
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled production of the elusive metastable form II of acetaminophen (paracetamol): a fully scalable templating approach in a cooling environment†
Lauren R.
Agnew
ab,
Dyanne L.
Cruickshank
c,
Thomas
McGlone
d and
Chick C.
Wilson
*ab
aEPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation, University of Bath, Bath, BA2 7AY, UK. E-mail: C.C.Wilson@bath.ac.uk
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
cDepartment of Chemistry, University of Cape Town, Rondebosch, 7701, South Africa
dEPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation, c/o Strathclyde Institute for Pharmacy and Biomedical Sciences, Technology and Innovation Centre, 99 George Street, Glasgow, G1 1RD, UK
First published on 19th February 2016
Abstract
A scalable, transferable, cooling crystallisation route to the elusive, metastable, form II of the API acetaminophen (paracetamol) has been developed using a multicomponent “templating” approach, delivering 100% polymorphic phase pure form II at scales up to 120 g. Favourable solubility and stability properties are found for the form II samples.
Enhancing physiochemical properties of active pharmaceutical ingredients (APIs) has been widely researched in recent years to aid in the downstream processing of many pharmaceuticals. For example, increased aqueous solubility results in higher bioavailability, the ability to engineer layered structures can result in improved compaction properties, and modifying the stability of APIs can be of critical importance in transportation and storage. Such property enhancement can be achieved both in polymorphic forms of the API, and in multicomponent molecular complexes containing the API (e.g. co-crystals). There have been numerous reports on the use of multicomponent crystallisation procedures, in particular co-crystallisation, to achieve such physical property enhancement.1–5 However, there are very limited reports targeting different polymorphic forms of single component APIs using a multicomponent approach. A range of API molecules have been shown to be polymorphic on crystallisation into the solid state; containing only the desired API, these polymorphs can display markedly different physical properties. Many of these polymorphic forms can be elusive and accessible under specialised conditions or in low yields, which hampers the potential to manufacture and utilise them in pharmaceutical products. Of relevance to the work presented here, a number of elusive, metastable polymorphic forms have been discovered serendipitously through attempted co-crystallisation experiments.6–8 Systems such as these have introduced the idea of using a second molecular component, termed here a templating molecule, in the crystallisation process to isolate and convert a previously stable polymorphic form of the target API to a new and often elusive metastable form, while not being present itself in the final product. Crucially, this has previously rarely been achieved by design and in this work such a designed process is not only achieved, but scaled to produce large quantities of an elusive polymorph with enhanced physical properties.
Acetaminophen (paracetamol; PCM) is a widely used analgesic and antipyretic which has been studied extensively in the solid state.9–12 It exists in five polymorphic forms (of which two can only be accessed at high pressure), two of which have relevance in this work (Fig. 1). Form I displays a herringbone arrangement of molecules within the crystal structure, whilst form II is layered. This layering gives form II the enhanced physical properties of increased solubility (assisting bioavailability) and compressibility upon tableting (assisting processing). Previous routes to obtaining metastable form II include reaction coupling,13 swift cooling,14 enforcing Ostwald's rule of stages,15 use of polymer additives,16 heterogeneous nucleation17 and multicomponent templating approaches.18 The last of these has shown the possibility to template form II with a variety of benzoic acid (BA) derivatives. However, this was only achieved in an evaporative crystallisation environment.18 The present work looks at the production of metastable form II, with 4-halobenzoic acid derivatives (4-bromobenzoic acid (4-BrBA), 4-chlorobenzoic acid (4-ClBA), 4-fluorobenzoic acid (4-FBA)) as templating molecules, in a cooling crystallisation environment. The 4-substituted benzoic acid derivatives were shown to produce form II most reliably in evaporative studies and so were chosen as initial additives. Here, we develop this designed approach by also using the structurally similar molecule metacetamol (MCM), only previously studied in relation to its effect on PCM-I morphology,19,20 as a templating molecule for PCM-II production. Use of this designed templating approach to access a range of elusive solid forms in a “discovery” context has also recently been discussed by Thomas et al. in a complementary paper in this issue.
