are held together by noncovalent bonds, has led to the buildup of diverse supramolecular architectures in a bottom-up approach. In a sense of employing noncovalent interactions, instead of covalent bonding, multidentate chelating ligands or enzymes are built in supramolecular chemistry by using one building block or a set of building blocks to form aggregates of them with the hope of achieving the chelating effect and/or the specificity in bio-recognition important for sensing.Indeed, supramolecular aggregation, driven by weak non-covalent interactions, such as van der Waals, π–π stacking, hydrogen/halogen bonding, metal coordination, electrostatic and even metallophilic interactions,4 has been utilized to construct sensing platforms with enhanced selectivity and sensitivity. Supramolecular aggregates as sensing ensembles are advantageous over the conventional small-molecule chemical sensors in the following aspects (Fig. 1). (i) The building blocks are assembled by means of relatively weak and dynamic noncovalent interactions, providing easy and facile approaches for manipulating supramolecular structures. (ii) The supramolecular sensing system can be easily made water compatible by introducing water-soluble substituents into the building blocks, which provides enormous possibilities for such recognition systems to be applied for biological analytes in the aqueous phase.5 (iii) The higher local concentration of the binding sites and the lower local concentration of water molecules around the binding sites in the aggregates substantially enhance the originally weak and water-susceptible interactions, which makes those otherwise insufficient binding events in water of the corresponding molecular sensors highly sensitive. Since in many cases the building block molecules are aromatic compounds with large π-systems, strong π⋯π stacking in the aggregates would make them conjugate-polymer like, affording an additional possibility for signal amplification.6 The relative orientations among the binding sites in the aggregates can be tuned good for analyte binding,7 resulting in high selectivity. (iv) In particular, the aggregation or disaggregation process may involve cooperative interactions that also result in signal amplification, leading to higher sensitivity and selectivity as well. Therefore, using supramolecular aggregates from building blocks with easy design and facile syntheses could achieve even better sensing performance than that of molecular sensory systems that rely on sensors with delicate structures.
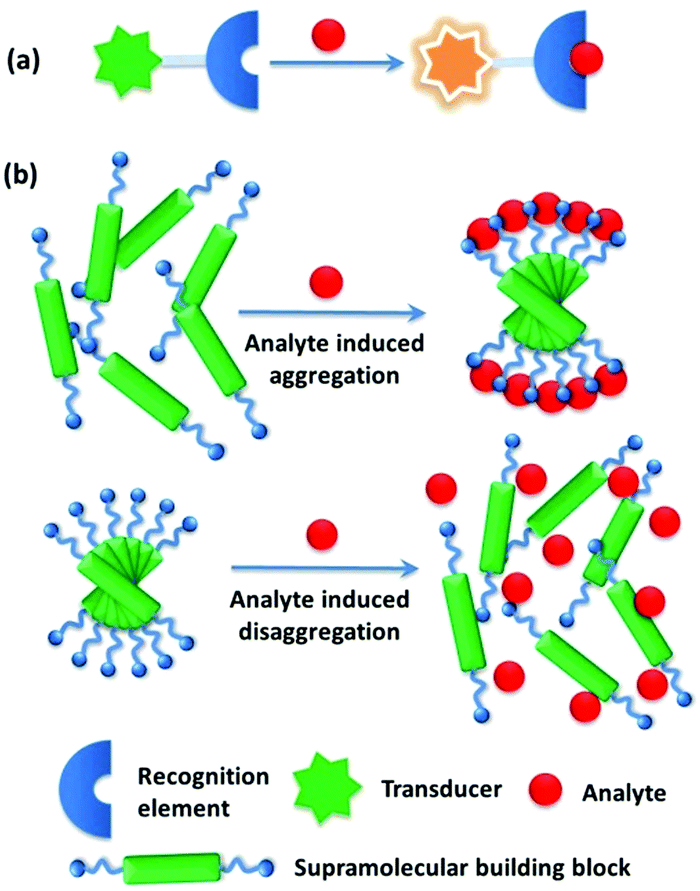 | ||
| Fig. 1 Comparison of (a) conventional sensory systems and (b) supramolecular sensory systems based on analyte induced supramolecular aggregation or disaggregation strategies. | ||
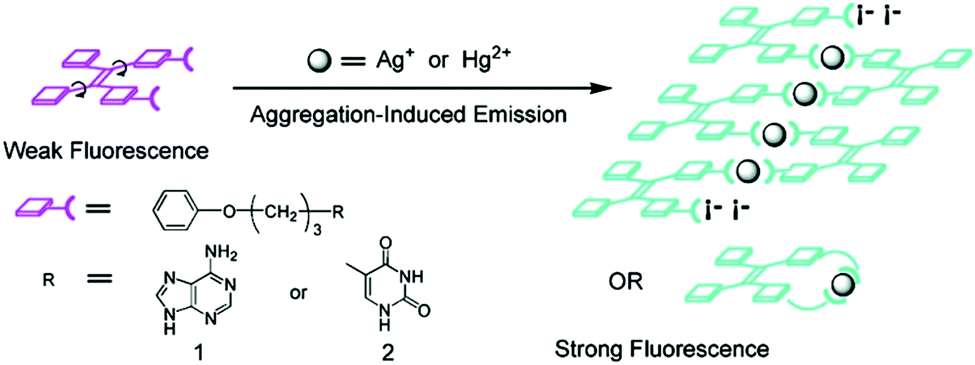
A variety of supramolecular sensing systems have been developed. Yet, regarding analyte induced aggregation or disaggregation strategies, considerable progress has been made only with limited categories of building blocks, which operate for sensing via π⋯π stacking modulated photophysics of π-conjugated dyes, aggregation induced emission, metallophilic interactions, and more recently those emerging from highly ordered metal–organic frameworks. We highlighted in 2012 supramolecular aggregation/disaggregation-based molecular sensing in China.8 With the rapid development of supramolecular sensing, it becomes necessary to update its current state of the art. In this Feature Article we accordingly review recent developments using supramolecular sensory ensembles consisting of the following five representative building blocks, i.e. perylene derivatives, pyrene derivatives, tetraphenylethylene derivatives, metallophilic species, and metal–organic frameworks (MOFs).
PBI derivatives
Derivatives of perylene dyes are extensively employed as molecular and supramolecular probes because of their strong absorptivity in the mid-visible spectral region, and thermal and photochemical stability.9 In particular, perylene-3,4:9,10-bis(dicarboximides) (PBIs) have attracted much attention as optimal fluorescent dyes and been intensively utilized in fluorescent switches, molecular transistors and sensors.10,11 They have been considered to be excellent fluorophores in sensor design because of their excellent electron accepting ability and strong fluorescence in the monomeric and small oligomeric states.12,13 Furthermore, the PBI structure and the planar π-electron deficient aromatic nature are known to strongly promote the formation of aggregates through stacking interactions between the π-conjugated cores in a face-to-face fashion, forming H-aggregates, which result in fluorescence quenching and a hypsochromic shift of the absorption.14,15 Thus, great efforts have been made to develop PBI based supramolecular sensing systems, by equipping a binding site into the PBI structures. Easy and versatile derivatization makes the PBI framework more attractive.Jiang's group in 2010 reported the Hg2+ selective PBI derivative 1 which bears an imide group similar to that in thymine (T) known to form the so-called “T–Hg2+–T” binding motif. Thus, Hg2+ was shown to be able to specifically induce aggregation of 1, leading to a dramatic fluorescence quenching of 1 and allowing for a highly sensitive and selective sensing of Hg2+. More importantly, it afforded a nonfluorescent chemosensing ensemble “(1-Hg)n” for thiol-containing species (Fig. 2).16 Addition of thiol-containing amino acids to the 1-Hg2+ ensemble solution induced dissociation of the aggregates and recovery of the fluorescence. For cysteine, a detection limit of 9.6 nM was achieved. It therefore represents a sensory system that works for both Hg2+ and thiol-containing species. Since both the aggregation and disaggregation processes are cooperative, high sensitivity and selectivity were observed, despite the otherwise weak interaction of Hg2+ with a simple imide group. This is the first report that shows that an imide group other than that from thymine could lead to a similar “T–Hg2+–T” binding motif for sensing applications.
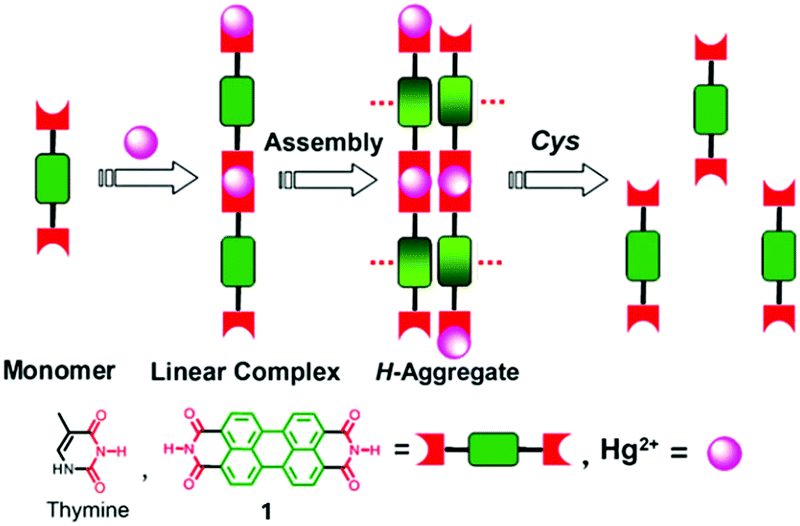 | ||
| Fig. 2 Hg2+ induced aggregation of 1 and dissociation of aggregates in the presence of cysteine. Reprinted with permission from Chem. Commun., 2010, 46, 4938–4940. Copyright 2010 RSC. | ||
An L-DOPA functionalized amphiphilic PBI 2 was reported in 2014 by Govindaraju et al.,17 as a reversible fluorescence switching probe for the detection and sensing of cationic surfactants and Fe3+/Cu2+ in aqueous solutions, following, respectively, the host–guest interactions driven aggregation and disaggregation of 2. Photophysical studies of 2 showed that the compound existed as a monomer in dimethyl sulfoxide (State-I), while in pure aqueous solutions in the form of self-assembled aggregates, displaying very weak fluorescence along with red-shifted broad absorption (State-II). Interestingly, the cationic surfactant cetyltrimethylammonium bromide (CTAB), but not neutral and anionic surfactants, could disassemble 2 under micellar conditions, restoring the bright yellow fluorescence and vibronically well-defined absorption bands of 2 (State-III). 2 was able to sense Fe3+ and Cu2+ owing to the metal chelating capacity of its DOPA moiety, so that metal binding drove the assembly of 2 (State-IV). Fluorescence of 2 was quenched by Fe3+ and Cu2+ by 3.5 and 2.5-fold, respectively. Such metallosupramolecular aggregates were found to revert back to the fluorescence “ON” state by the introduction of a metal ion chelator, diethylene triamine pentaacetic acid (DTPA, State-III), further signifying the role of metal ions toward the assembly of 2 (Fig. 3).
 | ||
| Fig. 3 Schematic representation of solvent driven molecular organization of 2 and transformation of aggregated State-II of 2 to molecularly dissolved State-III in the presence of CTAB micelles, and the transformation of State-III into State-IV via metal ion induced reorganization of 2 in the presence of CTAB micelles. Reprinted with permission from ACS Appl. Mater. Interfaces., 2014, 6, 21369–21379. Copyright 2014 ACS. | ||
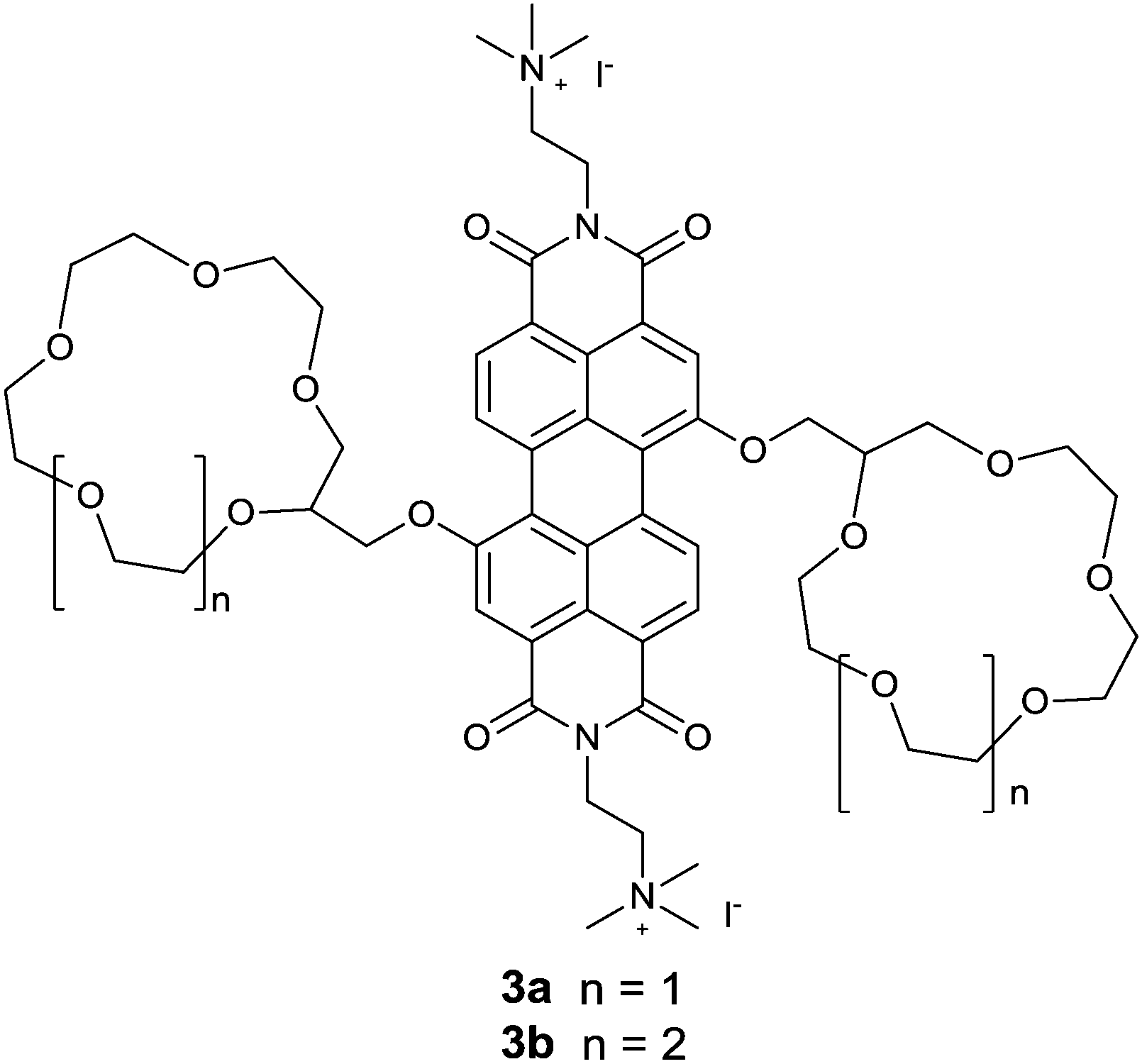
Würthner et al.18 reported an interesting metal templated assembly of crown ether functionalized PBIs 3a and 3b. The templated self-assembly of these PBIs was shown to be dependent on the size of the crown ether receptor and the nature of metal ions. Ba2+ selectively templates the assembly of 15-crown-5 functionalized PBI 3a into a dimeric PBI–Ba2+ complex through a positive homotropic cooperative binding, so that the fluorescence of 3a is efficiently and selectively quenched by Ba2+, whereas it is not quenched or quenched very little by the other alkali and alkaline earth metal ions such as Na+, K+, Rb+, Mg2+ and Ca2+. These results show the high selectivity of Ba2+ templating of the assembly of PBI, giving a binding constant of (1.51 ± 0.04) × 105 M−1 for Ba2+, despite the fact that 15-crown-5 is Na+ selective. In contrast, both absorption and emission of the 18-crown-6 functionalized PBI 3b were not influenced by those metal ions of up to 7 equivalents. This observation suggests the vital importance of the size of the crown ether receptor in the metal ion templated assembly of the PBI derivatives, actually implying the subtle balance of multiple interactions involved in the templating and advantages of the supramolecular sensing.
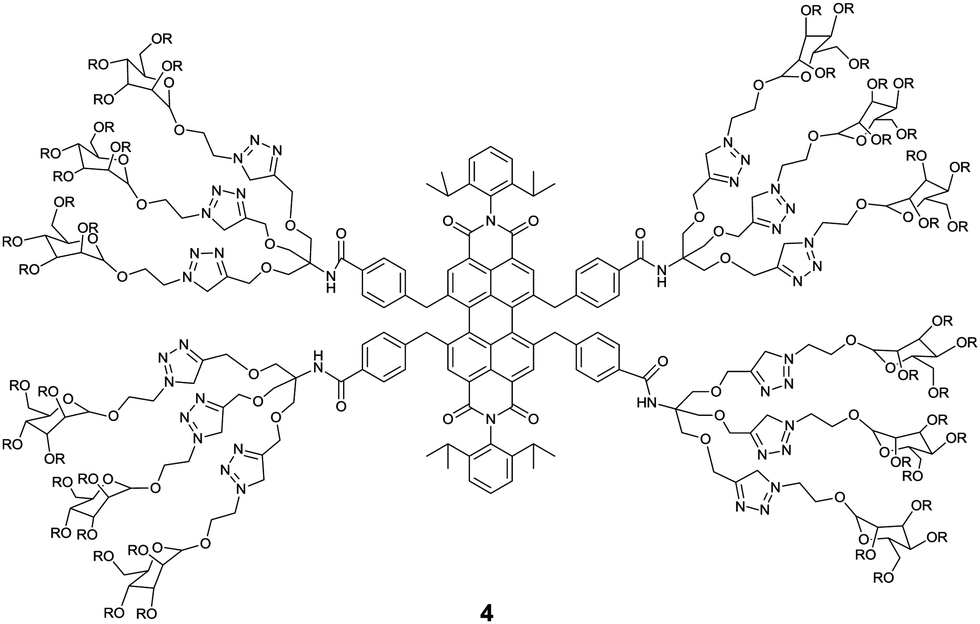
The essential role of carbohydrate–protein interactions in maintaining diverse biological processes has attracted much attention to develop carbohydrate-based sensors for these interactions. Among them, fluorescent glycoclusters hold great potential because of their intrinsic fluorescence, high sensitivity to minor external stimuli and good biocompatibility. A water-soluble mannose functionalized PBI 4, developed by Wang et al.,19 exhibited a specific response towards Concanavalin A (Con A) by ca. 3 times fluorescence enhancement, resulting from disaggregation of 4 in the presence of Con A. Whereas peanut agglutinin (PNA) and bovine serum albumin (BSA) led to no significant changes in the fluorescence of 4. The binding constant of 4 with Con A, (9.87 ± 0.3) × 106 M−1, is three orders of magnitude higher than that (8.2 × 103 M−1) with the monosaccharide ligand in Con A. Fluorescence imaging of 4 for macrophage cells also demonstrated that the sensor could be applied as a protein-specific probe for diagnosis and molecular imaging under live cell conditions.
Amphiphilic PBI dyes 5 and 6 functionalized with para- and ortho-phenylboronic acid moieties, for interacting with 1,2-/1,3-cis-diol to form a cyclic boronate ester, were designed as chirality sensors for α-hydroxycarboxylates.20 The achiral compounds 5 and 6 existed as dimers or small oligomers in the absence of an α-hydroxy carboxylate, yet formed large aggregates upon binding it, resulting in a decrease in the absorption of the perylene chromophore around 500 nm. Strong induced Cotton effects were observed in the absorption window of the achiral perylene chromophore and were utilized for enantiomeric excess (ee) determination and discrimination of seven α-hydroxy carboxylates that bear no chromophore, with an average absolute error of 2% in ee determination and 100% correctness in analyte identification. Note that the boronic acid group that interacts with the chiral analyte is located far away from the chromophore, yet an effective chirality induction was shown by the coupled exciton CD signals from the latter. This example highlights the potential of using induced helical aggregation of achiral dyes for CD measurement of the ee of those chiral analytes bearing no chromophore. In principle it should also work for a chiral species that bears a chromophore but is far away from the chiral centre. This expands the scope of applications in chirality sensing of classic CD spectroscopy that normally requires the presence of a chromophore next to the chiral centre for direct measurements. It is the aggregation of the achiral building blocks upon interacting with the chiral species that leads to the transformation of the molecular chirality into supramolecular structures.
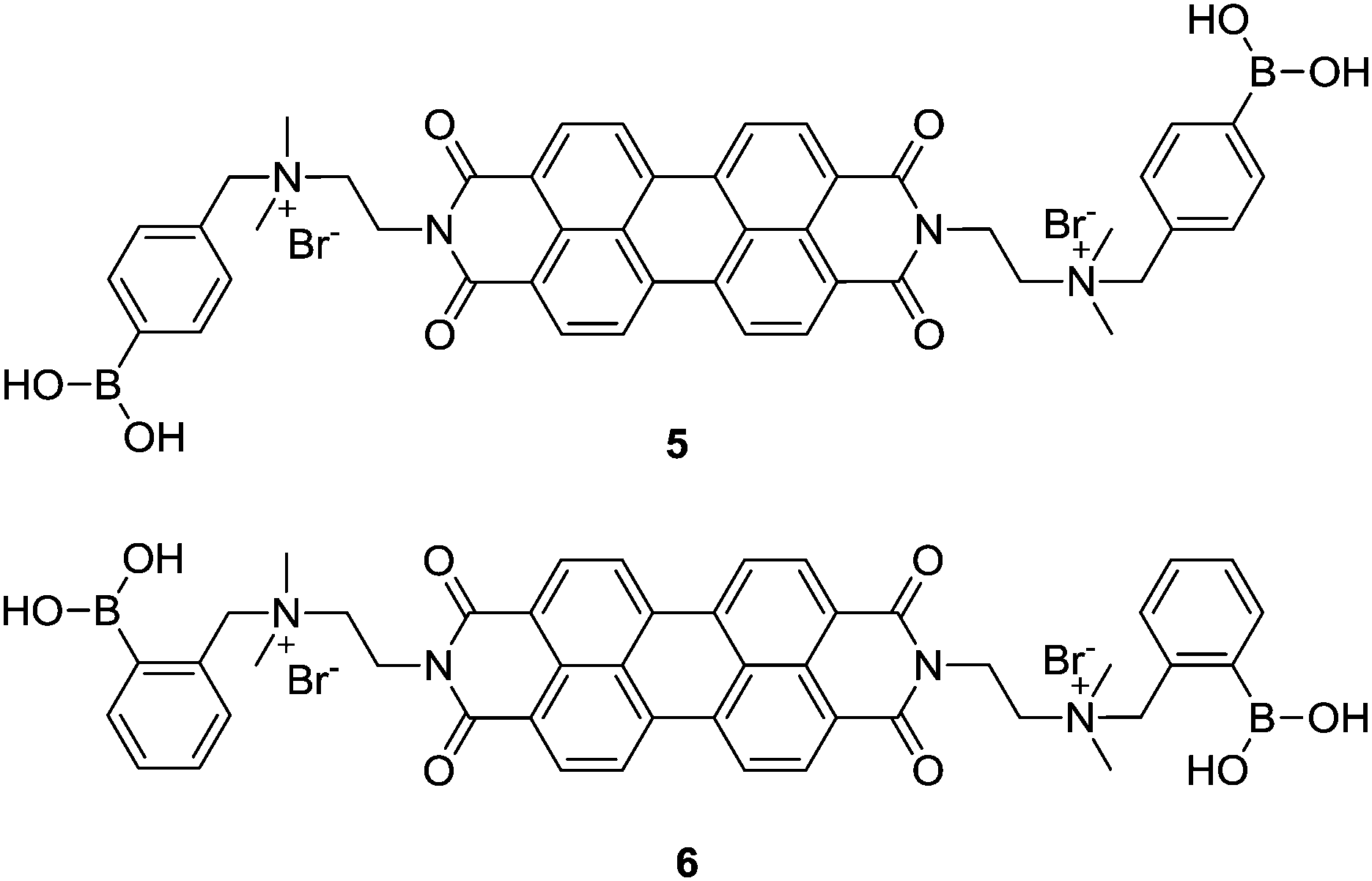
Molecular sensors for analytes with multiple functional groups are normally designed to have several binding sites that are well arranged to match the structures of the analytes, to achieve high sensitivity and selectivity. In terms of the supramolecular sensory ensemble, a set of monofunctional building blocks may form co-aggregates that thereby bear multiple binding sites as well and thus function similarly to the molecular sensors that contain a set of well-defined binding sites, yet greatly simplify the structure and syntheses of the building blocks. It should be emphasized that achieving a synergistic effect to overcome the entropic cost of forming such multicomponent co-aggregates is challenging. A successful example for sensing L-DOPA (7) was recently reported, employing the induced aggregation of a mixture of boronic acid and aldehyde functionalized PBIs 5 and 8.21 Here the boronic acid group in 5 was expected to interact with the cis-diol moiety in L-DOPA, while the aldehyde group in 8 was expected to form an imine bond with the amine group in L-DOPA, both being reversible and dynamic. The three-component 5–L-DOPA–8 complex formed at low concentrations in aqueous solutions underwent aggregation, exhibiting a strong synergistic effect between the two orthogonal dynamic covalent interactions. The CD-based sensing of 7 at a micromolar concentration level can be carried out by simply mixing with two simple building blocks 5 and 8. The high affinity towards 7 results from the stabilization by aromatic π-stacking between 5 and 8. The divalent binding of 7 to the 5–8 ensembles results in the formation of long fibrous PBI aggregates with a significantly larger size and stronger induced CD signals compared to the aggregates formed, respectively, by the 7–5 and 7–8 complexes in which only monovalent binding of 7 is possible. This report confirms that using a mixture of building blocks bearing different binding groups could indeed operate, at least to the same extent, as the molecular sensor that bears all those binding groups, since these much more easily designed and facilely synthesized building blocks may function cooperatively. This is important since many analyte targets may have more than one functional group so that the molecular sensors could be structurally complicated to ensure high binding efficiency.

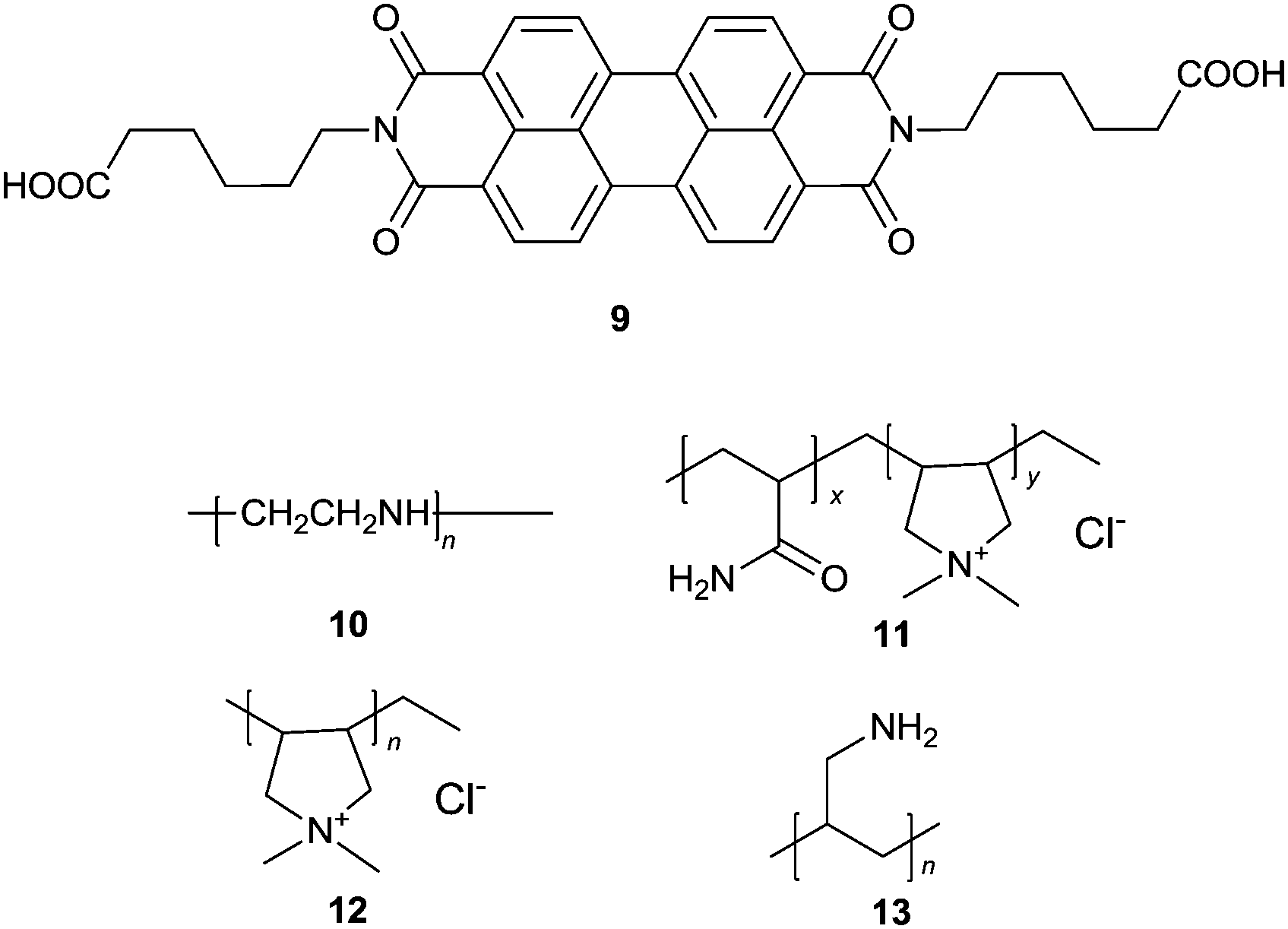
Wang et al.22 proposed a label free ultrasensitive fluorescence turn-on sensing scheme for DNA, using the cationic polyelectrolytes-induced aggregation of the negatively charged PBI dye 9 that is shown by the severe quenching of the emission of 9. The tested four cationic polymers 10–13 could induce almost complete quenching of the fluorescence of 9, but with varying efficiency as indicated by the concentrations of the polymers required to fully quench the emission, 300, 1150, 175, 50 nM, respectively. When a single stranded DNA (a polyanion) was added to the solution of the cationic polymer–9 aggregates, strong electrostatic interactions between the single stranded DNA and the cationic polymers released 9 from the aggregates, recovering the emission of 9. Aggregates 12–9 exhibited the best fluorescence recovery up to 89.8%, by only 1.5–2 nM DNA in ca. 5 minutes. This smart scheme allows a highly sensitive assay, with a limit of the detection of 2 pM for DNA. Based on the fact that alkaline phosphatase (ALP) could remove the phosphate group of the 5′-phosphorylated single-stranded DNA, thus preventing the DNA sample from being degraded by λ-exonuclease, an assay for the highly sensitive detection of ALP activity was established using the 12–9 supramolecular sensory ensemble. Upon mixing with the DNA samples, the fluorescence of the aggregates 12–9 was turned on, reaching a limit of detection down to 0.02 mU mL−1. This represents the rare example of induced disaggregation of sensory building blocks, releasing them in an amplified manner that ensures high sensitivity.
Pyrene derivatives
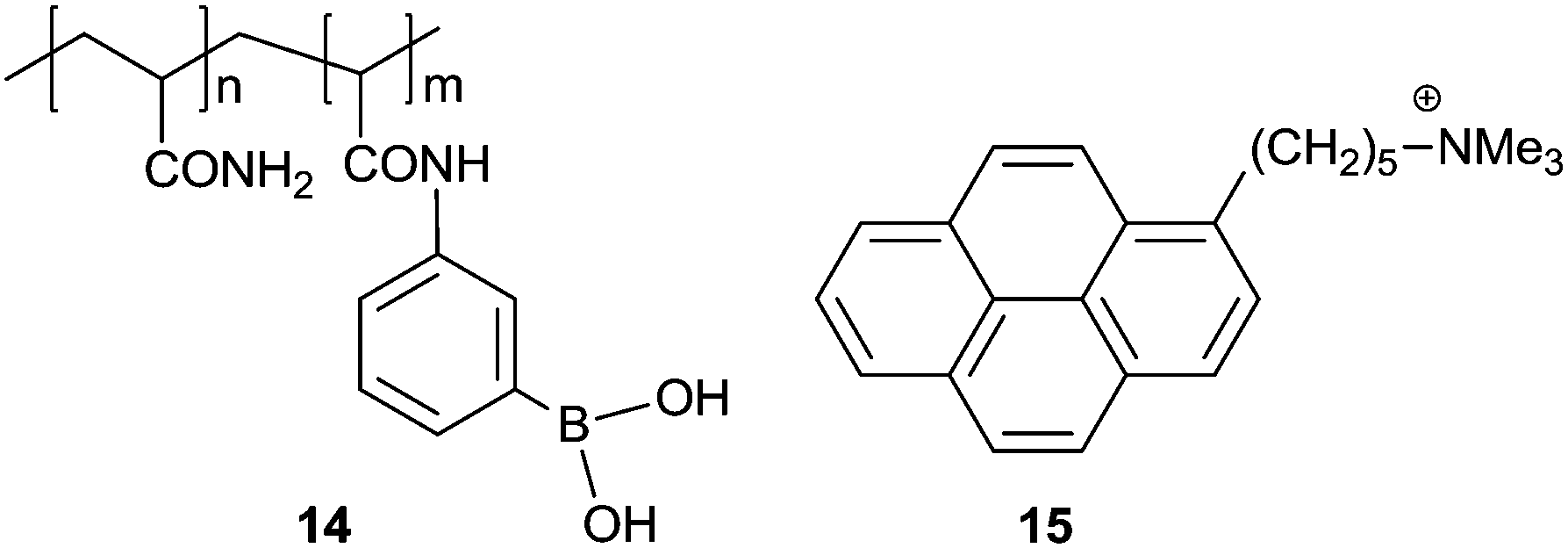
As a simple aromatic fluorophore, pyrene has a large extinction coefficient, high quantum yield and good stability in aqueous solutions. Pyrene is known to form an excimer, due to its long lifetime of 102 ns orders of magnitude and its strong tendency for π⋯π stacking, thereby emitting structureless excimer fluorescence red-shifted by ca. 100 nm from the monomer emission. Pyrene has thus been used for a long time to design building blocks for supramolecular aggregates that form in the presence of an analyte.Yam's group23 in 2009 developed a pyrene-based boronic acid fluorescence sensing ensemble for glucose, in which (i) the water-soluble boronic acid functionalized polymer 14 transformed into a polyanion upon binding with glucose and (ii) the positively charged pyrene derivative 15 electrostatically attached along the polymer backbone, in the aggregated form as indicated by the strong excimer emission at 492 nm. A linear response was found for glucose between 0 and 17 mM. Among a number of tested sugars, glucose produced the strongest excimer emission, which is likely due to the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 glucose–boron acid complexes that bring two pendant boronic acid moieties into close proximity, thus generating two adjacent negative charges which favour the aggregation of positively charged 15.
:2 glucose–boron acid complexes that bring two pendant boronic acid moieties into close proximity, thus generating two adjacent negative charges which favour the aggregation of positively charged 15.
An aggregation-based glucose sensor 16 developed by Jiang's group clearly manifests the multivalent effect, with higher apparent affinity to glucose than to fructose, in such a simple phenylboronic acid derivative containing a pyrene fluorophore (Fig. 4).24 In an alkaline (pH 10) aqueous solution 16 at 0.1 mM concentration exists in small aggregates that emit only pyrene monomer fluorescence at 390 nm. Glucose binding leads to more ordered aggregates of 16, since one glucose molecule binds two boronic acid groups, thus the resultant complex is more hydrophobic, enabling the observation of pyrene excimer emission. With fructose, 1:1 binding stoichiometry with the boronic acid group results in a neutral zwitterionic boronate ester of higher hydrophobilicity, however, electrostatic repulsions between the zwitterionic boronates lead to disordered aggregates of 16, unfavourable for excimer formation. Despite the simple phenylboronic acid structure, 16 in its aggregate state exhibits a highly selective and sensitive response toward glucose against fructose, opposite to what is known for phenylboronic acid that shows preferential binding to fructose.
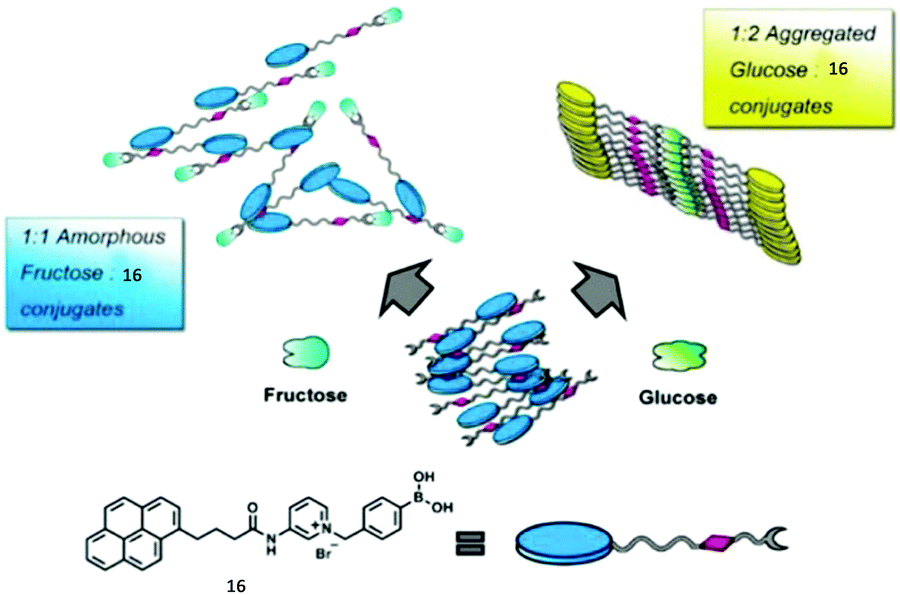 | ||
| Fig. 4 Cartoon illustration of the aggregates formed, respectively, from the 1:1 fructose complex and the 1:2 glucose complex with 16. Reprinted with permission from J. Am. Chem. Soc., 2013, 135, 1700–1703. Copyright 2013 ACS. | ||
Sensing of the fluoride ion in aqueous solutions using a boronic acid based sensor has been challenging because of the high hydration enthalpy of the fluoride ion and the severe interference from the hydroxide anion. Jiang's group reported25 a supramolecular amphiphilic pyrene-based boronic acid sensor for direct fluoride sensing in aqueous solutions (Fig. 5). Binding of the fluoride anion to the boronate ester of 16 and protocatechuic acid (PCA) triggered the formation of supramolecular aggregates of ester 17, leading to presumably lower local concentration of water and higher local density of binding sites, which enhance the fluoride affinity and therefore the selectivity against OH−. To enhance the Lewis acidity of boronic acid toward the fluoride ion and reduce the interference of OH−, PCA was chosen to interact with 16 to form ester 17 which aggregates under highly acidic conditions (pH ≤ 2), ruling out the interference of OH−. The excimer emission of 16 undergoes a dramatic enhancement upon addition of fluoride ions, together with an increase in solution turbidity, indicative of supramolecular aggregation, allowing sensitive detection of fluoride at the ppm level in aqueous solutions. The fact that the addition of CTAB micelles leads to a dramatic quenching of the excimer emission supports the conclusion that aggregation strengthens the boron–fluoride interaction in aqueous solutions. In the presence of 10 mM PCA, an apparent binding constant for fluoride at 103 M−1 orders of magnitude was obtained, a high value unprecedented for boronic acid sensors in water.
 | ||
| Fig. 5 Schematic representation of fluoride sensing by the 16-PCA boronate ester 17. Reprinted with permission from Chem. Commun., 2014, 50, 13987–13989. Copyright 2014 RSC. | ||
Tetraphenylethylene derivatives
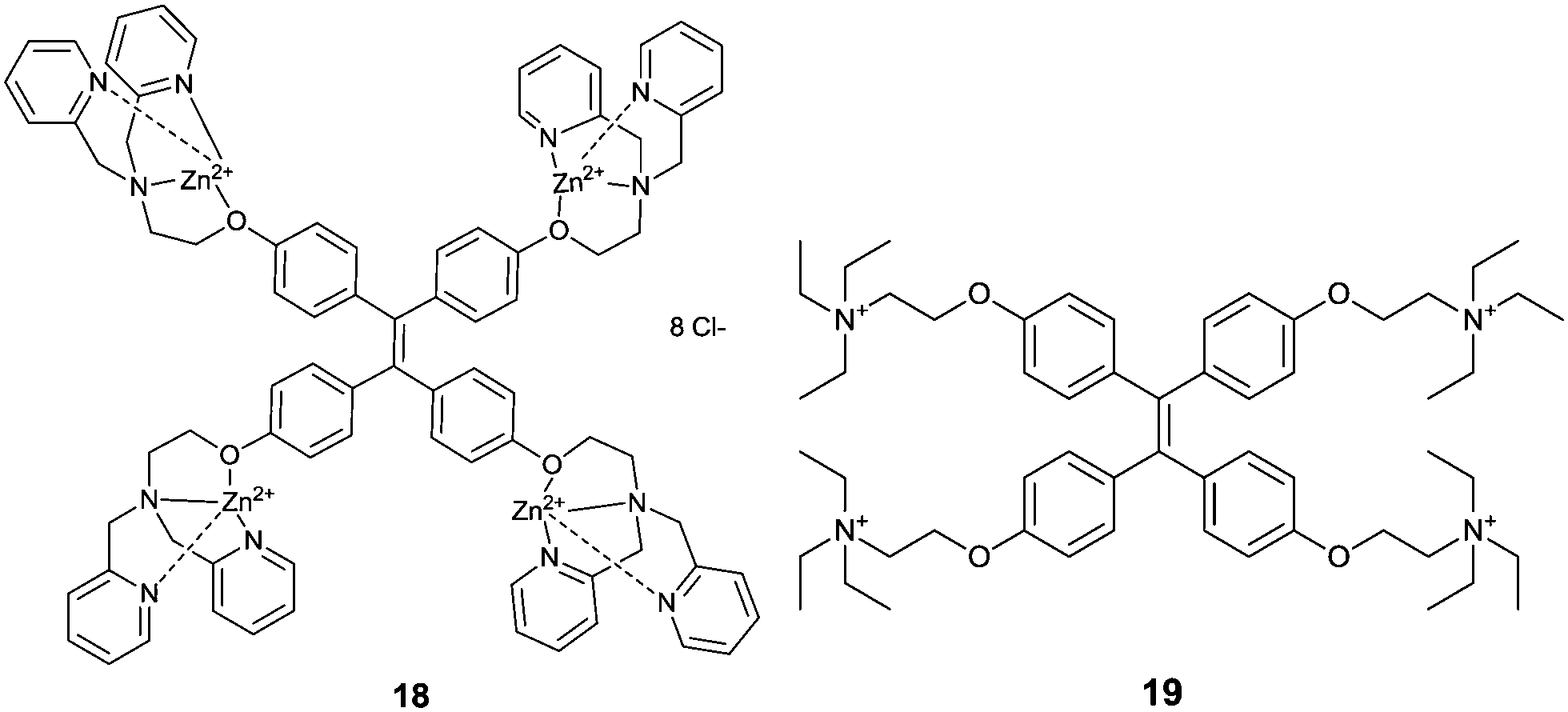
The concept of aggregation induced emission (AIE)26 or aggregation induced enhanced emission (AIEE)27 has been proposed and well received as a new promising strategy for creating potential fluorescence sensors. Compared with the typical aggregation-caused quenching (ACQ) effect which is unfavourable for chemical sensing in terms of sensitivity, AIE holds great potential in expanding the applications of traditional organic luminophores into new “turn-on” luminescence chemo-/bio-sensors. Tang's group28 was the first to report the AIE-active molecules, represented by the star molecule TPE, tetraphenylethene. TPE is non-emissive in the dissolved state, whereas it turns highly emissive in the aggregated state. Tang et al.26,28,29 attributed the AIE to the block of the radiationless pathways by the restriction of intramolecular rotations (RIR) in the aggregated state.Yang et al.30 prepared a TPE-based sensor 18 for DNA, employing the coordination of Zn2+ with the tridentate ligands substituted in the TPE core that results in a positively charged TPE derivative. Upon the addition of a 10 nt ssDNA (X10), a significant fluorescence enhancement in H2O/DMSO (99:1, v/v) was observed. The complementary sequence DNA of X10, Y10, and the hybridized dsDNA of X10/Y10 was also tested and fluorescence enhancement to a similar extent was shown for Y10 but slightly less for X10/Y10, despite the same emission maximum at 465 nm. The control TPE molecule 19, interacts with DNA in an electrostatic manner; its fluorescence enhancement is in general weaker than that observed for 18-Zn2+, suggesting that the coordination of Zn2+ with ligands in 18 contributes cooperatively with the electrostatic interactions of the cationic complex that forms to the total interaction of 18 with DNA.
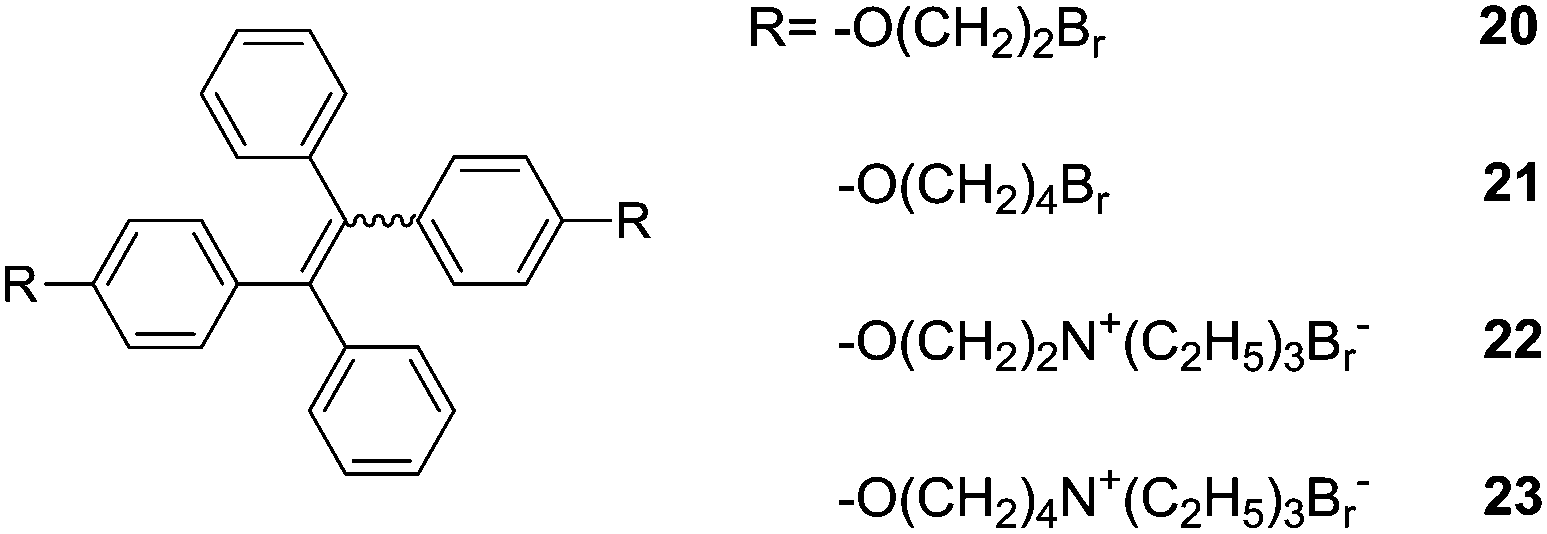
Tang et al.31 reported a set of AIE-active TPE derivatives 20–23. Cationic salts 22 and 23 are soluble in water and in DMSO and DMF, as well; 22 and 23 were tested as sensors for protein and DNA, by means of hydrophobic and electrostatic interactions. In dilute solutions of 20 and 21 in acetonitrile (AN) they are practically nonfluorescent, whereas they become highly fluorescent in 1:99 (v/v) AN/water solution. The intensity of 20 at 476 nm is increased by 240-folds and its absorption shifted to red from 310 nm in AN to 330 nm in the suspension. Similar observations were made with 21, therefore, both 20 and 21 are AIE-active. Because of the amphiphilic nature of 22 and 23, the addition of organic solvents such as methanol, AN, THF and 1,4-dioxane into their aqueous solutions did not cause salting-out or aggregation of them. However, their fluorescence quantum yields increased with increasing concentration, suggesting that 22 and 23 are AIE-active too. Calf thymus (ct) DNA or bovine serum albumin (BSA) were shown to enhance the fluorescence of 22 and 23 in aqueous phosphate buffer of pH 7.0 at 25 °C and red-shift its emission to 462 nm, which differs from its absorption maximum by 134 nm. 23 is more sensitive for the detection of DNA and BSA, for example, its fluorescence is enhanced by 16.3 and 23.8 times by 300 μg mL−1 DNA and 500 μg mL−1 BSA, respectively. These two systems of 18-Zn2+ and 22/23 can be similarly understood to that of 14/15 for the sensing of glucose.18
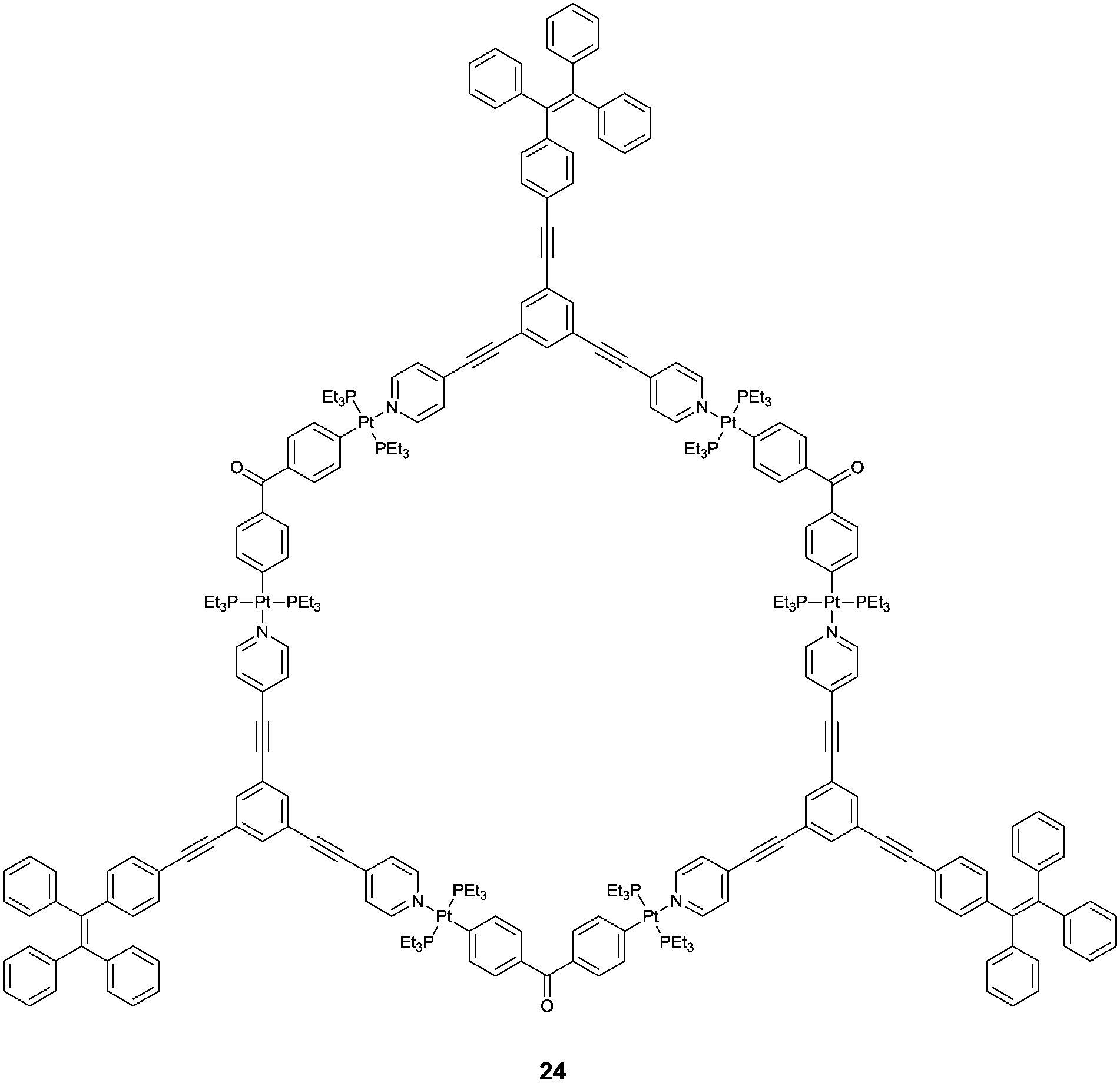
Hierarchical self-assembly of positively charged metallacycle 24 and negatively charged linear polysaccharides was observed by Yang et al.,32 in which they showed that multiple electrostatic interactions were driving forces. NMR, ESI-MS and morphology studies show that the elaborately designed tris-TPE metallacycle form entangled bead-like aggregates upon the threading of the linear heparin through the metallacycles, nicely supported by the enhanced emission of the TPE fluorophores that allow the detection of heparin at the clinical dosage level. The excellent selectivity for heparin over its analogues such as sodium polyacrylate and sodium poly(p-styrenesulfonate) is impressive, considering the multiple electrostatic interaction nature. The size matching appears to be a key factor which is amplified upon aggregation, explaining the observed selectivity for heparin.
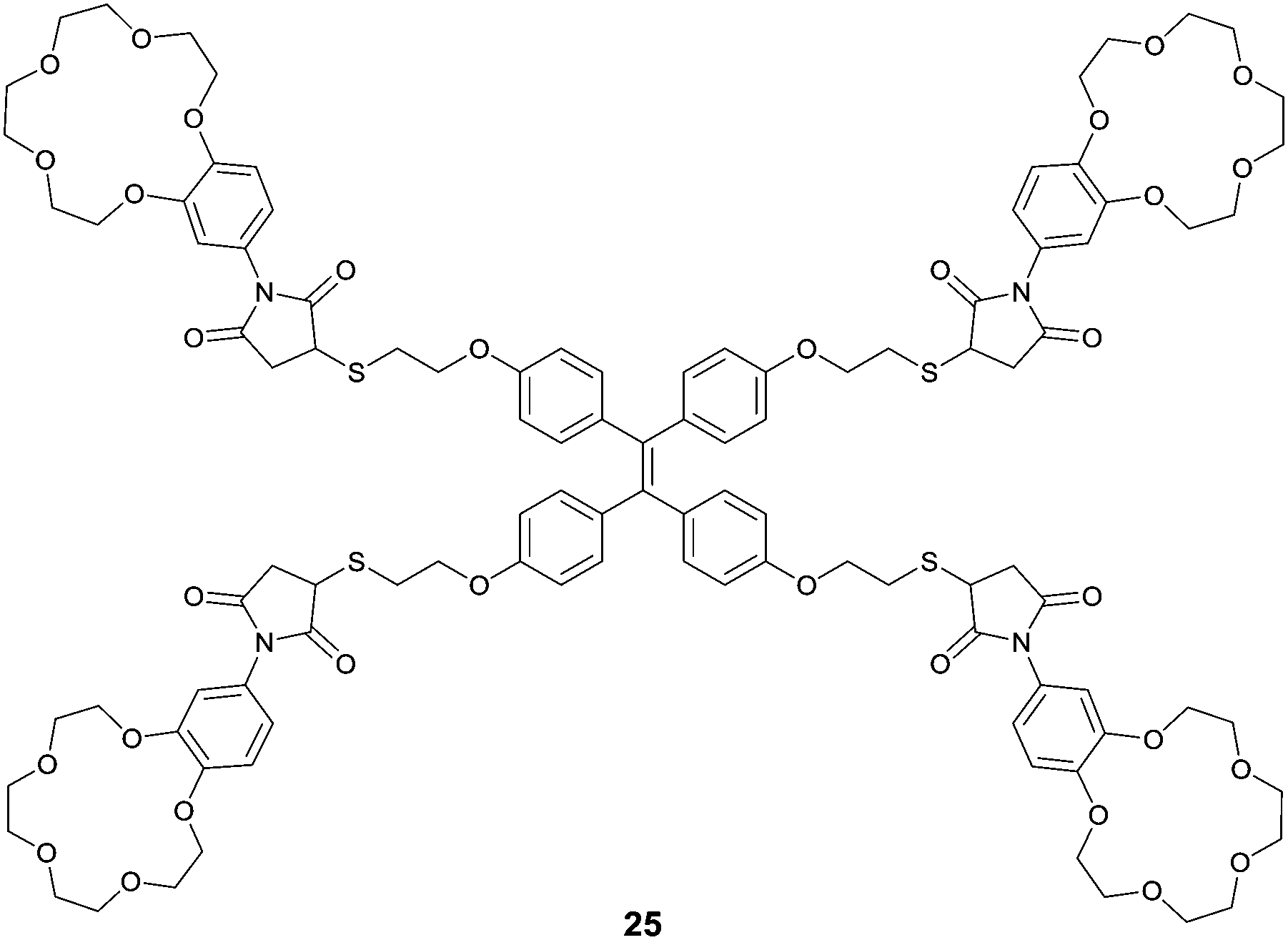
Integrating host–guest molecular recognition and the AIE effect, Liu et al.33 developed a highly sensitive and selective fluorescence sensor 25 for K+. As a crown-ether functionalized TPE derivative, the four pendant benzo-15-crown-5 moieties and the TPE core serve, respectively, as a K+ binding group and an AIE-active motif. 25 could dissolve well in THF, thus exhibiting faint fluorescence, whereas in aqueous THF solution, fluorescence of 25 was enhanced gradually with increasing water volume percentage until 70%, when a dramatic enhancement starts to happen because of the aggregation of 25. Meanwhile, the emission of 25 at 460 nm in THF also experienced a dramatic enhancement by 9.5 fold when K+ was introduced at 0.0–19.2 μM concentrations. A detection limit of 1.0 μM was obtained for K+ in THF at a concentration of 25 of 4.8 μM, demonstrating the high sensitivity of the sensing. The binding of K+ with the peripheral crown ether moieties in 25, forming K+/15-C-5 sandwich complexes, promoted the cross-linked networking of the TPE molecules and thus restricted the internal rotation, accounting for the observed fluorescence enhancement similar to that observed for 25 itself in water-rich THF solutions. Indeed, DLS data indicated the existence of the aggregate nanoparticles of size 200 nm when 25 was mixed with 1 equivalent of K+ ions in THF, and this was confirmed by the AFM observations. It also is worth pointing out that 25 is highly selective toward K+ in THF, since enhancement of the fluorescence of 25 was only observed by K+ among the tested metal cations of Li+, Na+, K+, NH4+, Ca2+, Mg2+ and Pb2+, despite the fact that 15-C-5 binds Na+ better.
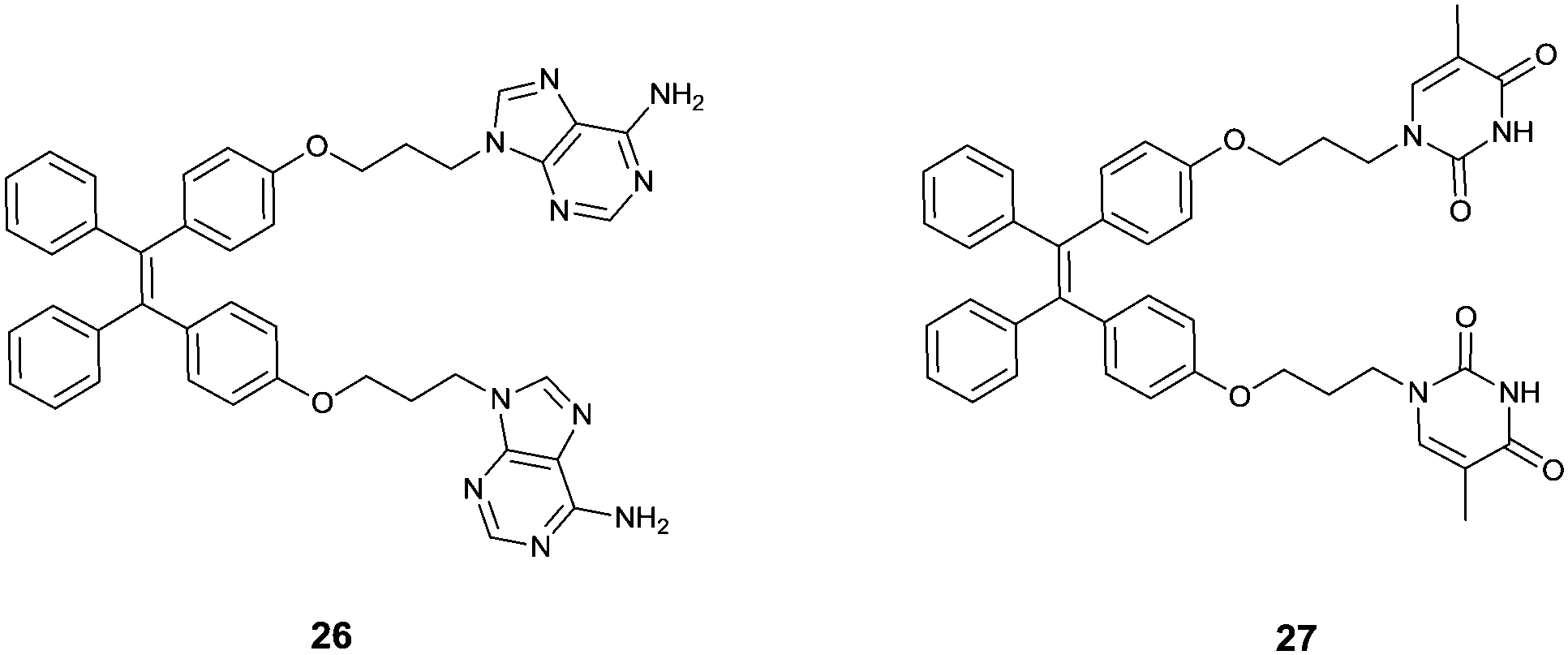
Attaching metal binding groups to the TPE core, Zhu's group34 developed two fluorescence chemosensors 26 and 27 for Ag+ and Hg2+, respectively. Here adenine (A) and thymine (T) are the respective binding groups for Ag+ and Hg2+, following the well-established “A–Ag+–A” and “T–Hg2+–T” binding motifs. Enhancement of the fluorescence of 26 and 27 occurs in the presence of Ag+ and Hg2+ due to the induced aggregation that restricts the intramolecular rotation in the TPE core (Fig. 5). 26 itself showed rather faint emission in H2O/THF (5:1, v/v), however, its emission at 470 nm increased linearly upon the addition of AgClO4 that can be visualized by the naked eye. This was attributed to the coordination of Ag+ with 26 to form the oligomer- or polymer-like complexes (Fig. 6), rather than the intramolecular coordination complex of 26 and Ag+. Despite the weak and less selective binding of free adenine with Ag+, the response of fluorescence of 26 in aqueous THF solutions was highly sensitive and highly selective as well since only Ag+, and slightly Hg2+, could induce emission enhancement, among the tested metal ions Ba2+, Ca2+, Cd2+, Co2+, Cu2+, Fe3+, Fe2+, Hg2+, Mg2+, Mn2+, Ni2+, Pb2+, Zn2+, Ag+, Cs+, and K+. Similar observations were made with fluorescence sensing by 27 for Hg2+.
 | ||
| Fig. 6 Design rationale for Ag+ and Hg2+ chemosensors 26 and 27. Reprinted with permission from Org. Lett., 2008, 10, 4581–4584. Copyright 2008 ACS. | ||
Ceroni and co-workers observed switchable phosphorescence upon complexation of Mg2+ to 28.35 Compound 28, another kind of AIE molecule, is nonluminescent in fluid solution, whereas it exhibits red-shifted absorption and phosphorescence in the presence of Mg2+. The assembled polymers display similar phosphorescence spectral profiles and lifetimes to those of their powder, suggesting that aggregation restricts the intramolecular rotations.36 Subsequent addition of fluoride disassembles the aggregates because of the formation of MgF2 and the vanishing of the phosphorescence. This process is reversible by adding Mg2+ and F− alternately, making it a tunable luminescence sensor for both Mg2+ and F−.

Tanaka et al.37 developed an interesting TPE-based sensor for Cu2+, by using an azide-modified TPE 29 that interacts with diethylene glycol dipropionate 30via click chemistry. This reaction between 29 and 30 in 1:1 (v/v) H2O/THF forms the intermolecular cross-linked products (like aggregates) in the presence of trace amounts of Cu+ which is in situ generated by reduction of Cu2+ by sodium ascorbate. Thus the fabricated assay establishes a sensitive and selective sensing scheme for Cu2+ with a limit of detection of 1 μM, sensitive enough for the level of Cu2+ in drinking water according to US EPA standard. Sample solutions containing various other metal ions (Li+, Na+, K+, Mg2+, Ca2+, Mn2+, Fe2+, Fe3+, Co2+, Ag+ and Zn2+) and a mineral water sample containing Na+, K+, Mg2+, and Ca2+ ions under the same treatment procedures produced no significant enhancement in the fluorescence, it only happened when Cu2+ ions were added. This opens a new avenue for employing the AIE phenomenon to achieve high selectivity, a character of chemodosimeters.38
Employing the click reaction, He and co-workers developed a new AIE fluorescence chemosensor 31 for Fe3+.39 The TPE derivative 31 started to aggregate in H2O/THF binary solvents containing 60% water, in which strong emission was observed with two bands that peaked at 408 nm and 432 nm. 31 in H2O/THF (2:1, v/v) exhibited significant fluorescence quenching only in the presence of Fe3+, whereas the tested other cations (Li+, Na+, K+, Mg2+, Ca2+, Ba2+, Mn2+, Ni2+, Fe2+, Pb2+, Al3+, Cr3+, Hg+, Ce2+ and Cd2+) did not. This was attributed to the coordination of Fe3+ to 31 that disfavours the aggregation of 31. Job's plot and 1H NMR titrations led to tentative assignment of the structure of the 2:1 Fe3+/31 complex which is multi-charged and thus more hydrophilic and tends to aggregate less in aqueous solutions. Again the system is sensitive with a limit of detection of 0.7 μM.
Hydrogen peroxide is the least reactive and mild oxidant among reactive oxygen species (ROS), which are toxic to cells if the level of ROS exceeds the tolerable physiological range. Developing sensitive sensors to monitor the level of H2O2 both in living cells and in the environment has attracted intensive attention. Zhang and co-workers40 designed a H2O2-selective fluorescence sensor 32 which contains a TPE core and a H2O2-reactive group, arylboronate ester, which are linked via a pyridinium bridge (Fig. 7). The pyridinium bridge makes 32 water-soluble, thus enabling it to exist in the monomer form and being nonfluorescent. Oxidative hydrolysis of 32 by H2O2 generates the much less water-soluble product 33, which undergoes aggregation and therefore becomes highly fluorescent, allowing turn-on fluorescence sensing of H2O2 with high selectivity as a chemodosimeter would normally exhibit. Indeed, among the tested reactive oxygen species such as H2O2, singlet oxygen, the hydroxyl radical, the superoxide anion and nitric oxide, enhancement of emission was only observed in the case of H2O2. The assay is highly sensitive as well, with a limit of detection for H2O2 down to 180 nM. More importantly, since D-glucose can be oxidized in the presence of glucose oxidase to produce H2O2, the scheme shown in Fig. 6 can be applied for D-glucose as well.
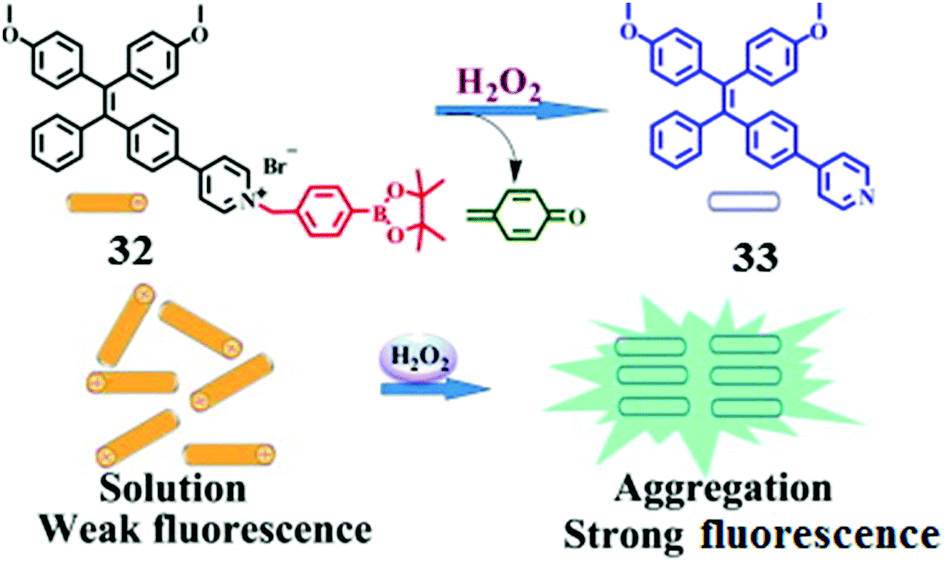 | ||
| Fig. 7 Rationale for the fluorescence turn-on detection of H2O2 using 32 which upon oxidative hydrolysis leads to the neutral TPE product 33 that undergoes aggregation. Reprinted with permission from Tetrahedron Lett., 2014, 55, 1471–1474. Copyright 2014 Elsevier. | ||
Metallophilic interactions
Two closed-shell metal cations (such as Au+, Ag+ and Cu+) tend to have attractive interactions rather than repulsion when their distance is smaller than the sum of the van der Waals radii, as is the so-called metallophilic interaction (metal⋯metal interaction). Metal-coordinated complexes with metallophilic interactions exhibit unique photochemical and photophysical properties, making them useful in many aspects, such as functional nanomaterials,18,22,41 luminescent materials for ion recognition and sensing,21,42,43 and chiral nanoclusters.26,27,44 Our group has been interested in establishing a series of coordination polymers facilitated by argentophilic interactions, with the aim of achieving versatile sensors, choosing zigzag structured (Ag+-SR)n backbones.A ratiometric luminescence sensor with outstanding sensitivity and selectivity for Ag+ was established by forming coordination polymers with the thiol ligand NCys, which is an N-(1-naphthylacetyl)cysteine derivative designed to bear an aromatic fluorophore (Fig. 8).45 The in situ formed coordination polymers were assumed to be facilitated by the Ag+⋯Ag+ interaction which is probed by a new absorption at 350 nm, assigned to ligand-to-metal charge transfer transition modified by the argentophilic interaction (LMMCT, S → Ag+). π⋯π stacking of the naphthyl moieties in the side chains may contribute additionally to the stabilization of the polymers. The naphthyl fluorophore was also designed to harvest light and emit fluorescence. Dual emission from the naphthyl and Ag+⋯Ag+ fluorophores allows establishing a sensitive ratiometric assay for Ag+ at the sub-μM level, in particular with high selectivity so that much more thiophilic Hg2+ exhibits little interference.
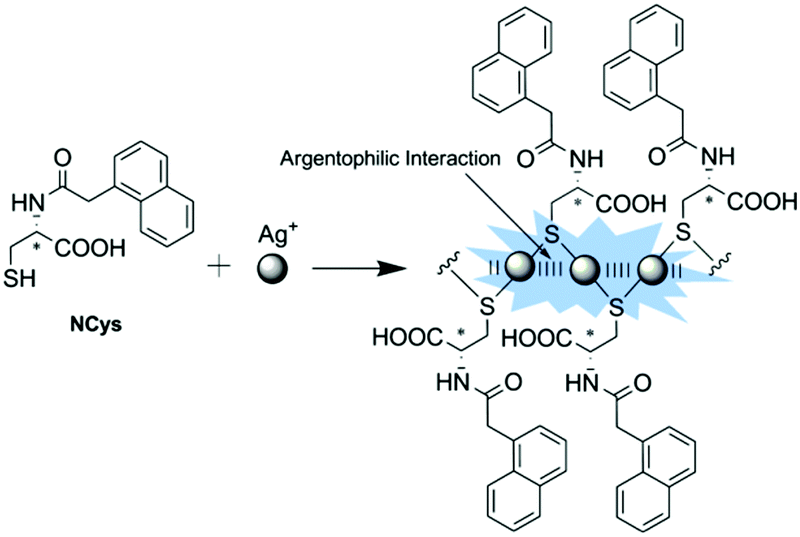 | ||
| Fig. 8 Formation of Ag(I)–NCys coordination polymers allowing for the ratiometric fluorescence sensing of Ag+. Reprinted with permission from Chem. Commun., 2011, 47, 5900–5902. Copyright 2011 RSC. | ||
CD signals corresponding to Ag+⋯Ag+ absorption were also observed and the signs of the CD signals are related to the chirality of the cysteine residue.42,45 A possible reason is that the Ag+⋯Ag+ interaction and the sidechain/sidechain interactions in the coordination polymers form a network including the Ag+⋯Ag+ chromophore and the chiral cysteine residues. We therefore extended the study to employ coordination polymers of Ag+ with an achiral thiol ligand that bears a binding group for chiral species, for chiral sensing. A first attempt was made with a thiophenol that bears a boronic acid group for interacting with saccharides (Fig. 9).42 CD spectra of varying profiles were observed with the tested saccharides such as glucose and fructose, confirming that the chirality of the saccharide is transferred into the Ag+⋯Ag+ chromophore in the supramolecular sensory ensembles upon interacting with the boronic acid group to form the cyclic boronate ester. Glucose that interacts with the supramolecular sensor in a multivalent manner results in stronger CD signals whereas the monovalent fructose induced practically no or very weak CD signals. The CD spectral profile could be acquired to correlate to the saccharide identity. The observation made here is of relevance for understanding signal transduction in biological systems. Applying coordination polymers featuring metal⋯metal interactions remains at its early stage and it deserves to be explored in a broad scope by designing new sensory ligands and employing other metal⋯metal interactions.
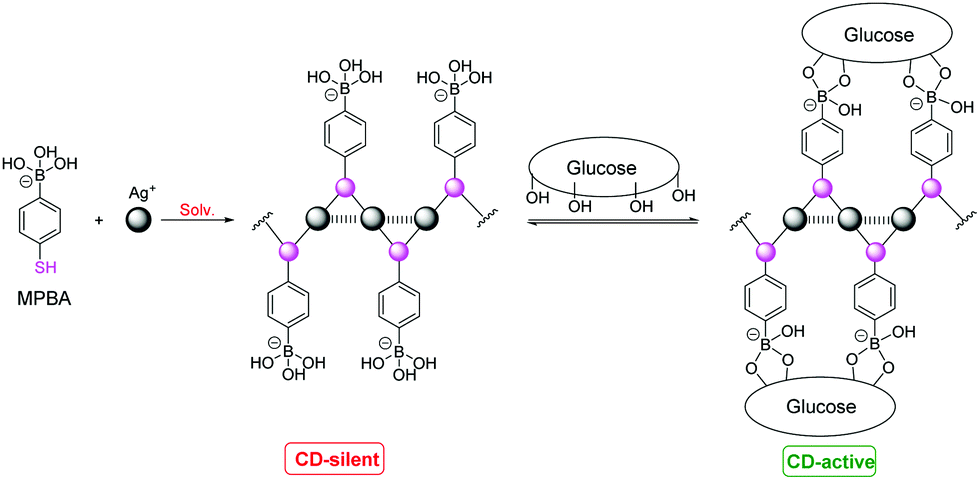 | ||
| Fig. 9 Interactions of glucose with Ag+–MPBA coordination polymers. Reprinted with permission from Chem. Commun., 2015, 51, 8017–8019. Copyright 2015 RSC. | ||
Metal–organic frameworks
Metal–organic frameworks (MOFs) with well-defined pore-structures have evolved into functional materials for many potential applications.46 Yet because of their poor stability in biological media, few of them have been applied to biosensing. Chen's group47 is among the first to construct a water-stable Cu2+-based zwitterionic carboxylate three-dimensional MOF (34, Fig. 10), for the detection of DNA and RNA. This elaborately fabricated MOF affords multiple interactions with probe DNAs attached with the fluorophore FAM (P-DNAs), forming two P-DNA@34 systems. Fluorescence of the P-DNA is efficiently quenched by Cu2+, presumably via a photo-induced electron-transfer (PET) mechanism. Upon introducing the human immunodeficiency virus (HIV) double-stranded DNA (ds-DNA) sequence (T0) or Sudan virus (SUDV) RNA sequence (T0′), relevant P-DNA gets rid of the MOF and hybridizes with the complementary sequence, resulting in fluorescence recovery up to 91% and ensuring high sensitivity with detection limits of 196 pM for T0 and 73 pM for T0′. In addition, the detection is hardly interfered by analogous DNA or RNA sequences with one base pair mismatching, demonstrating its high selectivity. The reasons for the excellent sensing performance remain undisclosed, while the size of the pores in MOF was proposed to be a possibility.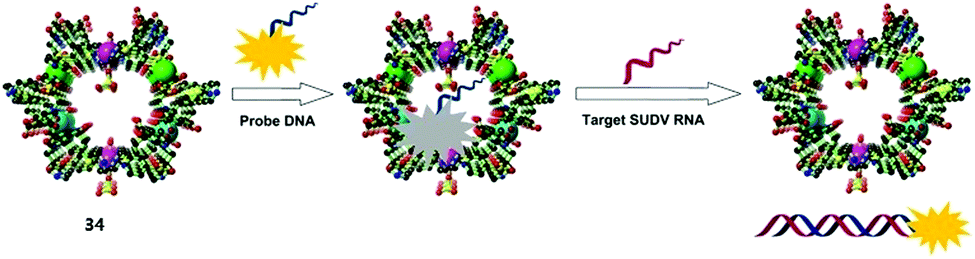 | ||
| Fig. 10 Proposed mechanism for the detection of target DNR or RNA based on a fluorescence biosensor formed from MOF 34 and fluorophore-labelled probe DNA. Reprinted with permission from Anal. Chem., 2015, 87, 12206–12214. Copyright 2015 ACS. | ||
Wang et al.44 carried out a noteworthy conceptual MOF-based “turn-on” fluorescence sensing using encapsulated AIE fluorophores for five-membered-ring energetic heterocyclic compounds (5MR-HCs, Fig. 11). 5MR-HCs are a kind of explosive, whose detection is urgently demanded to be explored. Adjusting metal nodes from Mg2+ to Ni+ and Co2+, the weak luminescence of MOFs declines to be nonfluorescent, arising from the ligand-to-metal charge transfer (LMCT) effect. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N or NN group in the 5MR-HCs could destroy the coordination bonds in MOFs through competitive coordination to the metal nodes, releasing the encapsulated AIE fluorophore TABD-COOH which aggregates spontaneously and emits enhanced fluorescence. The method is sensitive in that 5MR-HCs down to 40 nM can be detected via the fluorescence signal while that at 6.5 ng cm−2 can be identified by the naked eye in several seconds. Releasing the building components from the polymeric MOFs affords signal amplification by an exponential expansion of the amount of small components and by the subsequent AIE of these components.
N or NN group in the 5MR-HCs could destroy the coordination bonds in MOFs through competitive coordination to the metal nodes, releasing the encapsulated AIE fluorophore TABD-COOH which aggregates spontaneously and emits enhanced fluorescence. The method is sensitive in that 5MR-HCs down to 40 nM can be detected via the fluorescence signal while that at 6.5 ng cm−2 can be identified by the naked eye in several seconds. Releasing the building components from the polymeric MOFs affords signal amplification by an exponential expansion of the amount of small components and by the subsequent AIE of these components.
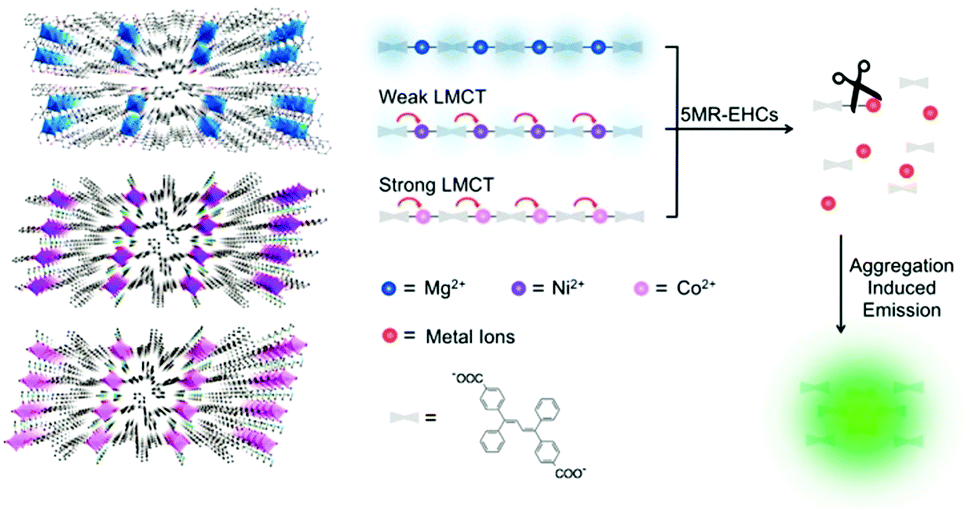 | ||
| Fig. 11 Crystal structures of Mg2+, Ni2+, and Co2+ constructed TABD-MOFs and a schematic representation of the mechanism for detection of the energetic heterocyclic compounds by the three TABD-MOFs. LMCT: ligand-to-metal charge transfer. Reprinted with permission from J. Am. Chem. Soc., 2014, 136, 15485–15488. Copyright 2014 ACS. | ||
Actually luminescent MOFs have become excellent candidates for the detection of explosives, thanks to their easy syntheses and structural diversity. Zheng et al.48 built two luminescent MOFs, whose photoluminescence is quenched only by picric acid (PA), a highly electron-deficient nitrobenzene derivative. Electron-rich π-conjugated fluorescent ligands are introduced as electron donors, which transfer their electrons to the electron-deficient analytes leading to fluorescence quenching. The resonance energy transfer mechanism is also operative, making it an effective sensor for PA. A similar mechanism of electron transfer is shown to operate in Lu's system with high thermostability up to 500 °C.49 The MOF material with high porosity allows its framework to accommodate guests of varying sizes. The highly stable MOF fluorescence sensor is able to detect a trace amount of nitroaromatic explosives, selectively with no interference from water. Fluorophore modified MOFs represent highly promising sensors as their porous structures accommodate guests of suitable size with the possibility of close contact, leading to highly effective electron and/or energy transfer that ensure high sensitivity, together with high selectivity.
Conclusions and perspectives
Despite the unique advantages of supramolecular sensors and the tremendous success they have achieved, they are still at a very early stage for practical applications. This Feature Article is thus hoped to provide an overview of the current state of the art; particular attention is given to how supramolecular aggregates have been applied for sensing to achieve a better performance, while the designing of chemosensor-like building blocks is much easier. Given the fact that supramolecular aggregates have been extensively studied for many decades and many of the outcomes of those efforts can be extended to the new applications outlined here, we thus wish to invite those working in the field of supramolecular aggregates to join together, bringing in their expertise to facilitate the exploration of aggregates for sensing. There are indeed several concerns that remain to be clarified, for example, the size distributions and concentrations, and even the morphology of the aggregates in complicated systems, to ensure whether they may or may not influence the sensing signaling. High enough local concentration of the building blocks needs to be loaded into bio-systems such as cells to allow aggregation and consequently to perform in vivo sensing and imaging. Future exploration of supramolecular aggregate based sensors is perhaps directed towards detecting diverse biologically important species, to meet the requirement for practical applications such as clinical detection and medical examinations. For example, the development of simple and reliable sensors for point-of-care personalized medicine holds great potential.Acknowledgements
We thank the NSF of China (91127019, 21275121, 21435003, and 21521004) and the Program for Changjiang Scholars and Innovative Research Team in University, the MOE of China (IRT13036) for their support.Notes and references
- B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- J. Sun, M. Cai and J. J. Lavigne, Supramol. Chem., John Wiley & Sons, Ltd, 2012 Search PubMed.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed.
- M. F. Geer and L. S. Shimizu, Supramol. Chem., John Wiley & Sons, Ltd, 2012 Search PubMed.
- D. Gorl, X. Zhang and F. Wurthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed.
- S. Rochat and T. M. Swager, ACS Appl. Mater. Interfaces, 2013, 5, 4488–4502 CAS.
- Z. Chen, Q. Wang, X. Wu, Z. Li and Y.-B. Jiang, Chem. Soc. Rev., 2015, 44, 4249–4263 RSC.
- J.-S. Shen, D.-H. Li, Y.-B. Ruan, S.-Y. Xu, T. Yu, H.-W. Zhang and Y.-B. Jiang, Luminescence, 2012, 27, 317–327 CrossRef CAS PubMed.
- C. Li, J. Schöneboom, Z. Liu, N. G. Pschirer, P. Erk, A. Herrmann and K. Müllen, Chem. – Eur. J., 2009, 15, 878–884 CrossRef CAS PubMed.
- T. Ye, R. Singh, H. J. Butt, G. Floudas and P. E. Keivanidis, ACS Appl. Mater. Interfaces, 2013, 5, 11844–11857 CAS.
- X. Feng, Y. An, Z. Yao, C. Li and G. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 614–618 CAS.
- B. A. Jones, M. J. Ahrens, M. H. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, Angew. Chem., Int. Ed., 2004, 43, 6363–6366 CrossRef CAS PubMed.
- C. Zhao, Y. Zhang, R. Li, X. Li and J. Jiang, J. Org. Chem., 2007, 72, 2402–2410 CrossRef CAS PubMed.
- T. Tang, K. Peneva, K. Mullen and S. E. Webber, J. Phys. Chem. A, 2007, 111, 10609–10614 CrossRef CAS PubMed.
- Y. Zheng, H. Long, G. C. Schatz and F. D. Lewis, Chem. Commun., 2005, 4795–4797 RSC.
- Y.-B. Ruan, A.-F. Li, J.-S. Zhao, J.-S. Shen and Y.-B. Jiang, Chem. Commun., 2010, 46, 4938–4940 RSC.
- A. K. Dwivedi, M. Pandeeswar and T. Govindaraju, ACS Appl. Mater. Interfaces, 2014, 6, 21369–21379 CAS.
- A. Weißenstein and F. Würthner, Chem. Commun., 2015, 51, 3415–3418 RSC.
- K.-R. Wang, H.-W. An, R.-X. Rong, Z.-R. Cao and X.-L. Li, Biosens. Bioelectron., 2014, 58, 27–32 CrossRef CAS PubMed.
- X. Wu, X.-X. Chen, B.-N. Song, Y.-J. Huang, Z. Li, Z. Chen, T. D. James and Y.-B. Jiang, Chem. – Eur. J., 2014, 20, 11793–11799 CrossRef CAS PubMed.
- X.-X. Chen, X. Wu, P. Zhang, M. Zhang, B.-N. Song, Y.-J. Huang and Y.-B. Jiang, Chem. Commun., 2015, 51, 13630–13633 RSC.
- Y. Wang, J. Chen, H. Jiao, Y. Chen, W. Li, Q. Zhang and C. Yu, Chem. – Eur. J., 2013, 19, 12846–12852 CrossRef CAS PubMed.
- C. Yu and V. W.-W. Yam, Chem. Commun., 2009, 1347–1349 RSC.
- Y.-J. Huang, W.-J. Ouyang, X. Wu, Z. Li, J. S. Fossey, T. D. James and Y.-B. Jiang, J. Am. Chem. Soc., 2013, 135, 1700–1703 CrossRef CAS PubMed.
- X. Wu, X.-X. Chen, B.-N. Song, Y.-J. Huang, W.-J. Ouyang, Z. Li, T. D. James and Y.-B. Jiang, Chem. Commun., 2014, 50, 13987–13989 RSC.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- Y. Hong, M. Haussler, J. W. Lam, Z. Li, K. K. Sin, Y. Dong, H. Tong, J. Liu, A. Qin, R. Renneberg and B. Z. Tang, Chem. – Eur. J., 2008, 14, 6428–6437 CrossRef CAS PubMed.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Z. Zhu, L. Xu, H. Li, X. Zhou, J. Qin and C. Yang, Chem. Commun., 2014, 50, 7060–7062 RSC.
- H. Tong, Y. Hong, Y. Dong, M. Haussler, J. W. Lam, Z. Li, Z. Guo, Z. Guo and B. Z. Tang, Chem. Commun., 2006, 3705–3707 RSC.
- L.-J. Chen, Y.-Y. Ren, N.-W. Wu, B. Sun, J.-Q. Ma, L. Zhang, H. Tan, M. Liu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2015, 137, 11725–11735 CrossRef CAS PubMed.
- X. Wang, J. Hu, T. Liu, G. Zhang and S. Liu, J. Mater. Chem., 2012, 22, 8622–8628 RSC.
- L. Liu, G. Zhang, J. Xiang, D. Zhang and D. Zhu, Org. Lett., 2008, 10, 4581–4584 CrossRef CAS PubMed.
- A. Fermi, G. Bergamini, M. Roy, M. Gingras and P. Ceroni, J. Am. Chem. Soc., 2014, 136, 6395–6400 CrossRef CAS PubMed.
- I. G. Occhiuto, M. Samperi, M. Trapani, G. De Luca, A. Romeo, R. F. Pasternack and L. M. Scolaro, J. Inorg. Biochem., 2015, 153, 361–366 CrossRef CAS PubMed.
- T. Sanji, M. Nakamura and M. Tanaka, Tetrahedron Lett., 2011, 52, 3283–3286 CrossRef CAS.
- A. T. Aron, K. M. Ramos-Torres, J. A. Cotruvo, Jr. and C. J. Chang, Acc. Chem. Res., 2015, 48, 2434–2442 CrossRef CAS PubMed.
- J. H. Ye, J. Liu, Z. H. Wang, Y. Bai, W. C. Zhang and W. J. He, Tetrahedron Lett., 2014, 55, 3688–3692 CrossRef CAS.
- F. Hu, Y. Y. Huang, G. X. Zhang, R. Zhao and D. Q. Zhang, Tetrahedron Lett., 2014, 55, 1471–1474 CrossRef CAS.
- F. Cheng, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2013, 135, 17286–17289 CrossRef CAS PubMed.
- Q. Zhang, Y. Hong, N. Chen, D. D. Tao, Z. Li and Y.-B. Jiang, Chem. Commun., 2015, 51, 8017–8019 RSC.
- Z. Yao, Y. Yang, X. Chen, X. Hu, L. Zhang, L. Liu, Y. Zhao and H.-C. Wu, Anal. Chem., 2013, 85, 5650–5653 CrossRef CAS PubMed.
- Y. Guo, X. Feng, T. Han, S. Wang, Z. Lin, Y. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485–15488 CrossRef CAS PubMed.
- D. H. Li, J. S. Shen, N. Chen, Y. B. Ruan and Y. B. Jiang, Chem. Commun., 2011, 47, 5900–5902 RSC.
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed.
- S.-P. Yang, S.-R. Chen, S.-W. Liu, X.-Y. Tang, L. Qin, G.-H. Qiu, J.-X. Chen and W.-H. Chen, Anal. Chem., 2015, 87, 12206–12214 CrossRef CAS PubMed.
- Z.-Q. Shi, Z.-J. Guo and H.-G. Zheng, Chem. Commun., 2015, 51, 8300–8303 RSC.
- Y.-N. Gong, L. Jiang and T.-B. Lu, Chem. Commun., 2013, 49, 11113–11115 RSC.
| This journal is © The Royal Society of Chemistry 2016 |