
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methodological evolutions of Raman spectroscopy in art and archaeology
Danilo
Bersani
a,
Claudia
Conti
b,
Pavel
Matousek
c,
Federica
Pozzi
d and
Peter
Vandenabeele
 *e
*e
aDepartment of Physics and Earth Sciences, University of Parma, Parco Area delle Scienze, 7/A, 43124 Parma, Italy
bConsiglio Nazionale delle Ricerche, Istituto per la Conservazione e la Valorizzazione dei Beni Culturali (ICVBC), Via Cozzi 53, 20125, Milano, Italy
cCentral Laser Facility, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, Harwell Oxford, OX11 0QX, UK
dDepartment of Scientific Research, Metropolitan Museum of Art, 1000 Fifth Avenue, New York, NY 10028, USA
eGhent University, Department of Archaeology, Sint-Pietersnieuwstraat 35, B-9000 Ghent, Belgium. E-mail: Peter.Vandenabeele@UGent.be
First published on 6th October 2016
Abstract
During the last decades, Raman spectroscopy has grown from research laboratories to a well-established approach that is increasingly often used in archaeometry and conservation science. When looking at these research fields, some novel trends can be detected and therefore we would like to review the recent literature on the technical aspects and new evolutions of Raman spectroscopy applied to art analysis. This article reviews Raman instrumentation, with a special focus on the use of mobile and portable instruments, recent developments in the field of surface-enhanced Raman spectroscopy (SERS), and the introduction of spatially offset Raman spectroscopy (SORS) in the field of art and archaeology.
1. Introduction: Raman spectroscopy in art and archaeology
During the last decades, Raman spectroscopy has grown from research laboratories to a well-established approach that is increasingly often used in archaeometry and conservation science.1–5 The technique is well-appreciated for its non-destructive character, its small spectral footprint and its ability to record molecular spectra of inorganic as well as organic materials. In general, Raman spectra are typically less complex and have narrower bands compared to infrared spectra.6–8Although the Raman effect was discovered in the first half of the 20th century, it is only in the 1980's and 1990's that the technique became increasingly accessible for art analysis. This is mainly the consequence of decades of technical improvements, such as the introduction of lasers and charge-coupled device (CCD) detectors, which allowed researchers to expand the range of possible applications. The coupling of spectrometers with microscope optics was an especially favourable evolution, enabling researchers to record Raman spectra of particles down to a diameter of 1 μm – the typical order of magnitude of pigment grains. Additional technical advances, including the introduction of fibre optics Raman spectroscopy, paved the way for new applications of the technique in the cultural heritage field. Overtime, typical uses of Raman spectroscopy evolved from pigment identification9–12 (by comparison against a reference database) to more complex cases, such as the study of degradation13 and corrosion processes,14,15 the analysis of organic materials16,17, complex microsamples,18etc.
When observing this research field, some novel trends can be detected and therefore we would like to review the recent literature on the technical aspects and new evolutions of Raman spectroscopy applied to art analysis. This article will review Raman instrumentation, with a special focus on the use of mobile and portable instruments, recent developments in the field of surface-enhanced Raman spectroscopy (SERS), and the introduction of spatially offset Raman spectroscopy (SORS) in the field of art and archaeology.
2. Laboratory experiments
Benchtop micro-Raman systems, designated for laboratory use, are usually equipped with multiple lasers, but their advantages, with respect to mobile instruments, are not only limited to the choice of the excitation wavelength. These added benefits will be discussed in the following. First, the stable environment allows researchers to use high magnification microscope objectives with large numerical apertures. In addition to the improved sensitivity (due to the large collection angle), high magnification objectives facilitate high spatial resolution.19–21Laboratory micro-Raman spectrometers, thanks to their larger focal length, usually also reach a better spectral resolution than their mobile counterparts, i.e. 1–2 cm−1 for standard micro-Raman setups (depending on the wavelength used) and even less, a few tenths of cm−1 for long focal systems. A good spatial and spectral resolution is fundamental when discriminating between similar phases or when a precise Raman shift is required to obtain information on the composition, or on temperature or pressure effects.22,23 As an example, in the study of ceramics, the precise knowledge of the position and width of the Raman bands of iron oxides is necessary to differentiate natural hematite from that obtained by heating goethite (α-FeOOH),24,25 to estimate the presence of Al in the hematite structure,26 to evaluate purity, disorder and degree of crystallinity27,28 as well as finite-size effects in nano-crystalline hematite29 and to relate them to raw materials' provenance and firing conditions. Also, in the study of modern pigments, such as azo dyes, a good spectral resolution is required to distinguish between similar spectra, very rich in fine bands.11,30
Benchtop micro-Raman spectrometers can be easily coupled with automatised micrometre XY (or XYZ) positioning stages, useful to collect Raman maps.31,32 Lofrumento et al.33 studied the distribution of feldspars in the ceramic body, and in particular around the body-coating boundary, by means of Raman imaging. In the study of the source of blue colour in blue enamels in Melfi castle (south of Italy), Caggiani et al. acquired maps of the S3− Raman signal of haüyne at different temperatures.34 Conti et al.35,36 proposed recently the use of SORS for studying opaque paint layers. More complex hyphenated techniques with interesting applications in art and archaeometry are also possible for benchtop Raman spectrometers. For instance, coupling with scanning electron microscopy/energy-dispersive X-ray (SEM/EDX) spectroscopy37,38 has shown great potential for simultaneous elemental and molecular analysis, while the combined use of Raman and atomic force microscopy (AFM) has been exploited for tip-enhanced Raman spectroscopy (TERS) applications.39
Many different combinations of laser sources40–48 are used in benchtop Raman systems, as reported in Table 1. Except when using large systems based on double or triple monochromators,49 the rejection of spurious wavelengths in the laser beam is achieved using notch filters or low-pass filters that are specific to each laser line. The most common filters, appearing on the first commercial micro-Raman spectrometers, are the holographic notch filters, allowing researchers to obtain both Stokes and anti-Stokes bands, with a low-wavenumber cut-off at 70–100 cm−1. The main problem is related to the aging of the notch filters, shifting and widening the rejection zone. Modern low-pass dielectric filters, sometimes called “edge” filters, are usually cheaper and more stable, with a low-wavenumber cut-off down to 30–40 cm−1 from the excitation line. They typically allow obtaining only the Stokes part of the spectrum. Their main problem is the presence of a periodic modulation of their transmittance. This can cause evident ripples in presence of a fluorescence background, sometimes highly disturbing. In the last years, a new kind of volume Bragg filters appeared on the market. They are notch filters in a glass matrix, very stable, with a very low cut-off, very near to the laser line (5–15 cm−1) allowing exploration of the low-wavenumber part of the Stokes and anti-Stokes spectra. Volume Bragg filters are more expensive than standard notch filters and require a very good alignment of the system. They have also been used in some studies related to cultural heritage or analysis of minerals.50–54
| Wavelength (nm) | Source |
|---|---|
| 413.1 | Ar+ ions |
| 457.9 | Ar+ ions |
| 473.1 | Doubled Nd:YAG |
| 488 | Ar+ ions |
| 514.5 | Ar+ ions |
| 532 | Doubled Nd:YAG |
| 632.8 | He–Ne |
| 647.1 | Kr+ ions |
| 780–785 | GaAlAs diode |
| 830 | GaAlAs diode |
| 1064 | Nd:YAG |
The main advantage of using different laser sources is the possibility to reduce the fluorescence background by the choice of laser excitation wavelength, as discussed later. In some cases, however, the detection of a characteristic fluorescence (or photoluminescence) emission while performing Raman measurements can be helpful in the characterization of the material.55 For example, when studying white ceramic pigments, for instance, alumina can be easily detected by its typical photoluminescence doublet at 694–693 nm due to Cr3+ impurities, appearing at the Stokes Raman shifts of 1367–1389 cm−1 when exciting with the 632.8 nm line.56 We would like to point out that the shifting of this photoluminescence doublet (when using a different excitation laser) is very often used to measure the pressure in high-pressure (HP) Raman studies performed in diamond anvil cells.57
The fluorescence detected during the Raman measurements was also used to study the presence and the local environment of Cr3+ ions in emeralds58,59 and selected silicates in unusual ceramics from Norway.42 The use of a 473.1 nm laser line enabled detection of very sharp photoluminescence bands ascribed to Dy3+, Er3+ and Eu3+ ions from baddeleyite inclusions in a natural ruby; this was crucial in identifying Madagascar as the provenance place of the gemstone.60
The possibility to choose the laser line can allow enhancement of the Raman signal by several orders of magnitude, reaching the resonance condition when the wavelength of the incoming or scattered light is close to an electronic absorption band. Fig. 1a shows this effect, as it occurs with the mineral crocoite. Another typical example of this, well-known since 1970, is represented by carotenoids (Fig. 1b), giving rise to an intense Raman enhancement when excited in the green and blue range.61,62 The resonance enhancement in carotenoids is so strong (up to 105), when excited at 488 nm, that it is possible to perform fast resonant Raman imaging of carotenoid pigments distribution even without a spectrometer, just using a band-pass filter centred on the resonant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C mode at 1525 cm−1.63 Many other pigments show strong resonance effects too: chromate yellow and red pigments, as well as lazurite, display strong enhancements when using the typical He–Ne laser emission at 632.8 nm for excitation.64,65
C mode at 1525 cm−1.63 Many other pigments show strong resonance effects too: chromate yellow and red pigments, as well as lazurite, display strong enhancements when using the typical He–Ne laser emission at 632.8 nm for excitation.64,65
 | ||
| Fig. 1 Raman spectra of crocoite (a) and carotenoids in coral (b) recorded with different excitation wavelengths (473.1 and 632.8 nm), illustrating the resonance Raman effect. | ||
Resonance effects can be combined with the powerful enhancements offered by SERS (discussed later) obtaining the so-called SERRS (surface-enhanced resonance Raman spectroscopy) effect, allowing extremely high sensitivity and enhancements up to 1014 times.66 Efremov et al.66 published a comprehensive review on resonance Raman spectroscopy that includes a few interesting examples of application of SERRS to the analysis of pigments and minerals.67–71 Colomban et al.43 used four different laser lines to study pigments and coloured glazes of ancient ceramics in order to obtain Raman enhancement, through resonance effects, in Cr2O3, SnO2, CoO, Sb2O5 and CuO. In a report published by Faurel et al., the authors used four different laser lines to examine Cr-doped Sn-based pink pigments by means of resonance Raman spectroscopy.72
When choosing a laser excitation wavelength, it is also important to consider that the intensity of the Raman scattering is proportional to the fourth power of the frequency of the incident light. This means that, using the same power for the incident light, the intensity of a Raman band at 1000 cm−1 measured with the 1064 nm line is only 3.4% of that measured at 488 nm.73,74
The efficiency of the CCD detector should also be taken into account. A typical silicon CCD increases its efficiency from 400 nm to nearly 650 nm. Then the efficiency decreases, becoming zero at ∼1100 nm. For that reason, in order to study hydrogen-related stretching modes as the OH stretching in water containing materials, a better choice would be the use of a short wavelength laser.32,58 When exciting with very long wavelengths (e.g. using 830 nm), the highest wavenumber part of the Stokes spectrum could be completely lost. It should be noted that Raman spectroscopy using a 1064 nm excitation wavelength is performed using a different type of detector, as discussed later on, and the above detector restrictions do not apply.
