
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular protection from the enzymatic tyrosine phosphorylation in a polypeptide†
Enrico
Faggi
a,
Yolanda
Pérez
b,
Santiago V.
Luis
*c and
Ignacio
Alfonso
*a
aDepartment of Biological Chemistry and Molecular Modelling, IQAC-CSIC, Jordi Girona 18-26, 08034, Barcelona, Spain. E-mail: ignacio.alfonso@iqac.csic.es
bNMR Facility, IQAC-CSIC, Jordi Girona 18-26, 08034, Barcelona, Spain
cDepartment of Inorganic and Organic Chemistry, ESTCE Universitat Jaume I, Avd. Sos Baynat, s/n, 12004, Castellón, Spain. E-mail: luiss@uji.es
First published on 31st May 2016
Abstract
Here we report two new artificial pseudopeptidic cages that bind the EYE peptide epitope in pure water at physiological pH (as studied by fluorescence and NMR spectroscopies). The supramolecular complexation of the Tyr residues efficiently precludes their subsequent PTK-catalysed phosphorylation. Our results show a supramolecular modulation of the PTK activity by competitive substrate caging.
The molecular recognition of peptides and proteins is a hot topic in supramolecular chemistry, with important implications in chemical biology.1 However, the binding of peptides in pure water and at neutral pH is still a challenging issue, due to the competitive effect of water and the flexibility of the peptides in solution.2 Some examples using cucurbituril,3 cyclodextrin4 or calixarene5 receptors have been reported mainly based on hydrophobic interactions that are favoured in pure water. Following our research with pseudopeptides,6 we have recently designed large pseudopeptidic cages as efficient receptors for dipeptides in organic or aqueous-organic solvents, showing a good selectivity for the Ac–EY–OH sequence.7 This selectivity is manifested in the differential binding of stereoisomers of the Ac–EY–OH dipeptide by CySer and CyThr cages (Fig. 1).8 Binding studies using several spectroscopic techniques with different dipeptides, combined with molecular mechanics calculations allowed defining a mode of binding.7,8 The corresponding host–guest complexes are stabilized by both polar (salt bridges and H-bonds) and non-polar (π–π stacking) interactions, where the encapsulation of the Tyr aromatic ring inside the cage cavity plays an important role. The Tyr residue in polypeptides is a biologically interesting target: for instance, the EYE sequence is a well-known substrate epitope of protein tyrosine kinases (PTKs),9 which catalyse the transfer of a phosphoryl group from ATP to the Tyr hydroxyl group. This phosphorylation is an important post-translational modification, being closely connected with cell regulation, signalling and growth.10 Thus, the abnormal function of PTKs is related to serious diseases, such as diabetes,11 cancer12 and neurodegenerative diseases.13 Therefore, there is growing interest in the design and study of new molecules and mechanisms to modulate kinase activity.‡
 | ||
| Fig. 1 Chemical structures of the pseudopeptidic cages, with the assumed labelling. | ||
Inspired by the highly regulated action of PTK enzymes in nature, we envisioned that artificial cages with good affinity for the corresponding peptidic substrates would form stable supramolecular complexes, protecting the Tyr residues from the phosphorylation action of the enzyme (Fig. 2). Here we show that this supramolecular approach of PTK modulation can be performed using synthetic pseudopeptidic cages as competitive substrate binders.
