DOI:
10.1039/C6CC04004G
(Feature Article)
Chem. Commun., 2016,
52, 12262-12277
Azobenzene photocontrol of peptides and proteins
Received
12th May 2016
, Accepted 1st August 2016
First published on 19th August 2016
Abstract
The last few years have witnessed significant advances in the use of light as a stimulus to control biomolecular interactions. Great efforts have been devoted to the development of genetically encoded optobiological and small photochromic switches. Newly discovered small molecules now allow researchers to build molecular systems that are sensitive to a wider range of wavelengths of light than ever before with improved switching fidelities and increased lifetimes of the photoactivated states. Because these molecules are relatively small and adopt predictable conformations they are well suited as tools to interrogate cellular function in a spatially and temporally contolled fashion and for applications in photopharmacology.
Introduction
The development of tools to control biomacromolecular function with light offers the potential to change the properties of defined molecules in biological systems with minimal perturbance to the rest of the system. This article presents highlights of current developments alongside their historical context to complement recent reviews of this field that have covered molecular switches,1,2 light-responsive materials,3,4 optogenetic tools5–7 and biological control.8
Principles of photocontrol
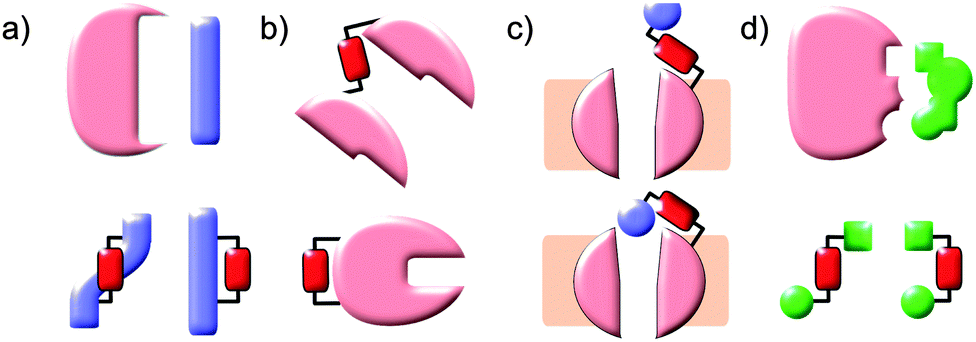
Protein–protein interaction networks allow cells to integrate chemical signals about their state and environment and respond by regulating protein production, enzyme activities and metabolism to suit their circumstances. Photocontrol of peptides, proteins or drugs provides a non-invasive way to perturb these networks to investigate their effect or cause a defined outcome. Achieving photocontrol requires the addition to a photoresponsive element to a protein, peptide or drug in such a way that its photochromism alters the properties of the conjugated molecule (Fig. 1). Some photochromic molecules respond to light by making or breaking bonds2 leading to changes in their geometry and polarity, but the increased degrees of freedom of one of the states can lessen conformational control. In contrast, molecules capable of acting as photoswitches that interconvert between E and Z double bonds offer highly predictable light-state structural properties. In such molecules isomerisation is induced by absorption of a photon to generate an electronically excited state, in which the barrier to rotation between the E and Z forms is reduced. Decay to the electronic ground state re-establishes the barrier, kinetically trapping the thermally less stable isomer. The barrier height relative to the available thermal energy then determines the rate of relaxation to the thermodynamically more stable isomer. Although stilbenes,9–12 hemithioindigos,11–23 thioxopeptides,24–28 selenoxopeptides,29 diarylethenes30,31 and acylhydrazones32 and rhodopsin-like molecules33,34 have been used as switches, azobenzenes35,36 currently offer the largest range of activation wavelengths and photoactivated state stabilities. In azobenzene-derived photoswitches rotation around the central double bond is transmitted with high fidelity through the rigid rings resulting in a predictable distance changes, particularly between substituents on the para positions. Azobenzenes are readily synthesised either non-symmetrically by the condensation of a nitroso-aryl compound and anilines or symmetrically by in situ nitroso formation under oxidising conditions. Classical aromatic chemistry allows access to various photo-switching wavelengths and thermal reversion characteristics tailored to suit a particular application (Fig. 2). The choice of azobenzene derivative is dependent on its spectral properties, the ratio of isomers at its photostationary state, its solubility, its resistance to reduction and the distance between the intended points of control.
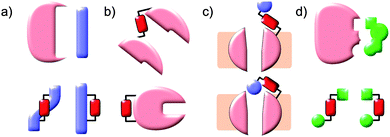 |
| | Fig. 1 Photochromic molecules can influence biomolecules by stabilising or destabilising their interacting conformations. This approach can be applied to (a) peptides corresponding to elements of protein–protein interaction partners, (b) loops or domain interfaces of proteins themselves, (c) tethered ligands to control ion channels or (d) incorporated into existing non-photochromic drugs. | |
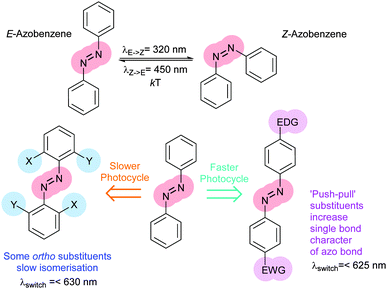 |
| | Fig. 2 Substituents can alter the photophysical properties of azobenzenes with wavelength ranges between 300 and 630 nm and provide the means to tune the half-life of the thermodynamically less stable Z-isomer. | |
Azobenzene photoswitches
Improving photophysical properties
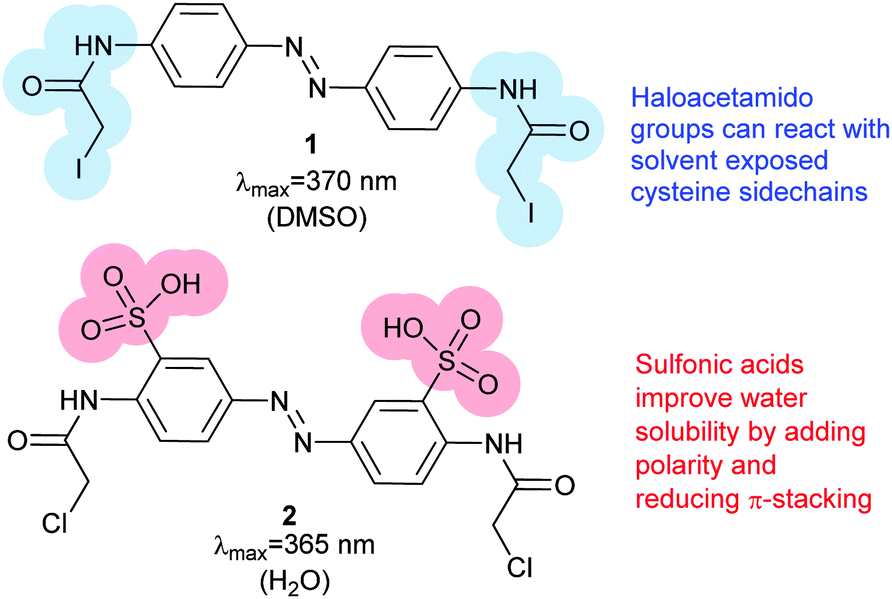
Photoswitches can be conveniently linked to proteins by selective reactions with the thiol side chains of cysteine residues. Surface accessible cysteine side chains react readily with e.g. para-haloacetamide substituted azobenzenes such as iodoacetamidoazobenzene (1) (Fig. 3).37 Early work demonstrated that with pairs of cysteines in i,i + 4 or i,i + 7 spacings along a peptide chain, an α-helical conformation is stabilised by the Z-isomer of 1 while the E-isomer is α-helix stabilising for i,i + 11 spacings.38 The water-soluble azobenzene 2 expanded the scope of this technique and allowed protein–protein, protein–DNA and protein–RNA interactions to be controlled through α-helical motifs.39,40 While the excitation wavelengths in the UV-range limited the biological applications of these early azobenenzene photoswitches, new photochromic molecules that absorb at higher wavelengths have recently been developed and can be actuated with non-toxic visible light.41
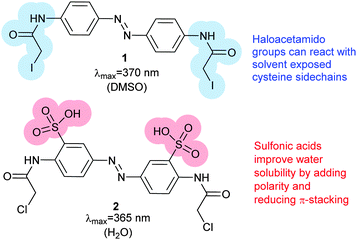 |
| | Fig. 3 Bis-para-(iodoacetamido)azobenzene (1) reacts with the sidechains of a pair of cysteines of a peptide to form a photo-responsive ‘staple’. The addition of sulfonic acid groups render 2 water-soluble. | |
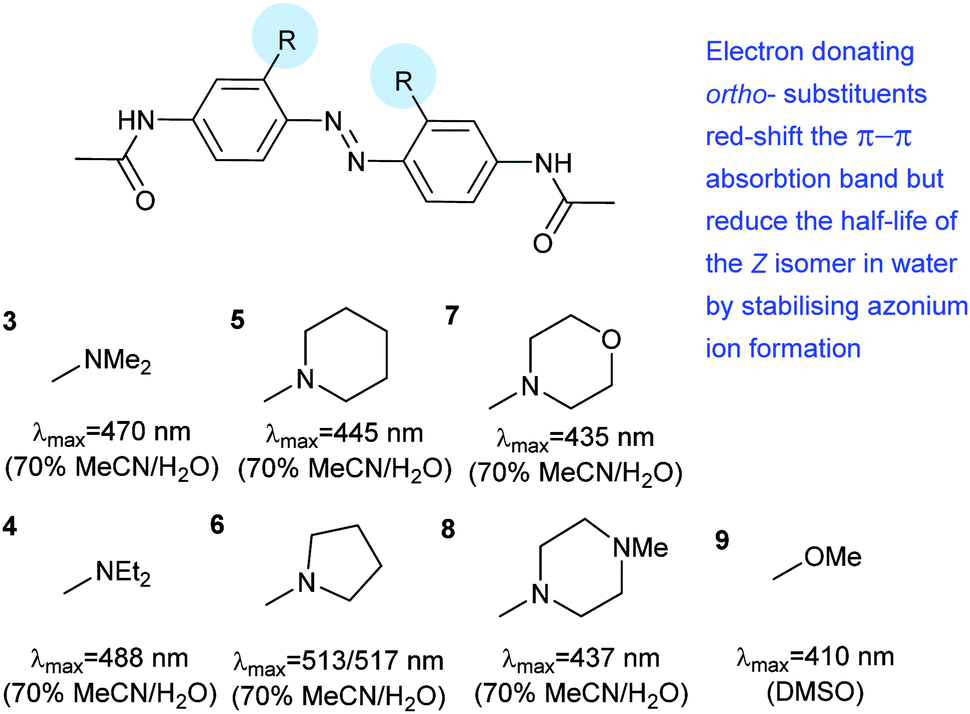
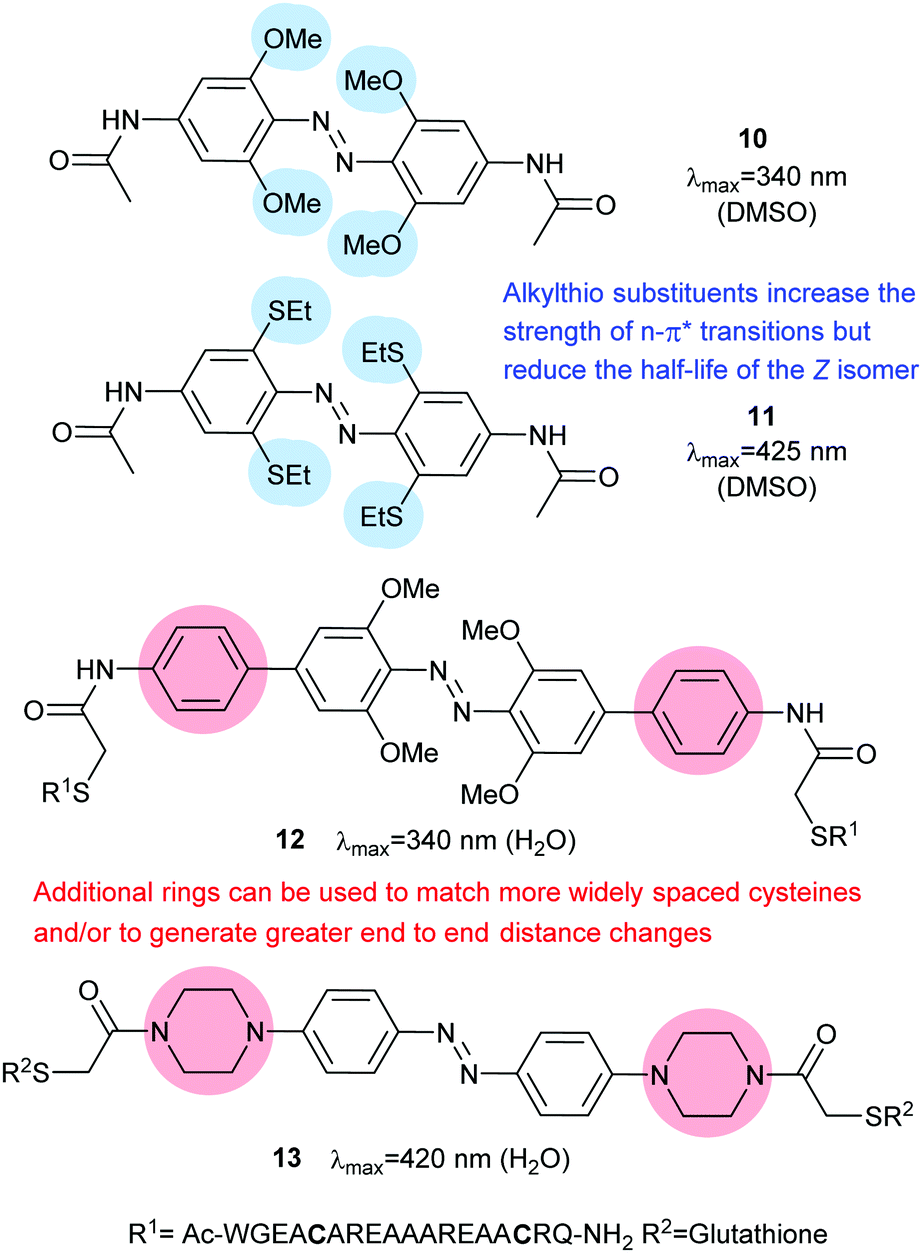
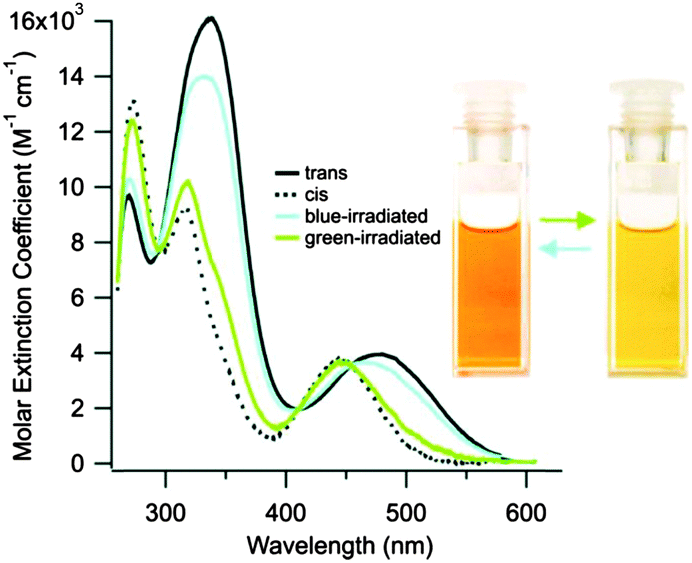
Introduction of ortho-amino groups yielded azobenzenes (3–8) with a wide range of photochemical properties (Fig. 4).42 Most have significantly red-shifted absorbance spectra (445–470 nm), but protonation of the amines produces forms that diminish the double bond character of the azo bond and lead to shorter relaxation half-lives of between 0.7 seconds and 5 minutes compared to ∼30 minutes for 2. A red-shifted π–π* absorbance maximum was also observed with a pair of ortho-methoxy substituents (9), but the introduction of four methoxy-groups ortho to the azo-bond (10) produced different properties (Fig. 5).43 The absorbance spectrum of Z-10 reveals a 36 nm shift of its n–π* band compared to E-10 (Fig. 6). This separation of the n–π* bands permits green light irradiation (530 to 560 nm) to convert E-10 to ∼80% Z-10, eliminating the need for damaging UV light altogether. Furthermore, the relaxation half-life of Z-10 is 12 hours in buffer at 40 °C, making it ideal for biological applications. However, the electron donating effect makes 10 vulnerable to reduction with a half-life of 90 min in glutathione-containing buffers that reproduce the cytosolic reduction potential.43 Replacing the methoxy groups with methylthioethers in 11 retained the separation between the n–π* transitions of the isomers, which were red shifted and strengthened compared to 10, but model bioconjugates of 11 had short half lives of the light activated state (3.3 minutes at 37 °C).44 In contrast, the extended analogue Z-12 had a thermal reversion half-life of several hours in water at 37 °C.45
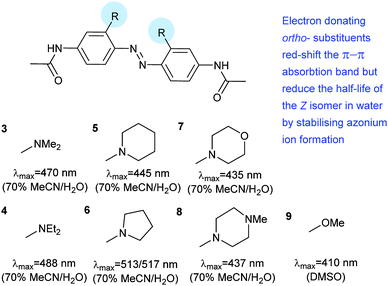 |
| | Fig. 4 Amino-substituted azobenzenes with red-shifted π–π* transitions and rapid relaxation. | |
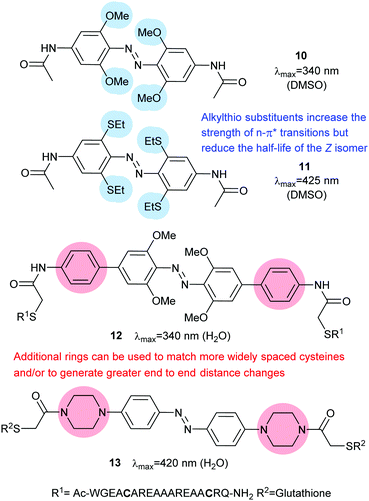 |
| | Fig. 5 Tetra-ortho ether or thioether substitution has the beneficial effect of shifting the n–π* transition bands so that at certain wavelengths the absorption spectra of the E- and Z-isomers do not overlap, allowing visible light switching. Extended azobenzenes may either be used with more widely spaced cysteine sidechains or to increase the change in end-to-end distance to provide greater photocontrol. | |
 |
| | Fig. 6 UV/visible spectra of 10 in dimethylsulfoxide show the separation of the n–π* bands at 450 and 500 nm. The separation is less pronounced in water, but bidirectional visible light switching is still possible. Reprinted with permission from Beharry et al., J. Am. Chem. Soc., 2011, 133, 19684. Copyright 2011 American Chemical Society. | |
Given the destabilising effect of amino (rather than amide) substituents of 3–8 on their Z isomers, it is understandable that installing piperidine as a more flexible spacer in the para position resulted in Z-13 having a half life of less than 4 seconds in buffer at 25 °C.46
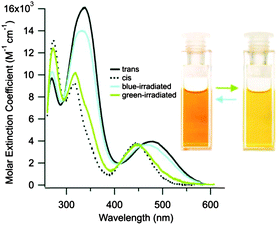
However, when this motif was combined with tetra-ortho-methoxy substitution, the resulting molecule (14) displayed unexpected properties.47 With methoxy and amino substituents in place the pKa of the protonated form of 14 rises to the physiologically accessible value of 7.548 and the absorption spectrum of the azonium species is broad enough for irradiation with 630 nm light to cause isomerisation. This effect was ascribed to a difference in pKa values between the protonated states of E-14 and Z-14, with the azonium species stabilised by a hydrogen bond in E-14 but not in Z-14 (Fig. 7).
