
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe soybean lipoxygenase-fluorescein reaction may be used to assess antioxidant capacity of phytochemicals and serum
M.
Soccio
,
M. N.
Laus
,
M.
Alfarano
and
D.
Pastore
 *
*
Dipartimento di Scienze Agrarie, degli Alimenti e dell'Ambiente, Università degli Studi di Foggia, Via Napoli, 25, 71122, Foggia, Italy. E-mail: donato.pastore@unifg.it
First published on 4th May 2016
Abstract
Evaluation of the putative beneficial effects of food antioxidants by in vitro assays of the antioxidant capacity (AC) of food extracts sometimes appears questionable. A more realistic evaluation of antioxidant effectiveness may derive from the integration of in vitro assays with ex vivo assays of blood/serum/plasma AC after food intake. The aim of this study was to develop the novel lipoxygenase-fluorescein (LOX-FL) method to assay the AC of both food extracts and serum/plasma. This method was applied on both extracts from the antioxidant-rich dietary cereal supplement, Lisosan G, and serum (seven subjects) for 240 min after the intake of 20 g of supplement. The widely used ORAC and TEAC methods were used for comparison. The new LOX-FL method is based on the reaction between the soybean LOX-1 isoenzyme and FL in the presence of linoleic acid, which undergoes hydroperoxidation, thus generating physiological reactive species, including LOO˙, LO˙, HO˙ and 1O2, which are able to quench FL. The quenching rate is slowed by antioxidants; this inhibition may be calibrated in terms of Trolox equivalents to assess the AC. Interestingly, the LOX-FL method discriminated the in vitro AC of four different Lisosan G extracts similarly to the ORAC and TEAC methods. In contrast, only the LOX-FL method was able to highlight a general increase in the serum AC (up to 40% after 30 min) after Lisosan G intake, thus confirming its physiological effectiveness by ex vivo serum assay. This quality of the LOX-FL method probably comes from the ability to highlight simultaneously different antioxidant mechanisms and to effectively show synergy among food phenols and endogenous serum antioxidants.
Introduction
A direct measurement of the antioxidant capacity (AC) of antioxidant-rich foodstuffs, in particular, fruits, vegetables, whole cereal grains, wine, tea and chocolate, has been the subject of a very large number of articles in the last few years. The AC has been evaluated by a lot of different in vitro systems (ORAC, TEAC, TRAP, FRAP, and DPPH).1–3 The interest in these measurements derives from the idea that food AC is related to prevention of diseases. Consistently, recent population studies found significant associations between the AC of the food consumed (obtained from food-frequency questionnaires) and healthy effects, including positive effects on chronic diseases and several tumours as well as a lower risk of heart failure and strokes.4–7 On the other hand, methods for AC measurement show many technical and conceptual limitations. The technical problems are mostly related to the chemistry behind the different AC assays making it difficult to compare AC values obtained using different methods and different experimental conditions.2 From a conceptual point of view, an important limitation is that the values indicating in vitro AC have not been demonstrated to be relevant to the biological effects of specific bioactive compounds.8 Moreover, these assays do not measure bioavailability, in vivo stability, retention of antioxidants by tissues, and reactivity in situ. Finally, there is growing evidence that the metabolic pathways associated with the prevention or amelioration of chronic diseases by bioactive compounds are often dependent on enzyme/protein and/or gene expression regulation rather than a true antioxidant effect.9,10 In the light of these considerations, suggesting that the AC values of foods have no relevance to the effects of specific bioactive compounds on human health, the U.S. Department of Agriculture (USDA) removed its ORAC database for selected foods from its nutrient data laboratory website in 2012.11 However, this choice was not fully shared and remains to a certain extent questionable.12A possible different, more physiological approach may be to move the focus from the analysis of the AC of foods to the analysis of the AC of serum/plasma after food intake. This measure may take into account bioavailability and metabolism, so giving integrated information on a true effect on blood antioxidant status, which is beyond the original AC of the ingested food. In addition, the assessment of the antioxidant status of blood may be per se an important target. In fact, blood plays a central role in the homeostasis of the cellular redox status by carrying and releasing antioxidants in the body; moreover, the maintenance of the blood physiological antioxidant status may preserve the endothelial function, which is thought to be an essential determinant of healthy aging.13 Given the simplicity of AC assays and the easy access to human blood, AC measurements of serum/plasma flourished in the last years, generating a large amount of data.14 Unfortunately, in short-term studies (minutes or hours after ingestion), foods very rich in antioxidants often induced a limited AC increase.15 These results may be, at least in part, dependent on some weakness in the analytical methods. For example, TEAC performs less well than other methods.14 Therefore, in this paper, we describe the use of the novel well-performing lipoxygenase (LOX)-fluorescein (FL) method. It essentially derives from the LOX/4-nitroso-N,N-dimethylaniline (LOX/RNO) method, which is advisable for use with food extracts,16–18 but it shows low sensitivity towards serum (unpublished data). Similarly to the LOX/RNO method, the LOX-FL assay utilizes the soybean LOX-1 reaction with linoleate to generate linoleate hydroperoxide (LOOH) and several physiologically reactive species, mainly the linoleate alkoxyl (LO˙) and peroxyl (LOO˙) derivatives, HO˙ and 1O2,19 which are able to cause the quenching of FL.
Here, we compared the LOX-FL method with the widely used ORAC and TEAC methods to measure the AC of food extracts from the antioxidant-rich dietary wheat grain supplement Lisosan G and of serum after Lisosan G intake. Lisosan G was chosen in the light of its well-documented bioactivity, including its protective role in carbon tetrachloride- and cisplatin-induced toxicity in rat tissues,20 the induction of antioxidant and detoxifying systems in rat hepatocytes,21 the antimutagenic and antioxidant activity in Saccharomyces cerevisiae22 and the improvement of human endothelial progenitor cell (EPC) function.23
Interestingly, the LOX-FL method, similarly to the ORAC and TEAC methods, was found to be very suitable for assessing the AC of hydrophilic and phenolic extracts of Lisosan G and was able to discriminate among the different extracts. On the other hand, only the new method was able to evaluate strong synergistic effects among phenols and serum and to effectively highlight an increase in serum AC after Lisosan G intake. So, the LOX-FL method may be successfully applied to both in vitro analysis of food extracts and ex vivo analysis of serum.