 | ||
| Fig. 2 Schematic representation of the proposed mechanism: the binding of the pseudopeptidic cage to the poly(EY) polypeptide protects the Tyr residues (blue hexagons in the cartoon) from the further phosphorylation mediated by PTK. | ||
To test our hypothesis, we used commercial poly(EY) as a model for the PTK substrate.9 Poly(EY) is a random 20–50 kDa copolymer with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Glu:Tyr molar ratio to maximize the occurrence of the EYE motif. Considering previous results,7,8 we designed two new hosts (CyOrn and CyLys, Fig. 1) with positively charged side chains at neutral pH, to increase the interaction with the anionic poly(EY). These new cages were synthesized following a convenient modification of a reported procedure14 (ESI†). The mechanism schematically proposed in Fig. 2 is specially challenging from the supramolecular perspective since it requires a strong interaction between a non-natural host (pseudopeptidic cages) and a highly solvated and flexible polypeptide in pure water at neutral pH (as shown by NMR and CD studies, see the ESI†). Moreover, the PTK–poly(EY) contact can be considered a protein–protein interaction (PPI), which is difficult to disrupt with synthetic small molecules.15
:1 Glu:Tyr molar ratio to maximize the occurrence of the EYE motif. Considering previous results,7,8 we designed two new hosts (CyOrn and CyLys, Fig. 1) with positively charged side chains at neutral pH, to increase the interaction with the anionic poly(EY). These new cages were synthesized following a convenient modification of a reported procedure14 (ESI†). The mechanism schematically proposed in Fig. 2 is specially challenging from the supramolecular perspective since it requires a strong interaction between a non-natural host (pseudopeptidic cages) and a highly solvated and flexible polypeptide in pure water at neutral pH (as shown by NMR and CD studies, see the ESI†). Moreover, the PTK–poly(EY) contact can be considered a protein–protein interaction (PPI), which is difficult to disrupt with synthetic small molecules.15
First, we studied the binding between poly(EY) and the cages by fluorescence spectroscopy in buffered water (60 mM Tris buffer at pH 7.3). The titration of poly(EY) with CyOrn produced the quenching of the Tyr emission at 304 nm, and the growth of an intense band at 385 nm, with a clear isoemissive point at 358 nm (Fig. 3A). This lower energy band was assigned to the formation of a CyOrn–poly(EY) complex through the Tyr residue encapsulation.16 The non-linear fitting of this band to a 1:1 binding mode rendered a dissociation constant of Kd = 920 μM (Table 1, entry 1). The commercial poly(EY) is a poly-disperse molecule containing many EYE binding sites and, in principle, all of them could bind to the cage. Accordingly, for the quantitative measurements, we considered the concentration of the repeating units (E4Y) as the binding species, since each repeating unit contains a Tyr residue. Therefore, the Kd values here reported reflect the average affinity for each EYE binding site, assuming a 1:1 host–guest stoichiometry. As a confirmation of the proposed interaction, we studied the binding of CyOrn to the tripeptide Ac–EYE–NH2 by fluorescence spectroscopy, also rendering the appearance of the band at 385 nm and a similar Kd = 1.17 mM (entry 2 in Table 1). This result suggests that most of the Tyr residues in poly(EY) are accessible to the cage and that they behave as nearly independent binding sites.
