
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCapsoplexes: encapsulating complexes via guest recognition†
Philipp J.
Altmann
and
Alexander
Pöthig
*
Catalysis Research Center & Department of Chemistry, Technische Universität München, Ernst-Otto-Fischer-Str. 1, 85747 Garching bei München, Germany. E-mail: alexander.poethig@tum.de
First published on 23rd February 2016
Abstract
A new dinuclear Ni–NHC complex is able to selectively recognise and self-assemble with guests via tennis-ball like encapsulation, exemplarily demonstrated employing halides. Addition of chloride or bromide leads to the formation of capsules with small cavities which are stabilised through non-classical hydrogen bonds and Coulomb interactions between the anion and the metal centres.
In the last two decades, sophisticated supramolecular concepts have been developed, which complement the classical tailoring of steric and electronic ligand-properties as a well-established principle in homogeneous catalysis.1 One pursued strategy is the special design of polydentate ligands which adopt a defined configuration upon complexation of multiple metal centres as is the case for the pacman complexes2 A (Scheme 1) as well as for the azacryptand complexes.3 Depending on the metal–metal distance, the properties of the coordination pockets as well as the flexibility of the hinge between the latter, these complexes can be optimised for a variety of applications, including reactions relevant for energy-conversion such as the O2-2a,4 or CO2-reduction.5 Cationic metallocages B, as reported by Fujita6 and Raymond & Bergman7 create defined cavities in a different way, namely using a coordination driven self-assembly of polydentate ligands and capped metal nodes. The resulting discrete polyhedra can be employed for a plethora of applications, e.g. acting as molecular flasks1a or sensors.8 The capsoplexes C presented in this work combine the advantages of a specially designed ligand system as in the case of A with the ability to create molecular capsules by self-assembly, in which a small guest is incorporated. In principle, similar tennis-ball like arrangements have been reported earlier,9 however, they do not allow for the encapsulation of small anions as presented in this work, since either larger organic molecules9a or cations9b were encapsulated or the formed cavity is too large.9c
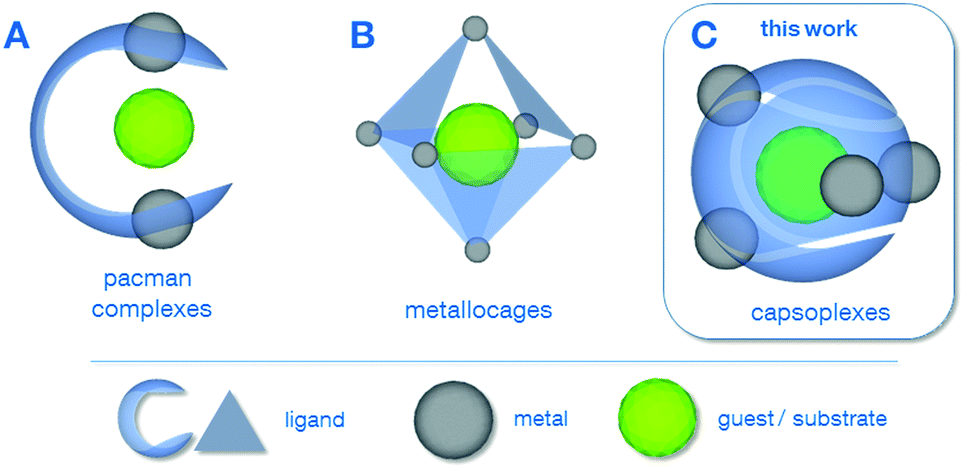 | ||
| Scheme 1 Different examples for supramolecular strategies employing molecular complexes: (A) pacman complexes2a–g (including azacryptates3), (B) metallocages6,7 and (C) capsoplexes (presented in this work). | ||
We recently introduced the family of calix[4]imidazolium[2]pyrazole cyclophanes, which can act as hydrogen bond receptors as well as ligand precursors for saddle-shaped dinuclear transition metal NHC-complexes,10 corresponding to category A. Herein we report the synthesis of the ethylene-bridged member of this compound class and its dinuclear nickel(II) complex. The putative small change of the ligand's ring size leads to crucial structural changes of the complex, which open the door to an exciting encapsulation chemistry exemplarily demonstrated by the selective recognition of halide anions.
