
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
X-ray structure of a lectin-bound DNA duplex containing an unnatural phenanthrenyl pair†
P.
Roethlisberger
a,
A.
Istrate
a,
M. J.
Marcaida Lopez
b,
R.
Visini
a,
A.
Stocker
a,
J.-L.
Reymond
a and
C. J.
Leumann
 *a
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: christian.leumann@dcb.unibe.ch
bEcole Polytechnique Fédérale Lausanne, Station 19, 1015 Lausanne, Switzerland
First published on 17th February 2016
Abstract
DNA duplexes containing unnatural base-pair surrogates are attractive biomolecular nanomaterials with potentially beneficial photophysical or electronic properties. Herein we report the first X-ray structure of a duplex containing a phen-pair in the center of the double helix in a zipper like stacking arrangement.
Besides its fundamental biological function, DNA has attracted considerable interest as a foldamer due to its well-ordered supramolecular association properties. In order to understand the self-association principles that guide DNA inspired polynucleotides into helical structures in more detail, a considerable number of artificial oligonucleotides have been prepared that are of interest in fundamental or applied material science.1,2 In particular DNA-like polymers in which DNA bases have been replaced by aromatic designer units, such as pyrenes, were recently found to have interesting photophysical properties if ordered in one or two dimensional arrays.3–6 In previous work we discovered that multiple replacements of natural bases by bipyridyl, biphenyl or phenanthrenyl residues, devoid of the capability of specific self-recognition, can smoothly be accommodated in the center of a DNA double-helix without compromising duplex stability.7–10 We hypothesized that interstrand intercalation of such aromatic units is responsible for the cohesion of the two strands. If tightly stacked, such aromatic units may improve and widen the scope of functionalities such as electron transfer. Consequently we demonstrated that excess electron transport through 1–3 phenanthrenyl pairs occurs efficiently.11,12
Of crucial importance to understand the functional properties in detail is the knowledge of the 3-D structure of such constructs. However, X-ray structures of oligonucleotide duplexes containing non-hydrogen bonding base surrogates are difficult to obtain due to their reluctance to crystallize, or to low scattering or phasing problems. A common strategy to overcome the phasing problem involves the incorporation of 5-halouracils, taking advantage of the anomalous signal. However, this approach potentially suffers from low signal intensity because of an unknown orientation of the brominated DNA within the crystal.13
An alternative is to co-crystallize DNA with a protein, the structure of which is known, and use molecular replacement techniques for structure solution. For co-crystallizing DNA with proteins, a non-covalent interaction between the two polymers is required. Non-functional enzymes or subunits of the transcription or replication machinery are known to be suitable for this purpose, since they have an intrinsic affinity to DNA and are structurally well described.14 Unfortunately the general need of a defined recognition sequence is limiting the application of this approach. Another way to facilitate crystallization of DNA invokes appending an affinity label to the desired DNA and co-crystallize it with its corresponding protein target. It was recently shown by Reymond et al. that the microbial lectin LecB, originally crystallized by Imberty et al.15 can serve as a template to solve the structure of intermediate size macromolecules that are difficult to crystallize, at the example of the first crystal structure of a second generation fucosylated peptide dendrimer.16 This suggested to us that the structure of short, fucosylated DNA duplexes might be similarly accessible as LecB complexes. A screen of the Nucleic Acid Structure Data Base (NDB) revealed that such an approach was not considered for DNA structures before.
A DNA 13-mer duplex incorporating a phenanthrenyl (phen) pair in the center of the helix and carrying an L-(−)-fucose analogue (FUC, for synthetic details see ESI† Scheme S1) was prepared (Fig. 1a). This construct was designed to bind to the fucose specific Pseudomonas aeruginosa lectin LecB (also called PA-IIL). The corresponding oligonucleotides were synthesized by standard phosphoramidite chemistry and purified by ion exchange HPLC. The duplex revealed an overall B-DNA conformation according to CD spectroscopy and showed an equal Tm compared to a natural duplex in which the phen-pair has been replaced by an A–T pair (see Table S2, ESI†). Crystals of the DNA–protein complex were obtained by mixing the duplex in two-fold excess with the LecB monomer, using the sitting drop technique. After three days at 18 °C, crystals were collected and stored in liquid nitrogen. The crystals were then analysed at the Paul Scherer Institute (PSI). A high-resolution data set was collected at 1.0 Å beam wavelength, integrated and scaled with XDS.17
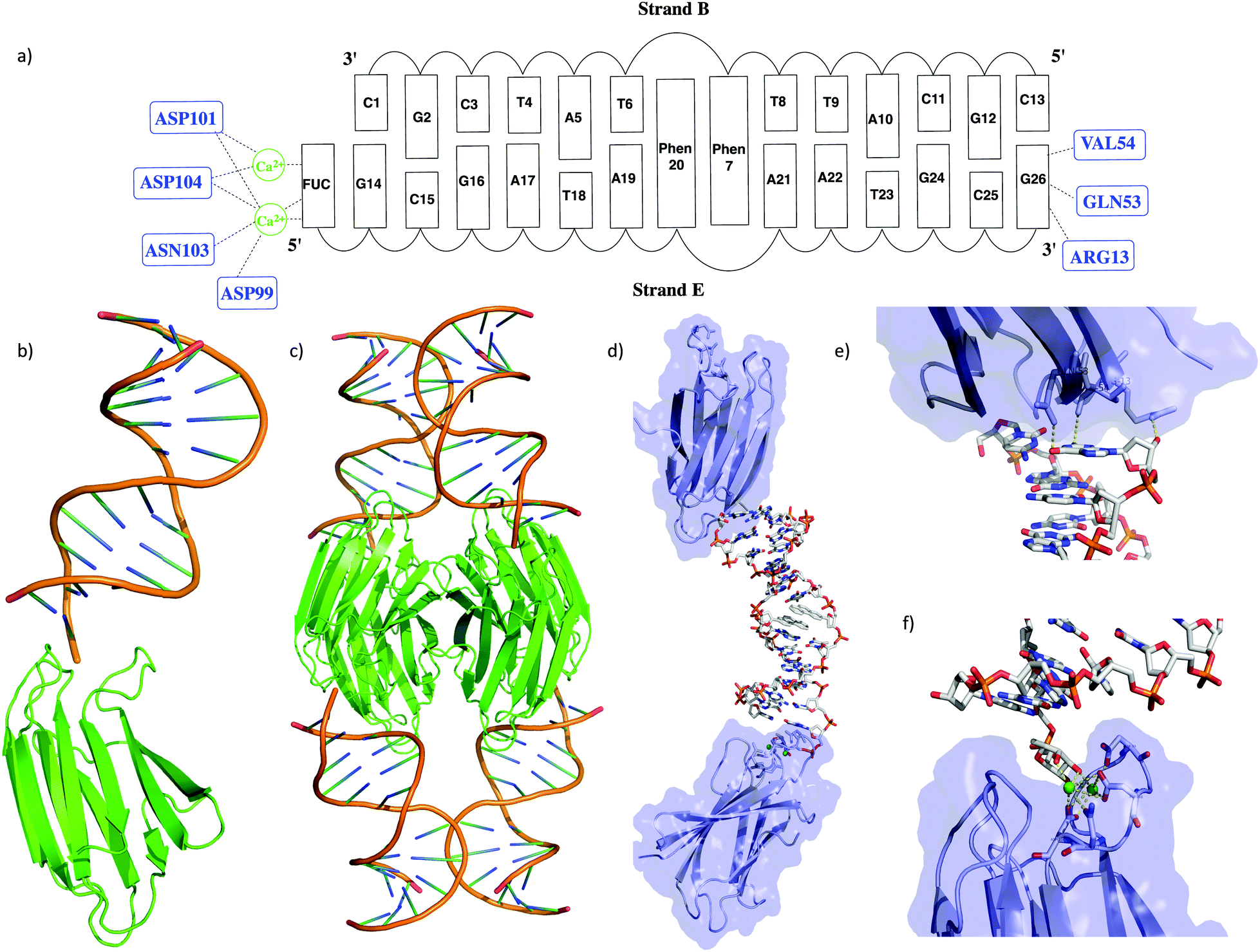 | ||
| Fig. 1 (a) Schematic representation of the interaction of the DNA duplex with the LecB; (b) representation of the monomeric unit consisting of the DNA duplex bound to the LecB via the specific FUC binding domain; (c) the native present tetrameric complex of the LecB; (d) visualization of the two distinct interactions of the DNA with the LecB monomeric unit; (e) non specific interactions of the FUC unit of the DNA with the carbohydrate binding site of the LecB; (f) binding site of the LecB with the fucose analog linked to the DNA. | ||
The structure was solved by molecular replacement with the high-resolution structure 1OXC, a LecB tetramer, as the starting model. Based on the superimposed Fourier sum (2Fo − Fc) and the difference (Fo − Fc) electron density, the DNA scaffold was built progressively and stepwise into the electron density space. Rigid-body, B-factors, occupancies and positional refinements were performed during this iterative process in order to optimize the electron density at each step. Also, the restrained anisotropic temperature factors were corrected. After parameterization, the restraints were optimized with respect to the model input and the data obtained experimentally (X-ray/stereochemistry weight and X-ray/ADP weight) and lastly the bulk solvent was updated. The Rfree was monitored, setting aside 10% of the reflections as a test set. In this way the structure of the DNA part could be refined to 2.9 Å resolution (PDB code 5HCH, see also ESI†).