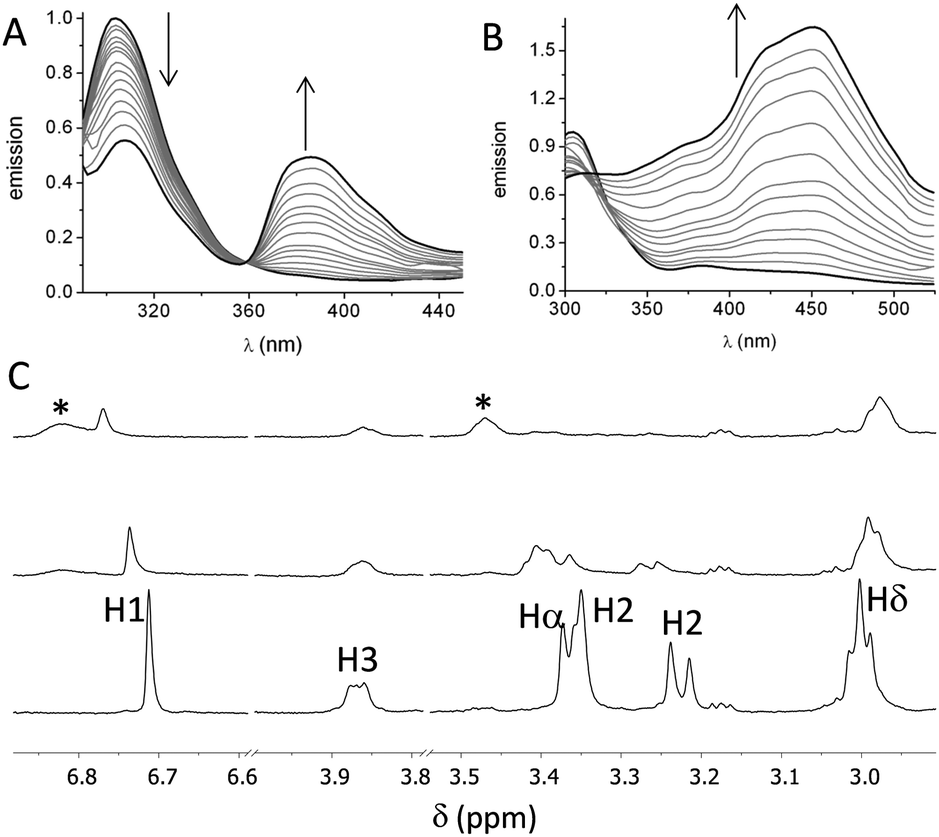 | ||
| Fig. 3 Binding of cages to EYE peptides: normalized fluorescence spectra (λexc = 276 nm) of poly(EY) (60 mM Tris buffer, pH 7.3 at 25 °C) upon the addition of the cages: (A) 2 × 10−4 M poly(EY) titrated with 0–1.33 × 10−3 M CyOrn or (B) 4 × 10−5 M poly(EY) titrated with 0–3.3 × 10−4 M CyLys. (C) Partial T1ρ-filtered 1H NMR spectra of CyOrn (0.4 mM in D2O, 75 mM Tris buffer, pD 7.4 at 25 °C) alone (lower trace) and with increasing amounts of poly(EY) (upper traces, 0.45 mM and 0.75 mM poly(EY), respectively). Signals marked with (*) correspond to the poly(EY). In all the cases the concentration of poly(EY) refers to the concentration of the repeating unit, E4Y. | ||
| Entry | Cage | Peptide | pH | K d (μM) |
|---|---|---|---|---|
| a Estimated statistical errors <15%. b Measured by fluorescence titration of the Tyr containing peptide upon addition of the cage. c Measured by NMR titration of the cage by addition of the peptide. | ||||
| 1 | CyOrn | Poly(EY) | 7.3 | 920b |
| 2 | CyOrn | Ac–EYE–NH2 | 7.3 | 1170b |
| 3 | CyLys | Poly(EY) | 7.3 | 450b |
| 4 | CyLys | Ac–EYE–NH2 | 7.3 | 570b |
| 5 | CyLys | Poly(EY) | 5.3 | 140b |
| 6 | CyLys | Poly(EY) | 8.7 | 440b |
| 7 | CyOrn | Poly(EY) | 7.4 (pD) | 403c |
| 8 | CyLys | Poly(EY) | 7.4 (pD) | 395c |
| 9 | CyLys | Poly(EY) | 7.3 (NaCl) | 342b |
| 10 | CyLys | Ac–YEEI–NH2 | 7.3 | 526b |
| 11 | CyLys | Ac–EEEIYEEFD–NH2 | 7.3 | 500b |
The fluorescence titration of poly(EY) with CyLys produced different fluorescence emission spectra (Fig. 3B). A more complex band at longer wavelength was observed upon the addition of the cage. Thus, the emission spectra showed several maxima (375 nm, 420 nm and 450 nm), probably corresponding to the co-existence of different complexes. The non-linear fitting of the fluorescence data rendered a slightly stronger interaction (Table 1, entry 3), which was also confirmed by performing the titration experiments with a minimal expression of the binding epitope Ac–EYE–NH2 (Table 1, entry 4). The different shape of the spectra with CyLys could be due to a different protonation degree of the complexes, since CyLys is more basic than CyOrn (see the ESI†). This hypothesis was confirmed by performing additional titration experiments at different pH (Table 1, entries 5 and 6), which showed similar binding constants but different shapes of the emission spectra (Fig. S33 in the ESI†). Thus, the maximum at 450 nm is more intense at lower pH while the band at 375 nm increases at higher pH, suggesting that the differences in the titration with CyOrn or CyLys must be due to a higher protonation degree of the complex formed with CyLys.