We first synthesized the ethylene-bridged calix[4]imidazolium[2]pyrazole macrocycle as a ligand precursor (H6LEt(PF6)2) by variation of our recently reported protocol10 employing ethanediyl bistriflate as the cyclisation reagent (Scheme 2, top). Interestingly, as it was the case for the methylene-bridged congener, the poly-imidazolium salt incorporates an acetonitrile molecule as a solvent of crystallisation which was confirmed by NMR, elemental analysis and SC-XRD studies (see ESI†). Subsequent deprotonation of the azoles with caesium carbonate in the presence of anhydrous nickel(II) acetate leads to the formation of the corresponding NHC complex Ni2LEt(PF6)2 (Scheme 2, bottom). The molecular structure of Ni2LEt was determined by single-crystal X-ray diffraction and is shown in Fig. 1.
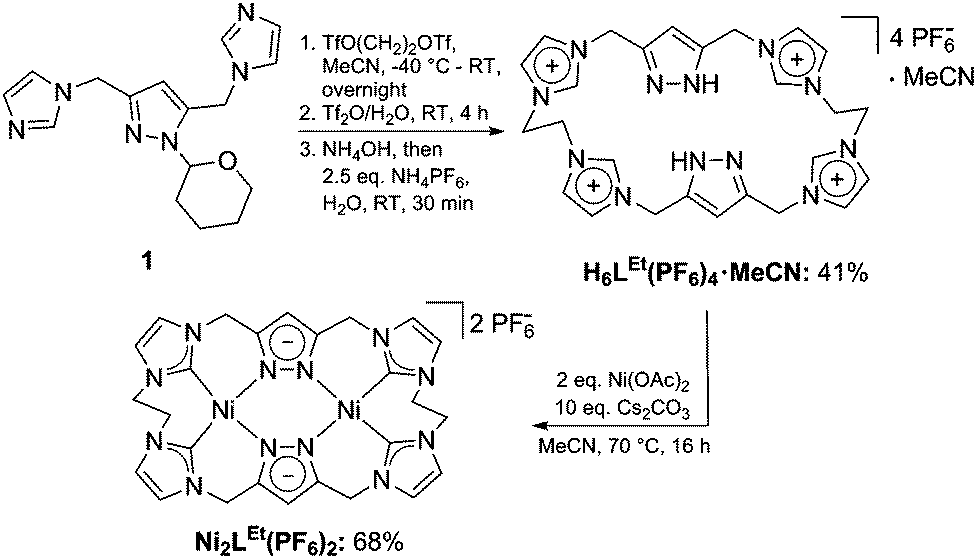 | ||
| Scheme 2 Synthesis of ligand precursor H6LEt(PF6)4 and dinuclear NHC-complex Ni2LEt(PF6)2. | ||
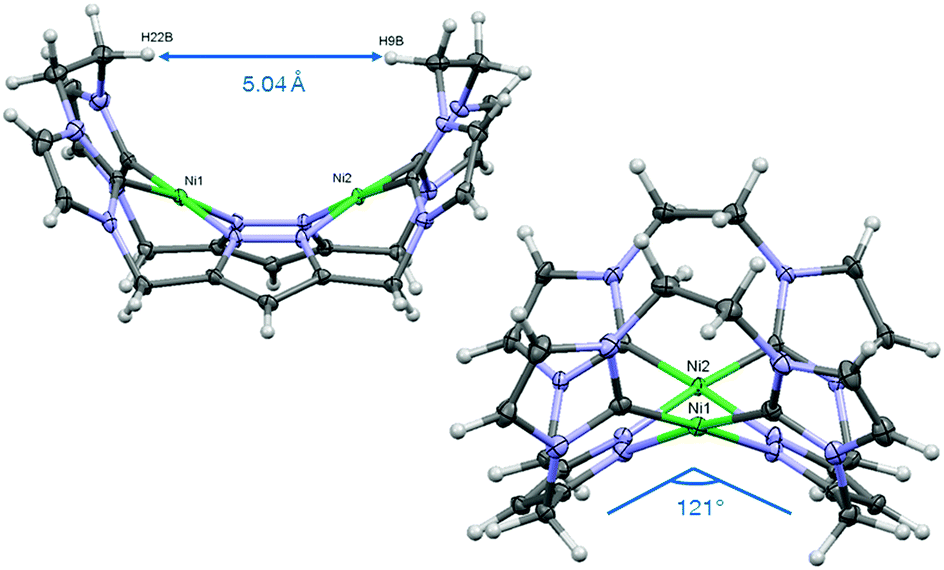 | ||
| Fig. 1 Molecular structure of the cation of Ni2LEt(PF6)2 in two perspectives showing the saddle shape. Ellipsoids are shown at 50% probability. Counter ions, solvent molecules and disorders are omitted for clarity. Selected geometrical parameters: Ni1–Ni2 3.6453(6) Å, C19–Ni1 1.8570(2) Å, C24–Ni1 1.9047(2) Å, N1–Ni1 1.9060(2) Å, N8–Ni1 1.9004(2) Å, C19–Ni1–C24 92.456(7)°, N1–Ni1–C24 90.066(9)°, C19–N8–N1–C24 3.092(7)°. | ||
Similarly to the C1-bridged complex Ni2LMe(PF6)2,10 the dinuclear complex adopts a saddle-shaped structure, with the two nickel atoms in a slightly distorted square planar coordination environment. However, with increasing length of the alkylene-bridge, the curvature of the saddle rises. As a consequence the Ni–Ni distance amounts to 3.645(2) Å and therefore is significantly longer than that observed in the methylene-bridged complex (3.5957(8)/3.6043(8) Å).10 Additionally, the angle between the pyrazolates decreases from 138.10(1)°/138.81(1)° to 121.13(1)°.11 The cationic complex molecule Ni2LEt exhibits C2-symmetry in the solid state due to the conformation of the ethylene groups. Hereby, for each bridge one methylene group is rotated towards (endo) and one away (exo) from the central Ni–Ni moiety – namely the opposing pairs with regard to the equatorial plane through the Ni atoms which intersects the complex in two bis-NHC-pyrazolato hemispheres. One hydrogen atom of each of the endo CH2-groups points towards the inner cavity of the bowl (H22B, H9B). The distance between these two hydrogen atoms amounts to 5.035(1) Å, such that the cavity should be of ideal size to host an anion or even a small molecule. Additionally, the presence of such hydrogen atoms might be advantageous for the pre-alignment of guests/reactants via hydrogen bonding. To prove the capability of Ni2LEt(PF6)2 to act as a host, we conducted a series of 1H NMR titration experiments. To a solution of Ni2LEt(PF6)2 in CD3CN we successively added 0.5 equivalents of the corresponding tetrabutylammonium halide salts, also as a stock solution in CD3CN. In case of fluoride and iodide, no change of the proton signals was observed, whereas for chloride significant shifts occurred when increasing the anion-to-host ratio (Fig. 2). A dinuclear pacman-Zn-complex was shown to selectively recognise halides by Love,12 such that we expected a similar mode of anion recognition with our system. Upon addition of chloride ions, the backbone signals of the imidazole moieties at 7.23 and 7.11 ppm significantly shift to 7.08 and 6.37 ppm, whereas the singlet signal of the pyrazolate essentially remains at an identical position (6.27 to 6.23 ppm). The doublet for the methylene resonance at 5.50 ppm broadens until 0.3 equivalents chloride are added and then increasingly sharpens again to a doublet at 5.40 ppm when 0.5 equivalents of the halide are present. In contrast, the second methylene resonance at 5.10 ppm is not affected at all. The most apparent effect can be observed for the proton signals of the ethylene bridge. The multiplets at 5.20 and 4.46 ppm drastically broaden and shift to higher field (4.31 and 2.81 ppm). Apparently the addition of the chloride has the strongest effect on these protons, which indicates that a resulting structural change should affect the alkylene bridges most. Addition of more than 0.5 equivalents of chloride does not lead to a change of the signal pattern (see ESI†) indicating that no further uptake of halide anions takes place. As in the case of chloride we observed significant shifts when NMR titrating a stock solution of NBu4Br to Ni2LEt(PF6)2 (see ESI†). Hereby, the resonance at 5.50 ppm does not sharpen again and the signals for the ethylene bridge completely disappear upon addition of 0.5 equivalents of bromide. Interestingly, the addition of 0.5 equivalents of AgPF6 reverts the changes in the 1H NMR spectra which occurred during the titration, strongly indicating that the halides are removed and free Ni2LEt(PF6)2 is recovered (Scheme 3).