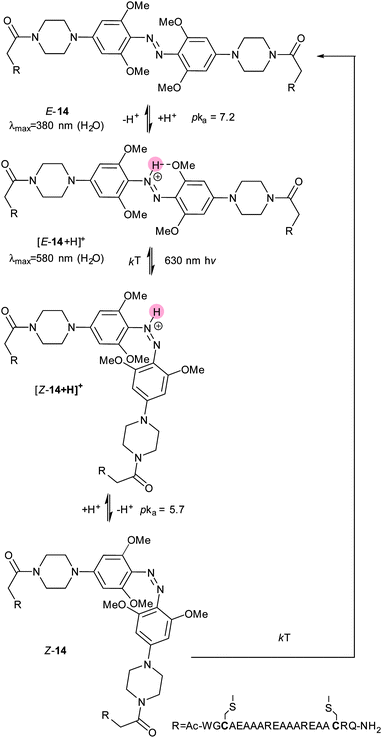 |
| | Fig. 7 The combination of tetra-ortho methoxy ether and para amino substitution increases the pKa of the diazo bond, so that azonium species are formed at neutral pH. However, the pKa of E-14 is approximately 1.5 units lower, resulting in deprotonation and slower relaxation to Z-14 than would be expected from the azonium ion. | |
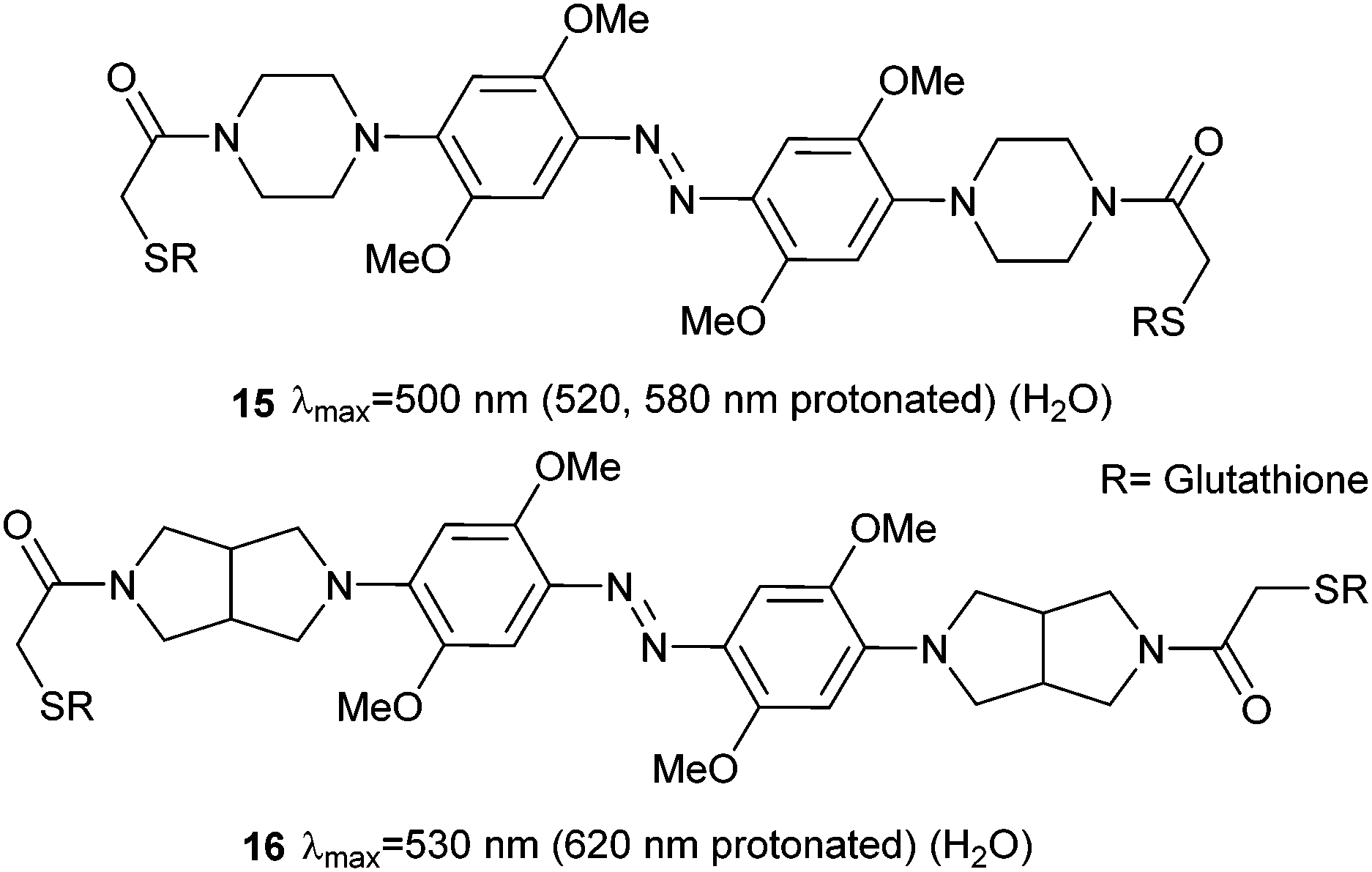
Therefore, at pH 7, 14 is protonated, photoswitches and then deprotonates. Whilst the half-life of Z-14 is only 13 seconds, it can reliably and repeatedly be photoswitched by red light in the presence of whole blood, although it is sensitive to reduction by glutathione.47 A second generation of azonium ion precursors (Fig. 8) were designed to determine the optimal methoxy substitution pattern to donate electrons to the ring without sterically hindering azonium ion formation.48 These efforts resulted in 15, which absorbs significantly at 650 nm. However, the meta methoxy group increases steric hindrance and forces the piperazine ring to rotate into a conformation that is less able to stabilise azonium ion formation, reducing the apparent pKa to 2.6. This effect is alleviated in 16 by the less bulky bicyclic pyrrollidine substituent and the apparent pKa is increased to 5.4. The absorption spectrum of 16 stretches into the near IR region with significant absorbance up to 750 nm.
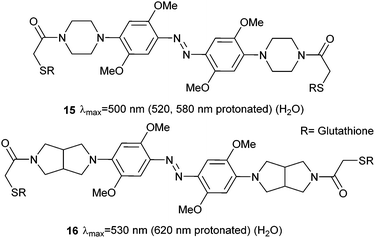 |
| | Fig. 8 Moving one methoxy group to the meta position maintains the electron rich nature of the azobenzene ring system, but removes steric congestion from the azonium ion formed on protonation. | |
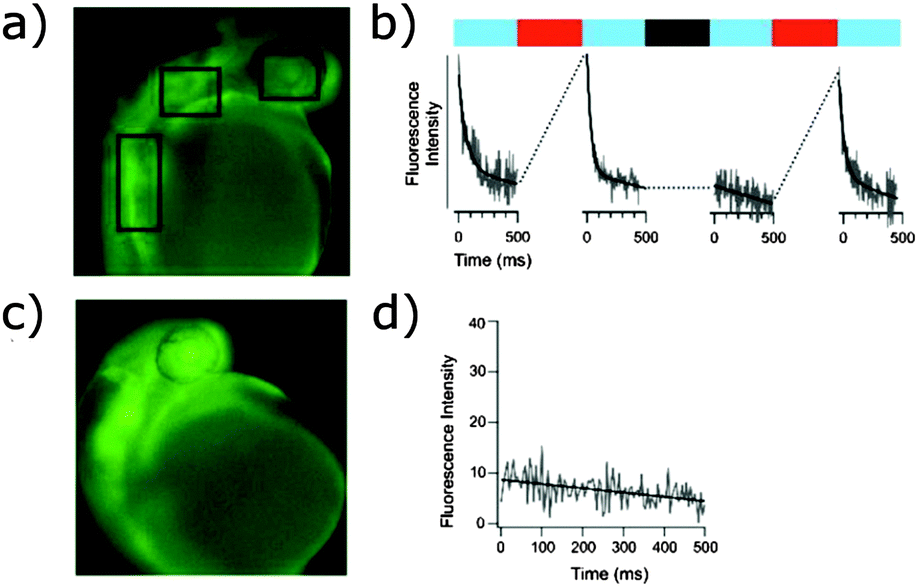
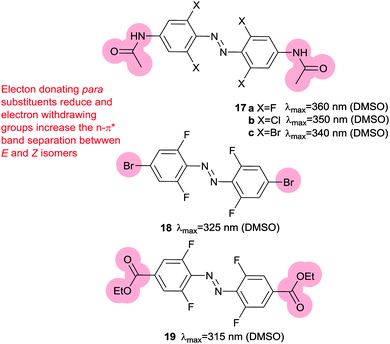
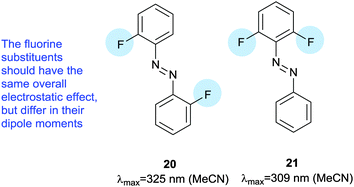
Electron rich azobenzenes can support protonation prior to or concomitant with nucleophilic attack by thiols and are vulnerable to reduction by glutathione. In contrast, tetra-ortho-halogen substituents (17) including tetra-ortho-fluoro49 and chloro variants45 are all resistant to glutathione reduction. Tetra-ortho-fluorodi-para-bromo azobenzene 18 has a clear (42 nm) separation between the n–π* transitions of its E and Z isomers, which allows visible light switching (Fig. 9).49 A mildly electron releasing group in the para position (17a) reduced the n–π* band separation to 22 nm compared to 18, whilst a more electron withdrawing ester substituent (19) increased it to 50 nm. Detailed studies of fluorinated azobenzene derivatives with symmetrical or non-asymmetrical rings and various electron withdrawing groups in the para-position including fluorine, esters, nitriles and amides were carried out. Symmetrical (20) or non-symmetrical (21) difluoro azobenzenes (Fig. 10) exhibited a smaller (28–32 nm) n–π* band separation than 18 (42 nm), but the presence of further additional fluorine atoms did not result in increased separation in hexa-, octa- or deca-fluorinated azobenzenes (36–43 nm).50 Introduction of a symmetric pair of more electron withdrawing trifluoromethyl groups in the ortho positions decreased the n–π* band separation, an effect attributed to the increased steric demand of this substituent resulting in the E-isomers phenyl rings not being coplanar. This reduction in conjugation in the ground state is likely to reduce the difference in energy and hence the n–π* band separation between the E- and Z-isomers. Time dependant density functional theory calculations qualitatively reproduced the spectral properties for this series of compounds and further modelling suggested that the extended half-lives of their Z isomers are a consequence of the dipole moments in the transition states.50 Tetra-ortho-chloro azobenzene 17b had a similar n–π* band separation (20 nm) to 17a (22 nm) and was shown to photoswitch under irradiation by a 635 nm LED.45 Rapid reversion to E-17b was conveniently induced by irradiation with 450 nm light and neither isomer of 17b was degraded by exposure to 10 mM reduced glutathione. When the chloroacetamido analogue of 17b was attached to a test peptide, a thermal reversion half-life of 3.5 hours was observed at 37 °C. Subsequently, 17b was conjugated to a D-peptide labelled with fluorescein and microinjected into zebrafish embryos.45 Photoswitch-induced quenching of the fluorophore51 was possible for 30 hours with no obvious ill-effects (Fig. 11).45
 |
| | Fig. 9 Tetra-ortho halogen substituents separate the n–π* transition energies of E and Z isomers, impart resistance to glutathione reduction and lead to long lived Z-isomers. | |
 |
| | Fig. 10 Symmetrical and non-symmetrical difluoroazobenzenes used to examine the origins of the n–π* band shifts and extended Z-isomer half-lives. | |
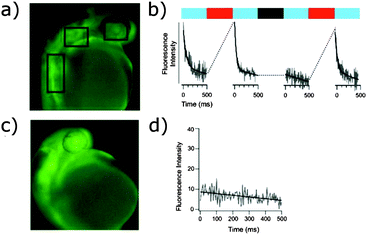 |
| | Fig. 11 (a) Fluorescence image of a zebrafish embryo (early pharyngula stage/∼30 hours post fertilisation) containing the fluorescent reporter peptide cross-linked with 4. (b) Measurements of fluorescence intensity at the zones indicated by black boxes in (a), as a function of time during blue light irradiation. A change in fluorescence emission was seen only if the peptide was first switched with red light (indicated by red bars). No pre-irradiation with red light (indicated by black bar) before blue light produces no exponential (switching) transient, only a background slope due to fluorescein reporter photobleaching. (c) Fluorescence image of a zebrafish embryo (∼30 hours post fertilisation) containing the fluorescent reporter peptide without a photoswitch. (d) Measurement of fluorescence intensity of the nonswitchable peptide as a function of time during blue-light irradiation after preirradiation with red light as in (b). Adapted with permission from Samanta, et al., J. Am. Chem. Soc., 2013, 135, 9777. Copyright 2013 American Chemical Society. | |
This system represents the current state of the art for a slow cycling switch in that it is stable against reductive and cytosolic reduction and offers the capability of bi-directional, visible-light switching in vivo. Further developments in slow-cycling photo-switches have been driven by potential uses for molecules that can respond to infrared activation to which the human body is mostly transparent. Such switches could be activated at defined points in 3D space using multiphoton techniques.