Spin-lock filtered NMR experiments17 were also employed for studying the binding process in solution. This NMR technique uses the spin–lattice relaxation time in the rotating frame (T1ρ) as the binding probe thanks to its dependence on the correlation time. Thus, the relaxation of the cage is highly enhanced upon binding to the poly(EY), leading to a decrease in the intensity of the NMR signals in the T1ρ filtered 1H NMR experiment. Accordingly, titration of CyOrn with poly(EY) in D2O (75 mM deuterated Tris, pD 7.4) induced a decrease in the intensity of many 1H NMR signals (Fig. 3C) implying an efficient interaction in solution. Moreover, some signals from the cage also shifted upon binding, mainly those corresponding to the aromatic tripodal ring (H1) and the methylene close to the primary amine of the side chains (Hδ). The non-linear fitting of the variation of the chemical shift of the H1 singlet rendered a Kd = 403 μM (Table 1, entry 7), which is within the same order of the value obtained by fluorescence spectroscopy. The experiments performed with CyLys rendered parallel results and a comparable binding constant (Table 1, entry 8 and ESI†). The 2D-NOESY experiment of a mixture of CyLys and poly(EY) (500 MHz, deuterated Tris buffer in D2O at pD 7.4, Fig. S49 in the ESI†) showed an intermolecular cross-peak between H1 of the cage and Hε of the Tyr residues of the peptide. This NOE further supported the proposed binding in solution. The CyLys cage was also able to bind to poly(EY) in the presence of 150 mM NaCl (Table 1, entry 9), a more challenging solvent for the interaction.
Considering that the prepared cages showed efficient binding of the EYE peptide sequence in pure water at neutral pH, we envisioned the possibility of the cages to protect the peptides from the action of the PTK enzyme. To that, we used an in vitro commercial assay for the evaluation of the effect of the cages on the phosphorylation reaction (assay description in the ESI†).18 We performed the experiments in the presence of the cages at different concentrations and the results were transformed into the remaining percent of kinase activity by comparison with the control experiment in the absence of cage (Fig. 4). Also CySer and CyThr (Fig. 1) were included as controls, since both showed very weak binding to the poly(EY) substrate in buffered water (by NMR and fluorescence spectroscopy, see the ESI†). All the cages were able to partially protect the substrates from the tyrosine kinase phosphorylation, showing differences depending on the cage structure and its concentration. The cages derived from Ser and Thr displayed a modest protection, requiring a high concentration (9 mM) to yield an ∼30% decrease of phosphorylation. Since they showed practically no effect at 1 mM, lower concentrations were not tested.
 | ||
| Fig. 4 In vitro enzymatic phosphorylation assays: PTK catalyzed phosphorylation of the poly(EY) substrate in the presence of different concentrations of the pseudopeptidic cages. The values show the average of two independent experiments, each one in three replicated measurements, with error bars showing the standard deviations. | ||
This low activity clearly correlates with their weaker binding to the EYE motif. However, CyOrn and CyLys displayed interesting competition with the PTK in the sub-millimolar range. At concentration values (0.26–1.10 mM) close to the corresponding Kd for the EYE-cage binding, the CyOrn host showed a 50–60% decrease in the phosphorylation, which can be directly ascribed to the cage-peptide recognition. Quite remarkably, a higher activity was observed for CyLys, with ∼70% protection from the kinase action in the 0.26–1.10 mM concentration range. These differences are more evident at lower concentrations of the cages: for instance, at 18 μM CyLys ∼50% protection was produced while CyOrn was practically inactive (∼100% of the remaining kinase activity, Fig. 4). The longer side chains of the Lys derivative lead to a more positively charged and more hydrophobic cage, which could produce a multivalent effect in the binding process, especially for this assay where the polypeptide substrate is anchored to a surface with possible clustering of the supramolecular complexes. Thus, the trends clearly reflect the validity of our proposal, since the cages with a higher affinity for the EYE motif showed a more efficient protection from the PTK-mediated phosphorylation reaction.
The modulation of the biochemical machinery with synthetic molecules has become a common procedure to study biological processes and, ideally, to design chemical tools for the treatment of diseases. In these cases, ad hoc designed synthetic molecules are prepared to bind large biomolecular systems (usually proteins). Within this rational, the inhibition of enzymatic activity has been normally faced by preparing chemicals able to bind the catalytic site of the enzymes.12a Here we demonstrated a complementary approach based on supramolecular chemistry. Thus, we have used the synthetic molecule to bind the substrate of the enzyme instead of the enzyme itself. This approach has been previously used to inhibit proteases3c,f and a demethylase5e but, to the best of our knowledge, it has never been used to modulate a kinase activity. This host–guest approximation shows the advantage of allowing a better implementation of substrate selectivity, in case the same enzyme is implicated in the modification of different substrates (as it is common for kinases). Despite the moderate potency of our cages for a formal PTK inhibition, the supramolecular mechanism of action targeting the substrate of the enzyme will open a new way for kinase activity modulation. The selectivity needed for improving this supramolecular kinase modulation can be tailored by structural modifications in the pseudopeptidic cages, thus possibly targeting known phosphorylation consensus sequences with specific characteristics.19 Thus, for instance, CyLys also recognized two Tyr-containing peptides (entries 10 and 11 in Table 1) that are target sequences of biologically relevant kinases,20 foreseeing the potential real applications of these cages.
In conclusion, the new pseudopeptidic cages described here efficiently recognize the EYE peptide sequence in buffered water, as shown by fluorescence and NMR spectroscopies. The binding occurs both with the isolated tripeptide and when the EYE motif forms part of a longer polypeptide chain as a model of a biomacromolecule. This supramolecular interaction efficiently protects the Tyr side chain from the enzymatic phosphorylation, as shown by in vitro kinase assays. Besides, the decrease in the phosphorylation degree can be roughly correlated with the cage-EYE sequence affinity. We believe that our results have wide implications, since they open a way to use supramolecular systems to modulate biochemical processes by substrate recognition, thus possibly hiding the substrates from their natural receptor or enzyme. A further generalization of this concept is under study in our group.
This work was supported by MINECO/FEDER (CTQ2012-38543-C03, CTQ2015-68429-R and CTQ2015-70117-R projects) and Generalitat de Catalunya (AGAUR, 2014 SGR 231).
Notes and references
- (a) W. C. Still, Acc. Chem. Res., 1996, 29, 155–163 CrossRef CAS; (b) M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2494 CrossRef CAS PubMed; (c) S. Tashiro, M. Tominaga, M. Kawano, B. Therrien, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2005, 127, 4546–4547 CrossRef CAS PubMed; (d) M. Wehner, D. Janssen, G. Schäfer and T. Schrader, Eur. J. Org. Chem., 2006, 138–153 CrossRef CAS; (e) J. E. Beaver and M. L. Waters, ACS Chem. Biol., 2016, 11, 643–653 CrossRef CAS PubMed.
- (a) C. Schmuck and L. Geiger, J. Am. Chem. Soc., 2004, 126, 8898–8899 CrossRef CAS PubMed; (b) C. Schmuck, Coord. Chem. Rev., 2006, 250, 3053–3067 CrossRef CAS; (c) F. Biedermann, U. Rauwald, M. Cziferszky, K. A. Williams, L. D. Gann, B. Y. Guo, A. R. Urbach, C. W. Bielawski and O. A. Scherman, Chem. – Eur. J., 2010, 16, 13716–13722 CrossRef CAS PubMed; (d) T. Gersthagen, J. Hofmann, F.-G. Klärner, C. Schmuck and T. Schrader, Eur. J. Org. Chem., 2013, 1080–1092 CrossRef CAS; (e) S. Yapar, M. Oikonomou, A. H. Velders and S. Kubik, Chem. Commun., 2015, 51, 14247–14250 RSC.
- (a) L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef CAS PubMed; (b) M. V. Rekharsky, H. Yamamura, C. Inoue, M. Kawai, I. Osaka, R. Arakawa, K. Shiba, A. Sato, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2006, 128, 14871–14880 CrossRef CAS PubMed; (c) A. Hennig, G. Ghale and W. M. Nau, Chem. Commun., 2007, 1614–1616 RSC; (d) M. V. Rekharsky, H. Yamamura, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Chem. Commun., 2008, 2236–2238 RSC; (e) J. M. Chinai, A. B. Taylor, L. M. Ryno, N. D. Hargreaves, C. A. Morris, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 8810–8813 CrossRef CAS PubMed; (f) L. A. Logsdon and A. R. Urbach, J. Am. Chem. Soc., 2013, 135, 11414–11416 CrossRef CAS PubMed; (g) L. C. Smith, D. G. Leach, B. E. Blaylock, O. A. Ali and A. R. Urbach, J. Am. Chem. Soc., 2015, 137, 3663–3669 CrossRef CAS PubMed; (h) C. Parente Carvalho, A. Norouzy, V. Ribeiro, W. M. Nau and U. Pischel, Org. Biomol. Chem., 2015, 13, 2866–2869 RSC.