Another effect due to the change of excitation wavelength is the variation in the spectral resolution. When using the same spectrometer with the same configuration (in particular, the same grating and the same slit), increasing the excitation wavelength will result in better spectral resolution. Fig. 2 illustrates this effect, where the same sample of aragonite is recorded with two different instruments. In the case of excitation with the 632.8 nm laser, where a higher spectral resolution is achieved, a doublet is observed.
 | ||
| Fig. 2 Raman spectra of the same sample of aragonite, recorded with a different spectral resolution with 632.8 and 473.1 nm excitation wavelengths. | ||
To evaluate the effect of using different lasers on the Raman spectra, Burrafato et al.75 assembled a spectral database of pigments, pure and with various binders, obtained at 532, 632.8 and 780 nm excitation. In a recent study, six different laser lines were used for the very challenging Raman identification of the copper resinate and verdigris pigments in works of art.48 This work clearly shows that it is not possible to define a priori an optimum excitation source for a given class of materials. On the other hand, in a multi-laser Raman study of natural ochres, Froment et al. found that the most useful excitation for this class of pigments was 532 nm.76 Pan et al. reported a detailed comparative Raman study aiming at optimizing the detection of walnut oil, used as a protective coating on Galician granite monuments,77 using five different laser wavelengths, taking advantage from the higher efficiency of the shorter wavelengths used (488 and 532 nm).
The main strategy to reduce the fluorescence is the use of low-photon energy (long wavelength) laser excitation lines. Near infrared (785–1064 nm) lasers are often used to study fluorescent molecules, in particular organics or samples with a biological origin.74,78 However, even when using the 785 nm excitation, it is possible to find samples where the fluorescence background can completely overwhelm the Raman spectrum. In these cases, the best way to quench the fluorescence is by using the 1064 nm fundamental emission of the Nd:YAG laser. A high radiance is often employed to compensate for the lower Raman cross-section,74 with the risk of undesired overheating of the sample.
The use of a 1064 nm laser line in the first Raman systems, however, required the adoption of the same interferometric technology used in Fourier-transform infrared (FTIR) spectrometers, leading to the birth of FT-Raman systems. This is because the CCDs used as multichannel detectors in dispersive Raman systems are made of silicon, whose band-gap (1.1 eV) causes a complete loss in sensitivity for wavelengths higher than 1100 nm. This made it impossible to detect the Raman Stokes spectrum at this wavelength; in rare cases, CCDs were used to collect anti-Stokes Raman spectra at 1064 nm excitation.79 The use of a single channel detector made of germanium or other low-bandgap semiconductors in a dispersive spectrometer would require a very long time to collect a Raman spectrum. Despite some disadvantages (low scattering efficiency and consequently high acquisition times, sample heating, bulky apparatus, difficult coupling with microscopes), FT-Raman spectrometers have been largely used, up to the present days, in the cultural heritage field. Reported applications include the analysis of inks – historical (iron-gall)80 and contemporary81 –, pigments from a Hindu statue82 and from an Augustinian friary,83 a red pigment from a mediaeval corbel,84 lacquerware pigments,85 a dye–mineral composite similar to Maya blue,86 the indigo dye,87 native American rock art,88 patinas in stone artefacts,89 ancient Egyptian pigments,90 and a painted Italian canvas dated to the 19th century.91 Comprehensive spectral databases of FT-Raman pigments have been published and widely used as a reference in identification studies.10,92
The first attempts at realizing a multichannel dispersive Raman spectrometer incorporating a 1064 nm laser date back to 1986,73 but only in the past few years, the development of multichannel indium gallium arsenide (InGaAs) detectors, along with their commercial availability at convenient prices, gave rise to a new generation of dispersive instruments, sometimes called dispersive near infrared (DNIR) Raman. The low bandgap of InGaAs detectors enables a good sensitivity up to 1650–1700 nm, with quantum efficiencies above 80%, much higher than that of germanium detectors93,94 that enabled detecting Raman signals up to 3400–3500 cm−1. Current commercial dispersive spectrometers show a S/N ratio that is more than one hundred times larger than that of FT-Raman systems at the same excitation wavelength.
The advantages of a dispersive NIR Raman system compared to FT-Raman are: higher robustness and compactness, better coupling with fibre optics, faster analysis, low laser power requirements, higher spatial resolution, easier coupling with microscopes, allowing for use with a confocal geometry (not possible with FT-Raman systems), and the possible integration of multiple laser lines within the same spectrometer.
In addition to 1064 nm, some companies proposed 980 nm as the excitation wavelength for dispersive NIR Raman systems. A dispersive Raman spectrometer working at 1064 nm and equipped with an InGaAs linear array detector was used by Schmidt and Trentelman to obtain in situ Raman spectra to be used in the identification of different red colorants highly fluorescent under visible excitation.95 The same experimental configuration was employed to compare normal Raman and SERS spectra obtained from the Cape Jasmine dye using dispersive Raman systems at visible and NIR excitation.96
3. Mobile instrumentation and in situ analysis
In the past, new applications in the field of Raman spectroscopy were often the result of technological evolutions, such as innovative developments in Raman instrumentation. The possibility to couple a Raman spectrometer with a fibre optics probe head paved the way for the development of mobile spectrometers for in situ analysis of art objects. Indeed, Raman spectroscopy can be considered as a non-destructive approach, provided the laser power at the sample is kept sufficiently low to avoid any thermal damage. Using fibre optics, it is possible to perform direct investigations of an artefact, preserving its appearance and physical integrity.‘In situ’ (on site) analysis means using mobile instrumentation that is transported to the art object, while ‘direct’ analysis, on the other hand, refers to the fact that the artefact is investigated without sampling.45,97,98 Also, researchers should be careful when speaking about ‘transportable’, ‘mobile’, ‘portable’, ‘handheld’ or ‘palm’ instruments.45 ‘Mobile’ spectrometers are designed in such a way that no complex alignment procedures are needed when set-up on site. ‘Portable’, ‘handheld’ and ‘palm’ devices are smaller and typically equipped with batteries. ‘Handheld’ and ‘palm’ Raman instruments are normally less useful for art analysis, due to difficulties associated with focusing, to the lower sensitivity and inability to modify the spectral acquisition settings. Typically, the latter types of instruments have pre-set spectral parameters (e.g. integration time and laser power) and a built-in spectral database for identification of unknowns. However, recently, a handheld instrument that combines two laser sources with sequentially shifted excitation was tested for applications in cultural heritage.99 By combining two different laser sources, it is possible to obtain spectra over a broad spectral range, whereas the sequentially shifted excitation algorithm100 allows for removal of the fluorescence background. This approach seems very promising, and all data processing (background removal and merging of spectra obtained at different excitations) is performed automatically. However, one should note that the so-called ‘photon shot noise’ associated with the fluorescence background and stemming from the fundamental nature of light cannot be removed by this method and remains imprinted on the resulting spectra. The method removes principally instrumental spectral artefacts, for example, due to filter ripples, CCD etaloning and channel to channel variation in detection sensitivity associated with high fluorescence backgrounds.
Handheld Raman spectrometers have proven useful for the identification of minerals and for the analysis of different gemstones in various collections.101–103 Also, portable instruments were used to investigate so-called glyphs104 – engraved stones and glass objects, used as stamp – or tesserae from Roman glass mosaics.105,106
Mobile Raman spectroscopy has been used for the investigation of several types of art objects. The suitability of the technique is well demonstrated for the direct analysis of wall paintings,45,107,108 rock art,109,110 statues111 or coloured glass in lead.112,113 Remarkable case studies involved the analysis of ‘sun’ rock art110,114 or the analysis of stained glass windows of ‘Sainte Chapelle’ in Paris.113,115 In the first case, transportation and positioning of the equipment (including an electrical generator) were challenging. In the case of the analysis of stained glass, avoiding interference from ambient light was also an issue.
The importance of stable placement of the equipment is often underestimated. On the one hand, successful and reliable analysis can only be performed upon positioning the instrumentation in a stable configuration, which however should also allow for a certain degree of flexibility to adapt to the situation on hand. Especially when several types of art objects (paintings, statues, manuscripts, etc.) are measured within the same measurement session, the set-up should be modifiable. Typically, such flexibility is needed when performing investigations in museums, where several types of artefacts (and storage or display cases) are found.111,116 Moreover, stability is often an issue, especially when investigations are carried out on a scaffolding.107,108 When studying mediaeval wall paintings on the ceiling of a church or chapel, or other artworks in outdoor environments, measurements can be performed at night to avoid interference of ambient light.
Often, in situ studies require using complementary methods.117–125 One of the analytical techniques most frequently used in association with Raman spectroscopy is X-ray fluorescence (XRF) spectroscopy. This technique reveals the elemental composition of the area under investigation, whereas Raman spectroscopy identifies the molecules contained in the artwork. The comparison and interpretation of the results, however, is not straightforward. Typically, the spectral footprint of handheld XRF measurements is much larger, with a beam size of ca. 0.5 cm in diameter. Also, while the area probed by Raman spectroscopy is limited to the top layers of the artwork, X-rays penetrate much deeper into the artefact. This was clearly shown126 in the analysis of the oil painting ‘Mad Meg’ by Flemish renaissance artist Pieter Brueghel. In this case, a combination of optical microscopy, mobile Raman spectroscopy, handheld XRF and mobile X-ray diffraction (XRD) was used in the investigation of specific art historical-related issues concerning this famous painting. Typically, analytical campaigns using multiple techniques require significant organization efforts, and all measurements need to be well coordinated.127 Other examples of such a multi-technique approach include an in situ campaign for the investigation of the tomb of Menna, in the Theban necropolis in Egypt, and the studies performed in Pompeii (Italy). Typically, during such campaigns, researchers are faced with many practical issues, such as transportation of the equipment, the need for providing power supply, protection against dust, etc.117
Some research groups go beyond simple pigment identification by using mobile Raman spectroscopy to evaluate the preservation state of certain objects and to examine degradation processes.128–132 Typically, this is done by combining Raman spectroscopy with physicochemical modelling in order to estimate the relative ratios of salts that may be found on the surface of the artefact and to evaluate possible degradation pathways of the original materials.
It is clear that mobile Raman instrumentation can be very useful for art analysis. Depending on the questions and artefacts in hand, handheld Raman instrumentation may be sufficient, although often portable/mobile devices are required. This is mainly the case when small details need to be analysed and focusing is of high importance. We foresee that the technical advances and implementation of algorithms for background removal may lead to a more widespread use of handheld instruments in archaeometry research. However, in the current state, some improvements (especially for what concerns the focusing and non-automatic setting of parameters) are needed to bring the Raman technique to the next level in the field of art and archaeology.
4. Microscale-spatially offset Raman spectroscopy (micro-SORS)
Although confocal Raman microscopy is a widely applicable technique in the areas of art and archaeology, its use in non-destructive analysis of stratified turbid (diffusely scattering) matrices is severely restricted by its inability to form direct optical images within such samples. Painted layers are by their nature highly turbid and typically only a few tens of micrometres thick, often spreading in multiple stratigraphy.36,133 Therefore, many analytical problems in the conservation field requiring deeper non-invasive probing in these turbid matrices are therefore not addressable by this conventional approach. The limitation often necessitates resorting to cross-sectional analysis to retrieve chemical information from such sub-layers. This is, however, often highly undesirable and, in some situations, not even permitted due to the uniqueness and high value of the object under analysis.Recently, a new technique in Raman spectroscopy capable of overcoming some of these limitations has emerged – micro-SORS.36,133,134 The method builds on earlier advances of a parent technique, spatially offset Raman spectroscopy (SORS).135 To date, two basic micro-SORS modalities have been proposed and demonstrated: defocusing and full micro-SORS.136 The defocusing micro-SORS36 is the simplest variant. Despite being less effective than full micro-SORS, as it does not involve fully separated laser illumination and Raman collection zones leading to lower contrast between individual layers derived from SORS, it is the most practical in existence. This is due to its suitability for use within existing conventional, unmodified Raman microscopes. The concept of defocusing micro-SORS is illustrated in Fig. 3. The basic measurement relies on the collection of at least two Raman spectra: (i) the first spectrum is acquired with the sample in a standard ‘imaged’ position where conventional Raman microscopy would traditionally be performed to analyse the surface of the sample, (ii) the second measurement is taken with the sample moved away from the microscope objective by a ‘defocusing distance Δz’. The result of the sample displacement is the enlargement (‘defocusing’) of both the laser illumination and Raman collection zones on the sample surface. The larger the defocusing distance, the greater the enlargement of both the laser illumination and Raman collection zones on the sample surface. The ‘imaged’ position measurement typically yields a Raman spectrum dominated by the surface layer35 and would correspond conceptually to a zero-spatially offset measurement in conventional SORS analysis. The measurement carried out in the ‘defocused’ position produces a Raman spectrum which has a significantly higher relative degree of Raman contribution from sublayers, in analogy with a non-zero spatially offset spectrum acquired in a conventional SORS measurement. The relative intensity change within spectral subcomponents typically signifies the presence of multiple layers in the sample.
 | ||
| Fig. 3 Schematic diagram of conventional backscattering Raman microscopy, defocusing micro-SORS and full micro-SORS modalities. | ||
A simple mathematical manipulation of the ‘imaged’ and ‘defocused’ spectra can be then used to retrieve a pure Raman spectrum of the sublayer, in the case of a two-layer system. This process involves a scaled subtraction of the ‘imaged’ spectrum from the ‘defocused’ spectrum, erasing the contribution of the top layer. More defocused spectra need to be acquired for the separation of a larger number of layers than two.134
Full micro-SORS utilises completely separated laser illumination and Raman collection zones (Fig. 3). As such, it exhibits much higher contrast enhancement between the surface and subsurface layers and larger penetration depths.136 However, as mentioned above, its deployment requires some modifications to the existing Raman microscopes in order to be practiced in an unrestricted manner. The principle of use is analogous to that described above with the measurement involving the collection of Raman spectra at zero and non-zero spatial offsets (the separation between the laser illumination and Raman collection zones on the sample surface – Δx) and subsequent numerical processing to reveal Raman signatures of individual layers as in conventional (macro-) SORS.135
In addition to establishing the chemical makeup of individual layers, the micro-SORS techniques, in general, are also capable of determining the order in which layers are laid. This is achieved by examining the relative rate of the decay of signals from individual layers.35 It is also possible to determine whether two chemical components detected by micro-SORS belong to two separate layers or if they are mixed within a single-layer.137 A study by Conti et al.138 provides further insight into micro-SORS versus the conventional confocal Raman microscopy, and emphasises differences and commonalities between the two techniques.
The practical applicability of micro-SORS has been demonstrated in isolating the chemical signatures of different layers of paint with both artificially assembled systems36 and the real objects of art.133 The latter included painted sculptures and plasters. Fig. 4 exemplifies the use of defocusing micro-SORS for the subsurface analysis of the blue mantle of a Christ's sculpture from ‘Ossuccio Sacred Mount’ (United Nations Educational, Scientific and Cultural Organization (UNESCO) World Heritage site), a prestigious devotional place in Northern Italy. The sculpture was repainted many times over the centuries and, consequently, its stratigraphy comprises a number of layers.133 The micro-SORS Raman measurements allowed researchers to elucidate its stratigraphy. The ‘imaged’ position spectrum exhibits only lazurite Raman bands, although the presence of traces of Prussian blue and azurite cannot be completely excluded. By displacing away the sample surface from the microscope objective, it was possible to discern the contribution from the most internal layer, Prussian blue, which increased dramatically relative to the surface layer Raman signal (lazurite). The findings were substantiated by cross-sectional Raman analysis.133
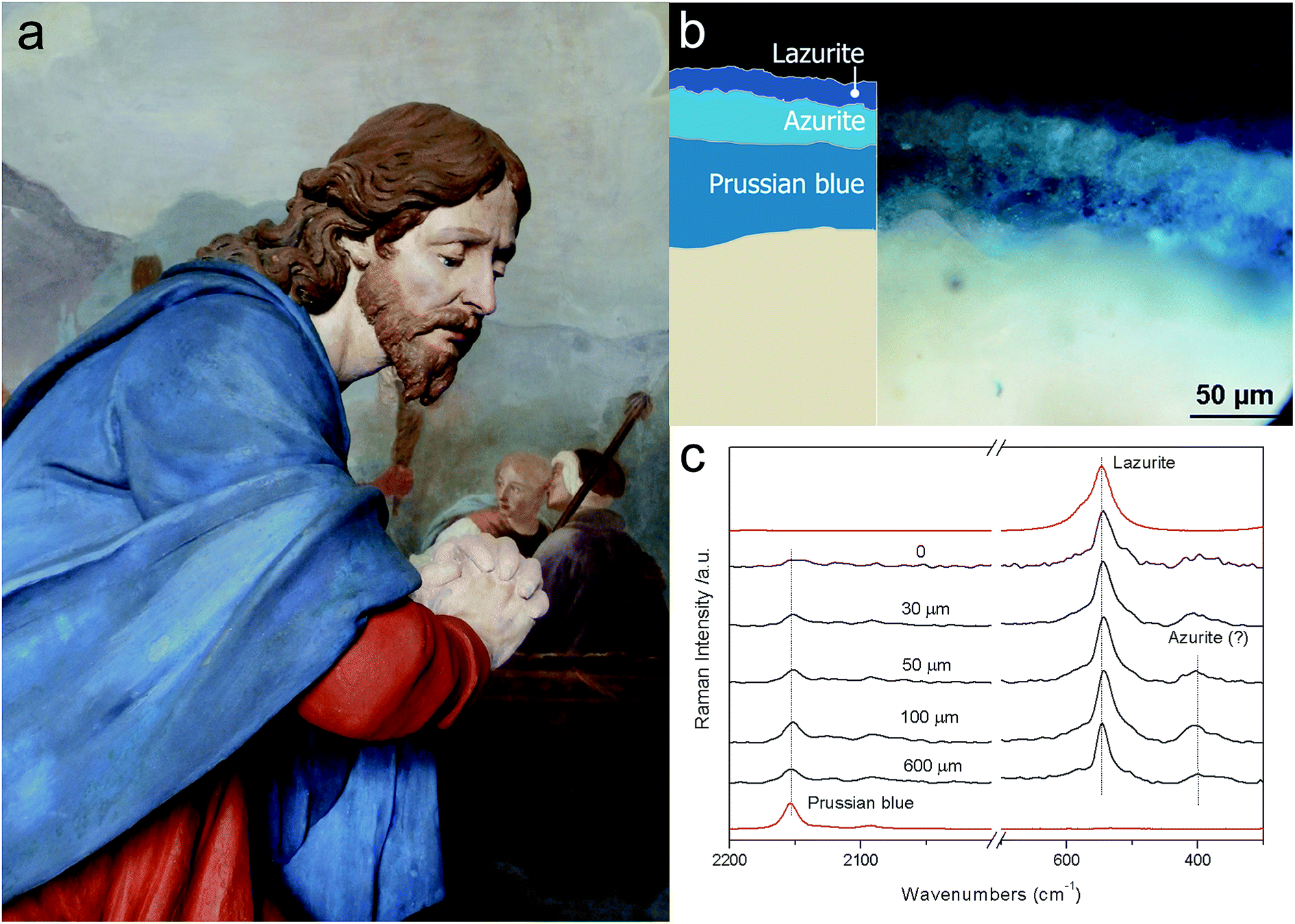 | ||
| Fig. 4 Micro-SORS on a painted sculpture of Christ from Ossuccio Sacred Mount (a); optical image and scheme of the stratigraphy (b); defocused spectra shown for different distances Δz from the ‘imaged’ plane indicated next to each spectrum (0 = ‘imaged’ position) (c). The spectra are offset for clarity. The dashed lines are provided for guidance to emphasise the varying relative Raman intensities of lazurite and Prussian blue with the defocusing distance; the reference spectra are shown in red.133 | ||
In general, the limitations of the micro-SORS technique include its inapplicability to highly absorbing top layers, extremely thin sublayers and compounds with low Raman cross-sections or highly fluorescent at the subsurface position. Recently, the ability of full micro-SORS to retrieve the chemical composition of a sublayer covered by a fluorescent top layer has been demonstrated on mock-up samples; micrometric layers of fluorescent pigments were spread over marble or different layers of pigments. A comparative study between the fluorescence suppression of the top layer achieved by defocusing and full micro-SORS was also carried out, highlighting that the latter modality is capable of recovering the sublayer signal, even when conventional Raman measurement yields only strong fluorescence.139
Samples possessing very high heterogeneity across their surface or within sublayers are also challenging and may require the use of a more complex data acquisition methodology.153 It should also be noted that the spatial resolution of micro-SORS is significantly inferior to that of conventional Raman microscopy and as such it should only be used when conventional confocal Raman microscopy is not applicable.
To enable micro-SORS to perform in situ measurements, a conventional portable Raman equipped with 785 nm excitation wavelength was adapted for defocusing micro-SORS analysis.140 The new set-up allowed obtaining significant results on artificial micrometric layer systems consisting of spray paint on stone and painted stratigraphy. Given the wide applicability of the technique and potential for further improvements by developing portable full micro-SORS, the concept is expected to find a number of application niches in several areas providing a beneficial complementary analytical capability to conventional Raman microscopy in situations where its accessible depths are inadequate.