Experimental section
Chemicals
All reagents used were of the highest commercially available purity and were purchased from SIGMA Chemical Co. (St. Louis, MO, USA).The chemicals, 3′,6′-dihydroxyspiro[isobenzofuran-1[3H],9′[9H]-xanthen]-3-one (fluorescein, FL), 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AAPH) and (±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) were dissolved in different media, depending on the assay used to determine the AC. The chemicals, 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), ascorbate, bilirubin, urate, and albumin were dissolved in deionized water. An ammonium sulfate suspension of soybean LOX type V (LOX-1 isoenzyme, E.C. 1.13.11.12) was used, properly diluted with 100 mM Na-borate buffer, pH 9.0. The linoleate solution was prepared and assayed as reported in Pastore et al.24
Antioxidant-enriched food
Lisosan G, an antioxidant-enriched wheat grain preparation produced by Agrisan Company (Larciano, PT, Italy), was kindly provided by Dr Vincenzo Longo from “Istituto di Biologia e Biotecnologia Agraria” (CNR, Pisa, Italy). It is registered by the Italian Ministry of Health as a dietary supplement. In the production process, the bran and germ are separated, collected, mixed with water and inoculated with selected microbial starter cultures, typically consisting of a mix of lactobacillus and natural yeast strains. Finally, once the product is fermented, it is dried and packaged.21Oxygen uptake catalyzed by soybean LOX-1
Oxygen uptake was monitored at 37 °C by means of a 5300A YSI (Yellow Spring, OH, USA) oxygraph equipped with a 5331 YSI Clark-type electrode. The reaction medium (2 mL) consisted of 100 mM Na-borate buffer, pH 9.0, 400 μM Na-linoleate, and 1 μL Tween 20 per μmol linoleate; the reaction was started by adding 0.5 enzymatic units (EU) of soybean LOX-1.FL quenching catalyzed by soybean LOX-1
FL quenching in the course of linoleate hydroperoxidation by soybean LOX-1 was fluorometrically monitored by measuring the FL fluorescence decrease (λex 485 nm; λem 515 nm) at 37 °C using a Perkin Elmer LS 55 fluorescence spectrometer. The reaction mixture (2 mL) contained 100 mM Na-borate buffer, pH 9.0, 400 μM Na-linoleate, 1 μL Tween 20 per μmol linoleate and 6.3 nM FL; the reaction was started by adding 0.5 EU of soybean LOX-1. The rate of the reaction, expressed as ΔArbitrary Units of Fluorescence (AUF)/s, was calculated as the highest slope to the experimental curve.As shown in Fig. 1C, FL quenching was monitored spectrophotometrically at 485 nm and 37 °C using the reaction medium reported above, with the exception of the FL concentration that, in this case, ranged from 1 to 12.5 μM. A Perkin Elmer LAMBDA 45 spectrophotometer was used. Rates of reaction were expressed as ΔA485/min.
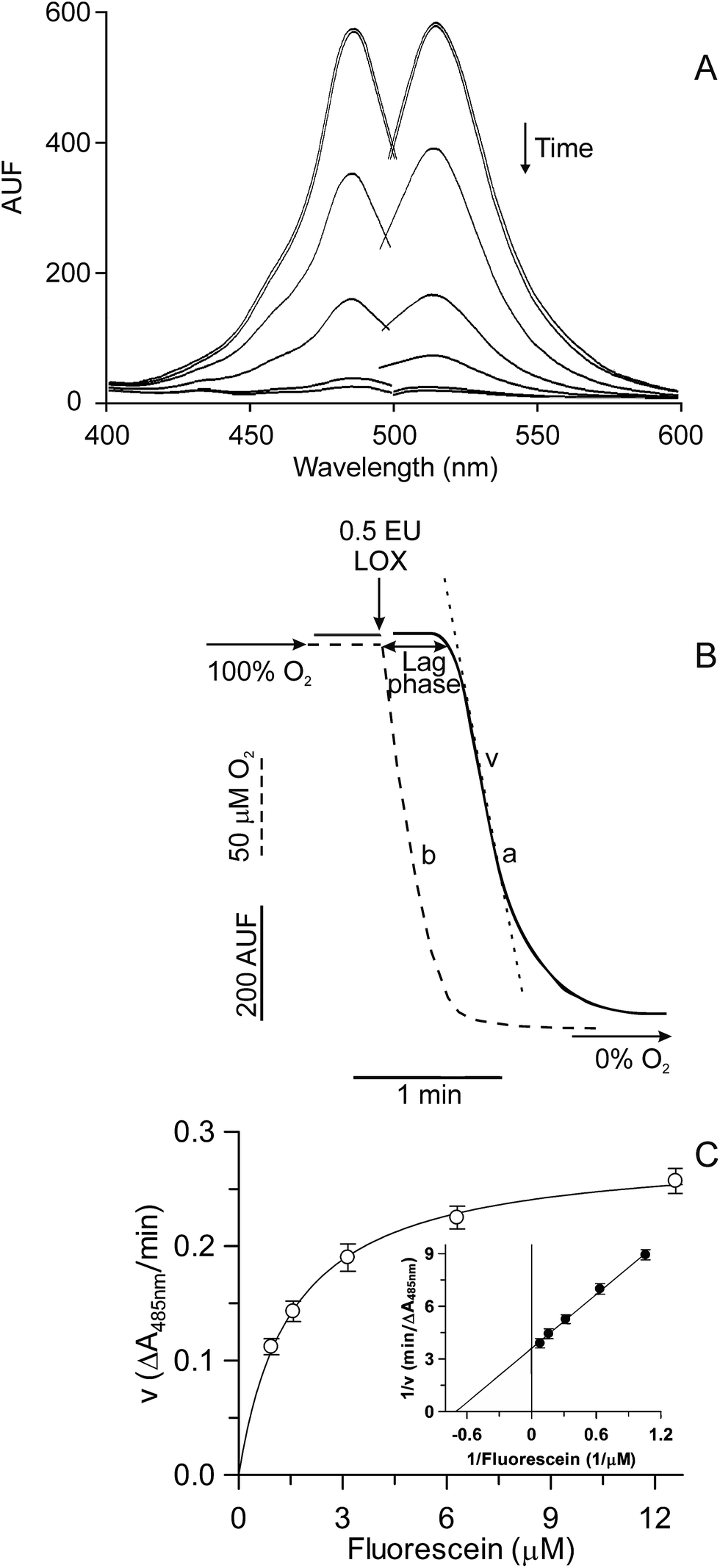 | ||
| Fig. 1 Kinetics of the FL quenching catalyzed by soybean LOX-1. The LOX-1-dependent FL quenching (A and B, trace a) was fluorometrically monitored (λex 485 nm; λem 515 nm) at 37 °C in 2 mL 100 mM Na-borate buffer, pH 9.0, containing 6.3 nM FL, 400 μM linoleate and 1 μL Tween 20 per μmol linoleate (see Experimental section). In (A) the reaction was started by adding 0.05 enzymatic units (EU) soybean LOX-1 and monitored by recording spectra every 90 s. In (B) the reaction was monitored as a time course and simultaneously oxygen uptake (dotted line, curve b) was oxygraphically measured (see Experimental section). At the time indicated by the arrow, 0.5 EU of LOX was added. The rate of the FL quenching (v) and the lag phase are also indicated. In (C), measurements were carried out spectrophotometrically at 485 nm by using the same reaction medium described in (B), but in the presence of the reported FL concentrations. The rates of FL quenching, expressed as ΔA485 nm/min, are reported as Michaelis–Menten and Lineweaver–Burk (inset) plots. | ||
Extraction and assay of antioxidant compounds
Sera collection
Sera were obtained from seven volunteers (4 men and 3 women with ages ranging from 24 to 33 years). Each volunteer provided informed written consent to participate in this study in accordance with the Declaration of Helsinki. The research was performed in compliance with relevant Italian laws and institutional ethical policies. In particular, the experiment was approved by both the Board of the SAFE Department of the University of Foggia, Italy, and the Scientific Committee of the Regional Technological District DARe-Puglia, Italy.For 2 days before analysis, the volunteers followed a low antioxidant diet, avoiding phenolic-rich foods, namely all fresh fruits and vegetables and derived products (tea, coffee, fruit juices, wine, and chocolate). The working window was free from interference by different variables (e.g. physical exercise and energy expenditure).