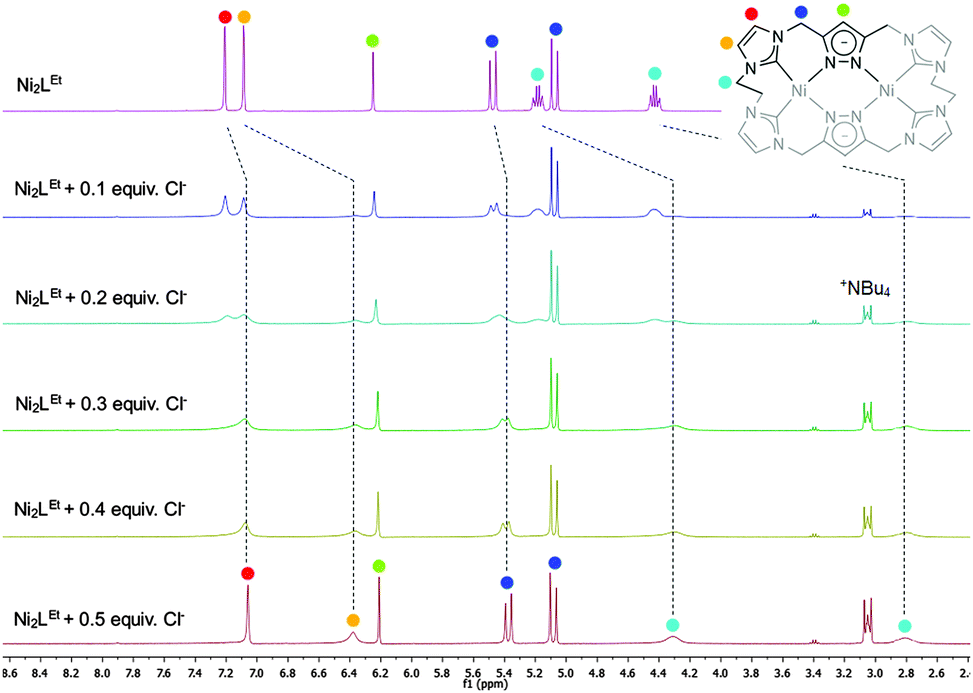 | ||
| Fig. 2 Titration experiments monitored by 1H NMR spectroscopy showing the signal changes upon stepwise addition of 0.5 equivalents NBu4Cl to complex Ni2LEt(PF6)2 in CD3CN. The dashed lines show the chemical shifts of the characteristic signals (marked with coloured dots) which were assigned by NOESY NMR experiments (see ESI†). | ||
 | ||
| Scheme 3 Top: Titration of tetrabutylammonium halides to Ni2LEt(PF6)2 leading to capsoplexes [(Ni2LEt)2X](PF6)3 (X = Cl, Br) and reverse reaction via precipitation of silver halides. Bottom: Direct synthesis of capsoplex [(Ni2LEt)2Cl](PF6)3via reaction of NiCl2(PPh3)2 and H6LEt(PF6)4 under basic conditions. | ||
As a matter of fact, the reaction between the ligand precursor and NiCl2(PPh3)2 yields the same product as the titration with NBu4Cl according to the resulting 1H NMR spectrum. The elemental analysis of the isolated product corresponds to a composition of two Ni2LEt cations, one chloride, and three PF6 anions. To gain further insight into the nature of the formed species, we conducted DOSY NMR experiments in CD3CN for complex Ni2LEt(PF6)2 as well as the products of the titration experiments (see ESI†). For Ni2LEt(PF6)2 a diffusion coefficient of 1.0 × 10−9 m2 s−1 and the respective solvodynamic radius 5.8 Å were determined. In contrast, diffusion coefficients of 7.3 × 10−10 m2 s−1 (Cl−) and 7.5 × 10−10 m2 s−1 (Br−) corresponding to significant larger radii of 8.0 Å (Cl−) and 7.8 Å (Br−) were observed for the titration products, indicating the formation of host–guest complexes that are significantly larger than Ni2LEt. Interestingly, the Ni2LMe congener previously reported by us10 does not show a comparable affinity towards halide anions as the addition of chloride does not lead to significant changes in the 1H NMR spectrum (see ESI†). We were able to crystallise these host–guest complexes by diffusion of diethyl ether into a dimethyl formamide solution (Cl−) or an acetonitrile solution (Br−) and determine their molecular structure by means of SC-XRD experiments (Fig. 3). The compounds crystallise in the space group C2/c and the crystal structures prove the formation of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest capsules. Both asymmetric units consist of one Ni2LEt molecule, 0.5 halide, 1.5 PF6 anions and two co-crystallised solvent molecules each. In both cases, the halide anion is located on the C2 axis encapsulated in a tennis-ball like fashion by two symmetry-equivalent cationic Ni2LEt complexes. In principle such an arrangement was observed earlier for pacman Mg–hydroxide cubanes,9c however the formed capsule is significantly larger in size compared to the capsoplex formed by Ni2LEt. The