
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Room temperature methoxylation in zeolites: insight into a key step of the methanol-to-hydrocarbons process†
Alexander J.
O'Malley
ab,
Stewart F.
Parker
bc,
Arunabhiram
Chutia
b,
Matthew R.
Farrow
a,
Ian P.
Silverwood
 c,
Victoria
García-Sakai
c and
C. Richard A.
Catlow
*abd
c,
Victoria
García-Sakai
c and
C. Richard A.
Catlow
*abd
aDepartment of Chemistry, Materials Chemistry, University College London, Third Floor, Kathleen Lonsdale Building, Gower Street, London, WC1E 6BT, UK. E-mail: c.r.a.catlow@ucl.ac.uk
bThe UK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Oxfordshire, OX11 0FA, UK
cISIS Facility, STFC Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire OX11 0QX, UK
dCardiff Catalysis Institute, School of Chemistry, Cardiff University, CF10 3AT, UK
First published on 8th December 2015
Abstract
Neutron scattering methods observed complete room temperature conversion of methanol to framework methoxy in a commercial sample of methanol-to-hydrocarbons (MTH) catalyst H-ZSM-5, evidenced by methanol immobility and vibrational spectra matched by ab initio calculations. No methoxylation was observed in a commercial HY sample, attributed to the dealumination involved in high silica HY synthesis.
Since the seminal publication of Chang and Silvestri,1 the zeolite catalyzed conversion of methanol to hydrocarbons (MTH) has been intensively studied, with the first commercialization of the MTH process in New Zealand in 1985.2 Recent demand for light olefins has also made conversions such as the methanol-to-olefins (MTO) process highly desirable.3–6
There has been a significant amount of research, theoretical and experimental studying mechanistic processes in this set of reactions.7–9 One early step in the reaction mechanism is the formation of framework methoxy species after initial physisorption of methanol through H-bonding to the zeolite Brønsted acid site.10–12 In the MTO process, methoxy groups have been proposed to form ethene directly,13–15 while other studies also support the methoxy group being the starting point for initial C–C bond formation, leading to intermediate alcohols and ethers before higher hydrocarbons.16 However, studying such species is difficult, as under reaction conditions secondary reactions dominate rapidly.17
Framework methoxylation at elevated temperatures has been observed by in situ FT-IR18–20 correlating with the onset of hydrocarbon formation. Solid state NMR21 has shown a variety of surface methoxy groups formed between 493–533 K, with other studies showing their role in dimethylether formation.22 Framework methoxy stability studies in H-ZSM-5, HY and SAPO-34 (with varying Si/Al ratios and thus different acidic content), concluded that formation was very favourable at 393–473 K upon constant removal of water,23 but rapid hydrolysis and reformation of methanol occurred under ambient conditions.
At ambient temperatures, only partial methoxylation of H-ZSM-5 has been proposed using IR24 and also in HY25 (Si/Al = 1.86) with maximum methoxylation occurring at ca. 403 K. Quantum mechanical simulations have studied framework methoxylation using small silicate clusters,26–29 with periodic DFT calculations in the ferrierite framework30 and ab initio molecular dynamics used in chabazite31 and H-ZSM-5.32 All of these measure at higher temperatures, or have concluded notable barriers to total methoxylation, suggesting that elevated temperatures are necessary, with the case further supported by other computational studies highlighting the part played by entropy in forming this intermediate at higher temperatures.33,34
Using quasielastic neutron scattering (QENS), inelastic neutron scattering (INS) spectroscopy, and density functional theory (DFT) calculations, we report the complete conversion of methanol and framework hydroxyls to framework methoxy species on adsorption into a commercial H-ZSM-5 sample at room temperature. This was not observed in zeolite HY which is less suitable for MTH catalysis (even though it is used extensively as an acidic fluid catalytic cracking catalyst); where despite the sample having the same composition (Si/Al = 30) the methanol remains intact, in contrast to partial methoxylation observed in a low silica (Si/Al = 1.86) sample.25 Unlike H-ZSM-5, to reach Si/Al = 30 the HY sample must be dealuminated by steaming to bring higher heat stability and Brønsted acidity, giving way to framework defects such as terminal hydroxyls and silanol nests (of lower Brønsted acidity).35–41 The rapid, complete conversion in H-ZSM-5 shows that elevated temperatures are not required for this step in active catalysts, but also suggests that the dealumination procedure necessary for heat stable faujasite zeolites inhibits this step.