After an overnight fast, the volunteers ingested 20 g of Lisosan G suspended in 500 mL of water. This is an amount able to induce a serum AC increase, as verified in preliminary experiments. Venous samples at baseline (T0) and at 30, 60, 90, 120 and 240 min after Lisosan G consumption were collected in vacutainers for serum collection. Sera were obtained by centrifugation (3000 × g, 5 min), distributed in portions and frozen at −80 °C until analysis.
AC determination by means of the LOX-FL, ORAC and TEAC methods
Since the rate of the LOX-FL reaction is affected by ethanol, when the lipophilic extract of Lisosan G, reconstituted in ethanol, was evaluated, a constant volume of ethanol was maintained in the assay mixture.
For all three AC methods, measurements were carried out in triplicate for three different amounts of sample and the AC was quantified by means of a dose–response curve obtained with Trolox.18
Results and discussion
Characterization and setting of the LOX-FL reaction as new tool to evaluate AC
A first characterization of the LOX-1-dependent FL quenching is reported in Fig. 1. The addition of the enzyme to the reaction mixture, containing an excess of linoleate (400 μM) and a limited amount of oxygen (200 μM at 37 °C, temperature of measurement), caused the disappearance of both excitation and emission FL fluorescence spectra in the vis region (Fig. 1A). The time course of this reaction can be easily monitored by continuously measuring the FL fluorescence decrease (Fig. 1B). In this experiment, the LOX-1-mediated FL quenching (curve a) and oxygen uptake (dotted curve, b) were simultaneously monitored. It should be noted that LOX-1 addition to the reaction mixture caused rapid oxygen consumption due to linoleate dioxygenation, whereas no simultaneous FL fluorescence change was observed. The FL quenching started about 20–25 s after enzyme addition, that is, only when a very low oxygen concentration, approaching anaerobiosis, was reached in the test sample. Therefore, the observed lag phase represents the time necessary to consume oxygen during the primary aerobic LOX-1 reaction of linoleate peroxidation.18,19 On the contrary, FL quenching is essentially a secondary anaerobic reaction.A study of the kinetics of the LOX-1-dependent FL quenching was also carried out; to do this, since the high FL concentration necessary in this experiment is incompatible with fluorescence measurements, the reaction was monitored spectrophotometrically at 485 nm. By plotting the rate of FL quenching vs the FL concentration, a hyperbolic relationship was obtained (Fig. 1C). Data were also plotted as Lineweaver–Burk (inset), Hanes, Eadie–Hofstee and Eadie–Scatchard plots (not shown). All plots were linear, thus confirming the hyperbolic kinetics, with Km and Vmax equal to 1.53 ± 0.08 (SD) μM and 0.28 ± 0.01 (SD) ΔA485 nm/min, respectively, calculated by means of GRAFIT 5.0 software.
The FL sensitivity to some relevant physiological reactive species, some of which were generated during the course of LOX-1-mediated anaerobic reactions,18,19 was investigated (not shown). In particular, FL was insensitive to H2O2 as well as to O2˙− and LOOH, generated via xanthine/xanthine oxidase28 and LOX/linoleate systems, respectively. On the contrary, FL was sensitive to LO˙ and HO˙ generated via the Fenton reaction from LOOH or H2O2, respectively, in the presence of CoCl2.29 Similarly, FL quenching was also observed in the presence of 1O2 generated via the Na2MoO4/H2O2 system.30
In another set of experiments, the sensitivity of the FL quenching reaction to antioxidants was evaluated. Firstly, the sensitivity of FL quenching to the standard antioxidant, Trolox, an α-tocopherol analogue with enhanced water solubility, was tested. In Fig. 2A, the experimental curves of the FL quenching reactions carried out in both the absence (control, trace a) and presence of Trolox at different concentrations (10, 15 and 20 μM; traces b, c and d, respectively) are shown. Trolox was found to inhibit FL quenching by causing a decrease in the reaction rate consistent with its antiradical activity. In the inset of Fig. 2A, Trolox-dependent inhibition, expressed as (%) decrease with respect to the control trace, is reported as a function of the standard antioxidant concentration: a linear dependence of the inhibition from about 15% to 45% on the Trolox concentration, ranging between 5 and 20 μM, was obtained (see the equation in the figure caption).