nickel centres form a distorted tetrahedron around the anion, exhibiting Ni–Cl distances of 2.9116(4)/2.9173(3) Å and Ni–Br distances of 2.9179(3)/2.9354(3) Å, respectively. These values are significantly higher than the sum of the respective ionic radii,13 indicating an exclusive attractive Coulomb interaction between metals and halides. This is in agreement with the solution studies: the nickel centres possess a square planar coordination geometry and are diamagnetic, that is, in a strong field environment. In contrast to the solid state structure of the cation in the crystal structure of Ni2LEt(PF6)2, each saddle-shaped Ni2LEt complex exhibits Cs-symmetry. The endo-CH2 groups now are located at the same bis-NHC-pyrazolato hemisphere of each dinuclear complex moiety at a C–C distance of 7.010(5) Å in case of the encapsulated chloride and 7.096(7) Å for the bromide. Apparently the dinuclear complex bowl has the ability to slightly adjust to the guest by opening the cavity. This is also reflected in the Ni–Ni distances (3.5790(4) (Cl−)/3.5985(3) Å (Br−)) and the angle between the planes of the coordinating atoms of each binding pocket which is 55.40° in case of Cl− and 56.02° in case of the larger Br−. As it was the case for Ni2LEt(PF6)2, one hydrogen atom of each endo-CH2 group is pointing towards the central cavity, thus stabilising the capsule via non-conventional H-bonding to the anion at a distance of 2.6163(3)/2.6313(3) Å [C⋯Cl 3.5437(4)/3.5839(4) Å] for chloride and 2.6556(2)/2.6948(2) Å [C⋯Br 3.5651(3)/3.6410(3) Å] for bromide (Fig. 3, right). VT NMR experiments in solution showed, that the ethylene bridge is flexible at room temperature in both cases since only one signal set is present in the respective spectra. However, for the bromide capsule one conformation can be trapped at low temperatures confirmed by the presence of a second signal set below 20 °C (see ESI†). This implies, that in the case of the cationic (Ni2LEt)2Br the activation barrier for the conformational change of the ethylene bridge is higher than in the case of (Ni2LEt)2Cl. To evaluate the relative thermodynamic stability of the capsules, we additionally performed competitive titration experiments. Similarly to the earlier titrations, we added up to 1 equivalent of tetrabutylammonium halide to the respective opposing halide containing capsules [(Ni2LEt)2X](PF6)3 (see ESI†). Hereby, the addition of bromide to a solution of the chloride capsule did not result in any change in the spectra. In contrast, the sequential addition of 1 equivalent of chloride to a solution of the bromide capsule eventually resulted in a complete transformation into the chloride capsule, which apparently is the thermodynamically favoured species.
:1 host–guest capsules. Both asymmetric units consist of one Ni2LEt molecule, 0.5 halide, 1.5 PF6 anions and two co-crystallised solvent molecules each. In both cases, the halide anion is located on the C2 axis encapsulated in a tennis-ball like fashion by two symmetry-equivalent cationic Ni2LEt complexes. In principle such an arrangement was observed earlier for pacman Mg–hydroxide cubanes,9c however the formed capsule is significantly larger in size compared to the capsoplex formed by Ni2LEt. The nickel centres form a distorted tetrahedron around the anion, exhibiting Ni–Cl distances of 2.9116(4)/2.9173(3) Å and Ni–Br distances of 2.9179(3)/2.9354(3) Å, respectively. These values are significantly higher than the sum of the respective ionic radii,13 indicating an exclusive attractive Coulomb interaction between metals and halides. This is in agreement with the solution studies: the nickel centres possess a square planar coordination geometry and are diamagnetic, that is, in a strong field environment. In contrast to the solid state structure of the cation in the crystal structure of Ni2LEt(PF6)2, each saddle-shaped Ni2LEt complex exhibits Cs-symmetry. The endo-CH2 groups now are located at the same bis-NHC-pyrazolato hemisphere of each dinuclear complex moiety at a C–C distance of 7.010(5) Å in case of the encapsulated chloride and 7.096(7) Å for the bromide. Apparently the dinuclear complex bowl has the ability to slightly adjust to the guest by opening the cavity. This is also reflected in the Ni–Ni distances (3.5790(4) (Cl−)/3.5985(3) Å (Br−)) and the angle between the planes of the coordinating atoms of each binding pocket which is 55.40° in case of Cl− and 56.02° in case of the larger Br−. As it was the case for Ni2LEt(PF6)2, one hydrogen atom of each endo-CH2 group is pointing towards the central cavity, thus stabilising the capsule via non-conventional H-bonding to the anion at a distance of 2.6163(3)/2.6313(3) Å [C⋯Cl 3.5437(4)/3.5839(4) Å] for chloride and 2.6556(2)/2.6948(2) Å [C⋯Br 3.5651(3)/3.6410(3) Å] for bromide (Fig. 3, right). VT NMR experiments in solution showed, that the ethylene bridge is flexible at room temperature in both cases since only one signal set is present in the respective spectra. However, for the bromide capsule one conformation can be trapped at low temperatures confirmed by the presence of a second signal set below 20 °C (see ESI†). This implies, that in the case of the cationic (Ni2LEt)2Br the activation barrier for the conformational change of the ethylene bridge is higher than in the case of (Ni2LEt)2Cl. To evaluate the relative thermodynamic stability of the capsules, we additionally performed competitive titration experiments. Similarly to the earlier titrations, we added up to 1 equivalent of tetrabutylammonium halide to the respective opposing halide containing capsules [(Ni2LEt)2X](PF6)3 (see ESI†). Hereby, the addition of bromide to a solution of the chloride capsule did not result in any change in the spectra. In contrast, the sequential addition of 1 equivalent of chloride to a solution of the bromide capsule eventually resulted in a complete transformation into the chloride capsule, which apparently is the thermodynamically favoured species.