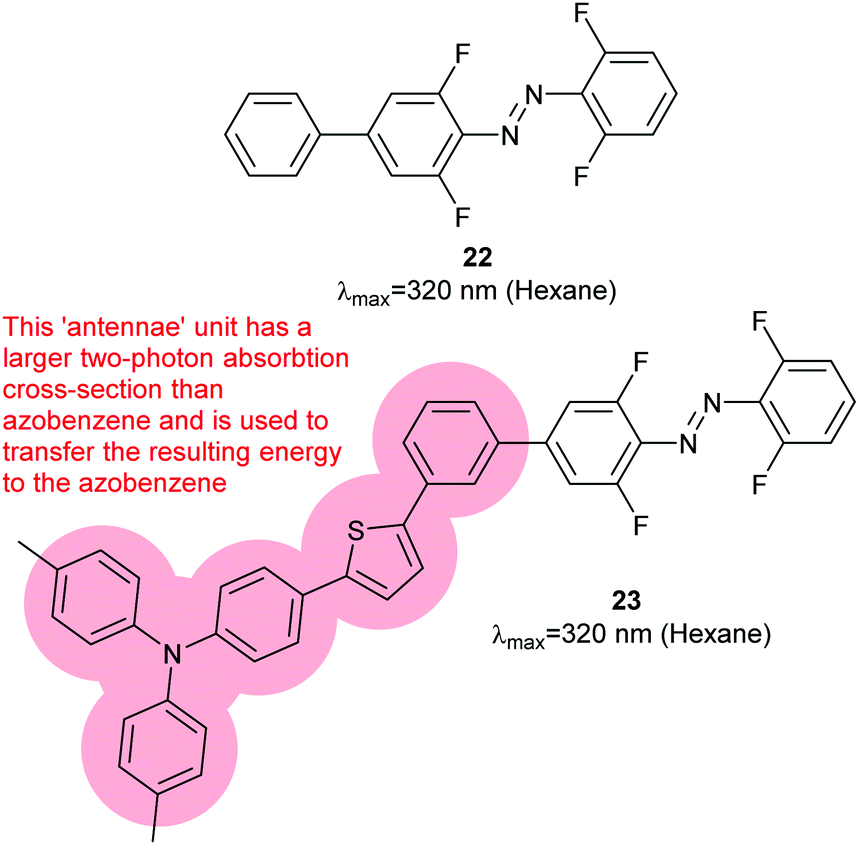
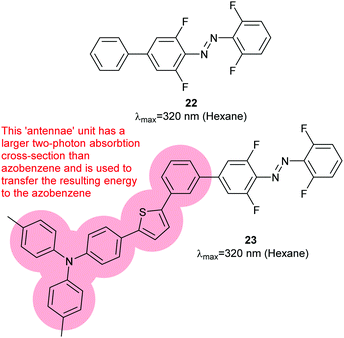
Two-photon photoswitching had previously been reported to be effective for 1 in blood lysates52 but not for tetra-ortho-methoxy dyes.45 However, a slightly modified tetra-ortho-fluoro azobenzene (22) was found to have a two-photon cross-section seven times greater than azobenzene alone; two-photon excitation of the π–π* band was possible at 640 or 720 nm.53 Installation of a larger aromatic system with extended conjugation to act as an antennae complex yielded 23 (Fig. 12), which can be converted to Z-23 by 510 nm light and back to E-23 by two-photon absorption at 750 nm.54
 |
| | Fig. 12 Tetra-ortho-fluoro substitution increases the two photon absorption cross section compared to azobenzene itself. A two photon antennae complex is excited, then transfers energy to the azobenzene. | |
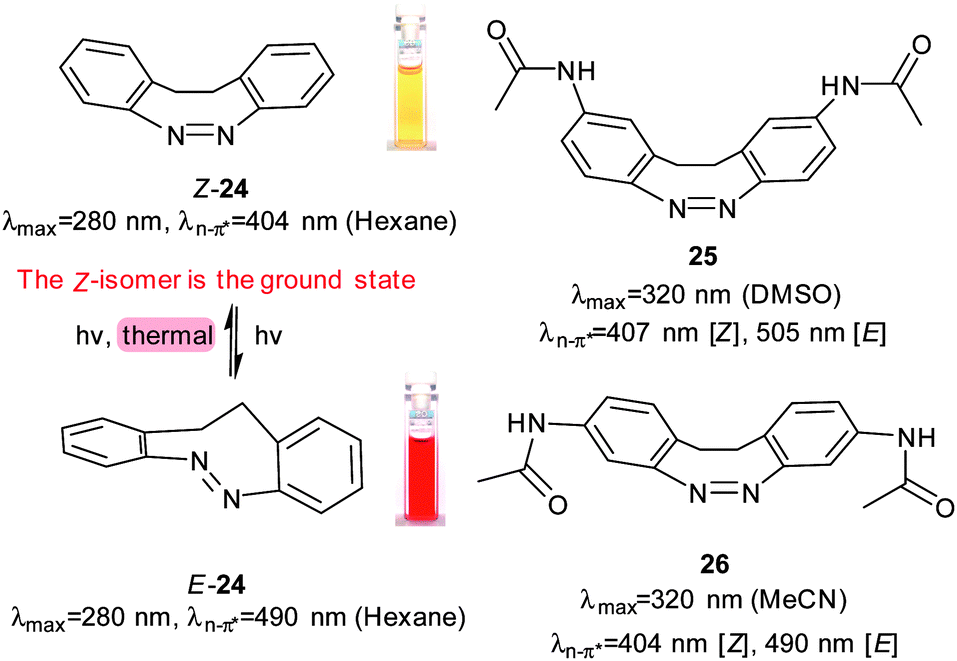
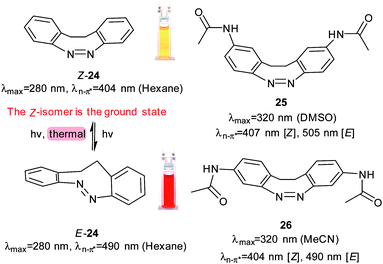
Another series of photoswitches employs different types of bridged azobenzenes (Fig. 13).55–58 The bridge has the effect of reversing the normal thermodynamic preference for the E-isomer so that 24 adopts almost exclusively the Z-form in its thermal equilibrium state. 24 is converted to >90% of the E-form on irradiation with 370–400 nm light. Reversion to Z-24 can be achieved with irradiation with 480–550 nm light. When a chloroacetimido analogue of 25 was attached to a test peptide, a photostationary state of 70% E-25 was formed upon irradiation with 407 nm light.59 Thermal relaxation from the E to the Z form occurred with a half-life of 8.3 hours at 20 °C, but reversion could also be driven with 518 nm light irradiation. Whilst Z-25 was completely resistant to glutathione reduction, E-25 degraded with a half-life of approximately 3 hours. A different ring substitution position resulted in 26, whose n–π* transitions were separated by almost 90 nm (Z-26: 404 nm, E-26: 490 nm) although only 53% E-26 was present in the photostationary state.60 Other examples of constrained azobenzenes are found in structures containing a borodifluoride bridge (27). The switching wavelengths for these molecules are strongly red shifted (570 nm) compared to conventional azobenzenes.61 Electron donating substituents can be used to push red absorbing 28 (630 nm) further towards the IR range in 29 (710 nm). An additional methoxy substituent ortho to the azo substituent in 30 caused a blue-shift (640 nm) because its steric demands twisted the phenyl ring out of the plane of the azo bond, thereby reducing its electronic influence (Fig. 14).62
 |
| | Fig. 13 The conformational constraints applied to bridged azobenzenes make their Z-isomers their thermodynamic ground states. Many bridged azobenzenes have very large separations between the n–π* band s of the E and Z isomers. Elements adapted with permission from Siewertsen, et al., J. Am. Chem. Soc., 2009, 131, 15594. Copyright 2009 American Chemical Society. | |
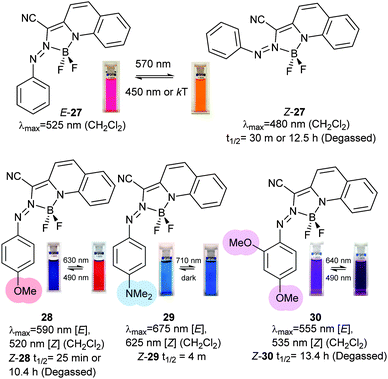 |
| | Fig. 14 Borodifluoride bridged azobenzenes are strongly red shifted and the half-lives of the Z isomers are strongly affected by dissolved oxygen. Elements adapted with permission from Yang, et al., J. Am. Chem. Soc., 2012, 134, 15221 (Copyright 2012 American Chemical Society) and Yang, et al., J. Am. Chem. Soc., 2014, 136, 13190. | |
New incorporation and conjugation approaches
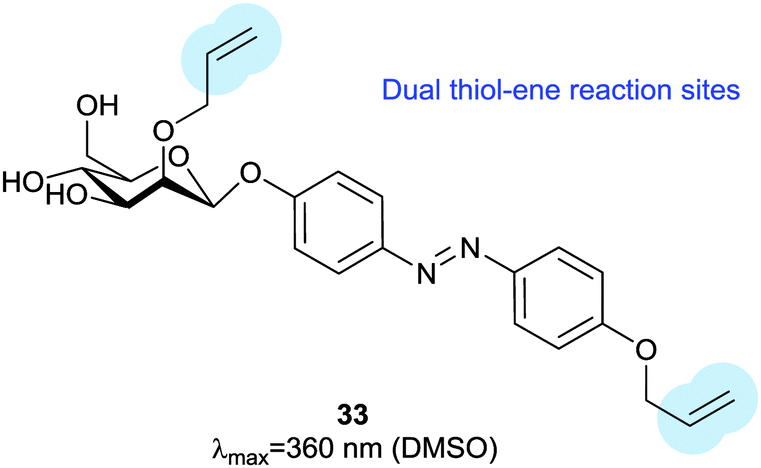
Thiol–ene reactions are gaining popularity for bioorthogonal functionalisation of peptides and proteins.63 The vinyl azobenzene amino acid 31 was incorporated into peptides by conventional solid phase synthesis and, when an i + 4 cysteine residue and an appropriate photoinitiator were present, irradiation with 360 nm light caused isomerisation and a thiol–ene click reaction between the cysteine sulfur and the pendant vinyl group.64 Consequent oxidation resulted in intramolecular crosslinking via a sulfone. Genetic code expansion can be used to introduce 31 into proteins expressed in E. coli or mammalian cells.65 Azobenzene 32 (Fig. 15) can also be incorporated into expressed proteins; its pentafluoro ring is electrophilic enough to undergo nucleophilic aromatic substitution by a proximal cysteine.66 The fluorine atoms of 32 provide sufficient n–π* band separation for bidirectional switching with 540 nm and 470 nm light to generate ∼70% of the desired isomer in the photostationary state. This switch was incorporated into calmodulin with an orthogonal tRNA synthetase. Whilst a degree of side reaction with glutathione was noted, cyclisation proceeded smoothly to give a visible light responsive calmodulin with distinct binding affinities for neuronal nitric oxide synthase under green and blue light irradiation. Thiol–ene chemistry was also investigated to introduce a bifunctional glycosylated azobenzene photoswitch.67 Using 2,2-dimethoxy-2-phenylacetophenone as a photoinitiator caused 33 to react with glutathione in 70% yield (Fig. 16).
