
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilisation of an amorphous form of ROY through a predicted co-former interaction†
Philip A.
Corner
a,
J. Jonathan
Harburn
a,
Jonathan W.
Steed
b,
James F.
McCabe
c and
David J.
Berry
*a
aDurham University, Division of Pharmacy, Queen's Campus, Stockton on Tees, TS17 6BH, UK. E-mail: d.j.berry@durham.ac.uk
bDepartment of Chemistry, Durham University, University Science Laboratories, South Road, Durham, DH1 3LE, UK
cPharmaceutical Development, AstraZeneca Macclesfield, SK10 2SN, UK
First published on 18th April 2016
Abstract
The highly polymorphic compound ROY, notorious for the colour of its crystals, was the subject of an optimised high-throughput ultrasound-based co-crystal screen. This screen involved a computational pre-screen which highlighted an interaction between ROY and the potential co-former pyrogallol. We have shown that the presence of pyrogallol stabilises the amorphous form of ROY, highlighting the potential for future prediction of co-amorphous behaviours.
The olanzapine precursor ROY (Fig. 1), 5-methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitrile, so called due to the red, orange and yellow crystal colours of its polymorphs, has been reported to exist in at least 10 polymorphic forms.1 With seven of these forms structurally characterised, it was until recently2 the compound with the most polymorphs recorded in the Cambridge Structural Database (CSD).3 Due to the large number of polymorphic forms of ROY, it was chosen as a model compound for co-crystal screening. In this work ROY was first synthesised following the procedure reported in the patent for the synthesis of Olanzapine.4 Analysis of the resulting material is present in the ESI† (S1–S4).
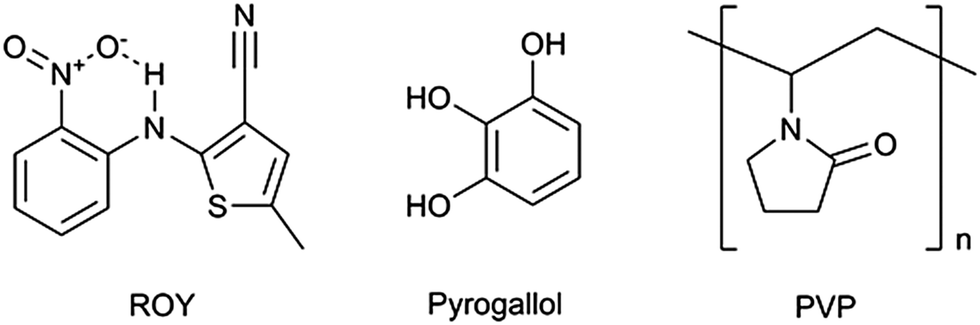 | ||
| Fig. 1 Structures of ROY, pyrogallol and PVP. | ||
It is the practice of the pharmaceutical industry to perform comprehensive polymorph screening during the development of a drug.5 Although a metastable crystalline polymorphic form may give benefits in terms of solubility, the risk of conversion to the less bioavailable form at ambient conditions means this option is seldom used. Solubility advantage can also be conferred by the amorphous form.6–9 These phases however also have the potential to convert to a thermodynamically more stable crystalline form. The timescale of this conversion dictates the degree of opportunity or risk associated with the phase.10 Stabilisation techniques such as formulating the product with the addition of a polymer have been widely studied11–13 and can yield suitable stability at a cost – financial in terms of greater development needed for the formulation, additional regulatory requirements and issues surrounding the quantity of active pharmaceutical ingredient (API) that is contained within a formulation (i.e. drug loading).14
Many of the polymers used in stabilising drugs in this way (such as PVP, PVPVA and HPMC6) are hygroscopic15 and can lead to faster re-crystallisation of the drug through modification of the glass transition (Tg), removing the solubility benefit. There are also issues associated with HPMC which can cause a laxative effect.16
These deficiencies have led to an expansion in research in co-amorphous materials.17,18 Co-amorphous phases have been the subject of significant study since 2009 as they have the potential to solve the problems of drug loading and toxicity. The co-amorphous approach has been defined as ‘the combination of two or more low molecular weight components that form a homogeneous amorphous single-phase system’.19 The stabilising interactions have been seen to occur via a number of mechanisms including hydrogen bonding, π–π stacking and salt formation. There are currently no purported means of predicting which small molecules will create such interactions and stabilise APIs in the desired fashion. This means many thousands of potential molecules could be used as the second entity, therefore some mechanism for practical selection would be beneficial.