We first probe the mobility of the methanol in the two zeolites at 298 K using QENS, which can provide detailed information on both translational and rotational motion, especially in the case of methanol in zeolites.42–44 The diffusivity is measured from the broadening of the QENS peak as a function of neutron momentum transfer vector Q. Experimental details can be found in the ESI.† The QENS spectra are shown in Fig. 1 at Q = 0.9 Å−1. The broadening at 298 K compared to the resolution spectra taken at 5 K in the HY system is consistent with significant movement. However, the distinct lack of broadening in H-ZSM-5 system at all Q values (shown in ESI†) suggests that the methanol molecules are not moving on the instrumental time scale. Indeed, the close fit to the resolution spectra suggests that virtually all the methanol protons remain static, indicating that the methanol is immobilized due to methoxylation. This immobility strongly supports the complete conversion to the immobilized framework bound methoxy species. Detailed analysis of the diffusion behavior of methanol in zeolite HY will be reported subsequently. The observations also suggest that water forms instantly and is removed in the gas stream, as any water inside the framework would give a strong quasielastic signal from rotational/translational motions.
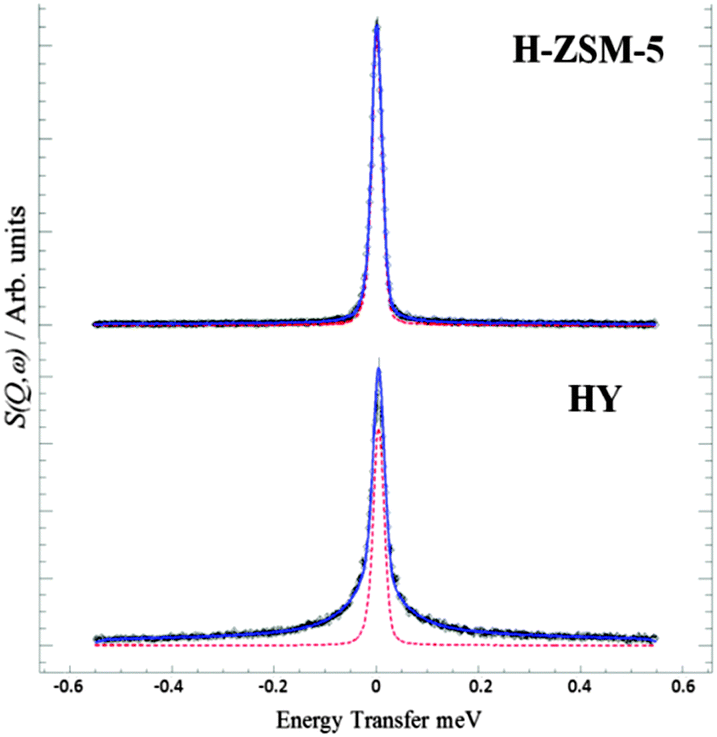 | ||
Fig. 1 QENS spectra obtained of methanol loaded in zeolites H-ZSM-5 and HY at Q = 0.9 Å at 298 K, ( ) represents the resolution data taken at 5 K. The close fit to the resolution function in H-ZSM-5 suggests immobilization, supporting framework methoxylation unlike the significant broadening in HY suggesting intact diffusing methanol. ) represents the resolution data taken at 5 K. The close fit to the resolution function in H-ZSM-5 suggests immobilization, supporting framework methoxylation unlike the significant broadening in HY suggesting intact diffusing methanol. | ||
INS, superior to optical techniques in this case due to the clear visibility of low energy vibrational modes and lack of absorbance by the framework structure, was then used to probe the species present in both zeolites at 298 K. Details on the zeolite samples, and INS experiments can be found in the ESI.† The vibrational spectra obtained are depicted in Fig. 2 showing high energy (a and c) and low energy (b and d) data for methanol in HY (top) and H-ZSM-5 (bottom). The methanol reference spectrum contains bands of the OH deformation at 747 cm−1, the CH3 rocking mode at 1160 cm−1, the CH bend at 1494 cm−1 and the CH and OH stretches at 2988 and 3259 cm−1 respectively. In the low energy spectra, the OH deformation is resolved into a doublet with bands at 713 and 777 cm−1. In the bare zeolites, the acidic OH stretching bands are observed at 3660 and 3720 cm−1 in H-ZSM-5 and HY respectively. OH deformations are observed at 1095 cm−1 in both zeolites. After loading the methanol into HY, the methanol OH deformation is slightly shifted (Fig. 2(a)) and exhibits a significant broadening in the lower energy vibrational spectrum (Fig. 2(b)) consistent with H-bonding, probably due to H-bonding with framework OH groups. The methanol OH stretch and the framework OH stretch bands appear combined with the CH stretch due to downshifting, again due to H-bonding of methanol with the framework. In contrast, on loading of methanol into H-ZSM-5, the methanol OH deformation is not present in either the high or low energy spectra. The methanol OH stretch at 3259 cm−1 is also not observed after adsorption as shown in Fig. 2(c); the framework OH stretch is also difficult to observe.
 | ||
Fig. 2 INS spectra obtained of methanol ( ), the empty dehydrated zeolite ( ), the empty dehydrated zeolite ( ) and methanol loaded into the zeolite ( ) and methanol loaded into the zeolite ( ) at incident energies of 650 meV (a and c) and 250 meV (b and d). Disappearance of the OH deformation at 747 cm−1 and stretch at 3259 cm−1 is observed upon adsorption of methanol into H-ZSM-5, whereas the CH3 rocking mode, CH bend and CH stretches persist. This disappearance is not observed upon adsorption into HY suggesting preferential methoxylation in H-ZSM-5. ) at incident energies of 650 meV (a and c) and 250 meV (b and d). Disappearance of the OH deformation at 747 cm−1 and stretch at 3259 cm−1 is observed upon adsorption of methanol into H-ZSM-5, whereas the CH3 rocking mode, CH bend and CH stretches persist. This disappearance is not observed upon adsorption into HY suggesting preferential methoxylation in H-ZSM-5. | ||
The disappearance of all OH groups upon adsorption into H-ZSM-5, and the persistence of the CH3 deformation and stretch modes would suggest the complete conversion of methanol to framework methoxy at 298 K. Notably, no absorptions corresponding to water are observed in either system. Upon condensation, a strong OH stretch at 3600 cm−1, bend at 1600 cm−1 and broad librational mode around 600 cm−1 would be expected to emerge. The absence of water is to be expected for the HY system as the zeolite is dehydrated and then loaded with methanol using a He/methanol gas stream. However the H-ZSM-5 system might be expected to show bands associated with water, the product of the condensation reaction. The absence of these bands suggests that the methoxylation is almost instantaneous on loading of methanol into the dehydrated zeolite, with the He/methanol stream carrying away any water formed, leaving behind the methoxylated zeolite, rather than the process taking place on a longer time scale inside the sealed can. This explanation indicates an unhindered methoxylation step in this catalyst at room temperature.
To confirm our assignment, DFT calculations were used to calculate the vibrational spectra of the empty protonated zeolite and the framework bound methoxy species. The calculated spectra were weighted for neutron scattering cross section to generate theoretical INS spectra. The calculations were performed for zeolite Y, which owing to its higher symmetry is more computationally tractable. The dependence of the spectroscopic features of the methylated framework oxygen are not expected to show significant sensitivity to the framework structure. The empty HY structure is shown in Fig. 3(a) and the methoxylated HY structure in Fig. 3(b). Full simulation details are in the ESI.†
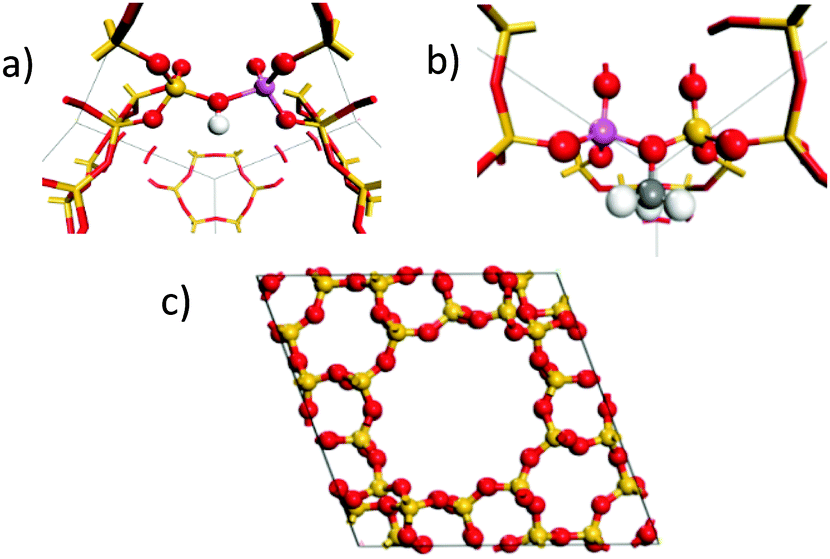 | ||
| Fig. 3 Active site configurations of the DFT calculation used of the protonated zeolite (a) and the methoxylated framework (b) in the primitive unit cell of HY (c). | ||
Fig. 4 shows the experimental and theoretical INS spectra. There is very close agreement for both systems with calculated adsorption bands of the OH stretch and deformation in the empty HY system calculated at 3624 and 1117 cm−1 respectively. The calculated methoxylated spectrum also gives close agreement with experiment, with the CH stretch at 3055 cm−1 and the CH bend and CH3 rocking mode calculated at 1428 and 1126 cm−1 respectively. The agreement coupled with the immobilisation in the QENS measurements confirms the room temperature methoxylation in the H-ZSM-5 framework, in contrast to the inactivity observed in HY.
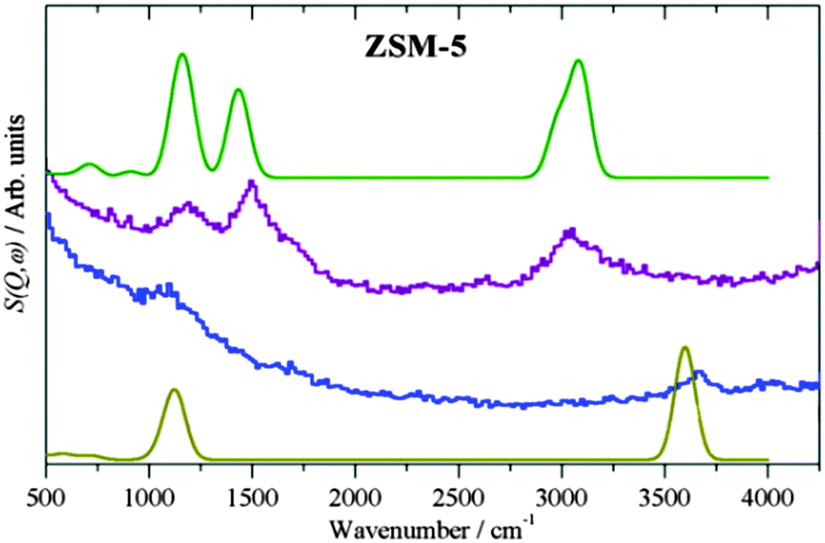 | ||
Fig. 4 Experimental spectra of the methoxylated ZSM-5 system ( ), and the empty zeolite system ( ), and the empty zeolite system ( ) with the calculated spectra of the methoxylated zeolite ( ) with the calculated spectra of the methoxylated zeolite ( ) and the empty zeolite ( ) and the empty zeolite ( ). Close agreement between the calculated experimental spectra support the case for ambient framework methoxylation. ). Close agreement between the calculated experimental spectra support the case for ambient framework methoxylation. | ||
In summary, using QENS, INS and DFT simulations we observed the total conversion of methanol and framework hydroxyls to framework methoxy, and therefore completion of an early step of the MTH/MTO reaction in H-ZSM-5 at room temperature. This conclusion was supported through the observed immobilization of methanol upon adsorption into H-ZSM-5 using QENS and the absence of hydroxyl related absorptions from the INS spectra. These spectra show agreement with DFT calculations of spectra generated of the methoxylated zeolite. Room temperature methoxylation was not observed in the catalytically less applicable zeolite HY of the same Si/Al ratio (brought to this composition through steam dealumination), where methanol was highly mobile upon adsorption and the hydroxyl bands persisted in the INS spectrum. The absence of water in the H-ZSM-5 system suggests very rapid methoxylation of the framework, with water removal in the He/methanol gas stream during dosing as opposed to methoxylation over an extended period in the sealed system.
These observations provide strong evidence that the initial framework methoxylation step of the MTH process takes place rapidly at ambient temperatures in an active catalyst, and that despite sharing the same Si/Al ratio, the defects produced through the necessary dealumination process of faujasites can diminish this methoxylation capability. Future work will elaborate on ZSM-5, probing the effect of differing Si/Al ratio in as-synthesised commercial samples on methanol reactivity. Our results also illustrate the power of the combination of neutron scattering with modelling techniques in probing dynamical and mechanistic aspects of catalytic processes.
The authors would like to acknowledge the Engineering and Physical Sciences Research Council (EPSRC): grant no. EP/G036675/1 for financial support under their Centres for Doctoral Training scheme, the STFC and the ISIS Neutron and Muon source for funding and access to beamline facilities. The UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/K014706/1, EP/K014668/1, EP/K014854/1EP/K014714/1 and EP/M013219/1). Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk).
References
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- C. Maiden, Stud. Surf. Sci. Catal., 1988, 36, 1–16 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef.
- M. Bjørgen, F. Joensen, M. S. Holm, U. Olsbye, K.-P. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43–50 CrossRef.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed.
- K. Hemelsoet, J. Van der Mynsbrugge, K. De Wispelaere, M. Waroquier and V. Van Speybroeck, ChemPhysChem, 2013, 14, 1526–1545 CrossRef CAS PubMed.
- V. Van Speybroeck, K. De Wispelaere, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet and M. Waroquier, Chem. Soc. Rev., 2014, 43, 7326–7357 RSC.
- U. Olsbye, S. Svelle, K. Lillerud, Z. Wei, Y. Chen, J. Li, J. Wang and W. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- R. Shah, J. D. Gale and M. C. Payne, J. Phys. Chem., 1996, 100, 11688–11697 CrossRef CAS.
- S. R. Blaszkowski and R. A. van Santen, J. Phys. Chem. B, 1997, 101, 2292–2305 CrossRef CAS.
- P. E. Sinclair and C. R. A. Catlow, J. Chem. Soc., Faraday Trans., 1997, 93, 333–345 RSC.
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128, 11679–11692 CrossRef CAS.
- W. Wang, Y. Jiang and M. Hunger, Catal. Today, 2006, 113, 102–114 CrossRef CAS.
- Y. Jiang, W. Wang, V. R. Marthala, J. Huang, B. Sulikowski and M. Hunger, J. Catal., 2006, 238, 21–27 CrossRef CAS.
- S. R. Blaszkowski and R. A. van Santen, J. Am. Chem. Soc., 1997, 119, 5020–5027 CrossRef CAS.
- Z.-M. Cui, Q. Liu, S.-W. Bain, Z. Ma and W.-G. Song, J. Phys. Chem. C, 2008, 112, 2685–2688 CAS.
- T. R. Forester, S.-T. Wong and R. F. Howe, J. Chem. Soc., Chem. Commun., 1986, 1611–1613 RSC.
- T. Forester and R. Howe, J. Am. Chem. Soc., 1987, 109, 5076–5082 CrossRef CAS.
- L. Kubelková, J. Nováková and K. Nedomová, J. Catal., 1990, 124, 441–450 CrossRef.
- F. Salehirad and M. W. Anderson, J. Catal., 1998, 177, 189–207 CrossRef CAS.
- C. Tsiao, D. R. Corbin and C. Dybowski, J. Am. Chem. Soc., 1990, 112, 7140–7144 CrossRef CAS.
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 2003, 125, 15260–15267 CrossRef CAS PubMed.
- Y. Ono and T. Mori, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2209–2221 RSC.
- P. Salvador and W. Kladnig, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1153–1168 RSC.
- C. Zicovich-Wilson, P. Viruela and A. Corma, J. Phys. Chem., 1995, 99, 13224–13231 CrossRef CAS.
- S. Blaszkowski and R. Van Santen, J. Phys. Chem., 1995, 99, 11728–11738 CrossRef CAS.
- S. R. Blaszkowski and R. A. van Santen, J. Am. Chem. Soc., 1996, 118, 5152–5153 CrossRef CAS.
- N. Tajima, T. Tsuneda, F. Toyama and K. Hirao, J. Am. Chem. Soc., 1998, 120, 8222–8229 CrossRef CAS.
- J. Andzelm, N. Govind, G. Fitzgerald and A. Maiti, Int. J. Quantum Chem., 2003, 91, 467–473 CrossRef CAS.
- F. Haase, J. Sauer and J. Hutter, Chem. Phys. Lett., 1997, 266, 397–402 CrossRef CAS.
- J. Van der Mynsbrugge, S. L. Moors, K. De Wispelaere and V. Van Speybroeck, ChemCatChem, 2014, 6, 1906–1918 CrossRef CAS.
- A. J. Jones and E. Iglesia, Angew. Chem., Int. Ed., 2014, 53, 12177–12181 CrossRef CAS PubMed.
- R. Y. Brogaard, R. Henry, Y. Schuurman, A. J. Medford, P. G. Moses, P. Beato, S. Svelle, J. K. Nørskov and U. Olsbye, J. Catal., 2014, 314, 159–169 CrossRef CAS.
- O. Cairon, ChemPhysChem, 2013, 14, 244–251 CrossRef CAS PubMed.
- O. Cairon and J.-C. Lavalley, J. Chem. Soc., Faraday Trans., 1998, 94, 3039–3047 RSC.
- O. Cairon, K. Thomas and T. Chevreau, Microporous Mesoporous Mater., 2001, 46, 327–340 CrossRef CAS.
- S. Li, S.-J. Huang, W. Shen, H. Zhang, H. Fang, A. Zheng, S.-B. Liu and F. Deng, J. Phys. Chem. C, 2008, 112, 14486–14494 CAS.
- M.-C. Silaghi, C. Chizallet and P. Raybaud, Microporous Mesoporous Mater., 2014, 191, 82–96 CrossRef CAS.
- A. Janin, M. Maache, J. Lavalley, J. Joly, F. Raatz and N. Szydlowski, Zeolites, 1991, 11, 391–396 CrossRef CAS.
- S. Menezes, V. Camorim, Y. Lam, R. San Gil, A. Bailly and J. Amoureux, Appl. Catal., A, 2001, 207, 367–377 CrossRef CAS.
- H. Jobic and D. N. Theodorou, Microporous Mesoporous Mater., 2007, 102, 21–50 CrossRef CAS.
- H. Jobic, A. Renouprez, M. Bee and C. Poinsignon, J. Phys. Chem., 1986, 90, 1059–1065 CrossRef CAS.
- N. M. Gupta, D. Kumar, V. S. Kamble, S. Mitra, R. Mukhopadhyay and V. Kartha, J. Phys. Chem. B, 2006, 110, 4815–4823 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc08956e |
| This journal is © The Royal Society of Chemistry 2016 |