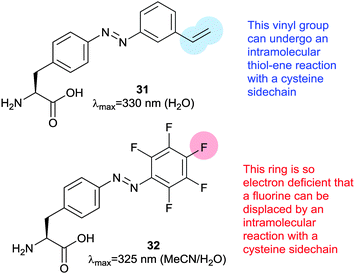 |
| | Fig. 15 A vinyl azobenzene (31) capable of intramolecular thiol–ene cyclisation with an appropriately placed thiol–ene sidechain and a pentafluoroazobenzene (32) whose para fluoro group can be displaced by a thiolate anion. | |
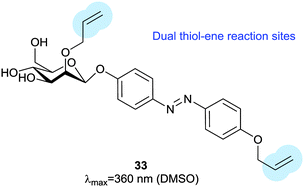 |
| | Fig. 16 Glycoazobenzene derivative for conjugation thiol–ene reactions. | |
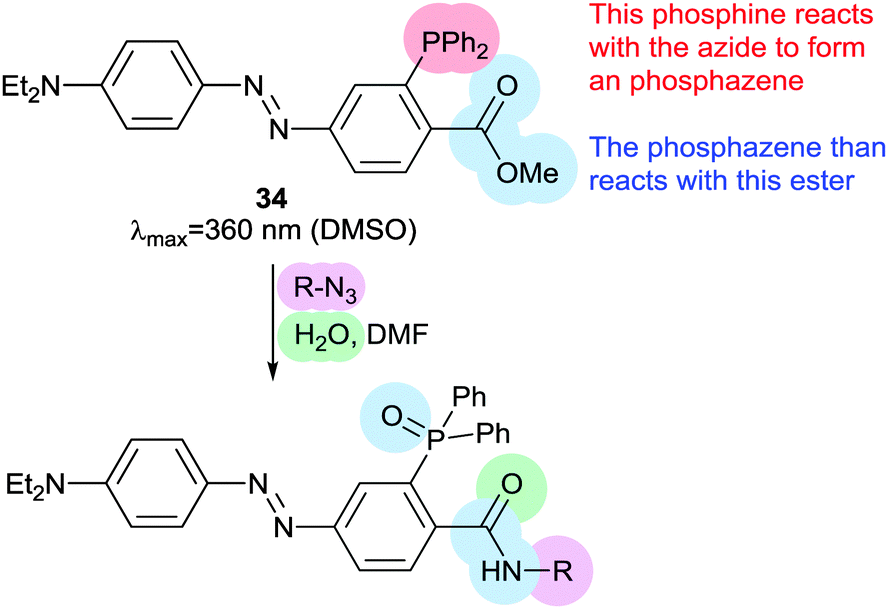
Staudinger ligations can be used to selectively functionalise synthetic biomolecules containing azide groups incorporated for in situ bioorthogonal labelling. The diphenylphosphoryl azobenzene 34 performs a variation of the Staudinger ligation, intramolecularly capturing the phosphazene intermediate formed by reaction of the phosphine with an azide-containing peptide. The resulting conjugate has a π–π* absorption maximum at 500 nm, but the reaction is relatively slow and the Z-isomer is short lived (0.5 ms) (Fig. 17).68
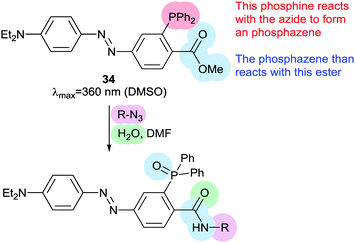 |
| | Fig. 17 Diphenylphorhinylazobenzene 34 can be used to introduce a pendant ‘push–pull’ azobenzene into peptides by Staudinger ligation chemistry. | |
In vitro translation and expanded genetic code techniques have been used to create a library of peptides containing the azobenzyl-lysine 35 (Fig. 18).69 By suppressing the stop codon and conducting the reaction at low temperature, neither the nascent peptide nor the coding mRNA dissociate from the ribosome. Streptavidin-coated beads were placed in the solution in the dark and the beads were washed and exposed to UV light, which resulted in dissociation of the peptide–ribosome–mRNA complex. The complexes were recovered, the mRNA dissociated and subjected to amplification by reverse transcriptase polymerase chain reaction; this identified peptide sequences that bound streptavidin 2.5-fold weaker in their photostationary state than in the dark. Libraries of peptides have also been generated using phage display.70 Peptides containing i,i + 7 spaced cysteines were expressed in E. coli, reduced with resin-bound tris-carboxyethylphosphine and treated with 2. Although the overall yield of modification with 2 was only 50%, peptides were identified with up to 22-fold decreases in affinity for streptavidin in their pure dark and light states. A later study also used i,i + 7 spaced cysteine residues alkylated with 2 and discovered peptides that had a greater affinity in for streptavidin in the light state with the best example found to bind 3-fold more tightly.71
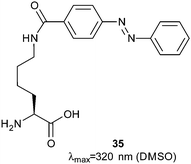 |
| | Fig. 18 Azobenzyl-lysine, a readily synthesised amino acid with a pendant azobenzene in its sidechain. | |
Selected applications
Control of nucleic acid binding peptides
Transcription factors act as ‘master switches’ in cells for the expression of genes in response to environmental cues. As such they are key targets to modulate downstream processes. Many common DNA binding motifs contain α-helices, which make direct and indirect contacts with the bases in the major groove of DNA. Such helices are often unstructured in the absence of DNA or when isolated from the rest of the protein, but are stabilised through interaction with other structural elements. Recognition helices in homeodomains, for instance, rely on a helix-turn-helix motif for stability (Fig. 19). The DNA recognition helix from the engrailed homeodomain has been modified to incorporate two i,i + 11 spaced cysteine residues to accommodate 2.72 When the peptide is folded into an α-helix, the intra-cysteine spacing matches the length of E-2 so that the cross-linked peptide is strongly helical and binds to target DNA 25 times more strongly than a control peptide without 2. Irradiation to form Z-2 diminished the DNA binding affinity 18-fold.
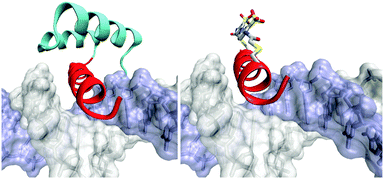 |
| | Fig. 19 Cartoon of the engrailed homeodomain DNA binding helix in the major groove of its target DNA and its N-terminal extension (red), which contacts the minor grove of the DNA (left) with two short α-helices (cyan) that stabilise it. The stabilising role of these α-helices can be replaced by introducing a pair of cysteine residues whose sidechains project opposite the DNA binding face to react with 2. | |
Basic helix-loop-helix proteins such as the transcription factor MyoD bind to DNA as homodimers (Fig. 20). The DNA-binding region is unstructured in the absence of DNA but adopts an α-helical conformation when it contacts the DNA. The recognition helix was modified to include i,i + 7 spaced cysteine residues and reacted with 2. The photo-activated state of the cross-linked protein not only bound DNA with an apparent dissociation constant for the complex three orders of magnitude higher than for the uncrosslinked protein, equivalent to a 60-fold decrease in the half-maximal binding concentration, but also displayed a significant increase in the specificity for its cellular target DNA.73
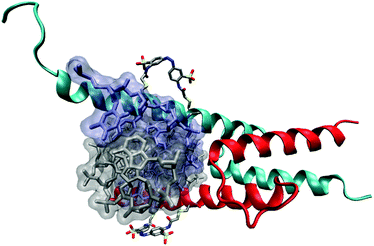 |
| | Fig. 20 Cartoon illustrating the use of 2 to control the conformation of the DNA recognition α-helix of MyoD. | |
bZIP proteins form a different family of transcription factors that rely on α-helical structures. They contain discrete DNA-binding and leucine zipper regions. DNA binding requires dimerization through the formation of a coiled coil (Fig. 21) so that photocontrol can be exerted through the leucine zipper, leaving the DNA-binding region unchanged. i,i + 7 spaced cysteines were used to create a GCN4-bZIP peptide, which reacted with 2 to give a photoswitchable DNA-binding peptide with increased α-helicity and a 20-fold decrease in dissociation constant towards a DNA target after irradiation.74
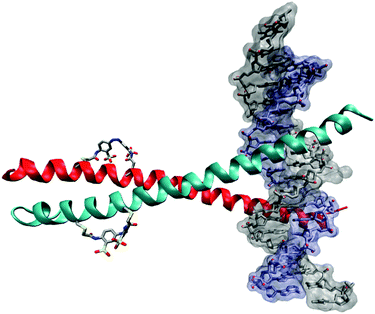 |
| | Fig. 21 Cartoon of a crosslinked bZIP DNA complex. The crosslinker 2 controls the α-helicity of the coiled-coil dimerization domains, which in turn controls the DNA binding avidity of the complex. | |
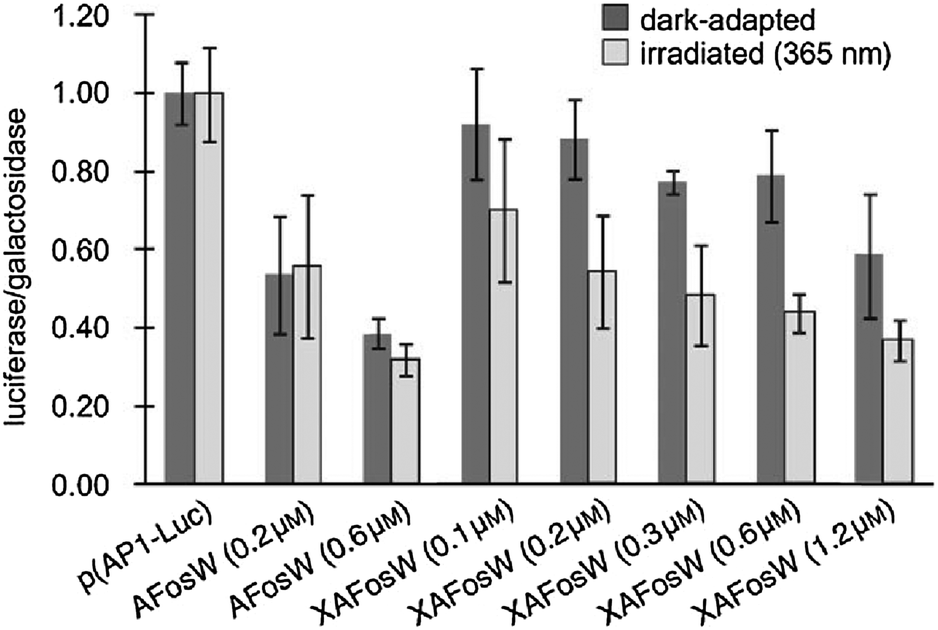
Similar methodology was also effective in controlling of the cyclic AMP response element-binding protein (CREB).75 Photocontrol of transcription in live cells was achieved by a modified bZIP helix with i,i + 7 spaced cysteines and a pair of additional acidic heptad repeats at its N-terminus.76 Once crosslinked with 2 and introduced into HEK293T cells alongside reporter DNA this molecule (0.1 to 1.2 μM) was able to alter the expression level 2-fold upon irradiation (Fig. 22).
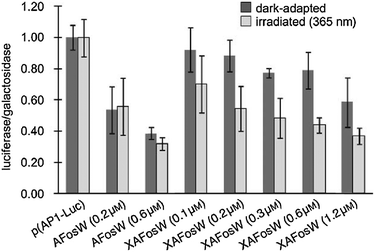 |
| | Fig. 22 293T cells were transfected with luciferase reporter plasmid p(AP1-Luc) and β-galactosidase plasmid pRSV-Gal. The ratio of the activities of the two enzymes was used to detect specific effects on AP1 promotor driven expression. Addition of non-crosslinked AFosW had a dose dependant effect as AFosW displaced native Fos from the Fos/Jun/AP-1 DNA complex. Addition of crosslinked XAFosW has less of an effect as the peptide is biased away from its active conformation by the crosslinker, UV irradiation removes this constraint and yields a 2-fold light-dependant inhibition of protein synthesis. Reproduced with permission from Zheng, et al., Angew. Chem., Int. Ed., 2010, 49, 3943. Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA. | |
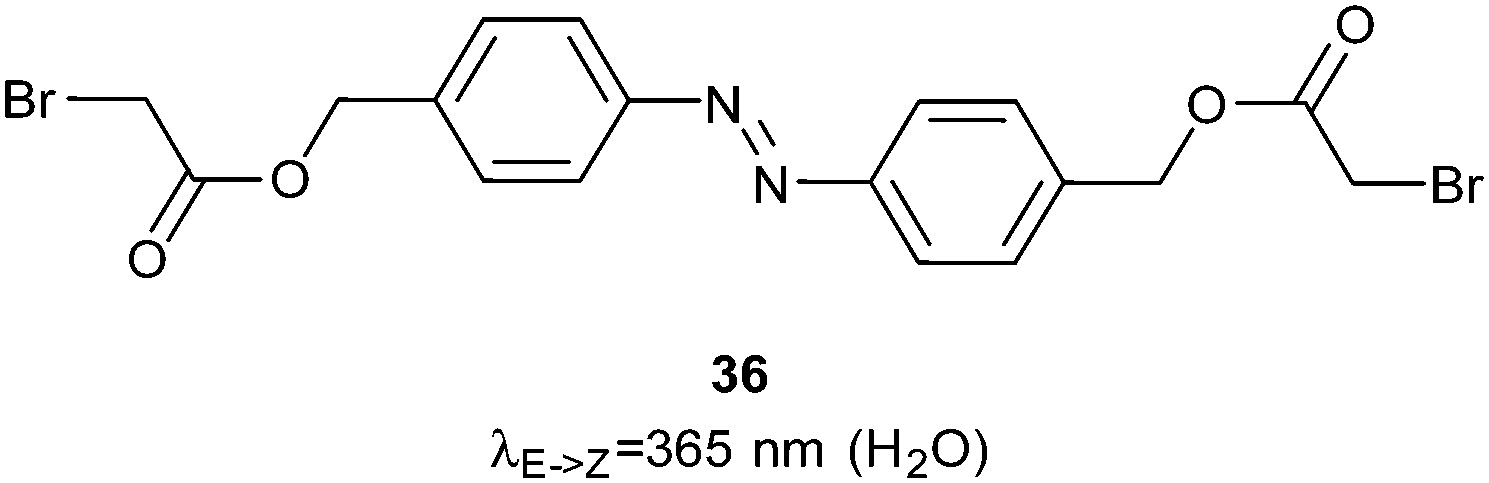
In a second approach to controlling the bZIP family, an azobenzene linker (36) was bound to two DNA-binding helices by forming thioethers with cysteine residue side chains (Fig. 23).77 When the azobenzene was switched into the Z-conformation these helices aligned scissor-like into the major groove of a DNA strand and bound DNA 60–70 times more strongly than in the dark state. Single stranded RNA presents a greater variety of binding geometries than double stranded DNA because of its propensity to adopt different knot and loop structures. Nevertheless, good photocontrol of RNA binding was achieved using a peptide derived from the HIV1 Rev response element (RRE)78 crosslinked with 1 for both i,i + 7 and i,i + 11 spacings.
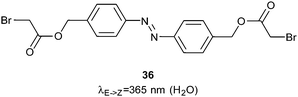 |
| | Fig. 23 A bromoacetate ester azobenzene used to control the relative orientation of two basic DNA-binding helices. | |
Control of protein–protein interactions
Although many examples of controlling the structure of β-hairpins79–86 and cyclic peptides87–94 are known, light is most frequently used to control protein–protein interactions by influencing the conformation of tethering loops or α-helices or by influencing the interactions of subunits (Fig. 24).
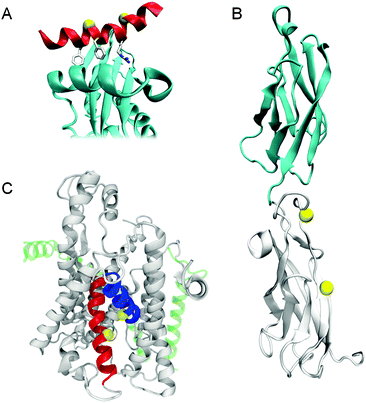 |
| | Fig. 24 Cartoons illustrating the positions changed to cysteines to accept azobenzenes in (A) AP2 β-appendage (PDB 2IV8). (B) Loop regions of cadherin (PDB 1FF5). (C) Trans-membrane helices 2 (blue) and 7 (red) of the SecYEG complex (PDB 1HRZ). | |
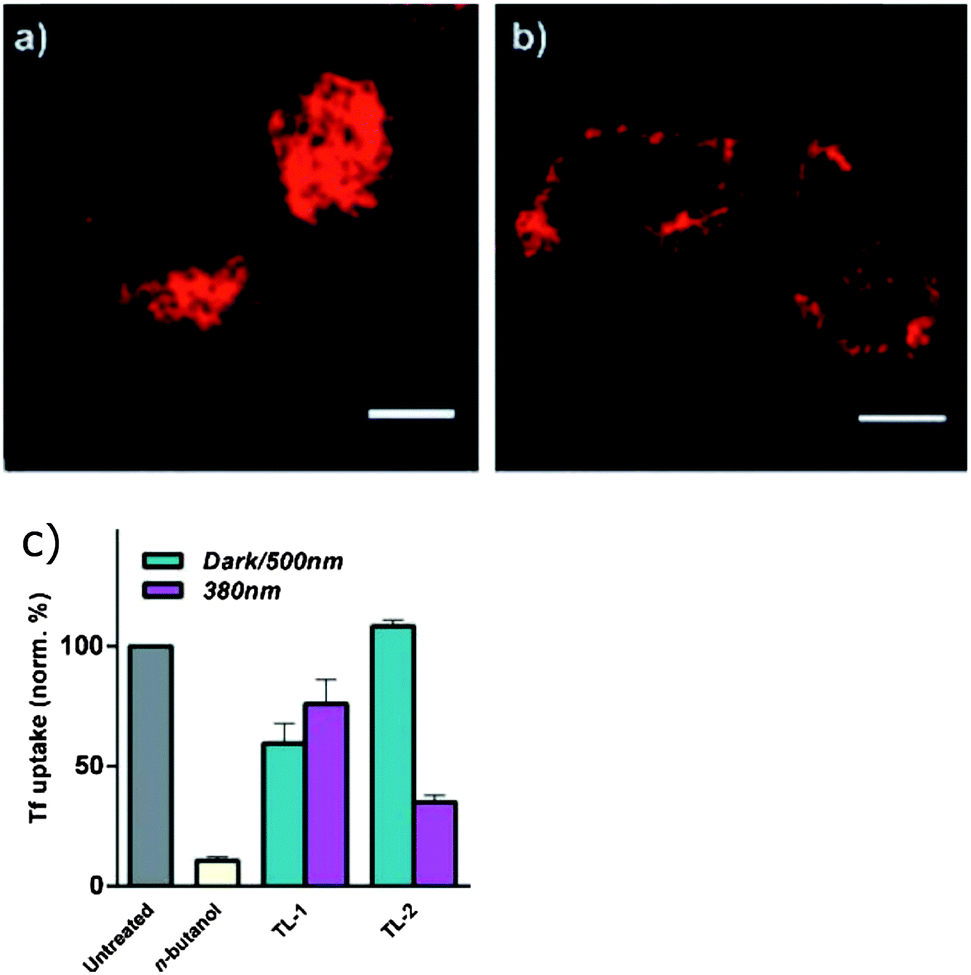
A photoswitchable peptide was designed so that its α-helical conformation bound to the adaptor protein AP2 (Fig. 24A).95,96 This protein is important for the generation of clathrin-coated pits for internalisation into cells. Peptides with i,i + 7 and i,i + 11 spaced cysteine residues, whose side chains were alkylated with 2, were able to enter cells and allowed photocontrol of fluorescent transferrin uptake. The i,i + 7 peptide was the most effective with a 3-fold decrease in intracellular fluorescence upon irradiation (Fig. 25). Peptide concentrations from 20 μM to 100 μM were employed depending on the cell type.
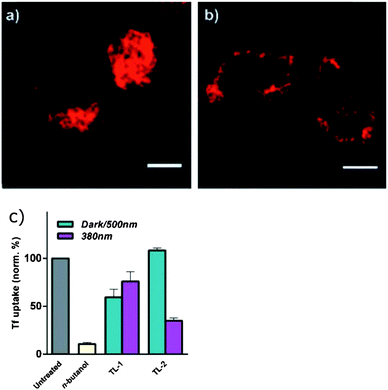 |
| | Fig. 25 HeLa cells incubated with Alexa647 labelled transferrin and acid-washed show internalisation in (a) the absence of peptide, but (b) the protein remains surface bound when the cells are treated with TL-1, an inhibitor peptide with i,i + 11 spaced cysteines crosslinked with 2. (c) Irradiation of TL-1 with 380 nm light decreases uptake inhibition, but the degree of photocontrol is significantly greater for TL-2 which has i,i + 7 spaced cysteines crosslinked by 2. The different cysteine spacing reverses the direction of the photocontrol with respect to TL-1. Reproduced with permission from Nevola, et al., Angew. Chem., Int. Ed., 2013, 52, 7704. Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA. | |
A light-responsive cadherin molecule was created by introducing a pair of cysteine residues into the EC1 and EC2 domains (Fig. 24B) and crosslinking them with 2.97 The resulting protein had different affinities for calcium ions under 365 nm or white light illumination and the half-life of the light state was strongly affected by the calcium ion concentrations. Experiments showed a light-dependent weakening of interactions between surface bound cadherin and irradiated crosslinked cadherins that indicated a degree of control over dimerisation. The B-cell leukemia-2 (Bcl-2) family of proteins, a vital part of the intrinsic apoptosis pathway, interact through conserved helical motifs. Photoswitchable peptides were designed based on the interacting helices of Bak and Bid.98 These peptides were cross-linked with 2 and their affinity to Bcl-xL was increased by 23-fold (i,i + 4), 20-fold (i,i + 7) and decreased by 7-fold (i,i + 11) upon irradiation. An NMR structure of Bak i,i + 11 in complex with Bcl-xL showed that the photoswitchable peptide bound to the canonical BH3 binding pocket.99,100 When introduced to permeabilised SU-DHL-4 cancer cells, the Bak i,i + 7 peptide (7.5 μM) proved capable of inducing light-dependent mitochondrial membrane depolarisation and cytochrome release.101
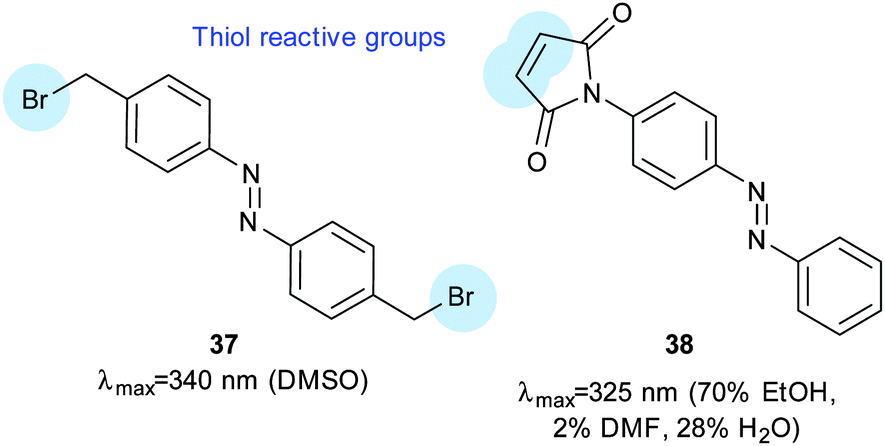
The SecYEG complex (Fig. 24C) translocates substrates from the cytosol to the lipid bilayer.102 Introduction of cysteine residues and a short dibromoalkyl azobenzene crosslinker (37, Fig. 26) facilitated control of the opening of a lateral gate from the translocation pore to the interior of the lipid membrane. Inner membrane vesicles containing this modified complex transported the proOmpA pre-protein in a reversible, light-responsive manner.
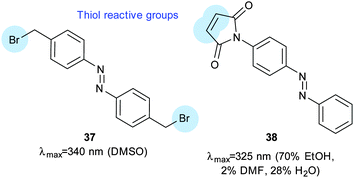 |
| | Fig. 26 A bisbromoalkyl azobenzene (37) used to control the SecYEG complex of E. coli and a maleimidoazobenzene (38) used to modify calmodulin to insert a pendant photoswitchable group. | |
Calmodulin moderates many calcium-dependent cellular signalling events. Ten calmodulin mutants incorporating cysteines at positions that might interfere with calcium binding sites were constructed and then reacted with 38 (Fig. 26).103 The most successful allowed 4-fold photocontrol of its binding affinity for its binding partner presented in a fluorescent fusion-protein. Control of subunit interactions was demonstrated using restriction endonucleases. Bis-maleimidoazobenzene 39 (Fig. 27) was used to photo-regulate the restriction enzyme PvuII with a 16-fold change in endonuclease activity upon irradiation; this was achieved through regulation of the turnover number rather than a change of the Michaelis constant.104,105
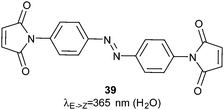 |
| | Fig. 27 A bis-maleimide used to control homodimeric PvuII complexes and mixed P2X receptor complexes. | |
The DNA methyltransferase SsoII was also modified to include cysteine residues and reacted with 39, which led to a 2-fold change of activity in response to light due to a 4-fold reduction in KM.106 The more subunits that are present in a complex, the greater the changes caused by irradiation; control of the relative orientations of the subunits of a group II chaperonin using 39 resulted in structural changes that were large enough to be detected by cryo-electron microscopy (Fig. 28).107
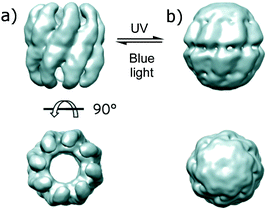 |
| | Fig. 28 15–19 Å single-particle cryo-electron microscopy reconstructions of an a homooligomeric chaparonin whose subunits are crosslinker with 39. Adapted by permission from Macmillan Publishers Ltd: Nat. Nanotechnol., 2013, 8, 928, copyright 2013. | |
Control of cellular uptake
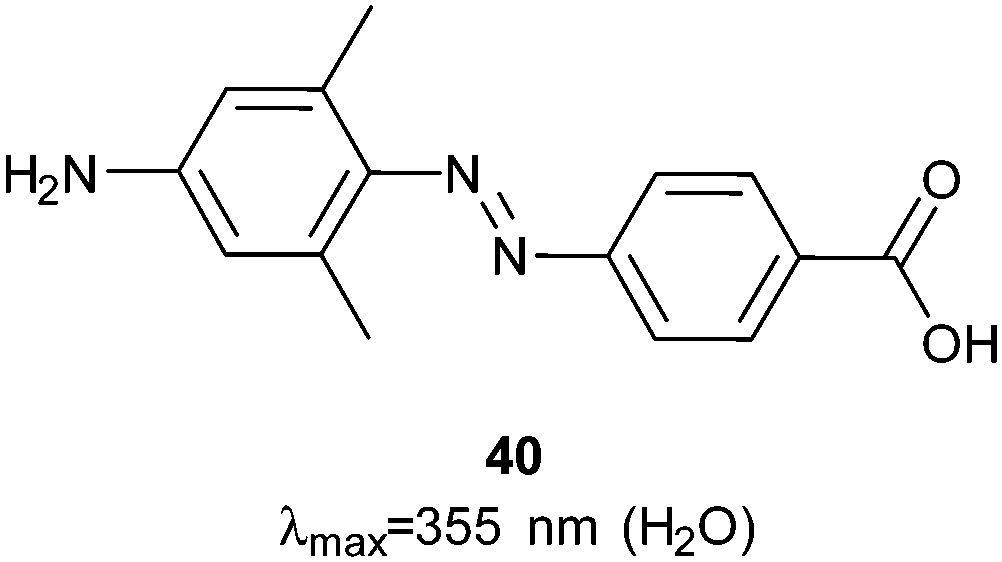
Photocontrol over the uptake of a peptide by cells was demonstrated by sandwiching azobenzene 40 (Fig. 29) between oligo-arginine and oligo-glutamate sequences.108Z-40 allows the glutamates to mask the cationic polyarginine cell transduction sequence while E-40 forces a conformational change that reveals the cell-penetrating sequence, resulting in a 10-fold increase in cellular uptake by HeLa cells incubated with the fluorescently labelled peptide (1 μM) (Fig. 30).
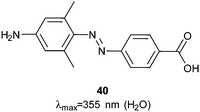 |
| | Fig. 29 A sterically hindered push–pull azobenzene used to photocontrol cell uptake of a fluorescently labelled peptide. | |
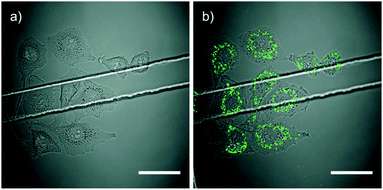 |
| | Fig. 30 HeLa cells incubated with rhodamine B (Asp)9-40-(Arg)9 after (a) UV light irradiation (1 μM) and (b) blue light irradiation. Adapted under CC-BY 3.0 from Prestel and Möller, Chem. Commun., 2016, 52, 701. | |
The para-alkylthio azobenzene 41 was included between two nuclear localising signal peptides during solid phase synthesis.109 The para-sulphur atom red-shifted the π–π* transition to 377 nm, which is higher than the wavelength observed with an equivalent ether (360 nm) but lower than for a para secondary amine substituent (429 nm). A polyproline spacer and fluorophore were added and when the resulting molecule (120 nM) was incubated with HeLa cells the dark state was taken up more efficiently than after light activation (Fig. 31).