5. Surface-enhanced Raman spectroscopy (SERS)
The fortuitous observation of a massive enhancement in the intensity of the Raman spectrum of pyridine at a roughened silver electrode, in 1974,141 paved the way for the development of SERS.142 In SERS, target organic molecules are adsorbed onto noble metal nanostructures, resulting in the simultaneous enhancement of their Raman signal intensity and quenching of their fluorescence emission via combined143,144 electromagnetic145 and charge-transfer146 mechanisms. The discovery of SERS, along with the subsequent demonstration of single-molecule analyte detection,147,148 prompted researchers to pursue possible applications of this technique in biochemistry, medical diagnostics, threat detection, forensics, and in all those areas where the ability to probe trace levels of target specimens is crucial to the success of the analysis.In the field of art and archaeology, SERS has been highly valued for its sensitivity to organic colourants, i.e. materials that are typically present in artworks in minute concentrations and within complex matrixes, and whose successful detection by normal Raman spectroscopy is often prevented by their inherent fluorescence properties. The first application of SERS to the study of cultural heritage materials, dating back to 1987, is by Guineau and Guichard, who reported spectra of synthetic alizarin and extracts of the plant dye madder, the source of alizarin, and related anthraquinoid derivatives, from an 8th century textile sample.149 With the exception of a report published in 1990 also focusing on madder,150 it is not until 2004 that SERS found increasing applications in the art conservation field. Since then, the great potential of this technique for the ultrasensitive detection and identification of historical dyes has been explored at length and described in several comprehensive reviews.154–158
Much of the research initially conducted in the field of SERS for cultural heritage focused on the characterization of reference substances and interpretation of their spectral patterns, on the study of their physicochemical properties and degradation processes catalysed by various nanoplasmonic substrates, and on the development of analytical strategies tailored to their ultrasensitive detection in samples from artworks and museum objects. While synthetic dyes have been featured in a limited number of publications,81,159–164 a substantial amount of work to date has dealt with natural organic colourants, especially red anthraquinones such as alizarin, purpurin, carminic and laccaic acids, and related compounds;151,152,165–169 yellow dyes mostly belonging to the flavonoid,170–176 alkaloid,177–179 carotenoid,96 and curcuminoid176,180 molecular classes; as well as blue and purple colourants including indigo,181,182 Tyrian purple,182,183 orcein,184 and anthocyanins.185
The availability of individual references186–188 and comprehensive SERS libraries of natural dyes is crucial to ensure positive identification of query spectra from works of art and historical artefacts; however, manually matching spectra from unknowns with series of references from a given database often turns out to be a time-consuming, not always fruitful process. A recent study has explored the use of statistical techniques and library search approaches to provide rapid, trustworthy classification of unknown samples by automatically highlighting their similarities and differences with respect to data from a reference database.189 In addition to proposing a clear library search protocol, this work has addressed the most commonly raised concern on the SERS technique's lack of reliability by showing that, in spite of spectral variations arising from chemical and instrumental factors, it is indeed possible to identify unknown spectra by automatically searching them against a reference library.
Most of the SERS work on reference dyes and cultural heritage materials to date has been carried out on silver nanoparticles obtained via chemical reduction of silver nitrate with sodium citrate, according to the synthesis protocol proposed by Lee and Meisel.190 A second type of colloids becoming increasingly popular in the conservation science field is produced by microwave-supported glucose reduction of silver sulfate in the presence of sodium citrate as a capping agent.191 The latter nanoparticles, stable and efficient over the course of several months, display a considerably narrower absorption band and particle size distribution compared to the traditional citrate-reduced ones, contributing to more reproducible SERS performances. Alternative approaches for the preparation of silver colloids include laser ablation192 and photoreduction,193–196 whose main advantage lies in the absence of unreacted species or oxidation byproducts in the suspension that may give rise to spurious bands in the SERS spectra, a phenomenon that is well documented for citrate-reduced colloids.183,197,198 The convenience of use of silver nanoparticles produced with the methods described above, as well as their enhancement efficiency, stability, and other interfacial characteristics such as the pH, surface availability and potential, has been compared in a recent review.192
Among the solid-state SERS substrates most commonly used in conservation science, silver island films (AgIFs) and silver films over nanospheres (AgFONs) are also present. Either directly deposited199,200 onto the pigment particles under study or thermally evaporated onto an Ag–Al2O3 support prior to adsorption of the analyte,165 AgIFs have been used to characterise anthraquinoid dyes. The unevenness of these substrates, however, resulting in a substantial lack of spectral reproducibility, caused them to be gradually replaced by AgFONs, whose excellent stability and tunability properties are yet counterbalanced by the possible occurrence of carbon contamination issues during the silver deposition phase.159,201,202
While distinctive spectra of pure commercial colourants can be acquired relatively effortlessly with SERS, the analysis of dyes in artworks poses additional challenges due to the fact that, in paints and fabrics, these materials are typically precipitated onto an inorganic substrate (lake pigments) or bound to textile fibres through bridging metal ions (mordant dyes). For this reason, the application of hydrolysis pretreatments is often required to isolate the dyes from their chemical environment and favor their adsorption on the metal surface. Initially exploited for their high dye extraction yield, heated solutions of strong acids or alkali, such as hydrochloric acid and sodium hydroxide,153,165,203 were gradually replaced by alternative procedures involving milder reagents and experimental conditions.177,201,204,205 Noteworthy among the latter is a non-extractive gas–solid hydrolysis based on the use of hydrofluoric acid (HF)203 that has enabled rapid identification of natural dyes in samples as small as 20 × 20 μm from a great variety of polychrome historical objects and works of art with increased sensitivity.206,210
The successful detection of alizarin, carminic and laccaic acids on filter paper coated with a thickness-controlled layer of silver nanoparticles165 paved the way for the direct identification of organic colourants without the need for pretreating the samples. Additional examples of in situ non-hydrolysis SERS include the analysis of reference dyed fibres on photoreduced silver nanoparticles,194,195 and the characterization of Greco–Roman cosmetics,46 coloured and plain canvas linen fibres91 and samples from 18th-century French objects on borohydride-, hydroxylamine- or citrate-reduced silver colloids.17 While in the last three instances samples were covered with a drop of colloid and analysis was performed before its complete evaporation, other research groups have found it more effective to collect SERS spectra upon drying of the droplet.211In situ non-hydrolysis SERS has also been used in combination with concentrated pastes of Lee–Meisel colloids to examine natural dyes on various supports96,212 and for the identification of organic colourants from textile fibres, pastels and watercolours.155,201,213–215 Recent studies have also considered extending the applicability of colloidal pastes to the direct SERS identification of lake pigments in cross-sections,216 sometimes with the addition of hydroxypropyl cellulose as a stabilising agent and to achieve greater control over the sampled area.196 Among the main drawbacks of in situ on-the-specimen non-hydrolysis SERS with colloids and colloidal pastes are its poor spatial resolution and unevenness of sample coating, resulting in the competitive adsorption of species from adjacent paint layers on the silver substrate and in a substantial lack of spectral reproducibility.215,217 The difficulty to obtain a thin, uniform, well-adhered film of silver nanoparticles with this method was partly circumvented by the use of a hyphenated system interfacing a micro-Raman spectrometer with a scanning electron microscope (SEM),218 which aids the localization of evenly coated sample areas that are suitable for analysis. In general, it has been shown that the introduction of a hydrolysis step in the SERS analytical procedure not only avoids slower analysis compared to non-hydrolysis methodologies, but, in some cases, is also the only way to ensure highly reproducible, conclusive dye identification in samples from coloured textiles and lake-containing glaze with additional paint materials.186,206–208 Recently, several treatment strategies using acids and organic solvents were integrated into a flowchart approach designed to enable the detection and identification of unknown blue and yellow pigments of varying resonance properties and surface affinities in a single microscopic sample.219
Recent efforts towards minimally invasive SERS have explored the use of various types of peelable gels that are able to deliver the SERS-active nanoprobe to the object under study while preserving its appearance and physical integrity. A first notable example consists of a polymer hydrogel loaded with a solution containing water, dimethylformamide and disodium ethylenediaminetetraacetic acid (EDTA).220 First, the gel is applied to the area of interest and the dye is extracted owing to the combined action of the organic solvent and chelating agent; upon removal of the gel, the target dye is analysed by SERS by focusing the laser beam directly onto the gel surface. It has been shown that the amount of dye transferred is so small that no fading can be perceived. A second type of cheap, environmentally friendly hydrogel, firstly tested on mock-up fabrics and a pre-Columbian textile dyed with anthraquinoid dyes, is based on Ag-agar.221 The efficiency of this substrate has been attributed to the shrinkage of its nanocomposite structure upon drying, which is thought to induce the interaction among silver nanoparticles by creating high plasmon density sites. The initial formulation was then improved by the addition of EDTA, which has been found to play a key role as a matrix stabiliser and for the improvement of the gel's micro-extractive performances.222 Other instances of SERS-active peelable gels include a methylcellulose gel matrix doped with glucose-reduced and -stabilised silver nanoparticles,223 tested on anthraquinones as such and in unvarnished mock-up tempera panel paintings by taking SERS measurements across the dried gel drop. An upgraded formulation in which both the viscosity and nanoparticle preparation were optimised compared to the initial recipe224 has been shown to significantly reduce substrate losses that were previously observed during the peeling process.
An alternative approach for the minimally invasive in situ identification of inks and colourants on questioned documents and artworks relies on inkjet nanoparticle delivery.225 This technique is based on the use of thermal and piezoelectric print heads to deliver minute volumes of silver colloid (≈60–220 picoliters, corresponding to impact diameters of ≈50–150 μm) onto substrates with great accuracy and precision. Successful examples of application include the analysis of textile fibres, gel pen ink writing on paper, and a Japanese woodblock print dating to the end of the 19th century. Among the drawbacks are the microscopic deposits of silver nanoparticles that are left on the surface of the object upon analysis, which, although nearly invisible by optical microscopy, may be deemed unacceptable for invaluable works of art. On the other hand, the higher spatial resolution compared to SERS-active gel substrates was proven beneficial to selectively investigate single features or parts of a feature of complex objects.
The recently proposed coupling with laser ablation (LA) micro-sampling has lent the SERS technique a degree of spatial resolution of a few micrometers and subpicogram sensitivity.226 In this hyphenated method, the sample under study is placed in a holder within a sealed vacuum chamber and ablated by an intense visible laser pulse emitted by a tunable optical parametric oscillator (OPO)227 adjusted to the resonance of the target molecule; the ablated sample is then collected onto a 10 nm SERS-active silver nanoisland film made by vapour deposition that decorates one side of the vacuum chamber's quartz window; after that, excitation of the ablated analytes at 488 nm is provided to produce SERS spectra. The LA-SERS approach has enabled researchers to detect and identify water-insoluble compounds, such as copper phthalocyanine, that are difficult to probe with regular silver colloids and other standard substrates. Moreover, the technique has afforded means to successfully resolve multiple-component mixtures, such as Pigment Orange 48 (a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of quinacridone and quinacridone quinone), as well as, upon HF treatment, to characterise dyes in ancient leather samples. In a second step of this research, the LA-SERS method was refined by ablating in the ultraviolet (UV) range, which has proven especially advantageous for the analysis of multilayered, heterogeneous samples, such as single pigment particles in paint cross-sections, due to a significantly lower contamination from the matrix and materials in adjacent layers.228 Recently, the use of a fast Fourier filtering approach in combination with UV-LA-SERS was key to the successful detection and identification of particularly challenging analytes such as flavonoid-based yellow lakes with an improved signal-to-noise ratio and cleaner background.229
:50 mixture of quinacridone and quinacridone quinone), as well as, upon HF treatment, to characterise dyes in ancient leather samples. In a second step of this research, the LA-SERS method was refined by ablating in the ultraviolet (UV) range, which has proven especially advantageous for the analysis of multilayered, heterogeneous samples, such as single pigment particles in paint cross-sections, due to a significantly lower contamination from the matrix and materials in adjacent layers.228 Recently, the use of a fast Fourier filtering approach in combination with UV-LA-SERS was key to the successful detection and identification of particularly challenging analytes such as flavonoid-based yellow lakes with an improved signal-to-noise ratio and cleaner background.229
Combining the sensitivity and ability to provide vibrational information of SERS with the outstanding spatial resolution of scanning probe microscopy, tip-enhanced Raman spectroscopy (TERS) was only recently introduced in the field of cultural heritage research. A proof-of-concept study39 described the development and application of AFM-TERS instrumentation to characterize indigo and iron gall ink directly on rice paper and on a fragment of historical document with handwritten text dating to the 19th century. Measurements are performed by irradiating a nanometer-scale silver scanning tip with a focused laser beam, leading to the specific excitation and enhanced detection of those molecules that are located within the tip apex region. In addition to further improving the SERS technique's spatial resolution, this approach is minimally invasive, as dyes were shown not to be subject to transferring after the tip is retracted from the object under study.