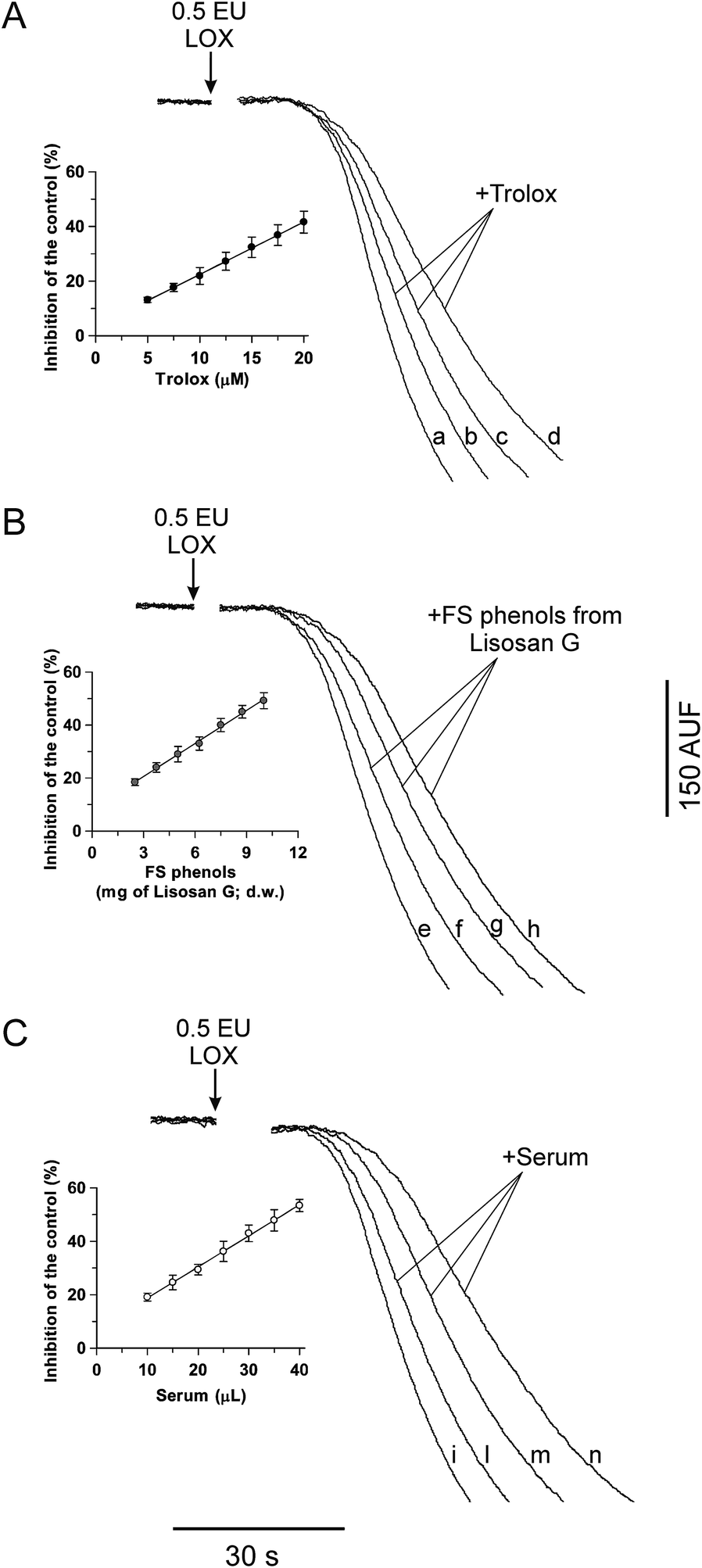 | ||
| Fig. 2 Inhibition of the LOX-1-dependent FL quenching by Trolox (A), FS phenols from Lisosan G (B) and human serum (C). The LOX-FL reaction was monitored as the FL fluorescence decreased at 37 °C as shown in Fig. 1B and in the Experimental section. Measurements were carried out in both the absence (control, traces a, e and i) and in the presence of (A) Trolox (10, 15 and 20 μM; traces b, c and d, respectively), (B) FS phenols from Lisosan G (obtained from 5, 7.5 and 10 mg of Lisosan G, d.w.; traces f, g and h, respectively) and (C) human serum (10, 20 and 35 μL; traces l, m and n, respectively). The rates of the FL quenching, expressed as percentage decrease with respect to the control, are reported as a function of the Trolox concentration (inset of (A)), FS phenols from Lisosan G (inset of (B)) and volume of serum (inset of (C)). The resulting equations are: inhibition (%) = 1.920[Trolox] + 3.23 (r = 0.9996, p < 0.001) (inset of (A)); inhibition (%) = 4.154(Lisosan G) + 8.12 (r = 0.9985, p < 0.001) (inset of (B)); and inhibition (%) = 1.165(serum) + 7.10 (r = 0.9989, p < 0.001) (inset of (C)), where Trolox, Lisosan G and serum are expressed in μM, mg d.w. and μL, respectively. | ||
Interestingly, kinetic analysis highlights the competitive inhibition mechanism of Trolox on the LOX-1-mediated FL quenching with a Ki value equal to about 5 μM (not shown). This result implies that the antioxidant/oxidant interaction occurs at the level of the enzyme macromolecule rather than in the bulk phase of the solution.
Since the LOX-1-mediated FL quenching reaction is sensitive to reactive species and also shows inhibition by Trolox, which may be easily quantified, we adopted this reaction as a new tool to measure the AC. Interestingly, this new LOX-FL method shows several advantages. The hyperbolic dependence of the reaction rate on the FL concentration indicates that the reaction cannot occur in the bulk phase of the reaction mixture, but at the LOX surface, in particular, at the level of a definite number of FL binding sites, generating a ternary enzyme-radical-FL complex. Therefore, the effect of an antioxidant should be considered to be more physiological, as it refers to a reaction taking place within a biological macromolecule. In addition, the LOX-FL assay evaluates the oxidant–antioxidant competition at low oxygen concentration, which often occurs in human cells. Moreover, the LOX-FL method may evaluate antioxidants acting in a different manner: as scavengers of different physiological radicals, including LO˙ and LOO˙ radical derivatives of linoleate, HO˙ as well as 1O2, but also as reducing agents or chelators of the non-heme Fe3+ necessary for LOX catalysis or as direct inhibitors of the peroxidation reaction catalysed by LOX, thus providing a comprehensive AC evaluation.18
A first application of the new LOX-FL method was carried out on the dietary supplement, Lisosan G. The capability of the LOX-FL assay to evaluate the AC in FS and IB phenols, hydrophilic and lipophilic extracts of Lisosan G, was tested. In particular, in Fig. 2B, typical experimental traces of FL quenching in the presence of FS phenols of Lisosan G are shown. Small amounts of extract (obtained from 2.5 to 10 mg dry weight, d.w., of Lisosan G) were found to inhibit the LOX-FL reaction. In this experiment, the FS phenols inhibited the rate of the FL quenching from about 20% to 50% with a linear relationship between the inhibition and the amount of sample (inset of Fig. 2B). Comparison with the linear regression of Trolox, carried out as reported in Pastore et al.,18 gave an AC value of 4.3 ± 0.4 (SD) μmol Trolox eq. per g (d.w.) (see also Table 2).