Co-crystal formation can also convey advantageous properties to pharmaceuticals.20,21 Without extensive screening it is not currently possible to know with certainty which co-formers, if any, will form a co-crystal with a given API. Crystal structure prediction (CSP) is a developing field with the potential to correctly predict structure improving year on year, as evidenced by the blind tests organised by the Cambridge Crystallographic Data Centre (CCDC).22 There is still much work to be done regarding prediction of the result of introducing a co-former into the crystal structure, in terms of physical and chemical properties. It is therefore necessary to perform empirical screening and analysis to obtain co-crystals for a given API and determine their properties. With the predictive advances in mind it is prudent, prior to physical screening for co-crystals, to conduct a computational pre-screen. This involves the use of computational chemistry such as utilising molecular quantum-mechanical calculations to predict drug-co-former pairs that are likely to form co-crystals.23 Different methods are available from simple energy minimisation to full crystal structure prediction24 and the decision on which is most appropriate will be based on the size of the screen and the resources available. COSMOtherm software, which performs single molecule gas phase calculations based on polarisation charge densities, was chosen as the method of pre-screening for this work.25 Here ROY was screened against a list of 342 potential co-formers following a similar screening process to that detailed by Grecu et al.26 based on the previous work by Musumeci et al.23 This modelling led to a list of energetically favourable combinations of co-former to ROY and those with the greatest enthalpic driver for interaction were taken forwards to the physical screen. The top ten from this list are shown in Table 1 ranked by excess enthalpy, obtained by subtracting the sum of calculated enthalpy of interaction of the pure components from that of the API-co-former mixture in the gas phase.
| Potential co-formers | Excess enthalpya (ΔH/kJ mol−1) | Wavenumber of peaks associated with bonds affected by hydrogen bonding within ROY polymorphs | Crystallisation in cycleb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ∼3300 peak (acetone) | ∼2220 peak (acetone) | ∼3300 peak (ethanol) | ∼2220 peak (ethanol) | ∼3300 peak (hexane) | ∼2220 peak (hexane) | ∼3300 peak (LAG) | ∼2220 peak (LAG) | |||
a Excess enthalpy calculated for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 and 1:2 stoichiometric ratios of API to co-former and ranked by lowest.
b Cycle in which crystallisation occurs during DSC of the grind of the respective co-former with ROY. :1, 2:1 and 1:2 stoichiometric ratios of API to co-former and ranked by lowest.
b Cycle in which crystallisation occurs during DSC of the grind of the respective co-former with ROY.
|
||||||||||
| Pentafluorophenol | −1.953 | 3280 | 2209 + 2230 | 3280 | 2229 | 3003 + 3281 | 2210 + 2230 | 3281 | 2230 + 2240 | 2nd heating |
| Acesulfame | −1.509 | 3280 | 2209 + 2230 | 3280 | 2210 + 2230 | 3280 | 2210 + 2230 | 3281 | 2230 | Cooling |
| Oxalic acid | −1.346 | 3282 + 3302 | 2209 + 2230 | 3279 | 2228 | 3281 | 2221 + 2230 | 3280 | 2229 | Cooling |
| Quercetin | −1.296 | 3280 | 2230 | 3279 | 2230 | 3280 | 2222 + 2230 | 3281 | 2230 | 2nd heating |
| Sulfamic acid | −1.222 | 3280 | 2229 | 3280 | 2229 | 3283 | 2221 | 3280 | 2230 | Cooling |
| 3,5-Dinitrobenzoic acid | −1.193 | 3280 | 2210 + 2229 | 3280 | 2230 | 3281 | 2223 + 2229 | 3281 | 2230 | Cooling |
| Pyrogallol | −1.145 | 3278 | 2229 | 3279 | 2229 | 3280 | 2230 | 3279 | 2230 | None |
| 2,4-Dihydroxybenzoic acid | −1.044 | 3280 | 2229 | 3280 | 2230 | 3282 | 2222 + 2230 | 3279 | 2230 | Cooling, 2nd heating |
| 5-Nitroisophthalic acid | −1.037 | 3278 | 2229 | 3278 | 2229 | 3281 + 3300 | 2210 + 2230 | 3280 | 2230 | Cooling |
| Gallic acid | −1.032 | 3277 | 2209 + 2229 | 3276 | 2228 | 3278 | 2209 + 2230 | 3279 | 2230 | Cooling |
Physical high-throughput screening of potential co-formers with the desired API using ultrasonication has been proven to be feasible for this purpose with researchers employing variations such as SonicSlurry™ and sonic bath processes to introduce the sonic energy to the samples in 96-well plates.27,28 Having adopted ultrasonication technology for the screening process and adapted the current methods, key parameters for co-crystal screening were optimised to achieve a robust, cost effective process and this was applied to ROY. This was achieved by adding a DCM/ROY solution to an equimolar amount of solid potential co-former, which had been pre-weighed in a borosilicate glass 96-well plate. After leaving the initial solvent (DCM) to evaporate, acetone was added to 8 wells and the 8-tips of the ultrasonic probe were placed in these wells and sonicated. This process was repeated for the remaining wells and the whole process repeated a further two times replacing the solvent used each time; thereby completing the screening process in three solvents for 48 potential co-formers. The products from each of the wells were analysed using FTIR (after each solvent) and compared to the spectra for the two parent compounds.