Despite having significantly reduced the sample size and analysis time compared to chromatographic techniques, SERS has shown a substantial inability to resolve complex dye mixtures and tendency to preferentially detect one component due to resonance, solubility, affinity for the metal surface, or a combination of these factors.151 This constitutes a major limitation as, throughout history, colourants were often used in combination to obtain particular colour shades.230 Although recent studies have offered occasional proof of the simultaneous SERS detection of multiple colourants in commercial pigments and samples from paintings and textiles,208,209,215,226 in most cases when both HPLC and SERS were used for identification, while the presence of multiple dyes in a single sample had been determined by HPLC, only one could be detected by SERS.177,205 Recent research confirmed that, for instance, when alizarin and purpurin are present in mixtures with other colourants, they are always preferentially detected by the SERS technique even if present in lower concentration (up to 1:10) than the primary dyestuff.231 This has prompted researchers to pursue alternative ways to accomplish analysis of dye mixtures by SERS upon physical separation of the individual constituents. The coupling of thin-layer chromatography (TLC) with SERS has been proposed as a first, immediate solution exploiting an equipment- and cost-effective separation method. Examples of application of TLC-SERS include the separation and characterization of anthraquinones in reference solutions and extracts from a dyed wool fibre;201 of dye components in ballpoint pen inks of interest to forensic science;81 of a mixture of alkaloids found in the Syrian rue plant, which are relevant as dyes and drugs;179 and of the structurally related components of mauve, the first synthetic organic dyestuff.163 More recently, an online hyphenated system combining HPLC with photodiode array (PDA) and SERS detection was shown to provide a detailed electronic and vibrational characterization of natural dyes in mixtures.232 Ongoing research has also demonstrated the viable use of microfluidics devices, typically created by imprinting polymer structures of polydimethylsiloxane, to combine efficient separation with the fingerprinting ability typical of vibrational spectroscopies.233 Along with the substantial body of work discussed above, the relevance and incidence of the advances reported in this area of SERS demonstrate the great interest and high impact that this technique continues to generate in the field of cultural heritage research.
6. Conclusions
In this review, the recent evolutions and trends in Raman spectroscopy applied to art and archaeology are described. Many advances in Raman spectroscopy result from technical advances. These involve new hardware (e.g. fibre optics), software improvements (e.g. automated shifted baseline subtraction), new insights (e.g. micro-SORS) and new approaches (e.g. SERS). It is clear that, in light of the remarkable progresses made over the past years, new steps are set paving the way for more novel developments in this research domain.References
- Raman Spectroscopy in Archaeology and Art History, ed. H. G. M. Edwards and J. M. Chalmers, Royal Society of Chemistry, Cambridge, 2005 Search PubMed.
- P. Vandenabeele, H. G. M. Edwards and L. Moens, Chem. Rev., 2007, 107, 675 CrossRef CAS PubMed.
- F. Casadio, C. Daher and L. Bellot-Gurlet, Top. Curr. Chem., 2016, 374, 62 CrossRef PubMed.
- J. M. Madariaga, Anal. Methods, 2015, 7, 4848 RSC.
- F. Casadio and R. P. Van Duyne, Analyst, 2013, 138, 7276 RSC.
- R. L. McCreery, Raman Spectroscopy for Chemical Analysis, John Wiley & Sons, Inc., Hoboken, 2000 Search PubMed.
- Handbook of Raman Spectroscopy: From the Research Laboratory to the Process Line, ed. I. R. Lewis and H. G. M. Edwards, Marcel Dekker, New York and Basel, 2001 Search PubMed.
- P. Vandenabeele, Practical Raman Spectroscopy: An Introduction, John Wiley, Chichester, 2013 Search PubMed.
- I. M. Bell, R. J. Clark and P. J. Gibbs, Spectrochim. Acta, Part A, 1997, 53, 2159 CrossRef.
- L. Burgio and R. J. H. Clark, Spectrochim. Acta, Part A, 2001, 57, 1491 CrossRef CAS.
- P. Vandenabeele, L. Moens, H. G. M. Edwards and R. Dams, J. Raman Spectrosc., 2000, 31, 509 CrossRef CAS.
- W. Fremout and S. Saverwyns, J. Raman Spectrosc., 2012, 43, 1536 CrossRef CAS.
- R. Schütz, L. Bertinetti, I. Rabin, P. Fratzl and A. Masic, Analyst, 2013, 138, 5594 RSC.
- G. Bertolotti, D. Bersani, P. P. Lottici, M. Alesiani, T. Malcherek and J. Schluter, Anal. Bioanal. Chem., 2012, 402, 1451 CrossRef CAS PubMed.
- V. Hayez, V. Costa, J. Guillaume, H. Terryn and A. Hubin, Analyst, 2005, 130, 550 RSC.
- P. Vandenabeele, B. Wehling, L. Moens, H. Edwards, M. De Reu and G. Van Hooydonk, Anal. Chim. Acta, 2000, 407, 261 CrossRef CAS.
- C. Daher, L. Drieu, L. Bellot-Gurlet, A. Percot, C. Paris and A. S. Le Ho, J. Raman Spectrosc., 2014, 45, 1207 CrossRef CAS.
- A. Kaminska, M. Sawczak, M. Oujja, C. Domingo, M. Castillejo and G. Sliwinski, J. Raman Spectrosc., 2006, 37, 1125 CrossRef CAS.
- E. Basso, C. Invernizzi, M. Malagodi, M. F. La Russa, D. Bersani and P. P. Lottici, J. Raman Spectrosc., 2014, 45, 238 CrossRef CAS.
- G. Barone, D. Bersani, V. Crupi, F. Longo, U. Longobardo, P. P. Lottici, I. Aliatis, D. Majolino, P. Mazzoleni, S. Raneri and V. Venuti, J. Raman Spectrosc., 2014, 45, 1309 CrossRef CAS.
- G. Bertolotti, D. Bersani, P. P. Lottici, M. Alesiani, T. Malcherek and J. Schlüter, Anal. Bioanal. Chem., 2012, 402, 1451 CrossRef CAS PubMed.
- D. Bersani, S. Andò, P. Vignola, G. Moltifiori, I.-G. G. Marino, P. P. Lottici and V. Diella, Spectrochim. Acta, Part A, 2009, 73, 484 CrossRef PubMed.
- F. Ospitali, D. Bersani, G. Di Lonardo and P. P. Lottici, J. Raman Spectrosc., 2008, 39, 1066 CrossRef CAS.
- D. L. A. de Faria and F. N. Lopes, Vib. Spectrosc., 2007, 45, 117 CrossRef CAS.
- A. Rousaki, C. Bellelli, M. C. Calatayud, V. Aldazabal, G. Custo, L. Moens, P. Vandenabeele and C. Vazquez, J. Raman Spectrosc., 2015, 46, 1016 CrossRef CAS.
- E. Cantisani, M. Cavalieri, C. Lofrumento, E. Pecchioni and M. Ricci, Archaeol. Anthropol. Sci., 2012, 4, 29 CrossRef.
- M. J. Ayora-Cañada, A. Domínguez-Arranz and A. Dominguez-Vidal, J. Raman Spectrosc., 2012, 43, 317 CrossRef.
- A. Raškovska, B. Minčeva-Šukarova, O. Grupče and P. Colomban, J. Raman Spectrosc., 2010, 41, 431 Search PubMed.
- J. Zuo, C. Xu, C. Wang and Z. Yushi, J. Raman Spectrosc., 1999, 30, 1053 CrossRef CAS.
- N. C. Scherrer, Z. Stefan, D. Francoise, F. Annette and K. Renate, Spectrochim. Acta, Part A, 2009, 73, 505 CrossRef PubMed.
- C. Baita, P. P. Lottici, E. Salvioli-Mariani, P. Vandenabeele, M. Librenti, F. Antonelli and D. Bersani, J. Raman Spectrosc., 2014, 45, 114 CrossRef CAS.
- J. R. Petriglieri, E. Salvioli-Mariani, L. Mantovani, M. Tribaudino, P. P. Lottici, C. Laporte-Magoni and D. Bersani, J. Raman Spectrosc., 2015, 46, 953 CrossRef CAS.
- C. Lofrumento, A. Zoppi and E. M. Castellucci, J. Raman Spectrosc., 2004, 35, 650 CrossRef CAS.
- M. C. Caggiani, P. Acquafredda, P. Colomban and A. Mangone, J. Raman Spectrosc., 2014, 45, 1251 CrossRef CAS.
- C. Conti, M. Realini, C. Colombo, K. Sowoidnich, N. K. Afseth, M. Bertasa, A. Botteon and P. Matousek, Anal. Chem., 2015, 87, 5810 CrossRef CAS PubMed.
- C. Conti, C. Colombo, M. Realini, G. Zerbi and P. Matousek, Appl. Spectrosc., 2014, 68, 686 CrossRef CAS PubMed.
- I. Guerra and C. Cardell, J. Microsc., 2015, 260, 47 CrossRef CAS PubMed.
- D. Bersani, P. P. Lottici, S. Virgenti, A. Sodo, G. Malvestuto, A. Botti, E. Salvioli-Mariani, M. Tribaudino, F. Ospitali and M. Catarsi, J. Raman Spectrosc., 2010, 41, 1556 CrossRef.
- D. Kurouski, S. Zaleski, F. Casadio, R. P. Van Duyne and N. C. Shah, J. Am. Chem. Soc., 2014, 136, 8677 CrossRef CAS PubMed.
- C. Fotakis, D. Anglos, V. Zafiropulos, S. Georgiou and V. Tornari, Lasers in the Preservation of Cultural Heritage: Principles and Applications, CRC Press, New York, 2006 Search PubMed.
- M. Hoehse, A. Paul, I. Gornushkin and U. Panne, Anal. Bioanal. Chem., 2012, 402, 1443 CrossRef CAS PubMed.
- U. Zimmermann, E. S. Kristoffersen, P. D. Fredriksen, S. A. R. Bertolino, S. Ando and D. Bersani, Sediment. Geol., 2015, 336, 183 CrossRef.
- P. Colomban, G. Sagon and X. Faurel, J. Raman Spectrosc., 2001, 32, 351 CrossRef CAS.
- M. Bouchard and D. C. Smith, Spectrochim. Acta, Part A, 2003, 59, 2247 CrossRef CAS.
- D. Lauwers, A. G. Hutado, V. Tanevska, L. Moens, D. Bersani and P. Vandenabeele, Spectrochim. Acta, Part A, 2014, 118, 294 CrossRef CAS PubMed.
- E. Van Elslande, S. Lecomte and A. S. Le Hô, J. Raman Spectrosc., 2008, 39, 1001 CrossRef CAS.
- L. Medeghini, P. P. Lottici, C. De Vito, S. Mignardi and D. Bersani, J. Raman Spectrosc., 2014, 45, 1244 CrossRef CAS.
- C. Conti, J. Striova, I. Aliatis, E. Possenti, G. Massonnet, C. Muehlethaler, T. Poli and M. Positano, J. Raman Spectrosc., 2015, 45, 1186 CrossRef.
- L. Burgio, R. J. H. Clark and H. Toftlumd, Acta Chem. Scand., 1999, 53, 81 CrossRef.
- P. Schmidt, G. Porraz, L. Bellot-Gurlet, E. February, B. Ligouis, C. Paris, P.-J. Texier, J. E. Parkington, C. E. Miller, K. G. Nickel and N. J. Conard, J. Hum. Evol., 2015, 85, 22 CrossRef PubMed.
- J. Aramendia, L. Gomez-Nubla, L. Bellot-Gurlet, K. Castro, G. Arana and J. M. Madariaga, Int. Biodeterior. Biodegrad., 2015, 104, 59 CrossRef CAS.
- J. Aramendia, L. Gomez-Nubla, L. Bellot-Gurlet, K. Castro, C. Paris, P. Colomban and J. M. Madariaga, J. Raman Spectrosc., 2014, 45, 1076 CrossRef CAS.
- I. Aliatis, E. Lambruschi, L. Mantovani, D. Bersani, S. Andò, G. Diego Gatta, P. Gentile, E. Salvioli-Mariani, M. Prencipe, M. Tribaudino and P. P. Lottici, J. Raman Spectrosc., 2015, 46, 501 CrossRef CAS.
- L. Mantovani, M. Tribaudino, I. Aliatis, E. Lambruschi, P. P. Lottici and D. Bersani, Phys. Chem. Miner., 2014, 42, 179 CrossRef.
- S. Lowry, D. Wieboldt, D. Dalrymple, R. Jasinevicius and R. T. Downs, Spectroscopy, 2009, 24, 52 CAS.
- J. M. Pérez and R. Esteve-Tébar, Archaeometry, 2004, 46, 607 CrossRef.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673 CrossRef CAS.
- D. Bersani, G. Azzi, E. Lambruschi, G. Barone, P. Mazzoleni, S. Raneri, U. Longobardo and P. P. Lottici, J. Raman Spectrosc., 2014, 45, 1293 CrossRef CAS.
- I. Moroz, M. Roth, M. Boudeulle and G. Panczer, J. Raman Spectrosc., 2000, 31, 485 CrossRef CAS.
- G. Barone, D. Bersani, P. P. Lottici, P. Mazzoleni, S. Raneri and U. Longobardo, J. Raman Spectrosc., 2016 DOI:10.1002/jrs.4919.
- L. Bergamonti, D. Bersani, D. Csermely and P. P. Lottici, Spectrosc. Lett., 2011, 44, 453 CrossRef CAS.
- D. Gill, R. G. Kilponen and L. Rimai, Nature, 1970, 227, 743 CrossRef CAS PubMed.
- I. V Ermakov, M. Sharifzadeh, M. Ermakova and W. Gellermann, J. Biomed. Opt., 2005, 10, 64028 CrossRef PubMed.
- A. M. Correia, M. J. V. Oliveira, R. J. H. Clark, M. I. Ribeiro and M. L. Duarte, Anal. Chem., 2008, 80, 1482 CrossRef CAS PubMed.
- P. Colomban, J. Raman Spectrosc., 2003, 34, 420 CrossRef CAS.
- E. V. Efremov, F. Ariese and C. Gooijer, Anal. Chim. Acta, 2008, 606, 119 CrossRef CAS PubMed.
- C. Coupry, A. Lutiè, G. Sagon and F. Froment, in Raman Spectroscopy in Archaeology and Art History, ed. H. G. M. Edwards and J. M. Chalmers, Royal Society of Chemistry, Cambridge, 2005, p. 207 Search PubMed.
- S. E. J. Bell, in Raman Spectroscopy in Archaeology and Art History, ed. H. G. M. Edwards and J. M. Chalmers, Royal Society of Chemistry, Cambridge, 2005, p. 292 Search PubMed.
- S. Karampelas, E. Fritsch, J.-Y. Mevellec, J.-P. Gauthier, S. Sklavounos and T. Soldatos, J. Raman Spectrosc., 2007, 38, 217 CrossRef CAS.
- R. M. Seifar, J. M. Verheul, F. Ariese, U. A. T. Brinkman and C. Gooijer, Analyst, 2001, 126, 1418 RSC.
- G. Massonnet, P. Buzzini, G. Jochem, M. Stauber, T. Coyle, C. Roux, J. Thomas, H. Leijenhorst, Z. Van Zanten, K. Wiggins, C. Russell, S. Chabli and A. Rosengarten, J. Forensic Sci., 2005, 50, 1028 CrossRef CAS PubMed.
- X. Faurel, A. Vanderperre and P. Colomban, J. Raman Spectrosc., 2003, 34, 290 CrossRef CAS.
- M. Fujiwara, H. Hamaguchi and M. Tasumi, Appl. Spectrosc., 1986, 40, 137 CrossRef CAS.
- Y. Wang and R. L. McCreery, Anal. Chem., 1989, 61, 2647 CrossRef CAS.
- G. Burrafato, M. Calabrese, A. Cosentino, A. M. Gueli, S. O. Troja and A. Zuccarello, J. Raman Spectrosc., 2004, 35, 879 CrossRef CAS.
- F. Froment, A. Tournié and P. Colomban, J. Raman Spectrosc., 2008, 39, 560 CrossRef CAS.
- A. Pan, E. Rebollar, S. Chiussi, J. Serra, P. González and B. León, J. Raman Spectrosc., 2010, 41, 1449 CrossRef.
- A. Nevin, I. Osticioli, D. Anglos, A. Burnstock, S. Cather and E. M. Castellucci, Anal. Chem., 2007, 79, 6143 CrossRef CAS PubMed.
- M. L. Lewis, I. R. Lewis and P. R. Griffiths, Appl. Spectrosc., 2004, 58, 420 CrossRef CAS PubMed.
- A. S. Lee, P. J. Mahon and D. C. Creagh, Vib. Spectrosc., 2006, 41, 170 CrossRef CAS.
- I. Geiman, M. Leona and J. R. Lombardi, J. Forensic Sci., 2009, 54, 947 CrossRef CAS PubMed.
- H. G. M. Edwards, E. Beale, N. C. Garrington and J. M. Alia, J. Raman Spectrosc., 2007, 38, 316 CrossRef CAS.
- H. G. M. Edwards, E. M. Newton, S. O'Connor and D. Evans, Anal. Bioanal. Chem., 2010, 397, 2685 CrossRef CAS PubMed.
- H. G. M. Edwards, J. Mol. Struct., 2003, 661–2, 271 CrossRef.
- P. Colomban and D. Mancini, Arts, 2013, 2, 111 CrossRef.
- F. S. Manciu, A. Ramirez, W. Durrer, J. Govani and R. R. Chianelli, J. Raman Spectrosc., 2008, 39, 1257 CrossRef CAS.
- A. Baran, A. Fiedler, H. Schulz and M. Baranska, Anal. Methods, 2010, 2, 1372 RSC.
- H. G. M. Edwards, L. Drummond and J. Russ, Spectrochim. Acta, Part A, 1998, 54, 1849 CrossRef.
- M. Oujja, C. Vázquez-Calvo, M. Sanz, M. Á. De Buergo, R. Fort and M. Castillejo, Anal. Bioanal. Chem., 2012, 402, 1433 CrossRef CAS PubMed.
- R. David, H. G. M. Edwards, D. W. Farwell and D. L. De Faria, Archaeometry, 2001, 43, 461 Search PubMed.
- O. M. M. Gui, A. Fǎlǎmaş, L. Barbu-Tudoran, M. Aluaş, B. Giambra and S. Cîntǎ Pînzaru, J. Raman Spectrosc., 2013, 44, 277 CrossRef CAS.
- K. Castro, M. Pérez-Alonso, M. D. Rodríguez-Laso, L. A. Fernández and J. M. Madariaga, Anal. Bioanal. Chem., 2005, 382, 248 CrossRef CAS PubMed.
- J. R. Gilchrist, J. Rebello, D. Lanzisera and J. Noonan, Laser Focus World, 2000, 36, 149 CAS.
- M. W. W. Meyer, J. S. S. Lupoi and E. A. A. Smith, Anal. Chim. Acta, 2011, 706, 164 CrossRef CAS PubMed.
- C. M. Schmidt and K. A. Trentelman, e-Preserv. Sci., 2009, 6, 10 CAS.
- M. V. Canamares, M. Leona, M. Bouchard, C. M. Grzywacz, J. Wouters and K. Trentelman, J. Raman Spectrosc., 2010, 41, 391 CAS.
- P. Vandenabeele, H. G. M. Edwards and J. Jehlicka, Chem. Soc. Rev., 2014, 43, 2628 RSC.
- P. Vandenabeele and M. K. Donais, Appl. Spectrosc., 2016, 70, 27 CrossRef CAS PubMed.
- C. Conti, A. Botteon, M. Bertasa, C. Colombo, M. Realini and D. Sali, Analyst, 2016, 141, 4599 RSC.
- A. P. Shreve, N. J. Cherepy and R. A. Mathies, Appl. Spectrosc., 1992, 46, 707 CrossRef CAS.
- G. Barone, D. Bersani, J. Jehlicka, P. P. Lottici, P. Mazzoleni, S. Raneri, P. Vandenabeele, C. Di Giacomo and G. Larina, J. Raman Spectrosc., 2015, 46, 989 CrossRef CAS.
- C. Baita, P. P. Lottici, E. Salvioli-Mariani, P. Vandenabeele, M. Librenti, F. Antonelli and D. Bersani, J. Raman Spectrosc., 2014, 45, 114 CrossRef CAS.
- J. Jehlicka, P. Vitek, H. G. M. Edwards, M. Hargreaves and T. Capoun, Spectrochim. Acta, Part A, 2009, 73, 410 CrossRef CAS PubMed.
- D. Lauwers, A. Candeias, A. Coccato, J. Mirao, L. Moens and P. Vandenabeele, Spectrochim. Acta, Part A, 2016, 157, 146 CrossRef CAS PubMed.
- E. Basso, C. Invernizzi, M. Malagodi, M. F. La Russa, D. Bersani and P. P. Lottici, J. Raman Spectrosc., 2014, 45, 238 CrossRef CAS.
- P. Ricciardi, Ph. Colomban, A. Tournie, M. Macchiarola and N. Ayed, J. Archaeol. Sci., 2009, 36, 2551 CrossRef.
- A. Deneckere, W. Schudel, M. Van Bos, H. Wouters, A. Bergmans, P. Vandenabeele and L. Moens, Spectrochim. Acta, Part A, 2010, 75, 511 CrossRef PubMed.
- P. Vandenabeele, K. Lambert, S. Matthys, W. Schudel, A. Bergmans and L. Moens, Anal. Bioanal. Chem., 2005, 383, 707 CrossRef CAS PubMed.
- M. Olivares, K. Castro, M. S. Corchon, D. Garate, X. Murelaga, A. Sarmiento and N. Etxebarria, J. Archaeol. Sci., 2013, 40, 1354 CrossRef CAS.
- L. C. Prinsloo, A. Tournie, Ph. Colomban, C. Paris and S. T. Bassett, J. Archaeol. Sci., 2013, 40, 2981 CrossRef CAS.
- P. Vandenabeele, T. L. Weis, E. R. Grant and L. J. Moens, Anal. Bioanal. Chem., 2004, 379, 137 CrossRef CAS PubMed.
- M. Bouchard, D. C. Smith and C. Carabatos-Nedelec, Spectrochim. Acta, Part A, 2007, 68, 1101 CrossRef PubMed.
- P. Colomban and A. Tournie, J. Cult. Herit., 2007, 8, 242 CrossRef.
- A. Tournie, L. C. Prinsloo, C. Paris, P. Colomban and B. Smith, J. Raman Spectrosc., 2011, 42, 399 CrossRef CAS.
- Ph. Colomban, J. Raman Spectrosc., 2012, 43, 1529 CrossRef CAS.
- P. Vandenabeele, J. Tate and L. Moens, Anal. Bioanal. Chem., 2007, 387, 813 CrossRef CAS PubMed.
- P. Vandenabeele, R. Garcia-Moreno, F. Mathis, K. Leterme, E. Van Elslande, F. P. Hocquet, S. Rakkaa, D. Laboury, L. Moens, D. Strivay and M. Hartwig, Spectrochim. Acta, Part A, 2009, 73, 546 CrossRef PubMed.
- A. Deneckere, M. De Reu, M. P. J. Martens, K. De Coene, B. Vekemans, L. Vincze, P. De Maeyer, P. Vandenabeele and L. Moens, Spectrochim. Acta, Part A, 2011, 80, 125 CrossRef CAS PubMed.
- C. Miliani, F. Rosi, B. G. Brunetti and A. Sgamellotti, Acc. Chem. Res., 2010, 43, 728 CrossRef CAS PubMed.
- B. G. Brunetti, M. Matteini, C. Miliani, L. Pezzati and D. Pinna, MOLAB, a Mobile Laboratory for In Situ Non-Invasive Studies in Arts and Archaeology, in Proceedings From: Lasers in the Conservation of Artworks, M. Schreiner, Vienna, Austria, September 21–25 2005, p. 453 Search PubMed.
- K. S. Andrikopoulos, S. Daniilia, B. Roussel and K. Janssens, J. Raman Spectrosc., 2006, 37, 1026 CrossRef CAS.
- X. Zhu, T. Xu, Q. Lin and Y. Duan, Appl. Spectrosc. Rev., 2013, 49, 64 CrossRef.
- R. Bruder, V. Detalle and C. Coupry, J. Raman Spectrosc., 2007, 38, 909 CrossRef CAS.
- M. Hoehse, D. Mory, S. Florek, F. Weritz, I. Gornushkin and U. Panne, Spectrochim. Acta, Part B, 2009, 64, 1219 CrossRef.
- I. Osticioli, M. Wolf and D. Anglos, Appl. Spectrosc., 2008, 62, 1242 CrossRef CAS PubMed.
- L. Van de Voorde, J. Van Pevenage, K. De Langhe, R. De Wolf, B. Vekemans, L. Vincze, P. Vandenabeele and M. P. J. Martens, Spectrochim. Acta, Part B, 2014, 97, 1 CrossRef CAS.
- L. Van de Voorde, M. Vandevijvere, B. Vekemans, J. Van Pevenage, J. Caen, P. Vandenabeele, P. Van Espen and L. Vincze, Spectrochim. Acta, Part B, 2014, 102, 28 CrossRef CAS.
- O. Gomez-Laserna, I. Arrizabalaga, N. Prieto-Taboada, M. A. Olazabal, G. Arana and J. M. Madariaga, Anal. Bioanal. Chem., 2015, 407, 5635 CrossRef CAS PubMed.
- J. Aramendia, L. Gomez-Nubla, K. Castro and J. M. Madariaga, Microchem. J., 2014, 115, 138 CrossRef CAS.
- O. Gomez-Laserna, M. A. Olazabal, H. Morillas, N. Prieto-Taboada, I. Martinez-Arkarazo, G. Arana and J. M. Madariaga, J. Raman Spectrosc., 2013, 44, 1277 CrossRef CAS.
- J. A. Carrero, N. Goienaga, M. Olivares, I. Martinez-Arkarazo, G. Arana and J. M. Madariaga, J. Raman Spectrosc., 2012, 43, 1498 CrossRef CAS.
- M. Maguregui, A. Sarmiento, I. Martinez-Arkarazo, M. Angulo, K. Castro, G. Arana, N. Etxebarria and J. M. Madariaga, Anal. Bioanal. Chem., 2008, 391, 1361 CrossRef CAS PubMed.
- C. Conti, M. Realini, C. Colombo and P. Matousek, J. Raman Spectrosc., 2015, 46, 476 CrossRef CAS.
- P. Matousek, C. Conti, M. Realini and C. Colombo, Analyst, 2015, 141, 731 RSC.
- P. Matousek, I. P. Clark, E. R. C. Draper, M. D. Morris, A. E. Goodship, N. Everall, M. Towrie, W. F. Finney and A. W. Parker, Appl. Spectrosc., 2005, 59, 393 CrossRef CAS PubMed.
- C. Conti, C. Colombo, M. Realini and P. Matousek, Analyst, 2015, 140, 8127 RSC.
- C. Conti, M. Realini, A. Botteon, C. Colombo, S. Noll, S. R. Elliott and P. Matousek, Appl. Spectrosc., 2016, 70, 156 CrossRef CAS PubMed.
- C. Conti, M. Realini, C. Colombo, A. Botteon and P. Matousek, J. Raman Spectrosc., 2016, 47, 565 CrossRef CAS.
- C. Conti, A. Botteon, C. Colombo, M. Realini and P. Matousek, Analyst, 2016, 141, 5374 RSC.
- M. Realini, A. Botteon, C. Conti, C. Colombo and P. Matousek, Analyst, 2016, 141, 3012 RSC.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163 CrossRef CAS.
- R. Aroca, Surface-Enhanced Vibrational Spectroscopy, John Wiley & Sons, Chichester, UK, 2006 Search PubMed.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2008, 112, 5605 CAS.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734 CrossRef CAS PubMed.
- D. L. Jeanmaire and R. P. van Duyne, J. Electroanal. Chem., 1977, 84, 1 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215 CrossRef CAS.
- S. Nie and S. E. Emory, Science, 1997, 275, 1102 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667 CrossRef CAS.
- B. Guineau and V. Guichard, ICOM Committee for Conservation: 8th Triennial Meeting, Preprints, The Getty Conservation Institute, Marina del Rey, CA, 1987, vol. 2, p. 659 Search PubMed.
- B. Guineau, Pigments et Colorants de l'Antiquitè et du Moyen Age: Teinture, Peinture, Enluminure Études Historiques et Physico-chimiques, Colloques International du CNRS, Departement des Sciences de l'Homme et de la Societè, Éditions du Centre National de la Recherche Scientifique, Paris, 1990, p. 249 Search PubMed.
- I. T. Shadi, B. Z. Chowdry, M. J. Snowden and R. Whitnall, J. Raman Spectrosc., 2004, 35, 800 CrossRef CAS.
- M. V. Cañamares, J. V. Garcia-Ramos, C. Domingo and S. J. Sanchez-Cortes, J. Raman Spectrosc., 2004, 35, 921 CrossRef.
- M. Leona, in Proceedings Volume of the Sixth Infrared and Raman Users Group Conference, ed. M. Picollo, Publisher Il Prato, Florence, TN, Padova, 2005 Search PubMed.
- K. Chen, M. Leona and T. Vo-Dinh, Sens. Rev., 2007, 27, 109 CrossRef.
- K. L. Wustholz, C. L. Brosseau, F. Casadio and R. P. Van Duyne, Phys. Chem. Chem. Phys., 2009, 11, 7350 RSC.
- F. Casadio, M. Leona, J. R. Lombardi and R. P. Van Duyne, Acc. Chem. Res., 2010, 43, 782 CrossRef CAS PubMed.
- F. Pozzi and M. Leona, J. Raman Spectrosc., 2016, 47, 67 CrossRef CAS.
- F. Pozzi, S. Zaleski, F. Casadio, M. Leona, J. R. Lombardi and R. P. Van Duyne, in Nanoscience and Cultural Heritage, ed. P. Dillmann, L. Bellot-Gurlet and I. Nenner, Springer, 2016, in press Search PubMed.
- N. G. Greeneltch, A. S. Davis, N. A. Valley, F. Casadio, G. C. Schatz, R. P. Van Duyne and N. C. Shah, J. Phys. Chem. A, 2012, 116, 11863 CrossRef CAS PubMed.
- M. V. Cañamares, C. Chenal, R. L. Birke and J. R. Lombardi, J. Phys. Chem. C, 2008, 112, 20295 Search PubMed.
- J. Chang, M. V. Cañamares, M. Aydin, W. Vetter, M. Schreiner, W. Xub and J. R. Lombardi, J. Raman Spectrosc., 2009, 40, 1557 CrossRef CAS.
- Y. Xie, Y. Li, Y. Sun, H. Wang, H. Qian and W. Yao, Spectrochim. Acta, Part A, 2012, 96, 600 CrossRef CAS PubMed.
- M. V. Cañamares, D. A. Reagan, J. R. Lombardi and M. Leona, J. Raman Spectrosc., 2014, 45, 1147 CrossRef.
- M. V. Cañamares and J. R. Lombardi, J. Phys. Chem. C, 2015, 119, 14297 Search PubMed.
- K. Chen, M. Leona, K.-C. Vo-Dinh, F. Yan, M. B. Wabuyele and T. Vo-Dinh, J. Raman Spectrosc., 2006, 37, 520 CrossRef CAS.
- M. V. Cañamares, J. V. Garcia-Ramos, C. Domingo and S. Sanchez-Cortes, Vib. Spectrosc., 2006, 40, 161 CrossRef.
- M. V. Cañamares and M. Leona, J. Raman Spectrosc., 2007, 38, 1259 CrossRef.
- A. Baran, B. Wrzosek, J. Bukowska, L. M. Proniewicz and M. Baranska, J. Raman Spectrosc., 2009, 40, 436 CrossRef CAS.
- D. C. Rambaldi, F. Pozzi, N. Shibayama, M. Leona and F. Preusser, J. Raman Spectrosc., 2015, 46(11), 1073 CrossRef CAS.
- Z. Jurasekova, J. V. Garcia-Ramos, C. Domingo and S. Sanchez-Cortes, J. Raman Spectrosc., 2006, 37, 1239 CrossRef CAS.