The LOX-FL assay was also tested to evaluate the AC in a human serum sample. Analogously, as shown in Fig. 2C, typical experimental traces in the presence of serum are reported. Also in this case, small volumes of serum (ranging from 10 μL to 40 μL) were found to linearly inhibit (from 20% to 50%) the LOX-FL reaction (inset of Fig. 2C). In this experiment, the AC value, in terms of Trolox, was 1.21 ± 0.03 (SD) μmol Trolox eq. per mL serum.
Interestingly, the LOX-FL method may be used on both serum and plasma (not shown). However, caution has to be paid when plasma is obtained using EDTA or citrate as anticoagulants, since they may chelate iron that is necessary to LOX activity. On the contrary, the LOX/RNO assay was found to require too high a volume of both biological fluids (unpublished data).
In order to check the capability of the LOX-FL assay to evaluate the AC of the main antioxidant compounds contained in serum samples, i.e. albumin, bilirubin, urate and ascorbate,2,11 they were individually investigated (Table 1). Trolox was used as an analogue of α-tocopherol. A comparison with the ORAC assay is also reported. All tested compounds were found to show the AC, as measured by both the LOX-FL and ORAC methods. Interestingly, the IC50 values are 4–160 fold higher for the LOX-FL than for the ORAC assay, thus indicating a general lower sensitivity of LOX-FL.
| LOX-FL | ||
|---|---|---|
| Antioxidant | Range (μM) | IC50a (μM) |
| a For LOX-FL, the IC50 values represent the antioxidant concentration able to halve the rates of the LOX-FL reaction. b As for ORAC, the IC50 values represent the antioxidant concentration able to double the area of the blank ORAC reaction. | ||
| Trolox | 5–20 | 26 ± 2 |
| Albumin | 7.5–30 | 25 ± 2 |
| Bilirubin | 1.5–6.0 | 7.6 ± 0.6 |
| Urate | 10–25 | 40 ± 2 |
| Ascorbate | 150–1000 | 1360 ± 10 |
| ORAC | ||
|---|---|---|
| Antioxidant | Range (μM) | IC50b (μM) |
| Trolox | 2.5–10 | 2.5 ± 0.2 |
| Albumin | 0.075–0.225 | 0.16 ± 0.02 |
| Bilirubin | 0.15–0.90 | 2.0 ± 0.2 |
| Urate | 1–4 | 1.6 ± 0.1 |
| Ascorbate | 1.5–18 | 11.1 ± 0.1 |
Consistently, AC values of sera measured by means of the LOX-FL method show a reference interval from about 1 to 1.6 μmol Trolox eq. per mL (see also caption of Fig. 4), about tenfold lower than that of ORAC as well as about twofold lower than TEAC. Interestingly, this ability to measure the lower basal AC in serum may help to better highlight AC changes after food antioxidant ingestion. In another set of experiments, the effect of the addition of an FS phenolic extract to serum (Fig. 3A and C) and the possible synergism among the antioxidant compounds reported in Table 1 (Fig. 3B and D) were also evaluated. Once again, LOX-FL and ORAC were compared. In each experiment, the AC of each sample was measured and the sum of the AC values of all tested compounds was calculated (ACsum). Then, the same samples were mixed and the AC was determined again (ACmix).
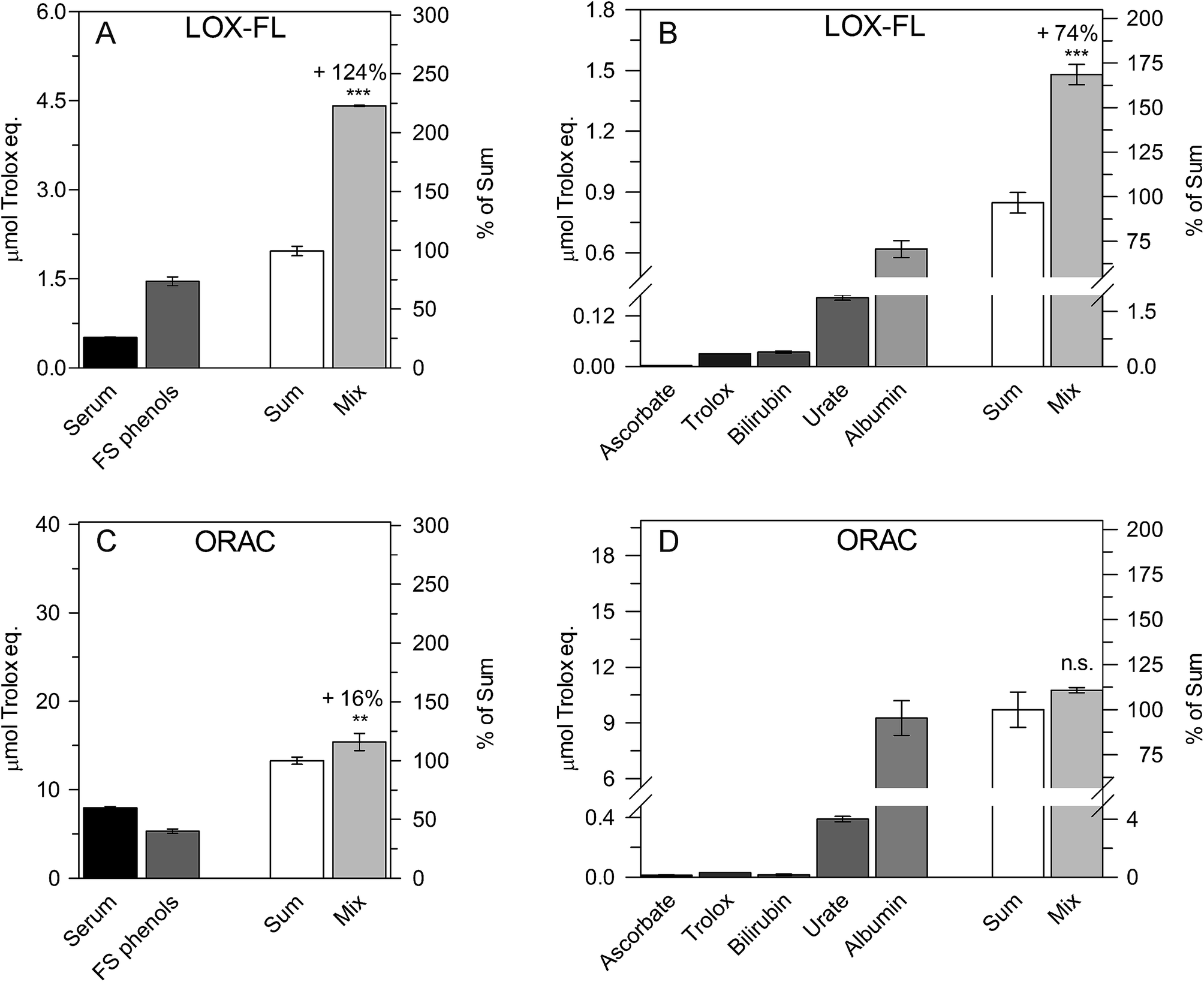 | ||
| Fig. 3 Synergism among human serum and FS phenols (A and C) and among ascorbate, Trolox, bilirubin, urate and albumin (B and D), evaluated by means of the LOX-FL (A and B) and ORAC (C and D) methods. In each plot, the AC values of each sample, as well as the sum (ACsum) and the AC of the Mix (ACmix) are shown as μmol Trolox eq. and as percentages of the ACsum. Synergism is reported as a percentage increase in the ACmix value with respect to the ACsum value according to the equation: synergistic increase (%) = [(ACmix/ACsum) − 1] × 100. Data are reported as mean value ± SD. The probability level (**, p ≤ 0.01; ***, p ≤ 0.001; n.s. = not significant) according to the Student's t-test is also reported. | ||
In the experiment shown in Fig. 3A, two equal amounts (0.5 mL) of serum and FS phenols were mixed. For the resultant mixture (1 mL), an ACmix value of 4.41 ± 0.01 μmol Trolox eq., 124% higher than the ACsum (1.97 ± 0.08 μmol Trolox eq.), was measured. In Fig. 3B, to mimic serum composition,31 the mix was a solution (1 mL) containing 250 μM urate, 50 μM ascorbate, 10 μM bilirubin, 600 μM albumin and 30 μM Trolox. Trolox was used instead of α-tocopherol due to its solubility in aqueous solution. Interestingly, an ACmix value equal to 1.48 ± 0.05 (SD) μmol Trolox eq. was obtained, 74% higher than the ACsum (0.85 ± 0.05 μmol Trolox eq.).