Further details of the methods used can be found in the ESI† (S5). Full optimisation of this screening method is on-going and will be reported elsewhere.
Here we report the discovery of a co-amorphous form of ROY, determined during follow up experimentation post co-crystal screening. Based on the analysis of the FTIR spectra, the screen applied to ROY found no evidence of the formation of co-crystals. This was not surprising as a degree of excess enthalpy from prediction is no guarantee of co-crystallisation in vitro, due to lack of consideration of the purported lattice in the adopted approach. The wavenumbers of the peaks of interest used in the analysis for the top 10 predicted co-formers are listed in Table 1, the results in their entirety can be found in the ESI† (S6). Further efforts to produce co-crystals of ROY from the top 10 predicted co-formers followed, this involved applying different methods of manufacture: liquid assisted grinding (LAG) and evaporative crystallisation. See ESI† for details of these methods.
The products of LAG were analysed by FTIR, the results again listed in Table 1. In addition, the products of LAG were analysed by DSC employing a heat/cool/heat method (see ESI† for full details). Nine of the 10 products of LAG displayed unremarkable thermal recrystallization behaviour, however the LAG product of ROY and pyrogallol stood out. Pure ROY and pyrogallol show poor amorphous stability. Pyrogallol crystallises on cooling from melt and ROY crystallises during the second heating cycle (class (I-A) and class (II) materials following the classification system as described by Baird et al.29 respectively), the product of the grind does not crystallise and remains in amorphous form throughout the temperature range tested. This suggests that an amorphous form was produced on cooling and this remained stable until at least 150 °C resulting in a material categorised as class (III); producing a co-amorphous material.9 The second heating cycles for the two parent components and the product of the 1:1 molar ratio grind are shown in Fig. 2. See ESI† (S12 and S13) for the initial heating and cooling cycles. This behaviour differs from all other ROY-co-former combinations, as they all crystallise either on cooling (class (I)), on the second heating phase (class (II)), or in one case both, as shown in Table 1.
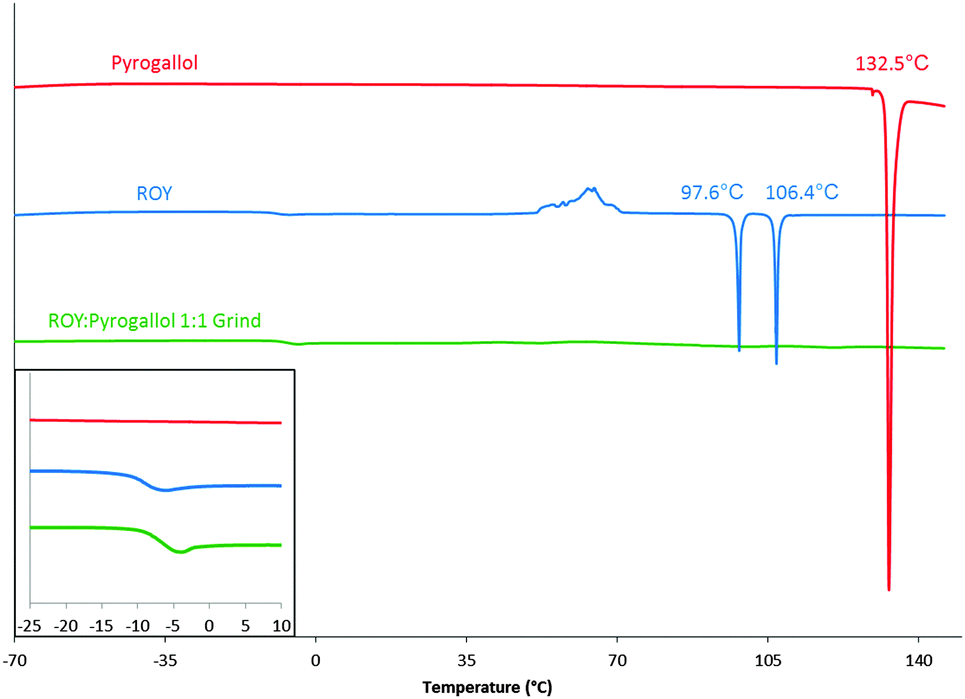 | ||
| Fig. 2 Second heating phase DSC curves of pyrogallol (red), ROY (blue) and ROY:pyrogallol 1:1 grind (green). Peak onset temperatures displayed. Presence of Tg highlighted in inset. | ||
The glass transition temperatures for ROY and for the ROY–pyrogallol 1:1 grind, shown in Fig. 2 and in more detail in the ESI† (S14), vary by less than 4 °C, and as such suggest that a change in molecular mobility great enough to affect molecular translational ability is not caused by the presence of pyrogallol. To determine whether the amorphous stability elicited by the presence of the pyrogallol was related to its stoichiometric ratio with ROY, further ROY:pyrogallol samples were produced at 5% w/w increments from 0% to 100%, representative thermograms of the second heating cycle are shown in Fig. 3 and further results detailing the initial heating cycle can be found in the ESI† (S17 and S18). The initial melting point varies very little over the range 5% to 95% w/w pyrogallol and therefore indicates that this is not a eutectic system, as, if it were, a single lower melting point at the eutectic composition would be expected.30 It is also worth noting that an endothermic melting peak at around 88.5 °C was detected during the initial heating cycle is present in all three samples (S17, ESI†), with decreasing intensity as the pyrogallol content is reduced.
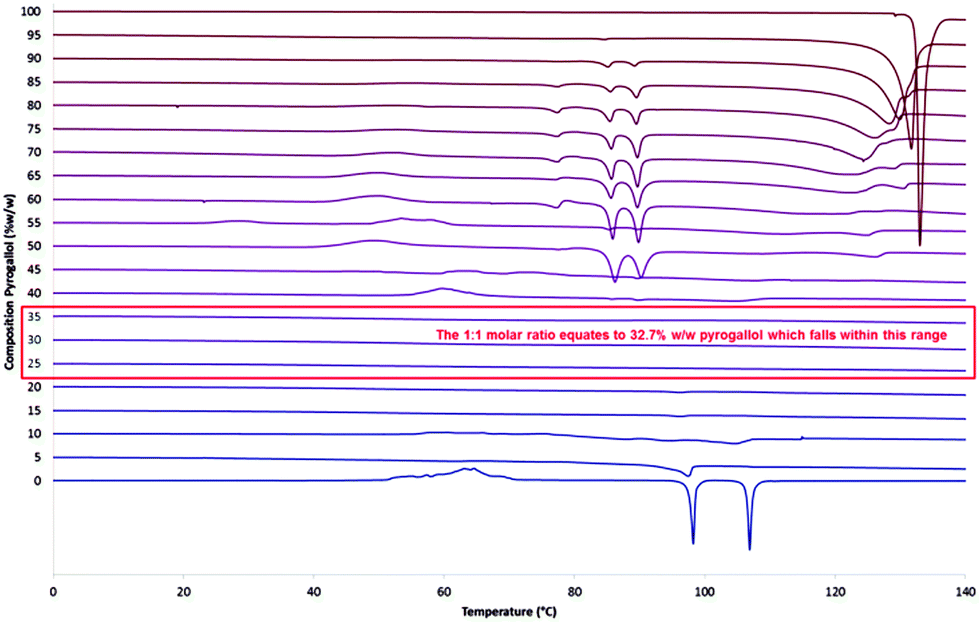 | ||
| Fig. 3 Second heating phase DSC curves of ROY (0%), pyrogallol (100%) and varying compositions of ROY:pyrogallol in 5% w/w increments. The red box highlights the range of compositions in which the amorphous form is stabilised. This equates to 1:1 stoichiometry. | ||
When looking at the second heating cycle (Fig. 3) at the 1:1 composition the lack of any endothermic or exothermic events in the given temperature range suggest the 1:1 stabilisation rather than a small amount of pyrogallol inhibiting the crystallisation of ROY. The approximate boundaries of this range (25% to 35% w/w pyrogallol content) equate to molar ratios (in the form 1 ROY to X pyrogallol) of 0.69 and 1.11 respectively. These data suggest that with a lower ratio of pyrogallol to ROY, there is excess ROY behaving as pure ROY and uninfluenced by the presence of pyrogallol. The formation of intermolecular interactions between individual molecules of ROY and pyrogallol in a one to one manner would give rise to such behaviour.
In order to explore this interaction the hydrogen bonding propensity of ROY, with the addition of aromatic hydroxyl to represent the potential pyrogallol interaction, was calculated using the logit model31 resulting in an area under ROC curve of 0.86 (see ESI,† S19–S22). This predicted strong hydrogen bonds between molecules of ROY only. Further modelling utilising a 200 molecule amorphous cell, generated in materials studio, displayed no change to the N–H bond of ROY, but changes to the environment around it (ESI,† S23–S25). IR and solution 1H NMR were conducted on a physical sample. Solution 1H NMR showed a 1:1 product with no thermal degradation from the mixture (ESI,† S26). Fig. 4 displays the FTIR spectra of an amorphous sample of the ROY:pyrogallol 1:1 grind and a peak shift corresponding to the N–H bond in ROY is apparent. A possible rationale, supported by the amorphous cell prediction (ESI,† S23–S25), could be that pyrogallol forms intermolecular bonds in proximity to the N–H bond of ROY causing the slight alteration in environment of the N–H bond. Such interactions in co-amorphous materials have previously been reported.17
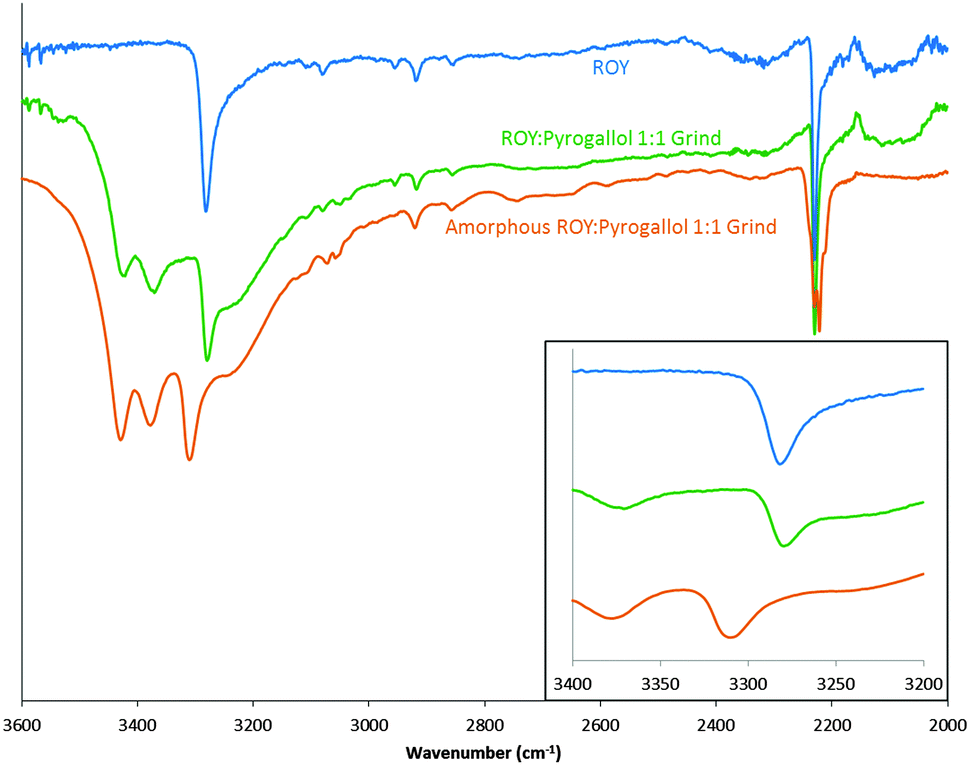 | ||
| Fig. 4 FTIR spectra of ROY (blue), ROY:pyrogallol 1:1 grind (green) and amorphous ROY:pyrogallol 1:1 grind (orange). Focused area of wavenumbers 3200–3400 shown in inset. | ||
In order to compare the pyrogallol-stabilised amorphous ROY form to a more traditional polymer stabilised form, a grind of ROY with polyvinylpyrrolidone (PVP) was created. This was at both a typical 10% w/w drug loading, with regards to ROY, and also at a 1:1 molar ratio, based on the molecular weight of the PVP monomer. Both of these ROY:PVP ratios led to stabilised amorphous forms being produced. These data are available in the ESI† (S27–S30). The similarity of the ROY:pyrogallol sample to the behaviour seen with the ROY:PVP grind adds further evidence to the possibility of replacing polymer with small molecule stabilising agents in some situations.
The timescale of the stability of the ROY:pyrogallol mix was also investigated by powder X-ray diffraction (PXRD) and DSC. This stability was seen to be highly dependent on atmospheric moisture conditions. Indeed samples stored under dry nitrogen flow, or in a lidded DSC pan were seen to be stable for up to 65 hours, with those exposed to atmospheric conditions only stable for around 30 minutes (ESI,† S33–S35). It was not possible to verify this through vapour sorption study due to the fast recrystallization that was seen on the instrument, but storage at 75% RH 25 °C showed immediate visual recrystallization, which was confirmed by DSC at 18 hours (ESI,† S33). As compared to pure ROY this is a significant improvement however as complete recrystallization from the amorphous form was seen after 15 minutes.
In this work the application of an optimised co-crystal screen, utilising computational tools to predict the most energetically favourable co-formers, has led to the discovery of no co-crystals, but has highlighted a 1:1 interaction between ROY and pyrogallol. This interaction stabilises ROY in the amorphous form, although this stability is moisture dependent. Although predictive technology exists for single component amorphous phases,32 currently no predictive method for co-amorphous phases has been suggested and all screening is by trial and error. The discovery of this behaviour stemmed from a screen of 342 co-formers, in three stoichiometries, in which predicted interaction had been ranked highly in the gas phase. This suggests that co-crystal screening approaches can be modified to enable study into co-amorphous phases and that ‘negative’ co-crystal hits should be investigated for alternative utility as co-amorphous materials. Such an approach would enable a broader palate of pharmaceutical development options and improve process efficiency.
We would like to thank AstraZeneca and Durham University for PhD funding to support the project. We would also like to thank the Royal Society (RG130663S) for equipment.
Notes and references
- L. Yu, Acc. Chem. Res., 2010, 43, 1257–1266 CrossRef CAS PubMed.
- V. López-Mejías, J. W. Kampf and A. J. Matzger, J. Am. Chem. Soc., 2012, 134, 9872–9875 CrossRef PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- J. K. Chakrabarti, T. M. Hotten and D. E. Tupper, EP0454436 B1, 1995.
- ICH Steering Committe, Specifications: Test Procedures and Acceptance criteria for new drug substances and new drug products: chemical substances Q6A, 1999.
- G. Van Den Mooter, Drug Discovery Today: Technol., 2012, 9, e79–e85 CrossRef CAS PubMed.
- S. Sotthivirat, C. McKelvey, J. Moser, B. Rege, W. Xu and D. Zhang, Int. J. Pharm., 2013, 452, 73–81 CrossRef CAS PubMed.
- D. E. Alonzo, Y. Gao, D. Zhou, H. Mo, G. G. Z. Zhang and L. S. Taylor, J. Pharm. Sci., 2010, 100, 3316–3331 CrossRef PubMed.
- H. Grohganz, K. Löbmann, P. Priemel, K. Tarp Jensen, K. Graeser, C. Strachan and T. Rades, J. Drug Delivery Sci. Technol., 2013, 23, 403–408 CrossRef CAS.
- B. C. Hancock and G. Zografi, J. Pharm. Sci., 1997, 86, 1–12 CrossRef CAS PubMed.
- T. Vasconcelos, B. Sarmento and P. Costa, Drug Discovery Today, 2007, 12, 1068–1075 CrossRef CAS PubMed.
- S. Janssens and G. Van den Mooter, J. Pharm. Pharmacol., 2009, 61, 1571–1586 CrossRef CAS PubMed.