- (a) H. Yamamura, M. V. Rekharsky, Y. Ishihara, M. Kawai and Y. Inoue, J. Am. Chem. Soc., 2004, 126, 14224–14233 CrossRef CAS PubMed; (b) Y. Liu, Y.-W. Yang, Y. Chen and F. Ding, Bioorg. Med. Chem., 2005, 13, 963–971 CrossRef CAS PubMed; (c) H. S. Christensen, B. W. Sigurskjold, T. G. Frihed, L. G. Marinescu, C. M. Pedersen and M. Bols, Eur. J. Org. Chem., 2011, 5279–5290 CrossRef CAS.
- (a) F. Sansone, L. Baldini, A. Casnati and R. Ungaro, New J. Chem., 2010, 34, 2715–2728 RSC; (b) A. D. Stancu, H.-J. Buschmann and L. Mutihac, J. Inclusion Phenom. Macrocyclic Chem., 2012, 75, 1–10 CrossRef; (c) R. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527–533 CrossRef CAS PubMed; (d) R. E. McGovern, A. A. McCarthy and P. B. Crowley, Chem. Commun., 2014, 50, 10412–10415 RSC; (e) H. Peacock, C. C. Thinnes, A. Kawamura and A. D. Hamilton, Supramol. Chem., 2016, 28, 575–581 CrossRef CAS.
- (a) S. V. Luis and I. Alfonso, Acc. Chem. Res., 2014, 47, 112–124 CrossRef CAS PubMed; (b) I. Alfonso, Chem. Commun., 2016, 52, 239–250 RSC.
- E. Faggi, A. Moure, M. Bolte, C. Vicent, S. V. Luis and I. Alfonso, J. Org. Chem., 2014, 79, 4590–4601 CrossRef CAS PubMed.
- E. Faggi, C. Vicent, S. V. Luis and I. Alfonso, Org. Biomol. Chem., 2015, 13, 11721–11731 CAS.
- D. M. Williams, D. Wang and P. A. Cole, J. Biol. Chem., 2000, 275, 38127–38130 CrossRef CAS PubMed.
- M. D. Haskell, J. K. Slack, J. T. Parsons and S. J. Parsons, Chem. Rev., 2001, 101, 2425–2440 CrossRef CAS PubMed.
- A. Fountas, L.-N. Diamantopoulos and A. Tsatsoulis, Trends Endocrinol. Metab., 2015, 26, 643–656 CrossRef CAS PubMed.
- (a) A. J. Bridges, Chem. Rev., 2001, 101, 2541–2572 CrossRef CAS PubMed; (b) J. B. Casaletto and A. I. McClatchey, Nat. Rev. Cancer, 2012, 12, 387–400 CrossRef CAS PubMed.
- (a) J. Ma, T. Jiang, L. Tan and J.-T. Yu, Mol. Neurobiol., 2014, 51, 820–826 CrossRef PubMed; (b) R. Abbassi, T. G. Johns, M. Kassiou and L. Munoz, Pharmacol. Ther., 2015, 151, 87–98 CrossRef CAS PubMed; (c) A. F. T. Arnsten, Nat. Neurosci., 2015, 18, 1376–1385 CrossRef CAS PubMed.
- A. Moure, S. V. Luis and I. Alfonso, Chem. – Eur. J., 2012, 18, 5496–5500 CrossRef CAS PubMed.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695–4748 CrossRef CAS PubMed.
- M. Shanmugam, D. Ramesh, V. Nagalakshmi, R. Kavitha, R. Rajamohan and T. Stalin, Spectrochim. Acta, Part A, 2008, 71, 125–132 CrossRef CAS PubMed.
- P. Śledź, C. Abell and A. Ciulli, NMR of Biomolecules, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 264–280 Search PubMed.
- Y. Jia, X.-j. Gu, A. Brinker and M. Warmuth, Expert Opin. Drug Discovery, 2008, 3, 959–978 CrossRef CAS PubMed.
- S. Lee, X. Lin, N. H. Nam, K. Parang and G. Sun, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14707–14712 CrossRef CAS PubMed.
- R. Amanchy, B. Periaswamy, S. Mathivanan, R. Reddy, S. G. Tattikota and A. Pandey, Nat. Biotechnol., 2007, 25, 285–286 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details, spectroscopic data, titration experiments and phosphorylation assays. See DOI: 10.1039/c6cc03875a |
| ‡ As an illustration of the current interest on kinases, see the American Chemical Society joined special issue on this topic at http://pubs.acs.org/page/vi/2014/kinases.html. |
| This journal is © The Royal Society of Chemistry 2016 |