By using the same approach, low synergism (16% increase, Fig. 3C) or no significant synergistic effect (Fig. 3D) was observed using the ORAC method.
Therefore, the LOX-FL assay may highlight the synergistic effect among food and serum antioxidants as well as among serum antioxidants better than ORAC.
This is consistent with the ability of the LOX-FL reaction to generate, as reported above, more than one oxidant species having relevant physiological significance and to evaluate simultaneously different antioxidant mechanisms. These findings are in accordance with the ability of the LOX/RNO method to effectively highlight antioxidant synergisms.17,18
An interesting result evident from Table 1 and Fig. 3 is the major role exerted by albumin as serum antioxidant, whatever the AC method used for the assay; this is in good agreement with previous literature findings.31
Ability of the new LOX-FL method to evaluate both the AC of food extracts (in vitro measurements) and the effect of food intake on the AC of serum (ex vivo measurements)
To better investigate the ability of the new LOX-FL method to measure the in vitro AC of food extracts, three different antioxidant extracts from Lisosan G were investigated in some detail. The AC was also evaluated by means of the ORAC and TEAC methods (Table 2). As expected, depending on the different chemistry of the assays, the absolute AC values measured by the different methods are very different from each other, but all three methods measured the highest AC values in the hydrophilic extract of Lisosan G, probably mainly attributable to proteins, as already demonstrated for wheat grain.17 With regard to the phenolic extracts of Lisosan G, all three methods measured much higher AC values in the IB phenolic extract with respect to the FS phenolic one; these data are in good agreement with numerous literature data reporting IB phenolic compounds as the major phenolic component in cereal whole grain.32 Consistently, the total phenolic and flavonoid contents were higher in the IB phenolic extract than in the FS phenolic one. The lipophilic extract was also evaluated, but only negligible activity was measured (not shown), with AC values equal to about 0.6%, 0.6% and 4% of the total activity as evaluated by the LOX-FL, ORAC and TEAC methods, respectively. On the whole, these results indicate a very high total AC of Lisosan G, which was about 2- to 10-fold higher than that of whole grain from different wheat species.16,17| Extract | AC (μmol Trolox eq. per g d.w) | Antioxidant compounds | ||||
|---|---|---|---|---|---|---|
| LOX-FL | ORAC | TEAC | Protein contenta | Phenolic contentb | Flavonoid contentc | |
| a Data are expressed as mg per g d.w. b Data are expressed as mg of gallic acid eq. per 100 g d.w. c Data are expressed as mg of catechin eq. per 100 g d.w. d Data are reported as mean value ± SD. n.d., not determined. | ||||||
| Hydrophilic | 81.7 ± 4.7d | 123 ± 6 | 48 ± 3 | 131.7 ± 2.6 | n.d. | n.d. |
| FS phenolic | 4.3 ± 0.4 | 25.6 ± 0.7 | 7.1 ± 1.2 | 34.5 ± 1.1 | 2.76 ± 0.15 | |
| IB phenolic | 7.1 ± 0.1 | 56.6 ± 4.8 | 29.5 ± 2.1 | 81.1 ± 0.8 | 26.6 ± 1.1 | |
This result is in agreement with the well-documented bioactivity of Lisosan G. In fact, it has been already shown that Lisosan G protects rats against toxicity induced in the liver by carbon tetrachloride and in the liver, kidney and testis by cisplatin, possibly by attenuating oxidative stress and by preserving antioxidant enzymes.20 Moreover, it induces a decrease in the intracellular ROS concentration and H2O2-dependent mutagenesis in Saccharomyces cerevisiae,22 activates the NAD(P)H:quinone oxidoreductase and heme oxygenase-1 in rat hepatocytes,21 glutathione peroxidase-1 and superoxide dismutase-2 expression in EPCs exposed to oxidative stress23 and decreases H2O2-induced toxicity in rat hepatocytes.21 Furthermore, activation of the nuclear Nrf-2/ARE in rat hepatocytes21 and EPCs exposed to oxidative stress23 has been reported. So, the bioactivity of phytochemicals from Lisosan G may be exerted in several different modes, possibly including AC properties, but also via direct modulation of metabolic pathways and/or gene expression.