The overall structure of the LecB/DNA complex is depicted in Fig. 1b–d. Each monomeric unit of LecB binds a DNA duplex via the FUC unit appended at the 5′ end of strand E. Moreover, a second contact of the DNA duplex at the 3′ end of strand E with the bottom side of the LecB monomer was observed. Thus an extended lattice is created where the DNA and the protein sheets are alternating (Fig. S7, ESI†). The specific binding of the FUC unit is facilitated by Ca2+ ions bound to ASN103, ASP104, ASP101 and ASP99 in the active site (Fig. 1e and f). The hydrogen bonding distances from the amino acids to the Ca2+ are in the range of 2.4–3.3 Å, while the distances from Ca2+ to the FUC unit only varies from 2.3 to 2.7 Å. The second interaction of the LecB with DNA occurs solely with strand E via the terminal G26, that forms direct hydrogen bonds with LecB (i.e. with residues GLN-53/G-O6, VAL-54/G-N2 and ARG13 G-O3′) within a range of 2.6 to 3.7 Å. Structural parameters of the duplex part were extracted with CURVES18 and x3DNA.19
In general it was found that the two oligonucleotide strands E and B form a B-duplex. The helical axis is bent by 60° at the position of the phenanthrenyl base surrogates (Fig. 2a). This is most probably due to the conformational stress conveyed to the backbone, caused by the intercalating zipper motif. The distance between the planes of the two phen residues is 3.6 Å and they are in a nearly perfect stacked arrangement (Fig. 2c). The top view of the zipper motif shows a parallel displacement of the phen units with that of strand B slightly shifted into the major groove (Fig. 2b). The electron density does only partially cover this phen residue. The phen unit of strand E appears to be within van der Waals (vdW) contact with A1 (distance 3.5 Å). The displaced phen unit in strand E is only in contact with its phen neighbor and shows no stacking with the neighboring natural base pair (vdWd > 4.0 Å). The distance between the natural base pairs surrounding the phen-pair was measured to be 11.1 Å, while only 6.8 Å would be expected if the phen units were replaced by a natural base-pair. This elongation of the DNA backbone is in agreement with other observed intercalation motifs.6,20,21 The average base-pair parameters were close to the averaged values found for B-DNA.22,23 The rise/base-pair amounts to 3.28 Å and a twist of 37.22 Å in the regular B-DNA part of the duplex. Perturbations in natural base pair structures were found in proximity to the phen units and close to the LecB. The most pronounced deviations could be found for the opening of base-pair A21–T8, which is more than 30° higher than average. The shear (1.80 Å, ave 0.09 Å) and stagger (0.9 Å, ave −0.10 Å) also deviate from standard values (Fig. S8, ESI†).
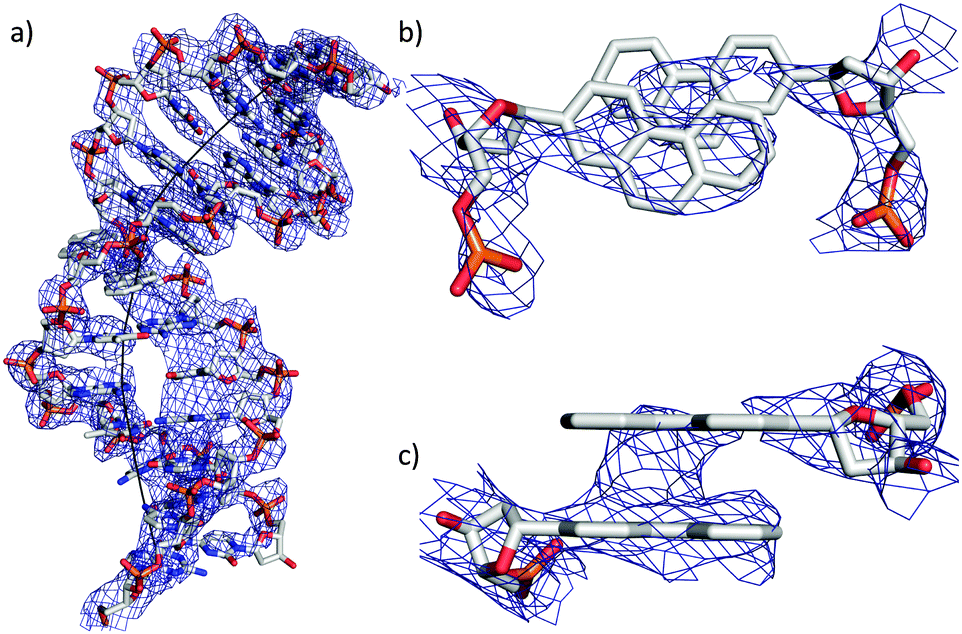 | ||
| Fig. 2 (a) Representation of the DNA duplex with the refined electron density (Fo − Fc) and the bent DNA axis computed with CURVES; (b) top view of the close up of the phenanthrenyl base surrogates; (c) side view of the close-up of the phenanthrenyl base surrogates. | ||
Additional structural information could be found at the level of the sugar-phosphate backbone. Indeed the sugar puckers adopt mainly the classical C2′-endo conformation but show variations to C3′-exo and even 04′-endo. Only two C3′-endo conformations could be found for T8 and C15 (Table S4, ESI†). Analysis of the backbone torsion angles, as expected, showed deviations from standard B-DNA mostly around the phen-pair (Table S3, ESI†). However, the values for δ and ε are largely unaffected by the phen modifications or the proximity to the LecB. Structural changes occur at the α and γ angles of C11 of strand B. At this position the γ angle changes from a +sc to a −sp conformation and the α angle transforms from a +sc to a −sp conformation. Similar features are detected for G24 of strand E, where the α angle changes form a −sc to a +sp conformation and the γ angle in opposite direction from a +sp to a −sc conformation. Intriguingly, the helix elongation induced by the intercalating phen modifications is compensated by the same angles α and γ. However, most of these changes were detected only in stand E which may be the origin for the observed kink in the helical axis. At the position phen20 the γ angle changes form a +sc to a −sc conformation and the α angle transforms form a +sc to a −ac conformation.
We note that similar deviations but to a lesser extent are also observed for the angles β and ζ for the two nearest neighbor A–T base-pairs flanking the phen modifications. At the same time the ζ angles change from the −ap to the −sc and the β angles undergo a transformation from the +ap conformation to the −ac conformation.
Analysis of the phosphate distances and grooves was performed according to El Hassan et al.24 It was found that the intercalated phen units not unexpectedly provoke a significant increase of the phosphate distances (+10.2 Å) between each phen residue and their adjacent natural nucleotides (Fig. S11, ESI†). Measurements of the major and minor groove widths at the phen modifications showed a widening from 21.8 Å (natural B-DNA)25 to 23.4 Å and of the minor groove width from 11.8 Å (natural B-DNA)25 to 13.4 Å (neglecting the vdR radii of 5.8 Å). This indicates that the interstrand-stacking motif widens both grooves significantly (Fig. 3).
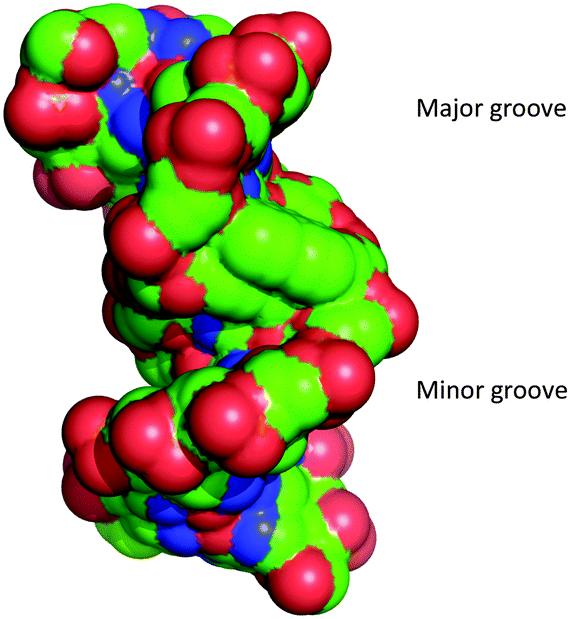 | ||
| Fig. 3 Space filling representation of the duplex showing the major and the minor groove. Phosphorous atoms are coloured in red. | ||
The fact that the phenanthrenyl residue of strand B was not clearly defined by the electron density Fc − Fo, evoked the use of molecular dynamics simulations to get an idea of how the residue could be arranged in an energy minimized structure using the AMBER03 force field26 within the GROMACS package (see ESI†).27 Molecular dynamics simulations on a 1 or 10 ns trajectory with or without position restraints at 300 K showed besides minor variations in backbone geometry and sugar pucker no remarkable changes compared to the crystal structure (see Fig. S12, ESI†). This observation is in agreement with the X-ray results and highlights the structural integrity of the zipper motif.
In summary it was successfully demonstrated that DNA structures can be co-crystallized within the FUC/LecB system and thus deliver important structural information. Furthermore the proposed intercalating zipper motif was confirmed for single phenanthrenyl pairs within a DNA duplex. This structure helps to understand energy and electron transport phenomena through such scaffolds in more detail. Moreover, the fact that such phen modifications induce a kink into the DNA helical axis may be exploited to characterize kink recognizing proteins28 or may be used to test how minor and major groove binding proteins interact with DNA.29
We thank K.-M. Bartels, Institute of Molecular Enzyme Technology, University of Düsseldorf, for providing us with the plasmid for LecB expression and the staff at the Swiss Light Source, Beamline X06DA (PXIII), Villigen, Switzerland, for support during data collection. Financial support from the Swiss National Science Foundation (grant-no. 200020_146646) is gratefully acknowledged.
Notes and references
- A. Chworos and L. Jaeger, Foldamers, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 291–329, DOI: http://10.1002/9783527611478.ch10 Search PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed.
- C. B. Winiger, S. Li, G. R. Kumar, S. M. Langenegger and R. Haener, Angew. Chem., Int. Ed., 2014, 53, 13609–13613 CrossRef CAS PubMed.
- M. Vybornyi, A. Rudnev and R. Haener, Chem. Mater., 2015, 27, 1426–1431 CrossRef CAS.
- Y. Vyborna, M. Vybornyi and R. Haener, J. Am. Chem. Soc., 2015, 137, 14051–14054 CrossRef CAS PubMed.
- C. B. Nielsen, M. Petersen, E. B. Pedersen, P. E. Hansen and U. B. Christensen, Bioconjugate Chem., 2004, 15, 260–269 CrossRef CAS PubMed.
- C. Brotschi, G. Mathis and C. J. Leumann, Chem. – Eur. J., 2005, 11, 1911–1923 CrossRef CAS PubMed.
- C. Brotschi and C. J. Leumann, Angew. Chem., Int. Ed., 2003, 42, 1655–1658 CrossRef CAS PubMed.
- C. Brotschi, A. Häberli and C. J. Leumann, Angew. Chem., Int. Ed., 2001, 40, 3012–3014 CrossRef CAS.
- N. A. Grigorenko and C. J. Leumann, Chem. – Eur. J., 2009, 15, 639–645 CrossRef CAS PubMed.
- N. A. Grigorenko and C. J. Leumann, Chem. Commun., 2008, 5417–5419 RSC.
- P. Roethlisberger, F. Wojciechowski and C. J. Leumann, Chem. – Eur. J., 2013, 19, 11518–11521 CrossRef CAS PubMed.
- R. Sanishvili, C. Besnard, F. Camus, M. Fleurant, P. Pattison, G. Bricogne and M. Schiltz, J. Appl. Crystallogr., 2007, 40, 552–558 CrossRef CAS.
- R. Sukackaite, S. Grazulis, M. Bochtler and V. Siksnys, J. Mol. Biol., 2008, 378, 1084–1093 CrossRef CAS PubMed.
- E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Perez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Mol. Biol., 2002, 9, 918–921 CAS.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- X. J. Lu and W. K. Olson, J. Mol. Biol., 1999, 285, 1563–1575 CrossRef CAS PubMed.
- X. J. Lu and W. K. Olson, Nucleic Acids Res., 2003, 31, 5108–5121 CrossRef CAS PubMed.
- C. Brotschi and C. J. Leumann, Angew. Chem., Int. Ed., 2003, 42, 1655–1658 CrossRef CAS PubMed.
- S. Matsuda, J. D. Fillo, A. A. Henry, P. Rai, S. J. Wilkens, T. J. Dwyer, B. H. Geierstanger, D. E. Wemmer, P. G. Schultz, G. Spraggon and F. E. Romesberg, J. Am. Chem. Soc., 2007, 129, 10466–10473 CrossRef CAS PubMed.
- N. C. Seeman, J. M. Rosenberg, F. L. Suddath, J. J. Parkkim and A. Rich, J. Mol. Biol., 1976, 104, 109–144 CrossRef CAS PubMed.
- J. M. Rosenberg, N. C. Seeman, R. O. Day and A. Rich, J. Mol. Biol., 1976, 104, 145–167 CrossRef CAS PubMed.
- M. A. El Hassan and C. R. Calladine, J. Mol. Biol., 1998, 282, 331–343 CrossRef CAS PubMed.
- A. H. J. Wang, G. J. Quigley, F. J. Kolpak, J. L. Crawford, J. H. van Boom, G. van der Marel and A. Rich, Nature, 1979, 282, 680–686 CrossRef CAS PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. M. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. M. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- R. Rohs, X. Jin, S. M. West, R. Joshi, B. Honig and R. S. Mann, Annu. Rev. Biochem., 2010, 79, 233–269 CrossRef CAS PubMed.
- K. Furuita, S. Murata, J. G. Jee, S. Ichikawa, A. Matsuda and C. Kojima, J. Am. Chem. Soc., 2011, 133, 5788–5790 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, Tm-data, CD-spectra, X-ray analysis and molecular dynamics calculations. See DOI: 10.1039/c6cc00374e |
| This journal is © The Royal Society of Chemistry 2016 |