- T. Teslova, C. Corredor, R. Livingstone, T. Spataru, R. L. Birke, J. R. Lombardi, M. V. Cañamares and M. Leona, J. Raman Spectrosc., 2007, 38, 802 CrossRef CAS.
- M. Wang, T. Teslova, F. Xu, T. Spataru, J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2007, 111, 3038 CAS.
- C. Corredor, T. Teslova, M. V. Cañamares, Z. Chen, J. Zhang, J. R. Lombardi and M. Leona, Vib. Spectrosc., 2009, 49, 190 CrossRef CAS.
- M. V. Cañamares, J. R. Lombardi and M. Leona, e-Preserv. Sci., 2009, 6, 81 Search PubMed.
- Z. Jurasekova, C. Domingo, J. V. Garcia-Ramos and S. Sanchez-Cortes, J. Raman Spectrosc., 2012, 43, 1913 CrossRef CAS.
- H. E. Mayhew, D. M. Fabian, S. A. Svoboda and K. L. Wustholz, Analyst, 2013, 138, 4493 RSC.
- M. Leona and J. R. Lombardi, J. Raman Spectrosc., 2007, 38, 853 CrossRef CAS.
- M. V. Cañamares, J. R. Lombardi and M. Leona, J. Raman Spectrosc., 2008, 39, 1907 CrossRef.
- F. Pozzi, N. Shibayama, M. Leona and J. R. Lombardi, J. Raman Spectrosc., 2013, 44, 102 CrossRef CAS.
- M. V. Cañamares, J. V. Garcia-Ramos and S. Sanchez-Cortes, Appl. Spectrosc., 2006, 60, 1386 CrossRef PubMed.
- L. H. Oakley, D. M. Fabian, H. E. Mayhew, S. A. Svoboda and K. L. Wustholz, Anal. Chem., 2012, 84, 8006 CrossRef CAS PubMed.
- E. Platania, C. Lofrumento, E. Lottini, E. Azzaro, M. Ricci and M. Becucci, Anal. Bioanal. Chem., 2015, 407, 6505 CrossRef CAS PubMed.
- S. Bruni, V. Guglielmi and F. Pozzi, J. Raman Spectrosc., 2010, 41, 175 CAS.
- B. Doherty, F. Gabrieli, C. Clementi, D. Cardon, A. Sgamellotti, B. Brunetti and C. Miliani, J. Raman Spectrosc., 2014, 45, 723 CrossRef CAS.
- C. Zaffino, B. Russo and S. Bruni, Spectrochim. Acta, Part A, 2015, 149, 41 CrossRef CAS PubMed.
- C. Zaffino, S. Bruni, V. Guglielmi and E. De Luca, J. Raman Spectrosc., 2014, 45, 211 CrossRef CAS.
- S. Bruni, V. Guglielmi and F. Pozzi, J. Raman Spectrosc., 2011, 42, 1267 CrossRef CAS.
- E. Casanova-González, A. García-Bucio, J. Luis Ruvalcaba-Sil, V. Santos-Vasquez, B. Esquivel, T. Falcón, E. Arroyo, S. Zetina, M. L. Roldán and C. Domingo, J. Raman Spectrosc., 2012, 43, 1551 CrossRef.
- F. Pozzi, S. Porcinai, J. R. Lombardi and M. Leona, Anal. Methods, 2013, 5, 4205 RSC.
- P. C. Lee and D. J. Meisel, J. Phys. Chem., 1982, 84, 3391 CrossRef.
- M. Leona, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14757 CrossRef CAS PubMed.
- M. V. Cañamares, J. V. Garcia-Ramos, S. Sanchez-Cortes, M. Castillejo and M. Oujja, J. Colloid Interface Sci., 2008, 326, 103 CrossRef PubMed.
- M. V. Cañamares, J. V. Garcia-Ramos, J. D. Gómez-Varga, C. Domingo and S. Sanchez-Cortes, Langmuir, 2007, 23, 5210 CrossRef PubMed.
- Z. Jurasekova, C. Domingo, J. V. Garcia-Ramos and S. Sanchez-Cortes, J. Raman Spectrosc., 2008, 39, 1309 CrossRef CAS.
- Z. Jurasekova, E. del Puerto, G. Bruno, J. V. Garcia-Ramos, S. Sanchez-Cortes and C. Domingo, J. Raman Spectrosc., 2010, 41, 1455 CrossRef.
- K. Retko, P. Ropreta and R. Cerc Korošec, J. Raman Spectrosc., 2014, 45, 1140 CrossRef CAS.
- S. Sánchez-Cortés and J. V. Garcia-Ramos, J. Raman Spectrosc., 1998, 29, 365 CrossRef.
- S. E. J. Bell and N. M. S. Sirimuthu, J. Phys. Chem. A, 2005, 109, 7405 CrossRef CAS PubMed.
- A. V. Whitney, R. P. Van Duyne and F. Casadio, Proc. SPIE, 2005, 5993, 117 CrossRef.
- A. V. Whitney, R. P. Van Duyne and F. Casadio, J. Raman Spectrosc., 2006, 37, 993 CrossRef CAS.
- C. L. Brosseau, A. Gambardella, F. Casadio, C. M. Grzywacz, J. Wouters and R. P. Van Duyne, Anal. Chem., 2009, 81, 3056 CrossRef CAS PubMed.
- A. V. Whitney, F. Casadio and R. P. Van Duyne, Appl. Spectrosc., 2007, 61, 994 CrossRef CAS PubMed.
- M. Leona, J. Stenger and E. Ferloni, J. Raman Spectrosc., 2006, 37, 981 CrossRef CAS.
- S. Bruni, V. Guglielmi, F. Pozzi and A. M. Mercuri, J. Raman Spectrosc., 2011, 42, 465 CrossRef CAS.
- F. Pozzi, G. Poldi, S. Bruni, E. De Luca and V. Guglielmi, Archaeol. Anthropol. Sci., 2012, 4(3), 185 CrossRef.
- F. Pozzi, J. R. Lombardi, S. Bruni and M. Leona, Anal. Chem., 2012, 84, 3751 CrossRef CAS PubMed.
- F. Pozzi, J. R. Lombardi and M. Leona, Heritage Sci., 2013, 1, 23 CrossRef.
- F. Pozzi, K. J. van den Berg, I. Fiedler and F. Casadio, J. Raman Spectrosc., 2014, 45, 1119 CrossRef CAS.
- R. Castro, F. Pozzi, M. Leona and M. J. Melo, J. Raman Spectrosc., 2014, 45, 1172 CrossRef CAS.
- F. Pozzi, L. K. Chang and F. Casadio, in ICOM-CC 17th Triennial Conference Preprints, Melbourne, ed. J. Bridgland, International Council of Museums, Paris, 15–19 September 2014, p. 8, art. 1808, ISBN 978-92-9012-410-8 Search PubMed.
- A. El Bakkali, T. Lamhasni, M. Haddad, S. Ait Lyazidi, S. Sanchez-Cortes and E. del Puerto Nevado, J. Raman Spectrosc., 2013, 44, 114 CrossRef CAS.
- M. V. Cañamares, S. Sanchez-Cortes and J. V. Garcia-Ramos, in IRUG7 Proceedings, Museum of Modern Art, New York, 28–31 March 2006, p. 19 Search PubMed.
- C. L. Brosseau, K. S. Rayner, F. Casadio, C. M. Grzywacz and R. P. Van Duyne, Anal. Chem., 2009, 81, 7443 CrossRef CAS PubMed.
- C. L. Brosseau, F. Casadio and R. P. Van Duyne, J. Raman Spectrosc., 2011, 42, 1305 CrossRef CAS.
- A. Idone, M. Gulmini, A.-I. Henry, F. Casadio, L. Chang, L. Appolonia, R. P. Van Duyne and N. C. Shah, Analyst, 2013, 138, 5895 RSC.
- A. Idone, M. Aceto, E. Diana, L. Appolonia and M. Gulmini, J. Raman Spectrosc., 2014, 45, 1127 CrossRef CAS.
- L. H. Oakley, S. A. Dinehart, S. A. Svoboda and K. L. Wustholz, Anal. Chem., 2011, 83, 3986 CrossRef CAS PubMed.
- S. V. Prikhodko, D. C. Rambaldi, A. King, E. Burr, V. Muros and I. Kakoulli, J. Raman Spectrosc., 2015, 46, 632 CrossRef CAS.
- J. Y. Roh, M. K. Mateck, S. A. Svoboda and K. L. Wustholz, Anal. Chem., 2016, 88, 2028 CrossRef CAS PubMed.
- M. Leona, P. Decuzzi, T. A. Kubic, G. Gates and J. R. Lombardi, Anal. Chem., 2011, 83, 3990 CrossRef CAS PubMed.
- C. Lofrumento, M. Ricci, E. Platania, M. Becuccia and E. Castellucci, J. Raman Spectrosc., 2013, 44, 47 CrossRef CAS.
- E. Platania, J. R. Lombardi, M. Leona, N. Shibayama, C. Lofrumento, M. Ricci, M. Becucci and E. Castellucci, J. Raman Spectrosc., 2014, 45, 1133 CrossRef CAS.
- B. Doherty, B. G. Brunetti, A. Sgamellotti and C. Miliani, J. Raman Spectrosc., 2011, 42, 1932 CrossRef CAS.
- B. Doherty, F. Presciutti, A. Sgamellotti, B. G. Brunetti and C. Miliani, J. Raman Spectrosc., 2014, 45, 1153 CrossRef CAS.
- D. P. Benedetti, J. Zhang, T. J. Tague Jr, J. R. Lombardi and M. Leona, J. Raman Spectrosc., 2014, 45, 123 CrossRef CAS.
- P. S. Londero, J. R. Lombardi and M. Leona, Anal. Chem., 2013, 85, 5463 CrossRef CAS PubMed.
- P. Londero, J. R. Lombardi and M. Leona, J. Raman Spectrosc., 2013, 44, 131 CrossRef CAS.
- A. Cesaratto, M. Leona, J. R. Lombardi, D. Comelli, A. Nevin and P. Londero, Angew. Chem., Int. Ed., 2014, 53, 14373 CrossRef CAS PubMed.
- A. Cesaratto, P. Londero, N. Shibayama, J. R. Lombardi and M. Leona, Microchem. J., 2016, 126, 237 CrossRef CAS.
- D. Cardon, Natural Dyes: Sources, Tradition, Technology and Science, Archetype Publications, London, 2007 Search PubMed.
- F. Pozzi, S. Zaleski, F. Casadio and R. P. Van Duyne, J. Phys. Chem. C, 2016, 120, 21017 CAS.
- C. Zaffino, G. D. Bedini, G. Mazzola, V. Guglielmi and S. Bruni, J. Raman Spectrosc., 2016, 47, 607 CrossRef CAS.
- S. Zaleski, A. Zrimsek, F. Casadio, N. C. Shah and R. P. Van Duyne, Microfluidics Combined With Surface-Enhanced Raman Spectroscopy for Multiple Analyte Dyestuff Identification, Presented at Scientific Methods in Cultural Heritage Research Gordon Research Conference, Sunday River Resort, Newry, ME, July 27–August 1, 2014 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |