
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amide-functionalized pillar[5]arenes as a novel class of macrocyclic receptors for the sensing of H2PO4− anion†
L. S.
Yakimova
,
D. N.
Shurpik
and
I. I.
Stoikov
 *
*
Kazan Federal University, A. M. Butlerov Chemical Institute, Kremlevskaya Street, 18, Kazan, 420008, Russian Federation. E-mail: ivan.stoikov@mail.ru
First published on 15th September 2016
Abstract
A novel class of amide-functionalized pillar[5]arenes, that is, alkyl and aryl amide derivatives, has been synthesized. Highly selective binding of the dihydrogen phosphate anion over a range of anions by the new synthetic receptors is reported. First, the “binding strength–selectivity” relationship has been decided for the pillar[5]arene/anion systems.
Selective binding of anions is one of the key problems frequently occurring in the design of supramolecular systems, in catalysis and materials sciences as well as in some analytical applications (sensors, extraction systems, etc.).1 Among others, recognition of tetrahedral oxoanions is of significant importance.2 Many of them are considered as environmentally dangerous (phosphates, sulfates) and hence require removal from industrial and agricultural wastewaters.3 Sensing phosphates and their derivatives is of special interest because they compose the backbone of nucleic acids and play significant roles in biological systems.4 Sensors for phosphate anions have been recently reported but they still need receptors with improved selectivity.5 Besides, H-bond receptors that show selectivity toward dihydrogen phosphate over acetate or fluoride anions are very scarce.2b,c,6
Charged and neutral polar fragments, e.g., ammonium, amide, urea, thiourea and hydroxyl groups, are commonly applied for the synthesis of effective and selective synthetic receptors toward anions.7 Amide fragments are more attractive due to the simultaneous presence of proton-donating N–H and proton-accepting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups capable of forming intra- and intermolecular hydrogen bonds and participating in recognition and self-assembling.8 Realization of intramolecular hydrogen bonds is unfavourable for anion binding. Therefore, the problem of creating receptors consists of minimization of the role of intramolecular H-bonds and expanding the contribution of intermolecular H-bonds between an anion and a receptor. The stronger these bonds and the greater their number, the more effective target binding. In addition, interactions between an anion and an amide fragment have rather high energy and can be multiplied using several H-bond donating groups. This approach can be realized by functionalization of the macrocyclic platform.9 For this reason, great attention of specialists working in this area has been recently attracted to macrocyclic structures, e.g., pillar[n]arenes.10 Pillar[5]arenes are polyfunctional macrocyclic compounds consisting of 1,4-hydroquinone fragments coupled by methylene bridges in a macrocycle and having ten hydroxyl groups which can be easily modified. Thus, functionalization of pillar[5]arenes allows carrying out synthesis of a macrocyclic host with desired properties.11 Importantly, hydrogen bonding is spatially sensitive, as it is formed only if the H-bond donor is positioned accurately with respect to the H-bond acceptor.12
O groups capable of forming intra- and intermolecular hydrogen bonds and participating in recognition and self-assembling.8 Realization of intramolecular hydrogen bonds is unfavourable for anion binding. Therefore, the problem of creating receptors consists of minimization of the role of intramolecular H-bonds and expanding the contribution of intermolecular H-bonds between an anion and a receptor. The stronger these bonds and the greater their number, the more effective target binding. In addition, interactions between an anion and an amide fragment have rather high energy and can be multiplied using several H-bond donating groups. This approach can be realized by functionalization of the macrocyclic platform.9 For this reason, great attention of specialists working in this area has been recently attracted to macrocyclic structures, e.g., pillar[n]arenes.10 Pillar[5]arenes are polyfunctional macrocyclic compounds consisting of 1,4-hydroquinone fragments coupled by methylene bridges in a macrocycle and having ten hydroxyl groups which can be easily modified. Thus, functionalization of pillar[5]arenes allows carrying out synthesis of a macrocyclic host with desired properties.11 Importantly, hydrogen bonding is spatially sensitive, as it is formed only if the H-bond donor is positioned accurately with respect to the H-bond acceptor.12
This feature allows designing receptors with high selectivity toward anions that match the size and shape of the binding pocket. As a result, optimization of the “rigidity” of the host skeleton and introduction of additional functional groups become important. Thus, in this paper, a new line of amide-functionalized pillar[5]arenes has been synthesized for achieving the twin goals of “binding strength–selectivity”.
In this paper, we have successfully synthesized three amide-functionalized pillar[5]arenes, bearing N-phenyl (1), N-octyl (2) and N-decyl (3) amide groups at both rims (Fig. 1). By reaction of perhydroxylated pillar[5]arene A and ethyl bromoacetate with KI as the catalyst,13 pillar[5]arene with ethoxycarbonyl fragments B was prepared in the presence of K2CO3 in CH3CN (Fig. 1). The base and the solvent were specified in accordance with their efficiency in alkylation of p-tert-butyl thiacalix[4]arene at the lower rim.14 Macrocycle C, which was obtained by treatment of B with NaOH in THF, reacted with SOCl2 in the presence of catalytic amounts of DMF, and in situ with amine derivatives (aniline, n-octylamine, and n-decylamine) in CH2Cl2 in the presence of N(C2H5)3 over 48 h. Macrocycles 1–3 were synthesized with high yields (76, 90 and 88%, correspondingly). The structure of the compounds obtained was characterized by 1H and 13C NMR, IR spectroscopy and mass spectrometry (MALDI TOF) (ESI,† Fig. S1–S12).
 | ||
| Fig. 1 Synthesis of the amide-functionalized pillar[5]arenes 1–3 and investigated anionic guests as tetrabutylammonium (TBA) salts. | ||
It is interesting that aryl units of pillar[5]arenes are conformationally mobile and able to rotate around their axis.15 The degree of inhibition of conformational transformations can be estimated by referring to signals of the oxymethylene protons in the macrocycle substituents using 1H NMR spectra. For –O–CH2–COOR groups, oxymethylene protons become diastereotopic at slow rotation or in its absence and appear as AB-quadruplets in 1H NMR spectra. Meanwhile, oxymethylene protons are observed during fast rotation as singlets (enantiotopic). Thus, oxymethylene proton signals for C and 1 are high-resolution AB-quadruplets, and hence the aryl units do not rotate (Fig. 2a and c). The singlet of oxymethylene protons in decaacid acyl chloride indicates free rotation of the aryl units (Fig. 2b). Rotation is slower with the introduction of long alkyl substituents in the structure of pillar[5]arenes: the AB system converts into a broad singlet (bs) in N-alkyl amides 2 and 3 (Fig. 2d and e). Obviously, the absence of rotation (AB system) or hindered rotation (bs) of aryl fragments around the axis is due to the formation of intramolecular H-bonds NH⋯OC, which stabilize the structure of decaacid C and amides 1–3. In the case of decaacid chloride, there are no intramolecular bonds. Thus, the system of intramolecular hydrogen bonds in amides 1–3 creates a pre-organized cavity for the anion binding.
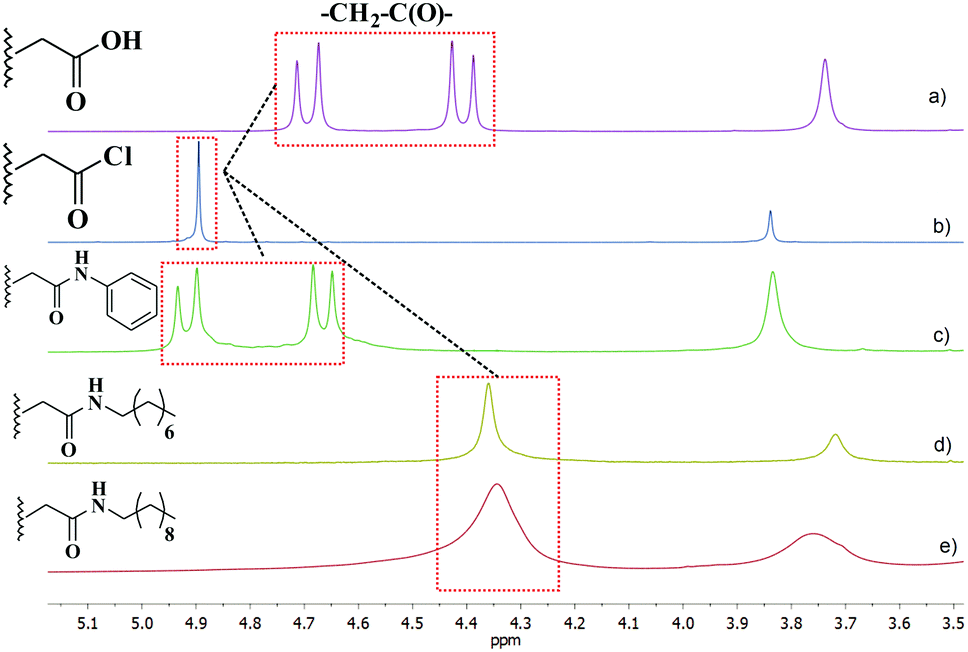 | ||
| Fig. 2 Fragments of 1H NMR spectra of pillar[5]arene derivatives: decaacid (a), decaacid acyl chloride (b), N-phenyl amide (c), N-octyl amide (d), and N-decyl amide (e) (DMSO-d6, 25 °C, 400 MHz). | ||
Anions (TBA+X−) with various sizes and shapes have been used to determine the binding capability of 1–3 (Fig. 1). Addition of the fluoride, dihydrogen phosphate and acetate anions to solution of 1 in CH2Cl2 resulted in a bathochromic (4.5 nm) and minor hyperchromic shift in the absorption maxima of macrocycle 1 at 240 nm (ESI,† Fig. S13 and S14). More abrupt changes were observed in the case of interaction with dihydrogen phosphate ions. In the case of pillar[5]arenes 2 and 3, changes in the electronic spectra were found only in the presence of TBA+H2PO4−, indicating the absence of interaction between the macrocycles and other anions.
To determine the association constants and stoichiometry of the 1–3 interaction with the anions, UV titration was carried out with the concentration of 1–3 kept constant at 1 × 10−5 M and that of tetrabutylammonium salts varied from 0 to 3.3 × 10−4 M at 25 °C. The lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kass values for F−, H2PO4− and AcO− associated with 1 were calculated to be 3.51 ± 0.18, 3.33 ± 0.23 and 3.17 ± 0.21, respectively, by using the equations described in the ESI.† The lgKass values for H2PO4− with 2 and 3 were calculated to be 2.43 ± 0.14 and 1.65 ± 0.08, respectively. In all cases, the stoichiometry was 1:1, confirmed by Job plots (ESI,† Fig. S23–S27). It could be assumed that one anion may bind to the amide fragment tethered to one side of the pillararene molecule, while the other side is left uncoordinated or protonated at oxygen atoms as observed in the oxygen-containing ligand.16
Kass values for F−, H2PO4− and AcO− associated with 1 were calculated to be 3.51 ± 0.18, 3.33 ± 0.23 and 3.17 ± 0.21, respectively, by using the equations described in the ESI.† The lgKass values for H2PO4− with 2 and 3 were calculated to be 2.43 ± 0.14 and 1.65 ± 0.08, respectively. In all cases, the stoichiometry was 1:1, confirmed by Job plots (ESI,† Fig. S23–S27). It could be assumed that one anion may bind to the amide fragment tethered to one side of the pillararene molecule, while the other side is left uncoordinated or protonated at oxygen atoms as observed in the oxygen-containing ligand.16
The measured association constants between 1 and F−, H2PO4− and AcO− were the highest. The binding strength between 1 and F− is the strongest, and with the reduction of basicity in this row, the association constants of complexation between 1 and the anions decrease gradually (F− > H2PO4− > AcO−). However, pillar[5]arenes with N-aliphatic amide fragments are most selective macrocycles because they bind only dihydrogen phosphate anions. Above mentioned, aryl units in macrocycle 1 do not rotate around its axis due to the formation of intramolecular H-bonds NH⋯OC, which stabilize the structure of amide 1 and form the pre-organized hard pocket for anions. Therefore, amide 1 is less selective than 2 and 3, which form complexes only with H2PO4−. Hindered rotation in 2 and 3 made their structure more labile and selectivity was higher. These results suggest two types of anion-binding centers in the macrocycle plane (NH and CO). The H2PO4− binding with macrocycles 1–3 is obviously due to the proton accepting carbonyl group (H-bonds CO⋯HO–P) in the structure of these pillar[5]arenes, which is capable of selective recognition of dihydrogen phosphate anions. Besides, the ligands have ten pairs of the NH protons that can form strong hydrogen bonds with negatively charged oxygen atoms (H-bonds NH⋯−O–P). The combination of these two factors has allowed creating effective and selective receptors for dihydrogen phosphate anions. The ability of macrocycle 1 to additionally bind fluoride and acetate anions is caused by the presence of the N-aromatic substituent at the amide group. The more acidic the donor or the more basic the acceptor, the stronger hydrogen bonding interaction (basicity varies in the series F− > AcO− ≥ H2PO4−).17 The functionalization of pillar[5]arenes by more number of electron donating alkyl groups (macrocycles 2 and 3) decreases the acidity of the NH protons and their complexation ability compared to those of 1.
The 1H and 2D 1H–1H NOESY NMR spectroscopy in DMSO-d6 was carried out to pinpoint the anion receptor sites and fully explore the interaction modes between some anions (F− > H2PO4− > AcO−) and 1–3. Anions form inclusion complexes with 1–3 in accordance with 1H NMR spectroscopy (ESI,† Fig. S28–S31). The downfield shift of the signals of NH protons (Δδ = 0.14 ppm) and signals of N-phenyl protons in the upfield region confirmed the complexation in the “pillar[5]arene 1/H2PO4−” system (Fig. 3 and ESI,† Fig. S28). It means involving secondary amide fragments in the complexation. The interactions between 1 and the dihydrogen phosphate anion were further confirmed using 31P NMR spectroscopy (ESI,† Fig. S31). After the addition of an equivalent amount of H2PO4−, the phosphorus signal significantly shifted to the downfield region (Δδ = 0.6 ppm) and broadened. In addition, the peak of phosphorus became broad during the addition of 1, indicating that the kinetics of the complexation were in the same time scale as the 31P NMR experiment, consistent with the stronger anion binding.
 | ||
| Fig. 3 Partial 1H NMR spectra of initial pillar[5]arene 1 and host/guest systems for 1 and TBA+H2PO4− and 1 and TBA+AcO− (1:1) (DMSO-d6, 25 °C, 400 MHz). | ||
More significant downfield shifts (0.31 ppm) in the NH peaks are seen in the case of the “pillar[5]arene 1/acetate anion” system compared to that of 1 (Fig. 3). The integral intensities of these two peaks are 4:1. It can be explained by different participation rates of ten NH fragments present in macrocycle 1 in complexation. It should be noted that the acetate ion, unlike other ions, is comprised of CH3 protons, and the spectrum can be observed for their shift signals. Namely, the signals of CH3 protons shifted to 0.15 ppm in the downfield region.
The 2D NMR NOESY 1H–1H spectroscopy of the complexes confirms host–guest complexation (ESI,† Fig. S31): the aromatic protons of the N-phenyl fragment (7.25 ppm) have cross-peaks with methyl protons (1.75 ppm) of the acetate anion. It clearly confirms that the interaction of the anion with the macrocycle is due to hydrogen bonding of NH protons in the macrocycle. Based on above described results for the host/guest systems, we can assume the structures of the complexes formed (Fig. 4).
 | ||
| Fig. 4 Proposed structures of the complexes pillar[5]arene 1 with H2PO4−, AcO− and F− ions. The structure of complex 1/F− is predicted according to ref. 18. | ||
So, we can conclude that complex formation of amide-functionalized pillar[5]arenes can be performed due to: (a) NH protons, which can form strong hydrogen bonds with negatively charged oxygen atoms; and (b) the proton accepting carbonyl group capable of selective recognition of dihydrogen phosphate anions. In addition, introduction of an N-aromatic substituent at the amide group allows simultaneously increasing the effectivity and decreasing the selectivity of binding. As a result, optimization of the “rigidity” of the host skeleton and the introduction of additional functional groups into the row of new amide-functionalized pillar[5]arenes allow achieving the twin goals of “binding strength–selectivity”.
In summary, we have successfully synthesized pillar[5]arenes bearing ten N-phenyl and N-alkyl amide fragments at both rims of a macrocycle. The presence of secondary amide fragments makes it possible for them to act as anion receptors. The anion affinity of the amide-functionalized receptors strongly depends on the acidity of the amide NH protons, which can be controlled by additional functional groups. The N-phenylamide macrocyclic receptor 1 showed superior affinity toward H2PO4− as compared to receptors 2 and 3 with a long N-alkyl amide fragment, but inferior selectivity compared to 2 and 3. These novel pillar[5]arene-based neutral anion receptors enrich the structural diversity of anion binding chemistry, and can be further used in the fabrication of sensing devices for the dihydrogen phosphate anion.
The work was supported by the Russian Science Foundation (No. 14-13-00058).
Notes and references
- (a) I. Stibor, Anion Sensing, Springer, 2005, p. 238 Search PubMed; (b) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 RSC; (c) N. Busschaert, C. Caltagirone, W. V. Rossom and P. A. Gale, Chem. Rev., 2015, 115(15), 8038 CrossRef CAS PubMed; (d) P. A. Gale and W. Dehaen, Anion Recognition in Supramolecular Chemistry, Springer, 2010, p. 372 Search PubMed.
- (a) H. K. Cho, D. H. Lee and J.-I. Hong, Chem. Commun., 2005, 1690 RSC; (b) D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Org. Biomol. Chem., 2005, 3, 1495 RSC; (c) V. Amendola, M. Boiocchi, D. Esteban-Gómez, L. Fabbrizzi and E. Monzani, Org. Biomol. Chem., 2005, 3, 2632 RSC; (d) C. Suksai and T. Tuntulani, Chem. Soc. Rev., 2003, 32, 192 RSC.
- (a) P. A. Gale, Chem. Commun., 2005, 3761 RSC; (b) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603 CrossRef CAS PubMed; (c) C. Bazzicalupi, A. Bencini and V. Lippolis, Chem. Soc. Rev., 2010, 39, 3709 RSC; (d) C. Warwick, A. Guerreiro and A. Soares, Biosens. Bioelectron., 2013, 41, 1 CrossRef CAS PubMed; (e) T. Lawal and S. B. Adeloju, Talanta, 2013, 114, 191 CrossRef PubMed.
- (a) W. Saenger, Principles of Nucleic Acid Structure, Springer, New York, 1984, p. 556 Search PubMed; (b) P. A. Furman, J. A. Fyfe, M. H. St Clair, K. Weinhold, J. L. Rideout, G. A. Freeman, S. N. Lehrman, D. P. Bolognesi, S. Broder and H. Mitsuya, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8333 CrossRef CAS PubMed; (c) A. Ojida, Y. Mito-oka, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 2454 CrossRef CAS PubMed.
- D. Zhang, J. R. Cochrane, A. Martinez and G. Gao, RSC Adv., 2014, 4, 29735 RSC.
- A. Aldrey, A. Macías, R. Bastida, G. Zaragoza, G. Rama and M. V. López, Org. Biomol. Chem., 2012, 10, 5379 CAS.
- (a) M. E. Moragues, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2011, 40(5), 2593 RSC; (b) P. A. Gale and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39(10), 3595 RSC; (c) B. P. Hay, Chem. Soc. Rev., 2010, 39(10), 3700 RSC; (d) P. A. Gale, Chem. Soc. Rev., 2010, 39(10), 3746 RSC.
- (a) S. Kondo and R. Takai, Org. Lett., 2013, 15, 538 CrossRef CAS PubMed; (b) Q. Y. Chen and C. F. Chen, Eur. J. Org. Chem., 2005, 2468 CrossRef CAS; (c) K. Ghosh, A. R. Sarkar and A. Patra, Tetrahedron Lett., 2009, 50, 6557 CrossRef CAS; (d) S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137(1), 499 CrossRef CAS PubMed; (e) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111(11), 6603 CrossRef CAS PubMed; (f) P. A. Gale, Acc. Chem. Res., 2011, 44(3), 216 CrossRef CAS PubMed; (g) J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198 RSC.
- (a) M. A. Ziganshin, L. S. Yakimova, K. R. Khayarov, V. V. Gorbatchuk, M. O. Vysotsky and V. Böhmer, Chem. Commun., 2006, 389 Search PubMed; (b) J. B. Puplampu, L. S. Yakimova, A. A. Vavilova, I. K. Rizvanov and I. I. Stoikov, Macroheterocycles, 2015, 8(1), 75 CrossRef; (c) V. V. Gorbachuk, L. S. Yakimova, A. A. Vavilova, R. V. Ziatdinova, I. K. Rizvanov, A. A. Trifonov, A. I. Samohina, V. G. Evtugyn and I. I. Stoikov, Silicon, 2014, 6, 215 CrossRef CAS; (d) J. B. Puplampu, L. S. Yakimova, A. A. Vavilova, D. A. Fayzullin, Y. F. Zuev and I. I. Stoikov, Macroheterocycles, 2014, 7(4), 337 CrossRef; (e) I. I. Stoikov, O. A. Mostovaya, L. S. Yakimova, A. A. Yantemirova, I. S. Antipin and A. I. Konovalov, Mendeleev Commun., 2010, 20(6), 359 CrossRef CAS; (f) L. S. Yakimova, M. A. Ziganshin, V. A. Sidorov, V. V. Kovalev, E. A. Shokova, V. A. Tafeenko and V. V. Gorbatchuk, J. Phys. Chem. B, 2008, 112, 15569 CrossRef CAS PubMed; (g) V. V. Gorbatchuk, M. A. Ziganshin, L. S. Savelyeva, N. A. Mironov and W. D. Habicher, Macromol. Symp., 2004, 210, 263 CrossRef CAS.
- (a) T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937 CrossRef CAS PubMed; (b) L. S. Yakimova, D. N. Shurpik, L. H. Gilmanova, A. R. Makhmutova, A. Rakhimbekova and I. I. Stoikov, Org. Biomol. Chem., 2016, 14, 4233 RSC; (c) D. N. Shurpik, P. L. Padnya, L. I. Makhmutova, L. S. Yakimova and I. I. Stoikov, New J. Chem., 2015, 39(12), 9215 RSC; (d) D. N. Shurpik, L. S. Yakimova, L. I. Makhmutova, A. R. Makhmutova, I. K. Rizvanov, V. V. Plemenkov and I. I. Stoikov, Macroheterocycles, 2014, 7(4), 351 CrossRef; (e) V. A. Smolko, D. N. Shurpik, R. V. Shamagsumova, A. V. Porfireva, V. G. Evtugyn, L. S. Yakimova and G. A. Evtugyn, Electrochim. Acta, 2014, 147, 726 CrossRef CAS; (f) D. N. Shurpik, L. S. Yakimova, I. K. Rizvanov, V. V. Plemenkov and I. I. Stoikov, Macroheterocycles, 2015, 8(2), 128 CrossRef; (g) Z. Zhang, B. Xia, C. Han, Y. Yu and F. Huang, Org. Lett., 2010, 12, 3285 CrossRef CAS PubMed; (h) Z. Zhang, B. Xia, C. Han, Y. Yu and F. Huang, Chem. Sci., 2012, 3, 3026 RSC; (i) Y. Yao, M. Xue, J. Chen, M. Zhang and F. Huang, J. Am. Chem. Soc., 2012, 134, 15712 CrossRef CAS PubMed.
- (a) X. Chi, X. Ji, D. Xia and F. Huang, J. Am. Chem. Soc., 2015, 137(4), 1440 CrossRef CAS PubMed; (b) D. Xia, G. Yu, J. Li and F. Huang, Chem. Commun., 2014, 50, 3606 RSC; (c) Z. Li, J. Yang, G. Yu, J. He, Z. Abliz and F. Huang, Org. Lett., 2014, 16(7), 2066 CrossRef CAS PubMed; (d) Y. Ma, X. Ji, F. Xiang, X. Chi, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Chem. Commun., 2011, 47, 12340 RSC.
- (a) V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889 RSC; (b) J. Jurczak, P. Dydio, P. Stepniak and T. Zielinski, RSC Adv., 2016, 6, 41568 RSC.
- I. I. Stoikov, L. I. Gafiullina, D. S. Ibragimova, I. S. Antipin and A. I. Konovalov, Russ. Chem. Bull., Int. Ed., 2004, 53(6), 1172 CrossRef CAS.
- (a) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106(12), 5291 CrossRef CAS PubMed; (b) A. A. Vavilova, R. V. Nosov, L. S. Yakimova, I. S. Antipin and I. I. Stoikov, Macroheterocycles, 2013, 6(3), 219 CrossRef CAS; (c) I. I. Stoikov, A. A. Vavilova, R. D. Badaeva, V. V. Gorbachuk, V. G. Evtugyn, R. R. Sitdikov, L. S. Yakimova and I. Zharov, J. Nanopart. Res., 2013, 15(5), 1617 CrossRef.
- (a) D. Cao and H. Meier, Asian J. Org. Chem., 2014, 3(3), 244 CrossRef CAS; (b) T. Ogoshi, M. Hashizume, T. A. Yamagishi and Y. Nakamoto, Chem. Commun., 2010, 46(21), 3708 RSC; (c) C. Li, X. Shu, J. Li, S. Chen, K. Han, M. Xu and X. Jia, J. Org. Chem., 2011, 76(20), 8458 CrossRef CAS PubMed; (d) H. Li, D. X. Chen, Y. L. Sun, Y. B. Zheng, L. L. Tan, P. S. Weiss and Y. W. Yang, J. Am. Chem. Soc., 2013, 135(4), 1570 CrossRef CAS PubMed; (e) T. Ogoshi, K. Masaki, R. Shiga, K. Kitajima and T. A. Yamagishi, Org. Lett., 2011, 13(5), 1264 CrossRef CAS PubMed.
- W. J. Wang, J. Lin, A. Wang, P. Zheng, M. Wang and B. Wang, Inorg. Chim. Acta, 1988, 149, 151 CrossRef CAS.
- R. M. Duke, J. E. O'Brien, T. McCabe and T. Gunnlaugsson, Org. Biomol. Chem., 2008, 6, 4089 CAS.
- G. Yu, Z. Zhang, C. Han, M. Xue, Q. Zhou and F. Huang, Chem. Commun., 2012, 48, 2958 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Compound characterization, synthetic details, determination of association constants, and other materials. See DOI: 10.1039/c6cc05797g |
| This journal is © The Royal Society of Chemistry 2016 |