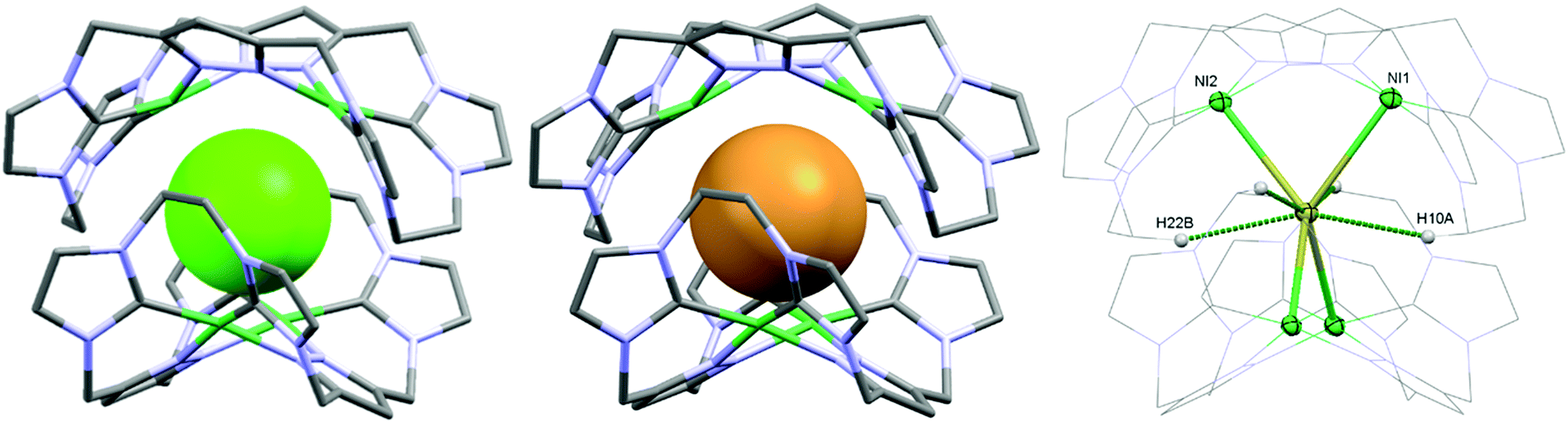 | ||
| Fig. 3 Molecular structures of capsoplexes [(Ni2LEt)2Cl](PF6)3 (left) and [(Ni2LEt)2Br](PF6)3 (middle). Non-encapsulated counter anions and solvent molecules are omitted for clarity. The dinuclear complexes are shown as capped sticks, the encapsulated anions in the space filling model, emphasising the different demand in volume of the anions. Right: Coordination environment around the halides (Cl−/Br−) including hydrogen bonding to the ethylene bridges. Selected geometrical parameters: Ni1–Ni2 3.5790(4) Å, Ni1–Cl 2.9217(3) Å, Ni2–Cl 2.9116(4) Å, Ni1–Cl–Ni2 75.694(4)°; Ni1–Ni2 3.5985(3) Å, Ni1–Br 2.9354(3) Å, Ni2–Br 2.9179(3) Å, Ni1–Br–Ni2 75.873(3)°. | ||
In summary, we successfully synthesized a new ethylene-bridged calix[4]imidazolium[2]pyrazole cyclophane as a ligand precursor and its corresponding dinuclear Ni(II) complex. NMR titration studies showed, that addition of certain halides leads to the formation of capsules with cavities of defined size, in which only chloride and bromide can be incorporated. The solid state structures suggest a stabilisation of the capsules through a combination of multiple hydrogen bonding and attractive Coulomb interactions between the guest and the metal centres. Competitive titration experiments revealed that the chloride containing capsule is thermodynamically more stable than its bromide containing congener. In contrast to most of the known metallocavitand systems, the formation of the capsules is not coordination driven and the guest plays an active role in the formation of the assembly. The unique arrangement of the metal centres and the hydrogen bond donors pointing into the cavity in principle allows for the encapsulation of small guests different than halides, e.g. dioxygen. This renders the presented capsules highly promising systems for the activation of small molecules via substrate recognition, encapsulation, catalytic transformation, and subsequent release of the product. Therefore, our future work aims on extending the scope of encapsulation to other anions and small molecules, employing different complexed metals and to further explore their reactivity.
The CRC, the TUM Faculty of Chemistry as well as the Fonds der Chemischen Industrie (FCI, Sachkostenzuschuss A. P.) are very much acknowledged for funding this work. P. J. A. thanks the TUM Graduate School for financial support. We thank Prof. W. A. Herrmann and Prof. F. E. Kühn for their continuous support and Johannes Richers for the help with the graphical design.
Notes and references
- Reviews: (a) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC; (b) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC; (c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- (a) A. M. Devoille and J. B. Love, Dalton Trans., 2012, 41, 65–72 RSC; (b) J. W. Leeland, A. M. Z. Slawin and J. B. Love, Organometallics, 2010, 29, 714–716 CrossRef CAS; (c) G. Givaja, M. Volpe, J. W. Leeland, M. A. Edwards, T. K. Young, S. B. Darby, S. D. Reid, A. J. Blake, C. Wilson, J. Wolowska, E. J. L. McInnes, M. Schröder and J. B. Love, Chem. – Eur. J., 2007, 13, 3707–3723 CrossRef CAS PubMed; (d) G. Givaja, A. J. Blake, C. Wilson, M. Schröder and J. B. Love, Chem. Commun., 2003, 2508–2509 RSC; (e) C. H. Lee, D. Villagran, T. R. Cook, J. C. Peters and D. G. Nocera, ChemSusChem, 2013, 6, 1541–1544 CrossRef CAS PubMed; (f) M. Schwalbe, R. Metzinger, T. S. Teets and D. G. Nocera, Chem. – Eur. J., 2012, 18, 15449–15458 CrossRef CAS PubMed; (g) Y. Deng, C. J. Chang and D. G. Nocera, J. Am. Chem. Soc., 2000, 122, 410–411 CrossRef CAS; (h) T. A. Betley, Q. Wu, T. Van Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849–1861 CrossRef CAS PubMed; (i) J. Rosenthal and D. G. Nocera, Acc. Chem. Res., 2007, 40, 543–553 CrossRef CAS PubMed.
- (a) J.-M. Lehn, S. H. Pine, E.-I. Watanabe and A. K. Willard, J. Am. Chem. Soc., 1977, 99, 6766–6768 CrossRef CAS; (b) J. Nelson, V. McKee and G. Morgan, Progress in Inorganic Chemistry, John Wiley & Sons, Inc., Hoboken, New Jersey, 2007, vol. 47, pp. 167–316 Search PubMed; (c) F. Möller, K. Merz, C. Herrmann and U. P. Apfel, Dalton Trans., 2016, 45, 904–907 RSC.
- (a) E. Askarizadeh, S. B. Yaghoob, D. M. Boghaei, A. M. Z. Slawin and J. B. Love, Chem. Commun., 2010, 46, 710–712 RSC; (b) M. Volpe, H. Hartnett, J. W. Leeland, K. Wills, M. Ogunshun, B. J. Duncombe, C. Wilson, A. J. Blake, J. McMaster and J. B. Love, Inorg. Chem., 2009, 48, 5195–5207 CrossRef CAS PubMed; (c) G. Givaja, M. Volpe, M. A. Edwards, A. J. Blake, C. Wilson, M. Schröder and J. B. Love, Angew. Chem., Int. Ed., 2007, 46, 584–586 CrossRef CAS PubMed.
- Reviews: (a) I. Siewert, Chem. – Eur. J., 2015, 21, 15078–15091 CrossRef CAS PubMed; (b) C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392–1399 RSC.
- D. K. Chand, K. Biradha and M. Fujita, Chem. Commun., 2001, 1652–1653 RSC.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 351–360 CrossRef PubMed.
- L. Xu, Y. X. Wang and H. B. Yang, Dalton Trans., 2015, 44, 867–890 RSC.
- (a) R. S. Meissner, J. Rebek and J. de Mendoza, Science, 1995, 270, 1485–1488 CAS; (b) C. O. Dietrich-Buchecker, P. A. Marnot, J. P. Sauvage, J. P. Kintzinger and P. Maltese, Multilingue, 1984, 8, 573–582 CAS; (c) J. W. Leeland, F. J. White and J. B. Love, J. Am. Chem. Soc., 2011, 133, 7320–7323 CrossRef CAS PubMed.
- P. J. Altmann, C. Jandl and A. Pöthig, Dalton Trans., 2015, 44, 11278–11281 RSC.
- Comment: The angle was calculated between the pyrazole-CH-carbon atoms and a central centroid placed between the Ni-centres.
- A. M. J. Devoille, P. Richardson, N. L. Bill, J. L. Sesser and J. B. Love, Inorg. Chem., 2011, 50, 3116–3126 CrossRef CAS PubMed.
- A. F. Hollemann, E. Wiberg and N. Wiberg, in Lehrbuch der Anorganischen Chemie, ed. W. de Gruyter, Berlin, 102nd edn, 2007, pp. 2002–2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1448008–1448011. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00507a |
| This journal is © The Royal Society of Chemistry 2016 |