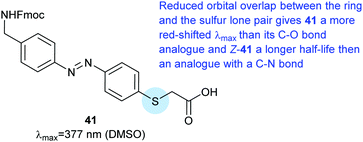 |
| | Fig. 31 Azobenzene amino acid derived from thioglycolic acid. The para-alkylthio substituent red-shifts the azobenzene absorbance to 377 nm. | |
Photopharmacology
Much work in photopharmacology is currently focused on creating molecules that can produce changes in cell behaviour.110 Such photopharmaceuticals allow increased activity at locations specified by irradiation to increase the effective therapeutic concentration range. For example, incretin glucagon-like peptide 1 can have beneficial effects on both the brain and the pancreas but can also cause numerous side effects elsewhere in the body. Introducing azobenzene 42 (Fig. 32) between two helical structural elements resulted in a peptide capable of controlling calcium ion fluxes in pancreatic beta cells and moderating insulin secretion in response to light (Fig. 33).111 Despite the power of modifying natural systems for photocontrol by identifying critical elements and controlling the conformation of the corresponding peptides, the properties of peptides are not always ideal for their use as therapeutics. An increasing number of small molecule drugs have been modified to include azobenzenes to provide photoswitching functionality. Small molecule 43 (Fig. 32) targets the same glucagon-like peptide 1 pathway as the peptide modified with 42111 and yields similar levels of photocontrol of insulin secretion without a being linked to a peptide.112
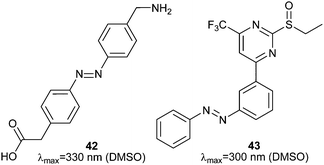 |
| | Fig. 32 Azobenzene amino acid that when incorporated into the backbone of a incretin peptide could control β-turns formation and insulin secretion and a small molecule capable of the same feat without a peptide component. | |
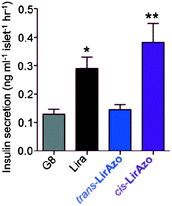 |
| | Fig. 33 (a) Insulin secretion from pancreatic beta cells in response to glucose (8 mM, G8), a liraglutide-derived peptide (Lira) and the same peptide with 42 incorporated in its dark (trans-LirAzo) and UV light irradiated (cis-LirAzo) states. Adapted under CC-BY from Broichhagen, et al., Angew. Chem., Int. Ed., 2015, 454, 15565. | |
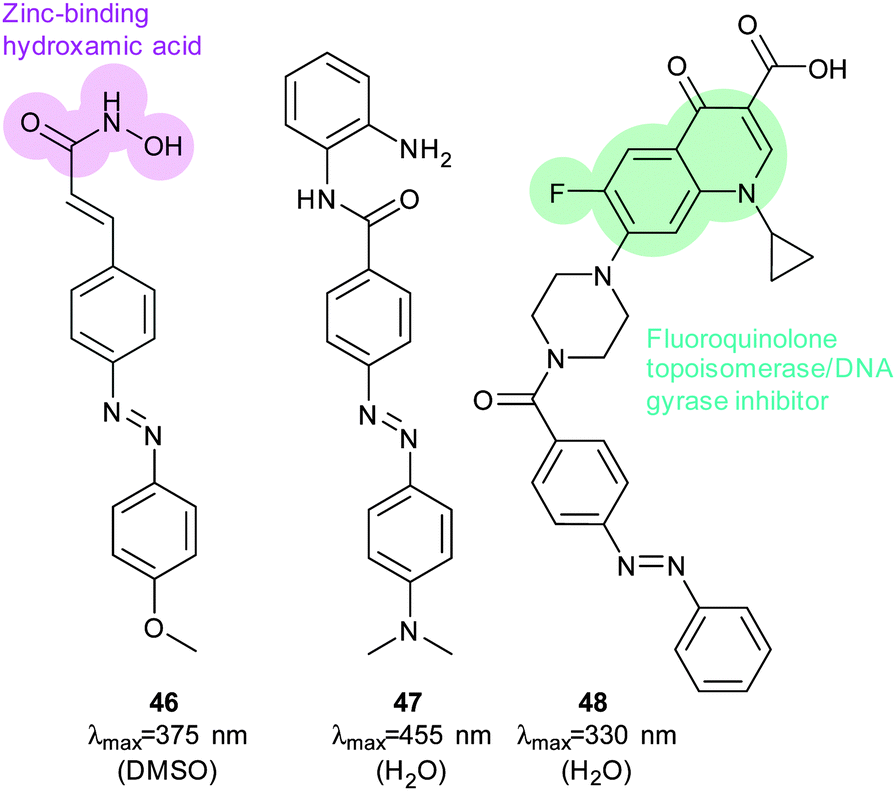
The stilbene element of combrestatin A4 (44), a cytotoxic experimental drug to treat advanced cancers, was replaced by azobenzene to yield 45.113–115 While E-45 is an extremely poor inhibitor of tubulin polymerisation (effective growth arrest at 25 μM in an cell proliferation assay), Z-45 (100 nM) is only tenfold less effective than 44 (10 nM) such that irradiation with 400 nm light causes effective photoswitching and a 200-fold change in cytotoxicity of the drug (Fig. 34 and 35).11445 and similar photostatins can induce mitotic arrest and cell death in Jurkat, HEK, HeLa and MDA-MB-231 cells.113 ‘Mitotic catastrophe’ could be selectively induced on an individual cell level in an impressive display of non-selective drug delivery coupled with light-driven activation. The same strategy was successfully applied to the synthesis of a range of histone deacetylase inhibitors exemplified by 46116 and 47117 and a photoresponsive version of the antibiotic ciprofloxacin 48 (Fig. 36).118
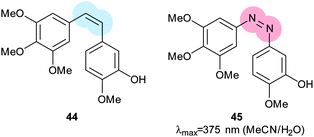 |
| | Fig. 34 The antitumour natural product combrestatin A4 and an azobenzene containing analogue. | |
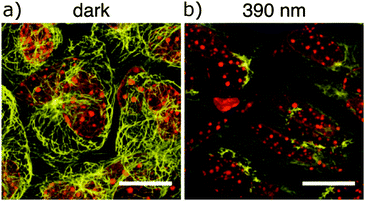 |
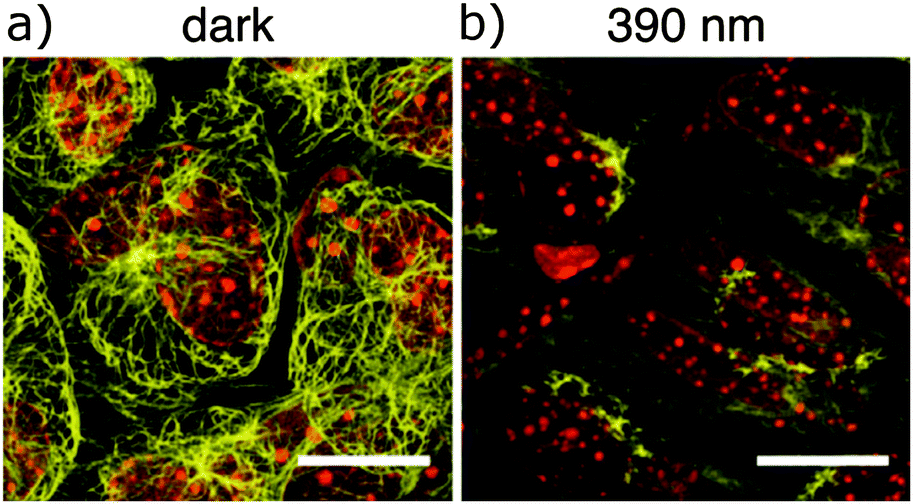
| | Fig. 35 Cremaster muscle cells of live C57BL/6 mice superfused for 40 minutes with 50 μM 45-O-phosphate (a soluble prodrug) (a) in the dark (left) or (b) under 390 nm light irradiation. Samples were then excised and stained for α-tubulin (yellow) and nuclei (red). Scale bars 15 μM. Adapted with permission from Borowiak, et al., Cell, 2015, 162, 403. Copyright 2015 Cell Press. | |
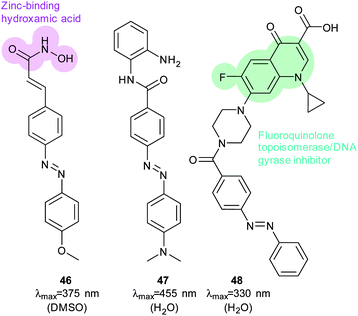 |
| | Fig. 36 An azobenene containing histone deacetylase inhibitor and a peripherally azobenzene-modified derivative of ciprofloxacin. | |
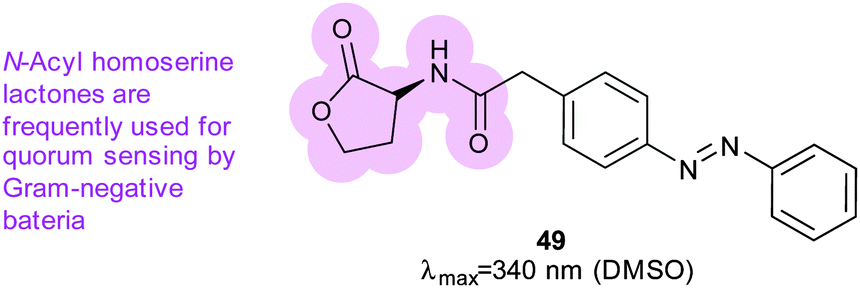
Azobenzene was also incorporated into acyl homoserine lactones to create a photoswitchable quorum sensing signalling molecule (49) (Fig. 37).119 When irradiated, 49 is five-fold more effective at activating the promoter of virulence genes in P. aeruginosa; its light-state has a half-life of 2.5 hours at 37 °C (Fig. 35).
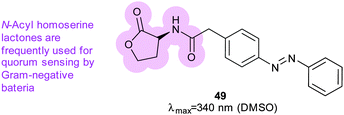 |
| | Fig. 37 Azobenzene-containing molecule used to mimic naturally occurring homoserine lactone molecules use by bacteria for quorum sensing. | |
Control of ion channels
The application of photoswitchable ligands to control ion channels is a remarkable success story.120 The first examples of photoswitchable channels were synthetic gramicidin derivatives,121–126 but attention soon turned to the many ligand-gated ion channels that adorn the outer membrane of cells. Initial experiments showed that azobenzene-containing molecules could mimic the natural ligands for such channels,127 leading to the development of two distinct methodologies. In the first a channel blocking ligand mimic group is tethered to an ion channel by an azobenzene-containing linker so that irradiation either leads to insertion or removal of a blocking group from the channel128,129 or binding at a distal site that induces structural changes in the channel. The second approach uses less specific freely diffusible ligand mimics. Tethering is most conveniently achieved through the introduction of cysteine residues at solvent exposed positions on receptor proteins and reacting their side chains with maleimides where the use of different cysteine positions allows the tuning of the response to photoswitching.130 The genes for individual subunits of heteromeric pores can be altered and heterologously expressed to create hybrid receptors. The function of individual receptor subtypes can then be mapped by their selective activation with light.5,131–135
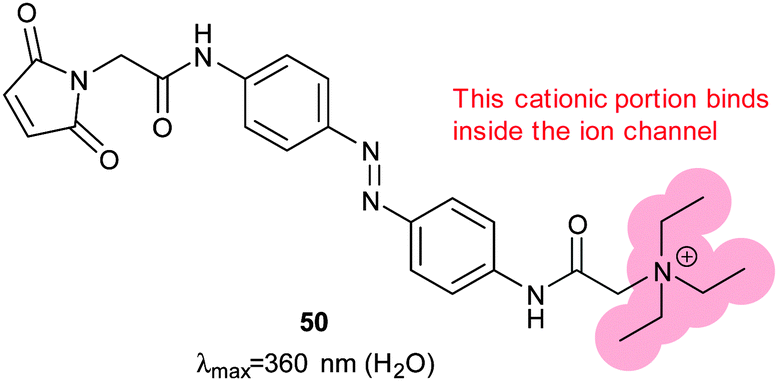
The tethered photoswitchable ligand 50 was applied to the voltage-gated potassium ion channel Shaker by selectively conjugating it to a loop on the rim of the pore. Whilst the extended E-50 form is long enough to allow the pendant ethylammonium group to block the pore, the photo-activated Z-50 is too short (Fig. 38 and 39). Z-50 leaves the pore open allowing action potentials to be silenced by blocking potassium ion transport136 or, with further mutations to allow sodium influx, action potentials to be triggered by irradiation.137 Use of less selective acrylamide, chloroacetate or epoxide agents allowed the non-selective labelling of endogenously expressed Shaker channels to create UV responsive cultured neurons.138 Viral co-transduction of DNA encoding mutant channels and GFP into the hippocampal region of mice caused no visible behavioural changes.139
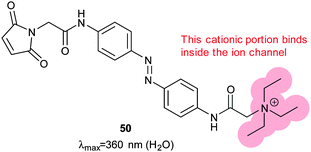 |
| | Fig. 38 A photochromic, tethering ligand optimised to control a mutant Shaker ion channel with a carefully positioned cysteine residue near the binding pocket for positively charged groups. | |
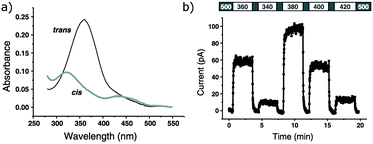 |
| | Fig. 39 (a) UV/visible absorbance spectra of 50 conjugated to glutathione. (b) Action spectra of inside-out membrane patches containing Shaker ion channels conjugated with 50. Adapted by permission from Macmillan Publishers Ltd: Nat. Neurosci., 2004, 7, 1381, copyright 2004. | |
To complement diffusible azobenzene-containing glutamate mimics,140,141 the photoswitchable tethered ligand 51 (Fig. 40) was applied to ionotropic glutamate receptors, which feature extracellular binding sites that cause conformational changes when populated by glutamate.142–146 Zebrafish larvae engineered to express modified channels were bathed in a solution of 51 for 30 minutes. Touching the treated larvae revealed reversibly altered behaviour according to the irradiation they had received; 365 nm light deactivated their escape reflex and 488 nm light reactivated it.147,148 Channels in five-day-old zebrafish were modified with 51 to elucidate the function of Kolmer–Agduhr cells, demonstrating the potential of this technique as an exquisitely selective investigative tool.149 In addition to pure ligand gating approaches, bis-maleimide (39) has been used to control the trimeric proteins that comprise P2X2/3 receptors moving the subunits apart to open the pore.150
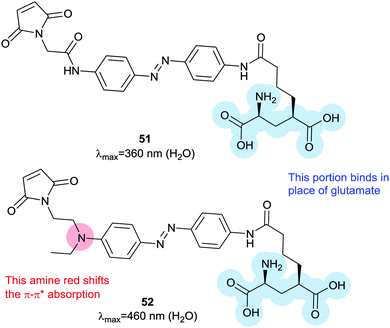 |
| | Fig. 40 Azobenzene-containing photoswitchable tethering ligands for ionic glutamate receptors. Replacing the amido substituent with an amino substituent creates a red-shifted, fast-relaxing switch. | |
An exciting long-term research goal is the use of these channels to achieve a degree of sight restoration for vision-impaired patients, which requires rapid relaxation and absorption at distinct wavelengths in the visible spectrum. A modified azobenzene (52) where one ring bears an electron-releasing amine substituent increases the channel activation wavelength from 380 nm to 445 nm, a wavelength better suited for vision restoration research. Channels modified with 52 returned to their ground state 2200-fold faster than channels modified with 51, allowing much higher temporal resolution control of neuron firing.151 Azobenzene 52 has a much-improved two-photon absorption cross-section that is comparable to that of fluorescent proteins.152 A tethered tetra-ortho-chloroazobenzene glutamate ligand (53) allowed triggering of a modified GluK2 kainate receptor with 560 nm light.153 High intensity 625 nm light could also be used, albeit at lower efficiency, but 440 nm irradiation induced no response so that 53 and 51 can work orthogonally with narrow wavelength light sources (Fig. 41).
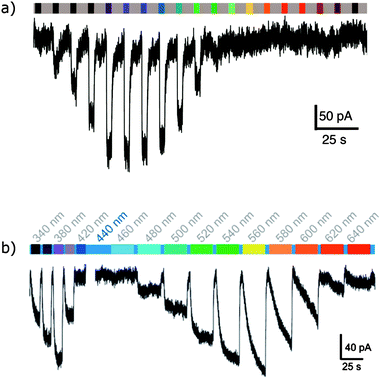 |
| | Fig. 41 (a) Representative action spectrum showing activation of the homotetrameric kainite receptor GluK2(439C) with 52 conjugated to the its ligand binding domain. (b) An action spectrum of the same protein conjugated with 53 showing how these two tethered ligands might be used in parallel. (a) Adapted with permission from Kienzler, et al., J. Am. Chem. Soc., 2013, 135, 17683 and Copyright 2013 American Chemical Society. (b) Adapted under CC-BY from Rullo, et al., Chem. Commun., 2014, 50, 14613. | |
A recent report describes late stage incorporation of ortho-chloro substituents to preformed azo compounds, making such molecules more accessible.154 Even longer wavelengths can be used to switch 54 (880 nm) and 55 (900 nm), which were designed for improved two-photon absorption (Fig. 42).155
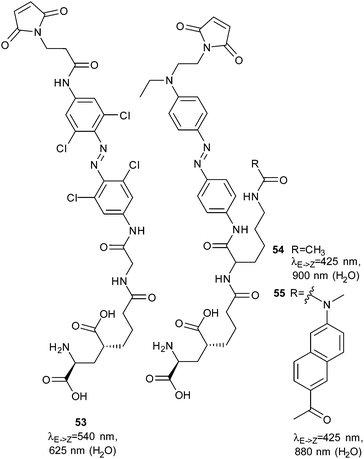 |
| | Fig. 42 A tetra-ortho-chloroazobenzene photoswitchable tethered ligand. The n–π* absorbance band separation allows this molecule to be switched with light up to 625 nm at high intensities. | |
Non-tethered, diffusible azobenzene ligands can also be used to control a wide variety of channels. Successful endeavours include analysing the behaviour of Purkinje cells using 56,156 modulating insulin secretion via ATP-sensitive potassium channels with 57 and 58,157,158 versions of the transient receptor potential pain receptor ion channel TRPV1,159 that respond to compounds including azo-capsaicin 59,160 metabotropic glutamate receptors (class C GPCR receptors) controlled by 60,161 trimeric amiloride-sensitive sodium channels gated by 61,162 G-protein coupled inwardly rectifying potassium channels regulated by 62,163 and endowment of retinal function on blind mice using 63 (Fig. 43).164
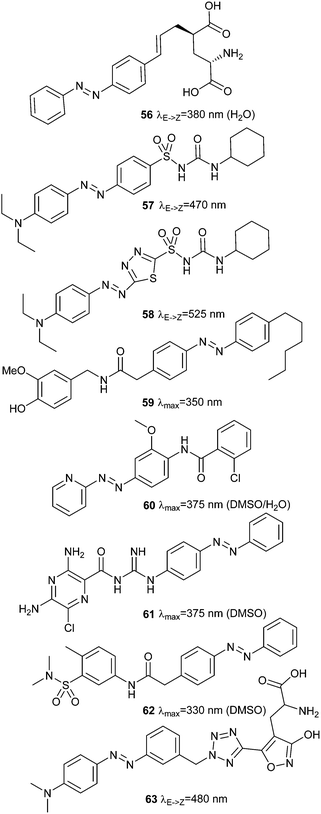 |
| | Fig. 43 Non-tethered azobenzene ligand for controlling various ion channels. | |
Conclusions
Recent breakthroughs in tuning the physical properties of photoswitches have improved the scope of existing tools, but have also opened up new opportunities. Azobenzenes with extended Z-isomer half-lives now cover the timescales of more complex biological events and allow in vivo kinetics experiments to be conducted. Control over the lifetime of active states is complemented by an expanded range of photoswitching wavelength and offers the possibility to combine two or more dyes to create multiplexed biophotonic systems. Such combinations of probes can lead to powerful tools to study thresholds in signalling events, as differentially timed light pulses could be used to simulate concentration gradients. The ever-growing number of photopharmaceutical tools could also be envisaged being used in pairs, with the effects of a pair of opposed molecules activated in different locations providing proportionally greater effects.