- A. Mahieu, J. F. Willart, E. Dudognon, F. Daneìde and M. Descamps, Mol. Pharmaceutics, 2013, 10, 560–566 CrossRef CAS PubMed.
- Y. Tian, J. Booth, E. Meehan, D. S. Jones, S. Li and G. P. Andrews, Mol. Pharmaceutics, 2013, 10, 236–248 CrossRef CAS PubMed.
- A. C. Rumondor and L. S. Taylor, Mol. Pharmaceutics, 2010, 7, 477–490 CrossRef CAS PubMed.
- CIR Expert Panel, Int. J. Toxicol., 1986, 5, 1–59 CrossRef.
- S. J. Dengale, H. Grohganz, T. Rades and K. Löbmann, Adv. Drug Delivery Rev. DOI:10.1016/j.addr.2015.12.009.
- N. Chieng, J. Aaltonen, D. Saville and T. Rades, Eur. J. Pharm. Biopharm., 2009, 71, 47–54 CrossRef CAS PubMed.
- R. Laitinen, K. Löbmann, C. J. Strachan, H. Grohganz and T. Rades, Int. J. Pharm., 2013, 453, 65–79 CrossRef CAS PubMed.
- J. W. Steed, Trends Pharmacol. Sci., 2013, 34, 185–193 CrossRef CAS PubMed.
- R. Thakuria, A. Delori, W. Jones, M. P. Lipert, L. Roy and N. Rodríguez-Hornedo, Int. J. Pharm., 2013, 453, 101–125 CrossRef CAS PubMed.
- D. A. Bardwell, C. S. Adjiman, Y. A. Arnautova, E. Bartashevich, S. X. M. Boerrigter, D. E. Braun, A. J. Cruz-Cabeza, G. M. Day, R. G. Della Valle, G. R. Desiraju, B. P. Van Eijck, J. C. Facelli, M. B. Ferraro, D. Grillo, M. Habgood, D. W. M. Hofmann, F. Hofmann, K. V. J. Jose, P. G. Karamertzanis, A. V. Kazantsev, J. Kendrick, L. N. Kuleshova, F. J. J. Leusen, A. V. Maleev, A. J. Misquitta, S. Mohamed, R. J. Needs, M. A. Neumann, D. Nikylov, A. M. Orendt, R. Pal, C. C. Pantelides, C. J. Pickard, L. S. Price, S. L. Price, H. A. Scheraga, J. Van De Streek, T. S. Thakur, S. Tiwari, E. Venuti and I. K. Zhitkov, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 535–551 CAS.
- D. Musumeci, C. A. Hunter, R. Prohens, S. Scuderi and J. F. McCabe, Chem. Sci., 2011, 2, 883–890 RSC.
- S. L. Price, Acc. Chem. Res., 2009, 42, 117–126 CrossRef CAS PubMed.
- Y. Abramov, C. Loschen and A. Klamt, J. Pharm. Sci., 2012, 101, 3687–3697 CrossRef CAS PubMed.
- T. Grecu, C. A. Hunter, E. J. Gardiner and J. F. McCabe, Cryst. Growth Des., 2014, 14, 165–171 CAS.
- H. Morrison, M. Mrozek-Morrison, J. Toschi, V. Luu, H. Tan and D. Daurio, Org. Process Res. Dev., 2013, 17, 533–539 CrossRef CAS.
- S. L. Childs, N. Rodríguez-Hornedo, L. S. Reddy, A. Jayasankar, C. Maheshwari, L. McCausland, R. Shipplett and B. C. Stahly, CrystEngComm, 2008, 10, 856–864 RSC.
- J. A. Baird, B. Van Eerdenbrugh and L. S. Taylor, J. Pharm. Sci., 2010, 99, 3787–3806 CrossRef CAS PubMed.
- S. Cherukuvada and A. Nangia, Chem. Commun., 2014, 50, 906–923 RSC.
- P. A. Wood, N. Feeder, M. Furlow, P. T. A. Galek, C. R. Groom and E. Pidcock, CrystEngComm, 2014, 16, 5839–5848 RSC.
- D. Mahlin, S. Ponnambalam, M. Heidarian Höckerfelt and C. A. Bergström, Mol. Pharmaceutics, 2011, 8, 498–506 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc02949c |
| This journal is © The Royal Society of Chemistry 2016 |