In order to evaluate the ability of the LOX-FL method to assess changes in the AC of blood, in comparison with ORAC and TEAC, ex vivo measurements of serum were carried out before and 30, 60, 90, 120 and 240 min after the intake of 20 g of Lisosan G (18 g in terms of d.w.). On the basis of data reported in Table 2, this implies the intake of about 1700, 3700 and 1600 μmol Trolox eq. as evaluated by means of the LOX-FL, ORAC and TEAC methods, respectively. The effect on the serum AC is reported as the percentage change in the initial (T0) AC (Fig. 4). As shown, the ingestion of Lisosan G caused an increase of about 40% in the AC, evaluated by means of the LOX-FL assay, after 30 min. Then, the AC decreased after 90 min, until it reached an AC value similar to T0, and increased again at 120 min. This behaviour might be compatible with a prompt release and absorption of FS phenols (see Table 2) within 30–60 min in the small intestine, followed by either a delayed release of IB phenols at 120 min, probably due to the activity of gut microbiota, or to metabolism-increasing antioxidant activity. Finally, the AC value gradually decreased and reached about the same value as that at T0 after 240 min. On the contrary, the ORAC method did not evaluate a significant increase in the AC after Lisosan G intake, even showing an incoherent decrease at 120 min (about 20%). The TEAC method also did not show relevant changes in the serum AC. Therefore, under the same experimental conditions, different methods may give very different results, thus suggesting that the use of different assays represents a main source of incoherence in a lot of literature data.
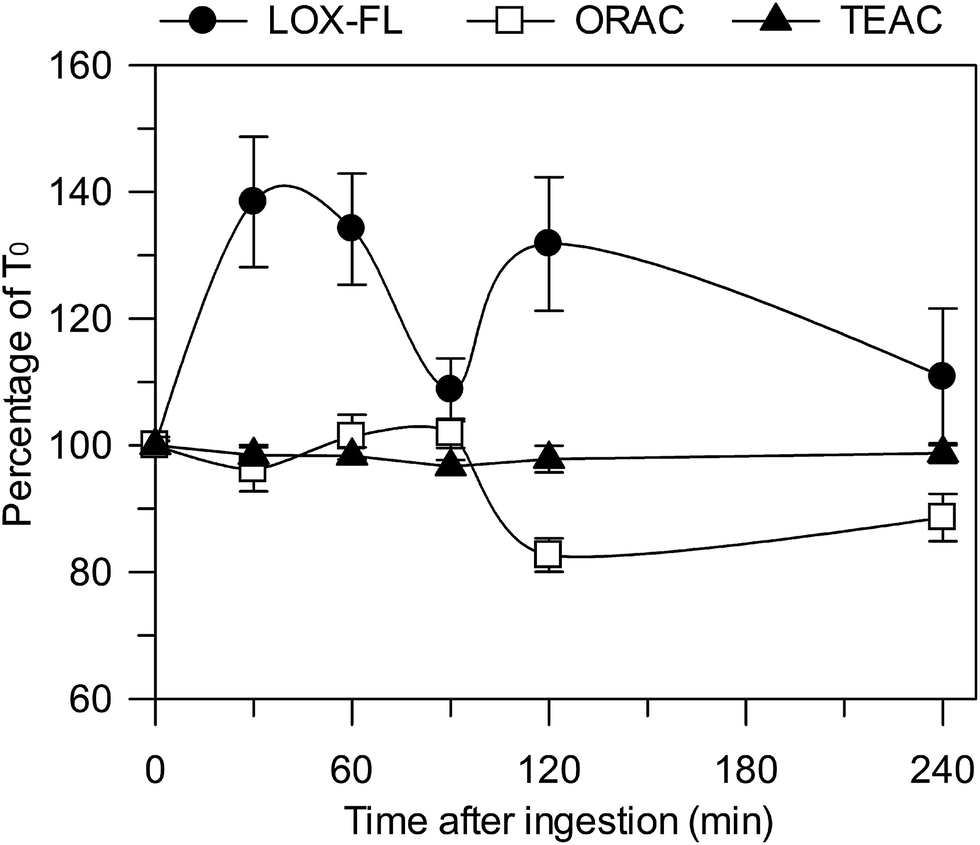 | ||
| Fig. 4 Percent changes of serum AC, evaluated by means of the LOX-FL, ORAC and TEAC methods, during 240 min after consumption of 20 g of Lisosan G in seven subjects. A different curve was obtained for each subject, having a T0 value ranging from 1.04 to 1.65, from 9.32 to 26.18 and from 1.78 to 3.71 μmol Trolox eq. per mL of serum, as calculated by LOX-FL, ORAC and TEAC methods, respectively. Then, for each AC method, the seven curves were mediated. Data are reported as mean value ± SE. | ||
Overall, it appears that the ORAC and TEAC methods perform less well with respect to LOX-FL; once again, this is probably due to the ability of this method to simultaneously evaluate different antioxidant mechanisms.
On the contrary, the ORAC assay only measures the chain-breaking AC against a peroxyl radical according to a hydrogen atom transfer (HAT) reaction, whereas the TEAC method evaluates the capability of antioxidants to induce the quenching of the non-physiological ABTS˙+ radical cation, mainly according to a single electron transfer (SET) redox reaction, this latter having a minor role in vivo.1 The different rationales for the different methods easily explain why the results of LOX-FL, ORAC and TEAC are unrelated to each other.
Conclusions
The LOX-FL method is able to give a physiological and comprehensive evaluation of the AC, therefore better highlighting the synergistic interactions among the different classes of antioxidants. This novel tool may be used to obtain both in vitro measurements of food extracts and ex vivo measurements of serum after food ingestion. This ability allows an integrated evaluation of food, using the same assay method, by testing whether foods showing a strong AC may give realistic physiological effects after ingestion. Interestingly, by using this approach on the antioxidant-rich dietary wheat grain supplement, Lisosan G, it was shown that its consumption may significantly improve the antioxidant status of serum.This information cannot be obtained when the ORAC and TEAC methods are used. In fact, although all methods are able to give coherent information about the AC of in vitro extracts of Lisosan G, LOX-FL was found to be able to highlight a remarkable AC increase in serum after Lisosan G ingestion, while under the same experimental conditions, ORAC and TEAC failed to do this.