Future challenges include improving the mechanisms researchers use to translate photoisomerisation energy into structural change.165–168 Natural systems have evolved to be delicately balanced to change conformation when a ligand or binding partner is present, but directed evolution is presently impractical for proteins containing non-proteinogenic amino acids. Computational studies may therefore offer the best chance of successfully (re)designing systems to achieve improved photocontrol over protein structure and function.
Acknowledgements
Financial support from the UK Biotechnology and Biological Research Council through grants B16000, BB/I021396/1 and BB/M006158/1, the UK Engineering and Physical Sciences Research Council through grants EP/F040954/1 and EP/L027240/1, the Royal Society (IIF-2007/R1) and Cardiff University is gratefully acknowledged.
References
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719 RSC.
- D. Bleger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338 CrossRef CAS PubMed.
- J. Zhang, J. Wang and H. Tian, Mater. Horiz., 2014, 1, 169 RSC.
- A. Priimagi, C. J. Barrett and A. Shishido, J. Mater. Chem. C, 2014, 2, 7155 RSC.
- T. Fehrentz, M. Schonberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156 CrossRef CAS PubMed.
- A. Gautier, C. Gauron, M. Volovitch, D. Bensimon, L. Jullien and S. Vriz, Nat. Chem. Biol., 2014, 10, 533 CrossRef CAS PubMed.
- P. R. O'Neill and N. Gautam, Photochem. Photobiol. Sci., 2015, 14, 1578 Search PubMed.
- W. Szymanski, J. M. Beierle, H. A. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114 CrossRef CAS PubMed.
- M. Erdelyi, M. Varedian, C. Skold, I. B. Niklasson, J. Nurbo, A. Persson, J. Bergquist and A. Gogoll, Org. Biomol. Chem., 2008, 6, 4356 CAS.
- C. Karlsson, M. Blom, M. Johansson, A. M. Jansson, E. Scifo, A. Karlen, T. Govender and A. Gogoll, Org. Biomol. Chem., 2015, 13, 2612 CAS.
- N. J. Lindgren, M. Varedian and A. Gogoll, Chem. – Eur. J., 2009, 15, 501 CrossRef CAS PubMed.
- M. Varedian, M. Erdelyi, A. Persson and A. Gogoll, J. Pept. Sci., 2009, 15, 107 CrossRef CAS PubMed.
- M. Varedian, V. Langer, J. Bergquist and A. Gogoll, Tetrahedron Lett., 2008, 49, 6033 CrossRef CAS.
- S. Herre, W. Steinle and K. Ruck-Braun, Synthesis, 2005, 3297 CAS.
- T. Cordes, D. Weinrich, S. Kempa, K. Riesselmann, S. Herre, C. Hoppmann, K. Ruck-Braun and W. Zinth, Chem. Phys. Lett., 2006, 428, 167 CrossRef CAS.
- S. Herre, T. Schadendorf, I. Ivanov, C. Herrberger, W. Steinle, K. Ruck-Braun, R. Preissner and H. Kuhn, ChemBioChem, 2006, 7, 1089 CrossRef CAS PubMed.
- M. Fullbeck, E. Michalsky, I. S. Jaeger, P. Henklein, H. Kuhn, K. Ruck-Braun and R. Preissner, Genome Inform., 2006, 17, 141 Search PubMed.
- T. Schadendorf, C. Hoppmann and K. Rueck-Braun, Tetrahedron Lett., 2007, 48, 9044 CrossRef CAS.
- T. Cordes, T. Schadendorf, B. Priewisch, K. Ruck-Braun and W. Zinth, J. Phys. Chem. A, 2008, 112, 581 CrossRef CAS PubMed.
- T. Cordes, T. Schadendorf, K. Ruck-Braun and W. Zinth, Chem. Phys. Lett., 2008, 455, 197 CrossRef CAS.
- T. Cordes, C. Elsner, T. T. Herzog, C. Hoppmann, T. Schadendorf, W. Summerer, K. Ruck-Braun and W. Zinth, Chem. Phys., 2009, 358, 103 CrossRef CAS.
- T. Cordes, B. Heinz, N. Regner, C. Hoppmann, T. E. Schrader, W. Summerer, K. Ruck-Braun and W. Zinth, ChemPhysChem, 2007, 8, 1713 CrossRef CAS PubMed.
- S. Wiedbrauk and H. Dube, Tetrahedron Lett., 2015, 56, 4266 CrossRef CAS.
- J. Z. Zhao, D. Wildemann, M. Jakob, C. Vargas and C. Schiene-Fischer, Chem. Commun., 2003, 2810 RSC.
- J. Helbing, H. Bregy, J. Bredenbeck, R. Pfister, P. Hamm, R. Huber, J. Wachtveitl, L. De Vico and M. Olivucci, J. Am. Chem. Soc., 2004, 126, 8823 CrossRef CAS PubMed.
- Y. Huang, Z. Y. Cong, L. F. Yang and S. L. Dong, J. Pept. Sci., 2008, 14, 1062 CrossRef CAS PubMed.
- Z. Y. Cong, L. F. Yang, L. Jiang, D. Ye and S. L. Dong, Chin. Chem. Lett., 2010, 21, 476 CrossRef CAS.
- J. M. Goldberg, S. Batjargal and E. J. Petersson, J. Am. Chem. Soc., 2010, 132, 14718 CrossRef CAS PubMed.
- Y. Huang, G. Jahreis, C. Lucke, D. Wildemann and G. Fischer, J. Am. Chem. Soc., 2010, 132, 7578 CrossRef CAS PubMed.
- K. Fujimoto, T. Maruyama, Y. Okada, T. Itou and M. Inouye, Tetrahedron, 2013, 69, 6170 CrossRef CAS.
- O. Babii, S. Afonin, M. Berditsch, S. Reibetaer, P. K. Mykhailiuk, V. S. Kubyshkin, T. Steinbrecher, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2014, 53, 3392 CrossRef CAS PubMed.
- D. J. van Dijken, P. Kovaricek, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982 CrossRef CAS PubMed.
- M. Blanco-Lomas, S. Samanta, P. J. Campos, G. A. Woolley and D. Sampedro, J. Am. Chem. Soc., 2012, 134, 6960 CrossRef CAS PubMed.
- A. Sinicropi, C. Bernini, R. Basosi and M. Olivucci, Photochem. Photobiol. Sci., 2009, 8, 1639 CAS.
- G. A. Woolley, Acc. Chem. Res., 2005, 38, 486 CrossRef CAS PubMed.
- C. Renner and L. Moroder, ChemBioChem, 2006, 7, 869 CrossRef PubMed.
- D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391 CrossRef CAS PubMed.
- S. H. Xia, G. Cui, W. H. Fang and W. Thiel, Angew. Chem., Int. Ed., 2016, 55, 2067 CrossRef CAS PubMed.
- Z. H. Zhang, D. C. Burns, J. R. Kumita, O. S. Smart and G. A. Woolley, Bioconjugate Chem., 2003, 14, 824 CrossRef CAS PubMed.
- D. C. Burns, F. Z. Zhang and G. A. Woolley, Nat. Protoc., 2007, 2, 251 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662 CrossRef CAS PubMed.
- O. Sadovski, A. A. Beharry, F. Z. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484 CrossRef CAS PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684 CrossRef CAS PubMed.
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos and G. A. Woolley, Chem. Commun., 2013, 49, 10314 RSC.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777 CrossRef CAS PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, Org. Biomol. Chem., 2008, 6, 4323 CAS.
- S. Samanta, A. Babalhavaeji, M. X. Dong and G. A. Woolley, Angew. Chem., Int. Ed., 2013, 52, 14127 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, M. J. Hansen, L. Kalman and G. A. Woolley, Chem. Commun., 2015, 51, 12981 RSC.
- D. Bleger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597 CrossRef CAS PubMed.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bleger, Chem. – Eur. J., 2014, 20, 16492 CrossRef CAS PubMed.
- A. A. Beharry, L. Wong, V. Tropepe and G. A. Woolley, Angew. Chem., Int. Ed., 2011, 50, 1325 CrossRef CAS PubMed.
- Y. X. Zhang, F. Erdmann and G. Fischer, Nat. Chem. Biol., 2009, 5, 724 CrossRef CAS PubMed.
- J. Moreno, M. Gerecke, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, D. Bleger, S. Hecht and S. A. Kovalenko, J. Phys. Chem. B, 2015, 119, 12281 CrossRef CAS PubMed.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Nather, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594 CrossRef CAS PubMed.
- O. Carstensen, J. Sielk, J. B. Schonborn, G. Granucci and B. Hartke, J. Chem. Phys., 2010, 133, 124305 CrossRef PubMed.
- M. Boeckmann, N. L. Doltsinis and D. Marx, Angew. Chem., Int. Ed., 2010, 49, 3382 CrossRef CAS PubMed.
- C. W. Jiang, R. H. Xie, F. L. Li and R. E. Allen, J. Phys. Chem. A, 2011, 115, 244 CrossRef CAS PubMed.
- S. Samanta, C. G. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51, 6452 CrossRef CAS PubMed.
- H. Sell, C. Nather and R. Herges, Beilstein J. Org. Chem., 2013, 9, 1 CrossRef CAS PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2012, 134, 15221 CrossRef CAS PubMed.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 13190 CrossRef CAS PubMed.
- Y. Wang and D. H. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931 CrossRef CAS PubMed.
- C. Hoppmann, P. Schmieder, N. Heinrich and M. Beyermann, ChemBioChem, 2011, 12, 2555 CrossRef CAS PubMed.
- C. Hoppmann, V. K. Lacey, G. V. Louie, J. Wei, J. P. Noel and L. Wang, Angew. Chem., Int. Ed., 2014, 53, 3932 CrossRef CAS PubMed.
- C. Hoppmann, I. Maslennikov, S. Choe and L. Wang, J. Am. Chem. Soc., 2015, 137, 11218 CrossRef CAS PubMed.
- A. Muller, H. Kobarg, V. Chandrasekaran, J. Gronow, F. D. Sonnichsen and T. K. Lindhorst, Chem. – Eur. J., 2015, 21, 13723 CrossRef PubMed.
- C. Poloni, W. Szymanski, L. Hou, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2014, 20, 946 CrossRef CAS PubMed.
- M. Liu, S. Tada, M. Ito, H. Abe and Y. Ito, Chem. Commun., 2012, 48, 11871 RSC.
- M. R. Jafari, L. Deng, P. I. Kitov, S. Ng, W. L. Matochko, K. F. Tjhung, A. Zeberoff, A. Elias, J. S. Klassen and R. Derda, ACS Chem. Biol., 2014, 9, 443 CrossRef CAS PubMed.
- S. Bellotto, S. Chen, I. Rentero Rebollo, H. A. Wegner and C. Heinis, J. Am. Chem. Soc., 2014, 136, 5880 CrossRef CAS PubMed.
- L. Guerrero, O. S. Smart, G. A. Woolley and R. K. Allemann, J. Am. Chem. Soc., 2005, 127, 15624 CrossRef CAS PubMed.
- L. Guerrero, O. S. Smart, C. J. Weston, D. C. Burns, G. A. Woolley and R. K. Allemann, Angew. Chem., Int. Ed., 2005, 44, 7778 CrossRef CAS PubMed.
- G. A. Woolley, A. S. I. Jaikaran, M. Berezovski, J. P. Calarco, S. N. Krylov, O. S. Smart and J. R. Kumita, Biochemistry, 2006, 45, 6075 CrossRef CAS PubMed.
- A. M. Ali, M. W. Forbes and G. A. Woolley, ChemBioChem, 2015, 16, 1757 CrossRef CAS PubMed.
- F. Z. Zhang, K. A. Timm, K. M. Arndt and G. A. Woolley, Angew. Chem., Int. Ed., 2010, 49, 3943 CrossRef CAS PubMed.
- A. M. Caamano, M. E. Vazquez, J. Martinez-Costas, L. Castedo and J. L. Mascarenas, Angew. Chem., Int. Ed., 2000, 39, 3104 CrossRef CAS.
- R. J. Mart, P. Wysoczanski, S. Kneissl, A. Ricci, A. Brancale and R. K. Allemann, ChemBioChem, 2012, 13, 515 CrossRef CAS PubMed.
- L. Ulysse and J. Chmielewski, Bioorg. Med. Chem. Lett., 1994, 4, 2145 CrossRef CAS.
- L. Ulysse, J. Cubillos and J. Chmielewski, J. Am. Chem. Soc., 1995, 117, 8466 CrossRef CAS.
- L. G. Ulysse and J. Chmielewski, Chem. Biol. Drug Des., 2006, 67, 127 CAS.
- A. Aemissegger, V. Krautler, W. F. van Gunsteren and D. Hilvert, J. Am. Chem. Soc., 2005, 127, 2929 CrossRef CAS PubMed.
- S. L. Dong, M. Loweneck, T. E. Schrader, W. J. Schreier, W. Zinth, L. Moroder and C. Renner, Chem. – Eur. J., 2006, 12, 1114 CrossRef CAS PubMed.
- T. E. Schrader, W. J. Schreier, T. Cordes, F. O. Koller, G. Babitzki, R. Denschlag, C. Renner, M. Loweneckt, S. L. Dong, L. Moroder, P. Tavan and W. Zinth, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15729 CrossRef CAS PubMed.
- T. E. Schrader, T. Cordes, W. J. Schreier, F. O. Koller, S. L. Dong, L. Moroder and W. Zinth, J. Phys. Chem. B, 2011, 115, 5219 CrossRef CAS PubMed.
- T. Podewin, M. S. Rampp, I. Turkanovic, K. L. Karaghiosoff, W. Zinth and A. Hoffmann-Roder, Chem. Commun., 2015, 51, 4001 RSC.
- R. Behrendt, M. Schenk, H. J. Musiol and L. Moroder, J. Pept. Sci., 1999, 5, 519 CrossRef CAS PubMed.
- C. Renner, J. Cramer, R. Behrendt and L. Moroder, Biopolymers, 2000, 54, 501 CrossRef CAS PubMed.
- C. Renner, R. Behrendt, S. Sporlein, J. Wachtveitl and L. Moroder, Biopolymers, 2000, 54, 489 CrossRef CAS PubMed.
- R. Behrendt, C. Renner, M. Schenk, F. Q. Wang, J. Wachtveitl, D. Oesterhelt and L. Moroder, Angew. Chem., Int. Ed., 1999, 38, 2771 CrossRef CAS.
- C. Renner, R. Behrendt, N. Heim and L. Moroder, Biopolymers, 2002, 63, 382 CrossRef CAS PubMed.
- M. Schutt, S. S. Krupka, A. G. Milbradt, S. Deindl, E. K. Sinner, D. Oesterhelt, C. Renner and L. Moroder, Chem. Biol., 2003, 10, 487 CrossRef CAS PubMed.
- A. G. Milbradt, M. Loweneck, S. S. Krupka, M. Reif, E. K. Sinner, L. Moroder and C. Renner, Biopolymers, 2005, 77, 304 CrossRef CAS PubMed.
- C. Hoppmann, S. Seedorff, A. Richter, H. Fabian, P. Schmieder, K. Ruck-Braun and M. Beyermann, Angew. Chem., Int. Ed., 2009, 48, 6636 CrossRef CAS PubMed.
- L. Nevola, A. Martin-Quiros, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52, 7704 CrossRef CAS PubMed.
- A. Martin-Quiros, L. Nevola, K. Eckelt, S. Madurga, P. Gorostiza and E. Giralt, Chem. Biol., 2015, 22, 31 CrossRef CAS PubMed.
- R. S. Ritterson, K. M. Kuchenbecker, M. Michalik and T. Kortemme, J. Am. Chem. Soc., 2013, 135, 12516 CrossRef CAS PubMed.
- S. Kneissl, E. J. Loveridge, C. Williams, M. P. Crump and R. K. Allemann, ChemBioChem, 2008, 9, 3046 CrossRef CAS PubMed.
- P. Wysoczanski, R. J. Mart, E. J. Loveridge, C. Williams, S. B. M. Whittaker, M. P. Crump and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 7644 CrossRef CAS PubMed.
- P. Wysoczanski, R. J. Mart, E. J. Loveridge, C. Williams, S. B. M. Whittaker, M. P. Crump and R. K. Allemann, Biomol. NMR Assignments, 2013, 7, 187 CrossRef CAS PubMed.
- R. J. Mart, R. J. Errington, C. L. Watkins, S. C. Chappell, M. Wiltshire, A. T. Jones, P. J. Smith and R. K. Allemann, Mol. BioSyst., 2013, 9, 2597 RSC.
- F. Bonardi, G. London, N. Nouwen, B. Feringa and A. J. M. Driessen, Angew. Chem., Int. Ed., 2010, 49, 7234 CrossRef CAS PubMed.
- H. Shishido, M. D. Yamada, K. Kondo and S. Maruta, J. Biochem., 2009, 146, 581 CrossRef CAS PubMed.
- B. Schierling, A. J. Noel, W. Wende, L. T. Hien, E. Volkov, E. Kubareva, T. Oretskaya, M. Kokkinidis, A. Rompp, B. Spengler and A. Pingoud, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1361 CrossRef CAS PubMed.
- B. Schierling, N. Dannemann, L. Gabsalilow, W. Wende, T. Cathomen and A. Pingoud, Nucleic Acids Res., 2012, 40, 2623 CrossRef CAS PubMed.
- L. T. Hien, T. S. Zatsepin, B. Schierling, E. M. Volkov, W. Wende, A. Pingoud, E. A. Kubareva and T. S. Oretskaya, Bioconjugate Chem., 2011, 22, 1366 CrossRef CAS PubMed.
- D. Hoersch, S. H. Roh, W. Chiu and T. Kortemme, Nat. Nanotechnol., 2013, 8, 928 CrossRef CAS PubMed.
- A. Prestel and H. M. Moller, Chem. Commun., 2015, 52, 701 RSC.
- S. Sawada, N. Kato and K. Kaihatsu, Curr. Pharm. Biotechnol., 2012, 13, 2642 CAS.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178 CrossRef CAS PubMed.
- J. Broichhagen, T. Podewin, H. Meyer-Berg, Y. von Ohlen, N. R. Johnston, B. J. Jones, S. R. Bloom, G. A. Rutter, A. Hoffmann-Roder, D. J. Hodson and D. Trauner, Angew. Chem., Int. Ed., 2015, 54, 15565 CrossRef CAS PubMed.
- J. Broichhagen, N. R. Johnston, Y. von Ohlen, H. Meyer-Berg, B. J. Jones, S. R. Bloom, G. A. Rutter, D. Trauner and D. J. Hodson, Angew. Chem., Int. Ed., 2016, 55, 5865 CrossRef CAS PubMed.
- M. Borowiak, W. Nahaboo, M. Reynders, K. Nekolla, P. Jalinot, J. Hasserodt, M. Rehberg, M. Delattre, S. Zahler, A. Vollmar, D. Trauner and O. Thorn-Seshold, Cell, 2015, 162, 403 CrossRef CAS PubMed.
- A. J. Engdahl, E. A. Torres, S. E. Lock, T. B. Engdahl, P. S. Mertz and C. N. Streu, Org. Lett., 2015, 17, 4546 CrossRef CAS PubMed.
- J. E. Sheldon, M. M. Dcona, C. E. Lyons, J. C. Hackett and M. C. Hartman, Org. Biomol. Chem., 2016, 14, 40 CAS.
- W. Szymanski, M. E. Ourailidou, W. A. Velema, F. J. Dekker and B. L. Feringa, Chem. – Eur. J., 2015, 21, 16517 CrossRef CAS PubMed.
- S. A. Reis, B. Ghosh, J. A. Hendricks, D. M. Szantai-Kis, L. Tork, K. N. Ross, J. Lamb, W. Read-Button, B. Zheng, H. Wang, C. Salthouse, S. J. Haggarty and R. Mazitschek, Nat. Chem. Biol., 2016, 12, 317 CrossRef CAS PubMed.
- W. A. Velema, M. J. Hansen, M. M. Lerch, A. J. Driessen, W. Szymanski and B. L. Feringa, Bioconjugate Chem., 2015, 26, 2592 CrossRef CAS PubMed.
- J. P. Van der Berg, W. A. Velema, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Chem. Sci., 2015, 6, 3593 RSC.
- A. Reiner, J. Levitz and E. Y. Isacoff, Curr. Opin. Pharmacol., 2015, 20, 135 CrossRef CAS PubMed.
- C. J. Stankovic, S. H. Heinemann and S. L. Schreiber, Biochim. Biophys. Acta, 1991, 1061, 163 CrossRef CAS.
- G. A. Woolley, A. S. I. Jaikaran, Z. H. Zhang and S. Y. Peng, J. Am. Chem. Soc., 1995, 117, 4448 CrossRef CAS.
- L. Lien, D. C. J. Jaikaran, Z. H. Zhang and G. A. Woolley, J. Am. Chem. Soc., 1996, 118, 12222 CrossRef CAS.
- S. Rudolph-Bohner, M. Kruger, D. Oesterhelt, L. Moroder, T. Nagele and J. Wachtveitl, J. Photochem. Photobiol., A, 1997, 105, 235 CrossRef.
- V. Borisenko, D. C. Burns, Z. H. Zhang and G. A. Woolley, J. Am. Chem. Soc., 2000, 122, 6364 CrossRef CAS.
- O. Babii, S. Afonin, L. V. Garmanchuk, V. V. Nikulina, T. V. Nikolaienko, O. V. Storozhuk, D. V. Shelest, O. I. Dasyukevich, L. I. Ostapchenko, V. Iurchenko, S. Zozulya, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2016, 55, 5493 CrossRef CAS PubMed.
- E. Bartels, H. Wasserman and B. F. Erlanger, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1820 CrossRef CAS.
- H. A. Lester, M. E. Krouse, M. M. Nass, N. H. Wassermann and B. F. Erlanger, J. Gen. Physiol., 1980, 75, 207 CrossRef CAS PubMed.
- M. E. Krouse, H. A. Lester, N. H. Wassermann and B. F. Erlanger, J. Gen. Physiol., 1985, 86, 235 CrossRef CAS PubMed.
- I. Tochitsky, M. R. Banghart, A. Mourot, J. Z. Yao, B. Gaub, R. H. Kramer and D. Trauner, Nat. Chem., 2012, 4, 105 CrossRef CAS PubMed.
- R. H. Kramer, J. J. Chambers and D. Trauner, Nat. Chem. Biol., 2005, 1, 360 CrossRef CAS PubMed.
- P. Gorostiza and E. Isacoff, Mol. BioSyst., 2007, 3, 686 RSC.
- P. Gorostiza and E. Y. Isacoff, Science, 2008, 322, 395 CrossRef CAS PubMed.
- R. H. Kramer, D. L. Fortin and D. Trauner, Curr. Opin. Neurobiol., 2009, 19, 544 CrossRef CAS PubMed.
- G. A. Woolley, Nat. Chem., 2012, 4, 75 CrossRef CAS PubMed.
- M. Banghart, K. Borges, E. Isacoff, D. Trauner and R. H. Kramer, Nat. Neurosci., 2004, 7, 1381 CrossRef CAS PubMed.
- J. J. Chambers, M. R. Banghart, D. Trauner and R. H. Kramer, J. Neurophysiol., 2006, 96, 2792 CrossRef CAS PubMed.
- D. L. Fortin, M. R. Banghart, T. W. Dunn, K. Borges, D. A. Wagenaar, Q. Gaudry, M. H. Karakossian, T. S. Otis, W. B. Kristan, D. Trauner and R. H. Kramer, Nat. Methods, 2008, 5, 331 CAS.
- D. L. Fortin, T. W. Dunn, A. Fedorchak, D. Allen, R. Montpetit, M. R. Banghart, D. Trauner, J. P. Adelman and R. H. Kramer, J. Neurophysiol., 2011, 106, 488 CrossRef CAS PubMed.
- M. Volgraf, P. Gorostiza, S. Szobota, M. R. Helix, E. Y. Isacoff and D. Trauner, J. Am. Chem. Soc., 2007, 129, 260 CrossRef CAS PubMed.
- L. Laprell, E. Repak, V. Franckevicius, F. Hartrampf, J. Terhag, M. Hollmann, M. Sumser, N. Rebola, D. A. DiGregorio and D. Trauner, Nat. Commun., 2015, 6, 8076 CrossRef CAS PubMed.
- M. Volgraf, P. Gorostiza, R. Numano, R. H. Kramer, E. Y. Isacoff and D. Trauner, Nat. Chem. Biol., 2006, 2, 47 CrossRef CAS PubMed.
- P. Gorostiza, M. Volgraf, R. Numano, S. Szobota, D. Trauner and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10865 CrossRef CAS PubMed.
- R. Numano, S. Szobota, A. Y. Lau, P. Gorostiza, M. Volgraf, B. Roux, D. Trauner and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6814 CrossRef CAS PubMed.
- J. Levitz, A. T. Popescu, A. Reiner and E. Y. Isacoff, Front. Mol. Neurosci., 2016, 9, 2 Search PubMed.
- S. Berlin, S. Szobota, A. Reiner, E. C. Carroll, M. A. Kienzler, A. Guyon, T. Xiao, D. Tauner and E. Y. Isacoff, eLife, 2016, 5, e12040 Search PubMed.
- S. Szobota, P. Gorostiza, F. Del Bene, C. Wyart, D. L. Fortin, K. D. Kolstad, O. Tulyathan, M. Volgraf, R. Numano, H. L. Aaron, E. K. Scott, R. H. Kramer, J. Flannery, H. Baier, D. Trauner and E. Y. Isacoff, Neuron, 2007, 54, 535 CrossRef CAS PubMed.
- H. Janovjak, S. Szobota, C. Wyart, D. Trauner and E. Y. Isacoff, Nat. Neurosci., 2010, 13, 1027 CrossRef CAS PubMed.
- C. Wyart, F. Del Bene, E. Warp, E. K. Scott, D. Trauner, H. Baier and E. Y. Isacoff, Nature, 2009, 461, 407 CrossRef CAS PubMed.
- L. E. Browne, J. P. Nunes, J. A. Sim, V. Chudasama, L. Bragg, S. Caddick and R. A. North, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 521 CrossRef CAS PubMed.
- M. A. Kienzler, A. Reiner, E. Trautman, S. Yoo, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2013, 135, 17683 CrossRef CAS PubMed.
- E. C. Carroll, S. Berlin, J. Levitz, M. A. Kienzler, Z. Yuan, D. Madsen, D. S. Larsen and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E776 CrossRef CAS PubMed.
- A. Rullo, A. Reiner, A. Reiter, D. Trauner, E. Y. Isacoff and G. A. Woolley, Chem. Commun., 2014, 50, 14613 RSC.
- D. B. Konrad, J. A. Frank and D. Trauner, Chem. – Eur. J., 2016, 22, 4364 CrossRef CAS PubMed.
- M. Izquierdo-Serra, M. Gascon-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, E. Ferrer, R. Alibes, F. Busque, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136, 8693 CrossRef CAS PubMed.
- Z. R. Abrams, A. Warrier, D. Trauner and X. A. Zhang, Front. Neural Circuits, 2010, 4, 13 Search PubMed.
- J. Broichhagen, M. Schonberger, S. C. Cork, J. A. Frank, P. Marchetti, M. Bugliani, A. M. Shapiro, S. Trapp, G. A. Rutter, D. J. Hodson and D. Trauner, Nat. Commun., 2014, 5, 5116 CrossRef CAS PubMed.
- J. Broichhagen, J. A. Frank, N. R. Johnston, R. K. Mitchell, K. Smid, P. Marchetti, M. Bugliani, G. A. Rutter, D. Trauner and D. J. Hodson, Chem. Commun., 2015, 51, 6018 RSC.
- A. Mourot, T. Fehrentz, Y. Le Feuvre, C. M. Smith, C. Herold, D. Dalkara, F. Nagy, D. Trauner and R. H. Kramer, Nat. Methods, 2012, 9, 396 CrossRef CAS PubMed.
- J. A. Frank, M. Moroni, R. Moshourab, M. Sumser, G. R. Lewin and D. Trauner, Nat. Commun., 2015, 6, 7118 CrossRef PubMed.
- S. Pittolo, X. Gomez-Santacana, K. Eckelt, X. Rovira, J. Dalton, C. Goudet, J. P. Pin, A. Llobet, J. Giraldo, A. Llebaria and P. Gorostiza, Nat. Chem. Biol., 2014, 10, 813 CrossRef CAS PubMed.
- M. Schonberger, M. Althaus, M. Fronius, W. Clauss and D. Trauner, Nat. Chem., 2014, 6, 712 Search PubMed.
- D. M. Barber, M. Schonberger, J. Burgstaller, J. Levitz, C. D. Weaver, E. Y. Isacoff, H. Baieer and D. Trauner, Chem. Sci., 2016, 7, 2347 RSC.
- L. Laprell, K. Hull, P. Stawski, C. Schon, S. Michalakis, M. Biel, M. P. Sumser and D. Trauner, ACS Chem. Neurosci., 2016, 7, 15 CrossRef CAS PubMed.
- F. Z. Zhang, A. Zarrine-Afsar, M. S. Al-Abdul-Wahid, R. S. Prosser, A. R. Davidson and G. A. Woolley, J. Am. Chem. Soc., 2009, 131, 2283 CrossRef CAS PubMed.
- A. A. Beharry, T. Chen, M. S. Al-Abdul-Wahid, S. Samanta, K. Davidov, O. Sadovski, A. M. Ali, S. B. Chen, R. S. Prosser, H. S. Chan and G. A. Woolley, Biochemistry, 2012, 51, 6421 CrossRef CAS PubMed.
- B. Buchli, S. A. Waldauer, R. Walser, M. L. Donten, R. Pfister, N. Blochliger, S. Steiner, A. Caflisch, O. Zerbe and P. Hamm, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11725 CrossRef CAS PubMed.
- Z. Yu and S. Hecht, Chem. Commun., 2016, 52, 6639 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a