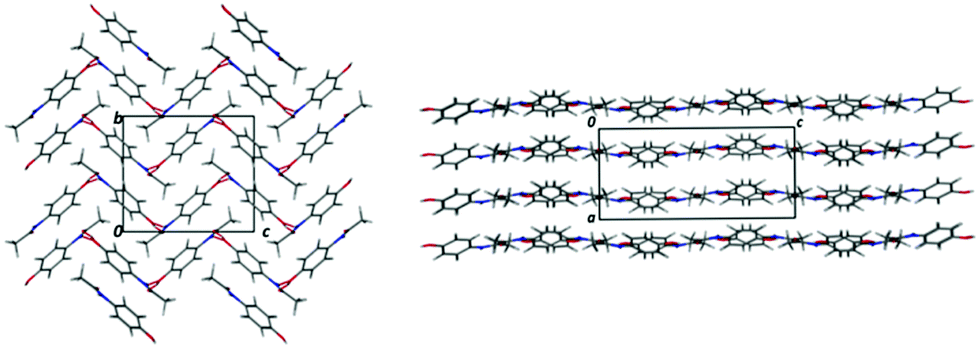 | ||
| Fig. 1 PCM-I (left) and PCM-II (right). | ||
Cooling crystallisation is becoming more widely used in industry, as it offers improved control of the supersaturation within the system and hence greater control of both polymorphic form and particle attributes, with many continuous manufacturing processes, such as those using the continuous oscillatory baffled crystalliser21 and mixed suspension mixed product removal platforms,22 employing it. There are a number of examples in the literature using cooling crystallisation methods with single component systems but to date obtaining polymorphic control of a single-molecule API system in a cooling environment with more than one (non-solvent) molecular component present has not been achieved. With regard to the PCM system, previous cooling crystallisation experiments, have shown the reproducible formation of only PCM-I.23 A solution-mediated phase transition from form II to form I does, however, occur during cooling, consistent with Ostwald's rule of stages.
In this work, the Polar Bear Plus Crystalliser from Cambridge Reactor Design is used to provide controlled cooling crystallisation conditions, in a systematic study of the production of PCM-II using a templating approach. 1.5 cm3 vials containing ca. 250 mg of paracetamol, varying weight percentages of BA templating molecules (0.4–2% for 4-BrBA and 4-ClBA and 1–4% for 4-FBA) and 1 g of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 v/v H2O:IPA solvent were subjected to a stepped cooling profile (Fig. 2) from 70 to 5 °C at a rate of 0.02 °C min−1. Solutions were cooled in 5 °C intervals and subsequently stirred for one hour to reach steady state conditions. Magnetic bottom stirring was used at a rate of either 800 rpm or 400 rpm. The experiments were designed to ensure that the BA templating molecule remained in solution at the end of the crystallisation (informed by solubility curves, ESI,† Fig. S1). A variety of cooling rates were investigated in order to optimise the crystallisation process, however production of PCM-II with BA co-former templating molecules was only achieved using the cooling conditions described above.
:40 v/v H2O:IPA solvent were subjected to a stepped cooling profile (Fig. 2) from 70 to 5 °C at a rate of 0.02 °C min−1. Solutions were cooled in 5 °C intervals and subsequently stirred for one hour to reach steady state conditions. Magnetic bottom stirring was used at a rate of either 800 rpm or 400 rpm. The experiments were designed to ensure that the BA templating molecule remained in solution at the end of the crystallisation (informed by solubility curves, ESI,† Fig. S1). A variety of cooling rates were investigated in order to optimise the crystallisation process, however production of PCM-II with BA co-former templating molecules was only achieved using the cooling conditions described above.
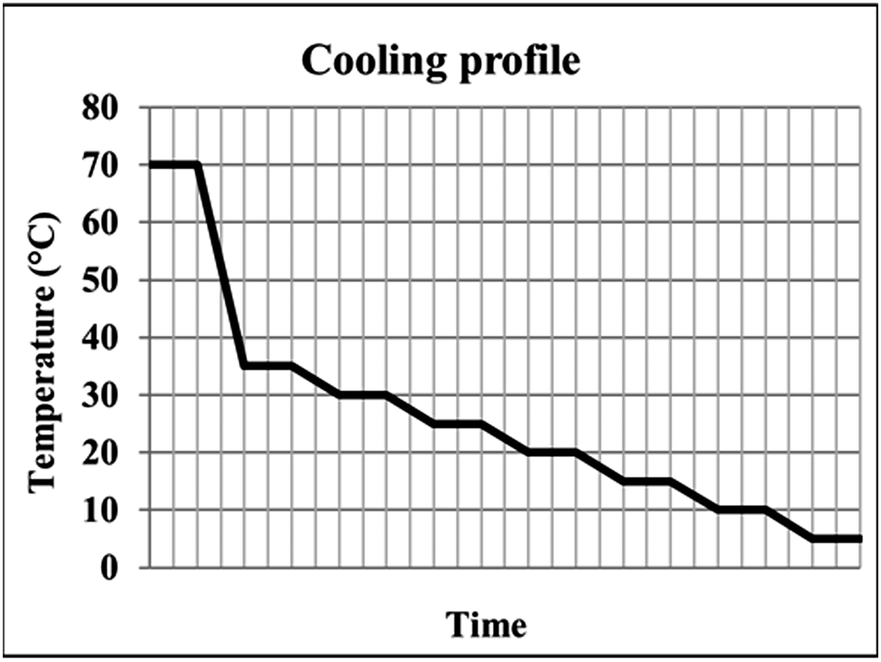 | ||
| Fig. 2 The stepped cooling profile employed, showing the six regions of dwelling employed to ensure steady state is reached. | ||
For MCM, varying weight percentages (1, 2, 5, 10 and 25%) as a proportion of ca. 250 mg PCM and 1 g of one of three solvents (ethanol, IPA or 60:40 H2O:IPA) were subjected to both a stepped cooling profile (Fig. 2) at three different cooling rates: 0.02 °C min−1, 0.2 °C min−1 and 1 °C min−1 and a linear cooling profile at 1 °C min−1; the latter, rapid cooling rate, was used to mimic the cooling conditions that may be more relevant to continuous crystallisation. Larger vials (20 ml) and varying sizes of round bottomed flasks (RBFs) were used to scale up the production of PCM-II in the presence of MCM once the optimal cooling conditions had been established.
Powder X-ray diffraction (PXRD) confirms the formation of PCM-II (ESI,† Fig. S2), with no additional peaks corresponding to the presence of co-former, PCM-I or any other solid form. Differential scanning calorimetry (DSC) measurements confirmed the absence of any templating molecule in lower concentrations than detectable by PXRD or Raman spectroscopy (ESI,† Fig. S3).
Table 1 highlights the specific conditions used to obtain PCM-II in a designed, reproducible manner. The production of PCM-II is highly dependent on crystallisation conditions as well as the concentrations of both the co-former molecules and PCM; more recently, design of experiments analysis on the PCM/MCM system has allowed these effects to be quantified, enhancing the design element of this study. The concentrations of the co-former with respect to the solvents were chosen so that the templating molecule was undersaturated at 5 °C, ensuring it remains in solution at the completion of the cooling regime. The higher stirring rates required for 4-BrBA can be attributed to the fact that these conditions provide the greater mass transfer required for lower concentrations of 4-BrBA. Investigations into the scaling potential of the templated crystallisation of PCM-II with 4-FBA have been largely unsuccessful. It can be suggested that this is due to poor mass transfer at higher scales and the low solubility of the co-former molecule.
| Stirring rate (rpm) | Co-former | Solvent | Conc. of PCM (mg g−1 of solvent) | Cooling rate (°C min−1) | Cooling profile | wt% co-former |
|---|---|---|---|---|---|---|
| a Vials containing higher concentrations of metacetamol contained some metacetamol in the solid product. This was mitigated by cooling to higher temperatures. | ||||||
| 800 | 4-BrBA | 60:40 H2O:IPA |
250 | 0.02 | Stepped | 0.4, 1.6, 2 |
| 400 | 4-FBA | 60:40 H2O:IPA |
260 | 0.02 | Stepped | 3.5 |
| 400 | Metacetamol | 60:40 H2O:IPA |
250 | 0.02 | Stepped | 10, 25 |
| 400 | Metacetamol | EtOH | 250 | 0.02 | Stepped | 5, 10, 25a |
| 400 | Metacetamol | 60:40 H2O:IPA |
250 | 0.2 | Stepped | 10, 15 |
| 400 | Metacetamol | EtOH | 250 | 0.2 | Stepped | 5, 10, 15 |
| 400 | Metacetamol | 60:40 H2O:IPA |
250 | 1 | Stepped | 10, 15, 20, 25a |
| 400 | Metacetamol | EtOH | 250 | 1 | Stepped | 10, 15, 20, 25a |
| 400 | Metacetamol | 60:40 H2O:IPA |
250 | 1 | Linear | 10, 20, 25a |
| 400 | Metacetamol | EtOH | 250 | 1 | Linear | 10, 20, 25a |
On the other hand, the use of MCM as a templating molecule has allowed for the reproducible scale-up to the 10 ml solvent scale (ca. 2.5 g PCM) at all three cooling rates and all cooling profiles currently explored. Scaled production of PCM-II using a rapid cooling rate of 1 °C min−1 is a significant step towards mimicking the conditions that would likely be experienced in operational continuous crystallisation platforms. Further scale-up attempts required increasing the concentration of MCM and truncating the cooling at a final temperature of 20 °C, allowing the system to be scaled into a 50 ml RBF, yielding large quantities of >10 g of 100% pure PCM-II in single runs. It can be suggested that the higher concentrations of MCM are needed to account for the poorer mass transfer experienced through magnetic bottom stirring at larger scales. Use of overhead stirring, and the greater associated mass transfer, has allowed for the further scaling of the system to volumes as high as 800 ml, with production on the 100 ml scale fully reproducible. Due to the widening of the metastable zone width with overhead stirring, a greater concentration of PCM was used; a concentration of 300 mg g−1 PCM coupled with 25 wt% MCM reproducibly gives 100% PCM-II. Currently, all samples of PCM-II produced are stable for periods of months (ESI,† Fig. S4), further confirming the absence of any form I, the presence of which would seed the conversion of form II back to the stable form I. The kinetics of this conversion are currently under investigation, with initial results showing that presence of 5 wt% or greater of form I in proportion to form II results in partial transformation, in the solid state, of the sample back to form I within a week. The stability of form II established here is significantly enhanced from that resulting from previously reported methods of obtaining PCM-II; the use of polymer additives gave samples that were only stable for five hours.16
It is worth noting that previous evaporative work emphasised the need to use stoichiometric ratios of PCM to co-former molecule to ensure the production of form II; the lower, at times catalytic, amounts used in this work under cooling conditions are in stark contrast to this.
A solubility curve for PCM-II has been previously reported,14 however the methodology or literature used to obtain these data were undisclosed. The aqueous solubility of PCM-II as determined using turbidimetric methods in a Technobis Crystal 16 device is thus reported here (Fig. 3; and in ESI†), confirming and quantifying its enhanced solubility.
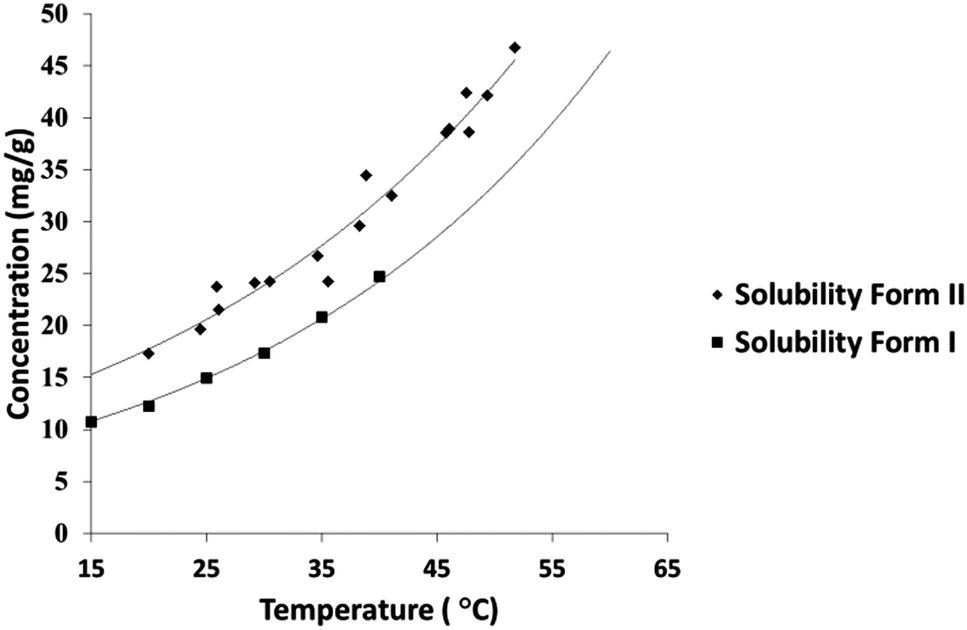 | ||
| Fig. 3 Solubility curve showing the aqueous solubility of PCM-II, compared with that of PCM-I. | ||
In summary, a multicomponent templating approach to paracetamol form II has been established in a cooling crystallisation environment using two templating molecules. Form II is produced reliably and in 100% polymorphic purity, with a 0.4–4% and 10–40% w/w co-former to PCM concentration for 4-fluorobenzoic acid and metacetamol respectively required to template this conversion. Metacetamol displays a higher solubility in the chosen solvent system than 4-fluorobenzoic acid and so higher concentrations were able to be used, aiding in the scale-up of the system to 800 ml volume, producing >100 g PCM-II in a single run. The crystallisation experiment was designed to ensure the templating co-former remains in solution thus reducing the need for further separation processes. The use of cooling crystallisation provides improved control of supersaturation within the system and is the first key step in transfer of the process to continuous crystallisation platforms. Although we have been able to design this templated crystallisation, the mechanism of this templating process is unknown; however it can be suggested that the templating co-former affects the solution-mediated phase transition that normally occurs during the cooling of PCM in the absence of a co-former molecule. This initial transfer into a cooling crystallisation environment provides a key start point for the transfer of this system into continuous platforms at industrial scale.
This work was supported by the EPSRC CMAC under grants EP/I033459/1 and EP/K503289/1.
Notes and references
- Y. Huang, B. Zhang, Y. Gao, J. Zhang and L. Shi, J. Pharm. Sci., 2014, 103, 2330 CrossRef CAS PubMed.
- J.-X. Song, Y. Yan, J. Yao, J.-M. Chen and T.-B. Lu, Cryst. Growth Des., 2014, 14, 3069 CAS.
- R. Thakuria, A. Delori, W. Jones, M. P. Lipert, L. Roy and N. Rodriguez-Hornedo, Int. J. Pharm., 2013, 453, 101 CrossRef CAS PubMed.
- D. P. Elder, R. Holm and H. L. de Diego, Int. J. Pharm., 2013, 453, 88 CrossRef CAS PubMed.
- N. Shan, M. L. Perry, D. R. Weyna and M. J. Zaworotko, Expert Opin. Drug Metab. Toxicol., 2014, 10, 1255 CrossRef CAS PubMed.
- J. Li, S. A. Bourne and M. R. Caira, Chem. Commun., 2011, 47, 1530 RSC.
- P. Sanphui, N. R. Goud, U. B. R. Khandavilli, S. Bhanoth and A. Nangia, Chem. Commun., 2011, 47, 5013 RSC.
- M. Karanam, S. Dev and A. R. Choudhury, Cryst. Growth Des., 2012, 12, 240 CAS.
- G. Nichols and C. S. Frampton, J. Pharm. Sci., 1998, 87, 684–693 CrossRef CAS PubMed.
- M.-A. Perrin, M. A. Neumann, H. Elmaleh and L. Zaske, Chem. Commun., 2009, 3181 RSC.
- G. G. Z. Zhang, D. Law, E. A. Schmitt and Y. Qiu, Adv. Drug Delivery Rev., 2004, 56, 371 CrossRef CAS PubMed.
- E. Joiris, P. Di Martino, C. Berneron, A. M. Guyot-Hermann and J. C. Guyot, Pharm. Res., 1998, 15, 1122 CrossRef CAS.
- H. L. Lee, H. Y. Lin and T. Lee, Org. Process Res. Dev., 2014, 18, 539 CrossRef CAS.
- C. Sudha and K. Srinivasan, CrystEngComm, 2013, 15, 1914–1921 RSC.
- J. C. Burley, M. J. Duer, R. S. Stein and R. M. Vrcelj, Eur. J. Pharm. Sci., 2007, 31, 271 CrossRef CAS PubMed.
- C. Sudha, R. Nandhini and K. Srinivasan, Cryst. Growth Des., 2014, 14, 705 CAS.
- K. Chadwick, A. Myerson and B. Trout, CrystEngComm, 2011, 13, 6625 RSC.
- L. H. Thomas, C. Wales, L. H. Zhao and C. C. Wilson, Cryst. Growth Des., 2011, 11, 1450 CAS.
- A. Saleemi, I. I. Onyemelukwe and Z. Nagy, Front. Chem. Sci. Eng., 2013, 7, 79 CrossRef CAS.
- C. Thompson, M. C. Davies, C. J. Roberts, S. J. B. Tendler and M. J. Wilkinson, Int. J. Pharm., 2004, 280, 137 CrossRef CAS PubMed.
- T. McGlone, N. E. B. Briggs, C. A. Clark, C. J. Brown, J. Sefcik and A. J. Florence, Org. Process Res. Dev., 2015, 19, 1186 CrossRef CAS.
- J. Garside and M. B. Shah, Ind. Eng. Chem. Process Des. Dev., 1980, 19, 509 CAS.
- I.-C. Wang, M.-J. Lee, D.-Y. Seo, H.-E. Lee, Y. Choi, W.-S. Kim, C.-S. Kim, M.-Y. Jeong and G. J. Choi, AAPS PharmSciTech, 2011, 12, 764 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Solubility curves, PXRD and DSC patterns. See DOI: 10.1039/c6cc01032f |
| This journal is © The Royal Society of Chemistry 2016 |