Acknowledgements
This work was supported by the following research projects: the Apulian Region Project “FutureInResearch – Study of antioxidant action of plant foods using innovative in vitro and ex vivo methods for valorisation of health properties of apulian products (code 2GE9US7)”, the MiUR projects PON01_01145 “ISCOCEM” and PON02_00186_2937475 “Pro.Ali.Fun.” and the Apulian Region Project “LAIFF-Laboratories Network for the Innovation on Functional Foods”. We thank Dr Gaetano Pio Spera who participated as a PhD student in the present work. Publication supported by funds “5 × 1000 dell'IRPEF a favore dell’Università degli Studi di Foggia” in memory of Gianluca Montel.References
- D. Huang, O. U. Boxin and R. L. Prior, J. Agric. Food Chem., 2005, 53, 1841–1856 CrossRef CAS PubMed.
- E. Niki, Free Radical Biol. Med., 2010, 49, 503–515 CrossRef CAS PubMed.
- F. Shahidi and Y. Zhong, J. Funct. Foods, 2015, 18, 757–781 CrossRef CAS.
- D. Del Rio, A. Rodriguez-Mateo, J. P. E. Spencer, M. Tognolini, G. Borges and A. Crozier, Antioxid. Redox Signaling, 2013, 18, 1818–1892 CrossRef CAS PubMed.
- S. Rautiainen, S. Larsson, J. Virtamo and A. Wolk, Stroke, 2012, 43, 335–340 CrossRef CAS PubMed.
- S. Rautiainen, E. B. Levitan, M. A. Mittleman and A. Wolk, Am. J. Med., 2013, 126, 494–500 CrossRef PubMed.
- S. Rautiainen, E. B. Levitan, M. A. Mittleman and A. Wolk, Eur. J. Heart Failure, 2015, 17, 20–26 CrossRef PubMed.
- C. G. Fraga, P. I. Oteiza and M. Galleano, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 931–934 CrossRef CAS PubMed.
- C. Bumke-Vogt, M. A. Osterhoff, A. Borchert, V. Guzman-Perez, Z. Sarem, A. L. Birkenfeld, V. Bähr and A. F. H. Pfeiffer, PLoS One, 2014, 9, e104321 Search PubMed.
- M. Marimoutou, F. Le Sage, J. Smadja, C. L. D'Hellencourt, M.-P. Gonthier and C. R. Da Silva, J. Inflammation, 2015, 12, 10 CrossRef CAS PubMed.
- A. Pompella, H. Sies, R. Wacker, F. Brouns, T. Grune, H. K. Biesalski and J. Frank, Nutrition, 2014, 30, 791–793 CrossRef CAS PubMed.
- R. L. Prior, J. Funct. Foods, 2015, 18, 797–810 CrossRef CAS.
- M. El Assar, J. Angulo and L. Rodríguez-Mañas, Free Radical Biol. Med., 2013, 65, 380–401 CrossRef CAS PubMed.
- D. Lettieri-Barbato, F. Tomei, A. Sancini, G. Morabito and M. Serafini, Br. J. Nutr., 2013, 109, 1544–1556 CrossRef CAS PubMed.
- M. S. Fernández-Panchón, D. Villano, A. M. Troncoso and M. C. Garcia-Parrilla, Crit. Rev. Food Sci. Nutr., 2008, 48, 649–671 CrossRef PubMed.
- M. N. Laus, A. Gagliardi, M. Soccio, Z. Flagella and D. Pastore, J. Food Sci., 2012, 77, C1150–C1155 CrossRef CAS PubMed.
- M. N. Laus, D. Tozzi, M. Soccio, A. Fratianni, G. Panfili and D. Pastore, J. Cereal Sci., 2012, 56, 214–222 CrossRef CAS.
- D. Pastore, M. N. Laus, D. Tozzi, V. Fogliano, M. Soccio and Z. Flagella, J. Agric. Food Chem., 2009, 57, 9682–9692 CrossRef CAS PubMed.
- D. Pastore, D. Trono, L. Padalino, N. Di Fonzo and S. Passarella, Plant Physiol. Biochem., 2000, 38, 845–852 CrossRef CAS.
- V. Longo, P. G. Gervasi and V. Lubrano, Food Chem. Toxicol., 2011, 49, 233–237 CrossRef CAS PubMed.
- M. La Marca, P. Beffy, A. Pugliese and V. Longo, PLoS One, 2013, 8, e83538 Search PubMed.
- S. Frassinetti, C. M. Della Croce, L. Caltavuturo and V. Longo, Food Chem., 2012, 135, 2029–2034 CrossRef CAS PubMed.
- D. Lucchesi, R. Russo, M. Gabriele, V. Longo, S. Del Prato, G. Penno and L. Pucci, PLoS One, 2014, 9, e109298 Search PubMed.
- D. Pastore, D. Trono, L. Padalino, S. Simone, D. Valenti, N. Di Fonzo and S. Passarella, J. Cereal Sci., 2000, 31, 41–54 CrossRef CAS.
- B. Ou, M. Hampsch-Woodill and R. L. Prior, J. Agric. Food Chem., 2001, 49, 4619–4626 CrossRef CAS PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- M. N. Laus, N. A. Di Benedetto, R. Caporizzi, D. Tozzi, M. Soccio, L. Giuzio, P. De Vita, Z. Flagella and D. Pastore, Plant Foods Hum. Nutr., 2015, 70, 207–214 CrossRef CAS PubMed.
- D. Trono, M. Soccio, M. N. Laus and D. Pastore, Plant Sci., 2013, 199–200, 91–102 CrossRef CAS PubMed.
- B. Ou, M. Hampsch-Woodill, J. Flanagan, E. K. Deemer, R. L. Prior and D. Huang, J. Agric. Food Chem., 2002, 50, 2772–2777 CrossRef CAS PubMed.
- J. M. Aubry and S. Bouttemy, J. Am. Chem. Soc., 1997, 119, 5286–5294 CrossRef CAS.
- G. Cao and R. L. Prior, Clin. Chem., 1998, 44, 1309–1315 CAS.
- N. Okarter and R. H. Liu, Crit. Rev. Food Sci. Nutr., 2010, 50, 193–208 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |