
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrahigh pressure liquid chromatography-atmospheric pressure photoionization-tandem mass spectrometry for the determination of polyphenolic profiles in the characterization and classification of cranberry-based pharmaceutical preparations and natural extracts†
Lidia
Parets
a,
Élida
Alechaga
a,
Oscar
Núñez
*abc,
Javier
Saurina
ab,
Santiago
Hernández-Cassou
ab and
Lluis
Puignou
ab
aDepartment of Chemical Engineering and Analytical Chemistry, University of Barcelona, Martí i Franquès, 1-11, E-08028 Barcelona, Spain. E-mail: oscar.nunez@ub.edu; Fax: +34-93-402-1233; Tel: +34-93-403-3706
bResearch Institute in Nutrition and Food Safety, University of Barcelona, Av. Prat de la Riba 171, Edifici Recerca (Gaudí), E-08901 Santa Coloma de Gramanet, Barcelona, Spain
cSerra Hunter Fellow, Generalitat de Catalunya, Spain
First published on 29th April 2016
Abstract
Ultrahigh pressure liquid chromatography-atmospheric pressure photoionization-tandem mass spectrometry (UHPLC-APPI-MS/MS) was applied to the analysis and authentication of fruit-based products and pharmaceutical preparations. Two sub-2 μm C18 reversed-phase columns, Syncronis (100 × 2.1 mm, 1.7 μm) and Hypersil Gold (50 × 2.1 mm, 1.9 μm), were proposed under gradient elution with 0.1% formic acid aqueous solution and methanol mobile phases for the determination of 29 polyphenols, allowing us to obtain polyphenolic profiles in less than 13.5 and 23.5 min, respectively. Several atmospheric pressure ionization (API) sources (H-ESI, APCI, and APPI) were compared. For dopant-assisted APPI, four organic solvents, toluene, acetone, chlorobenzene and anisole, were evaluated as dopants. Both H-ESI and acetone-assisted APPI were selected as the best ionization sources for the analysis of targeted polyphenols. Acceptable sensitivity (LOD values down to 0.5 μg kg−1 in the best of cases), linearity (r2 higher than 0.995) and good precision (RSD values lower than 15.1%) and trueness (relative errors lower than 10.2%) were obtained using both UHPLC-API-MS/MS methods. A simple extraction procedure, consisting of sample sonication with acetone/water/hydrochloric acid (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :29.9:0.1 v/v/v) and centrifugation, was used. The proposed UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods with both C18 reversed-phase columns were then applied to the analysis of 32 grape-based and cranberry-based natural products and pharmaceutical preparations. Polyphenolic profile data were then analyzed by principal component analysis (PCA) to extract information on the most significant data contributing to the classification of natural extracts according to the type of fruit.
:29.9:0.1 v/v/v) and centrifugation, was used. The proposed UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods with both C18 reversed-phase columns were then applied to the analysis of 32 grape-based and cranberry-based natural products and pharmaceutical preparations. Polyphenolic profile data were then analyzed by principal component analysis (PCA) to extract information on the most significant data contributing to the classification of natural extracts according to the type of fruit.
Introduction
Over the past years, researchers and food manufacturers, as well as the public in general, have become increasingly interested in polyphenolic compounds. Polyphenols are aromatic secondary metabolites that are ubiquitously spread throughout the plant kingdom. They comprise more than 8000 substances with highly diverse structures and molecular masses ranging from small molecules (<100 Da) such as phenolic acids to big molecules (>30000 Da) consisting of highly polymerized polyphenolic compounds. They can be divided into several classes, e.g., phenolic acids (hydroxybenzoic acids and hydroxycinnamic acids), flavonoids (flavonols, flavones, flavanols, flavanones, isoflavones, and proanthocyanidins (PACs)), stilbenes, and lignans.1 Our diet has a great abundance of these compounds which provide important health benefits mainly based on their antioxidant properties and their probable role in the prevention of various diseases such as skin pathologies, various types of cancer, cardiovascular disorders and other age-related degenerative pathologies.2–6 Moreover, polyphenols are important descriptors of fruit quality because of their contribution to taste, color and nutritional properties.7
Cranberry (Vaccinium macrocarpon) and its derived products, including juices and nutraceuticals, contain a great number of polyphenols and have shown some beneficial health effects, including antioxidant activity, antimicrobial activity against bacteria involved in a wide range of diseases (dental caries, gastritis, enteritis, and infections), anti-inflammatory activity in periodontal disease, and antiproliferative activity on human oral, colon, and prostate cancer cell lines, among others.8 However, the best known bioactivity of cranberry polyphenols deals with their capacity to inhibit the adhesion of pathogenic bacteria to uroepithelial cells of the urinary tract, thus contributing to the prevention of urinary tract infections. Such an activity has been attributed to the presence of A-type PACs.9,10 Almost 65% of proanthocyanidins present in cranberry are A-type ones.11 PACs are also present in common foods such as fruits (apples, pears, plums, strawberries, grapes, dates, many red fruits…), cereals (sorghum, barley…), seeds and nuts (beans, peas, and almonds), spices, aromatic plants, and more scarcely in vegetables.12 They can also be found in various foodstuffs of plant origin (wines, tea, ciders, beers, chocolates, jams, puree, etc.).13
The most common PACs are procyanidins, exclusively consisting of several (epi)catechin units, which are mainly linked through C4–C8 or sometimes C4–C6 bonds. Both these linkages are called B-type linkages (B-type PACs). When an additional ether linkage is formed (mainly) between C2-0-C7, these compounds are called A-type PACs. One of the most notable differences between the two families of PACs is that only the A-type is capable of inhibiting the adhesion of bacteria to urinary tract tissues.8 Recently, several commercial products which are claimed to be manufactured from cranberry-based extracts, such as some pharmaceutical preparations, have appeared in the market. These products are sold as if they have the same health properties of cranberries, but they do not contain the appropriate polyphenols for having the desired bioactivity, probably due to adulterations with more economic fruits, such as grapes. For this reason, it is important to develop analytical methodologies for the characterization of natural extracts to achieve unambiguous authentication regarding the type of fruit.
Liquid chromatography (LC) with UV detection or coupled to mass spectrometry (LC-MS) are the most common techniques described for the determination of polyphenols and the characterization of a great variety of plants and fruit-based products.14–26 High resolution mass spectrometry (HRMS) has also been proposed for the analysis and characterization of polyphenols in fruit products.27–30 For instance, recently Regueiro et al.30 reported a comprehensive identification of walnut polyphenols by LC-HRMS using a hybrid linear ion trap-Orbitrap mass spectrometer, being able to reveal for the first time the presence of 8 polyphenols never reported in this kind of sample. In general, most of the proposed LC-MS(/MS) or LC-HRMS methods for the analysis of polyphenols rely on electrospray ionization (ESI)16–21 or atmospheric pressure chemical ionization (APCI)22–26 as ionization sources. In 2000, Robb and co-workers31 developed atmospheric pressure photoionization (APPI) as a complementary ionization source for LC-MS to expand the application of these techniques to non-polar compounds and compounds which are difficult to ionize by ESI and/or APCI sources. Only a few studies have been reported in the literature employing APPI for the analysis of polyphenols. For instance, Bernard Jean-Denis et al.32 proposed the use of LC-APPI-MS/MS with an ion-trap mass analyzer for the identification of stilbenes in downy mildew-infected grapevine leaves, and Riffault et al.33 employed an UHPLC-APPI-TOF-MS/MS method for the analysis of some flavonoids in the characterization of a rose flower ethyl acetate extract. In both cases, acetone was the solvent selected as the dopant for APPI. The effect of the eluent on the ionization efficiency of five flavonoids, including (+)-catechin, by ion spray, APCI and toluene dopant-assisted APPI using a triple quadrupole mass spectrometer has also been reported.34
In our previous studies, chromatographic separation of 26 polyphenols was established by employing a kinetex C18 reversed-phase (100 × 4.6 mm, 2.6 μm particles) column and gradient elution with 0.1% formic acid aqueous solution and methanol mobile phases at a flow rate of 1 mL min−1.14,15 When coupling this chromatographic separation with tandem mass spectrometry, a 1:1 postcolumn split of the chromatographic eluent was necessary to make compatible chromatographic and MS conditions.15 Although an acceptable chromatographic separation of the 26 studied polyphenolic compounds was obtained under these conditions (last eluting compound at 21 min), the total elution program of this method required 36 min. Although coelution of several compounds occurred, ion suppression effects were not observed when electrospray ionization was used as the ionization source in LC-MS/MS, demonstrating that baseline chromatographic separation was not mandatory because MS, using the appropriate SRM transitions, could selectively resolve coelutions. Taking all these facts into consideration, the present work aimed to develop an UHPLC method for the analysis of the previous 26 polyphenols together with 3 proanthocyanidin (PAC) compounds (procyanidins A2, B2 and C1) which are expected to be more discriminant in the analysis of cranberry-based products, for the determination of polyphenolic profiles in the authentication and characterization of cranberry-based pharmaceutical preparations and foodstuffs. However, an important reduction on the total chromatographic elution program produced higher polyphenol overlapping than that previously reported, and matrix effects were expected to be a handicap when dealing with the polyphenolic profile treatment. In order to reduce matrix effect influence on polyphenolic characterization, several atmospheric pressure ionization (API) sources, such as H-ESI, APCI and APPI, were compared. Several kinds of cranberry-based and grape-based samples were analyzed, as well as commercial cranberry-based products such as pharmaceutical natural extracts, powder capsules, syrup and sachets. Data corresponding to the polyphenolic profile composition obtained using different LC-API-MS/MS methods were considered as a source of potential descriptors to be exploited for the authentication of fruit-based products.
Experimental
Materials and methods
Unless otherwise stated, all reagents were of analytical grade. Gallic acid, protocatechualdehyde, (+)-catechin hydrate, gentisic acid, p-salicylic acid, chlorogenic acid, caffeic acid, (−)-epicatechin, (−)-epigallocatechingallate, syringic acid, syringaldehyde, ethyl gallate, umbelliferone, p-coumaric acid, taxifolin, polydatin, ferulic acid, sinapic acid, resveratrol, quercitrin hydrate, fisetin, and kaempferol were obtained from Sigma-Aldrich (Steinheim, Germany). Homogentisic acid, protocatechuic acid, vanillic acid, procyanidin A2, procyanidin B2, and procyanidin C1 were purchased from Fluka (Steinheim, Germany), and quercetin dihydrate from Riedel-de Haën (Seelze, Germany). The structures and CAS numbers of all studied polyphenols are summarized in Table 1.| Peak | Polyphenol | Family | Structure | CAS number |
|---|---|---|---|---|
| 1 | Gallic acid | Phenolic acid |
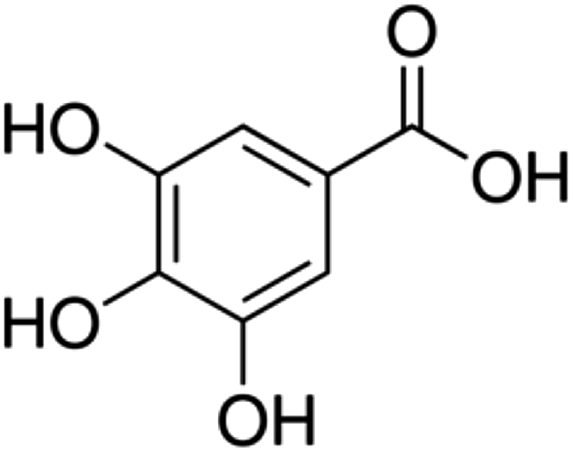
|
149-91-7 |
| 2 | Homogentisic acid | Phenolic acid |
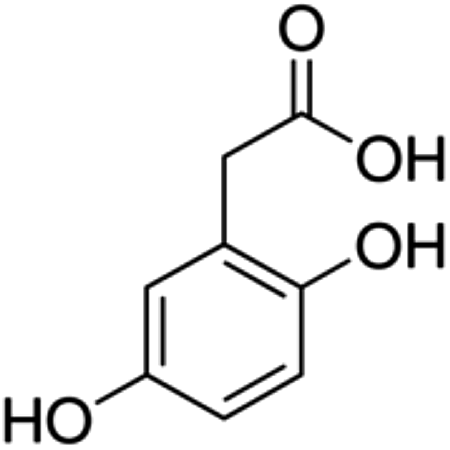
|
451-13-8 |
| 3 | Protocatechuic acid | Phenolic acid |
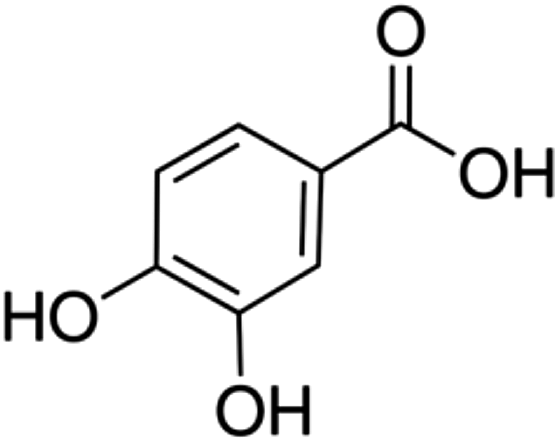
|
99-50-3 |
| 4 | Protocatechualdehyde | Phenolic aldehyde |
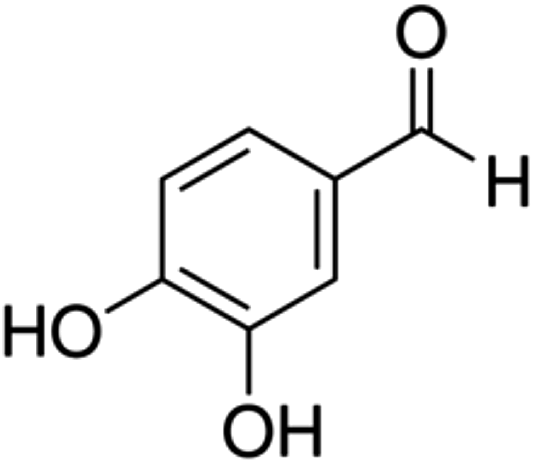
|
139-85-5 |
| 5 | (+)-Catechin hydrate | Flavanol |
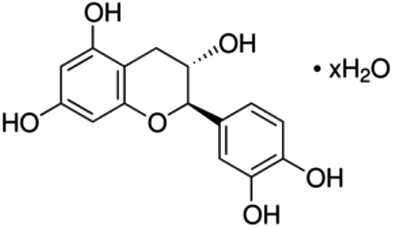
|
225937-10-0 |
| 6 | Gentisic acid | Phenolic acid |
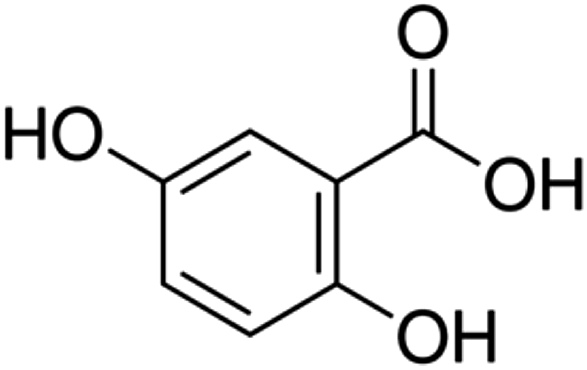
|
490-79-9 |
| 7 | p-Salicylic acid | Phenolic acid |
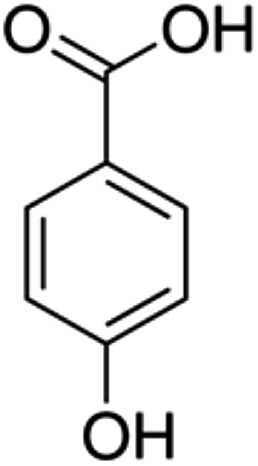
|
99-96-7 |
| 8 | Chlorogenic acid | Phenolic acid |
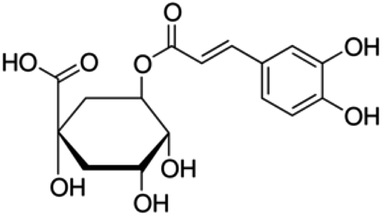
|
327-97-9 |
| 9 | Vanillic acid | Phenolic acid |
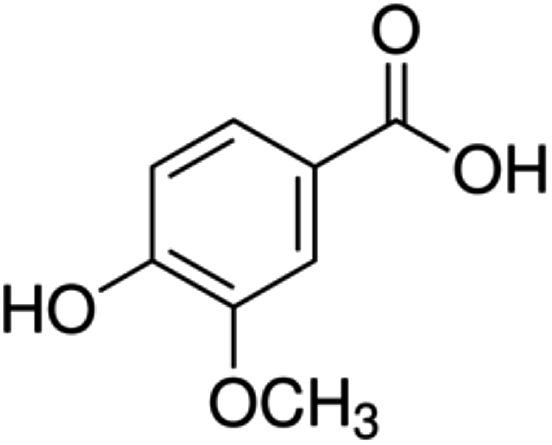
|
121-34-6 |
| 10 | Caffeic acid | Phenolic acid |

|
331-39-5 |
| 11 | (−)-Epicatechin | Flavanol |
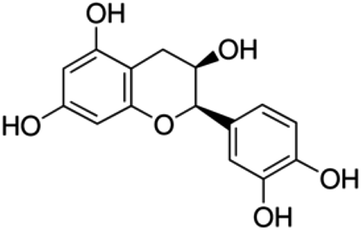
|
490-46-0 |
| 12 | (−)-Epigallocatechin gallate | Flavanol |
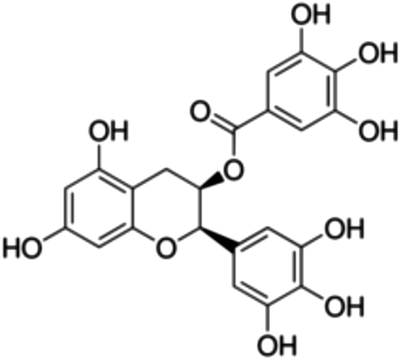
|
989-51-5 |
| 13 | Syringic acid | Phenolic acid |
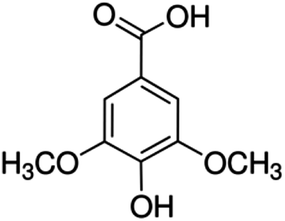
|
530-57-4 |
| 14 | Syringaldehyde | Phenolic aldehyde |
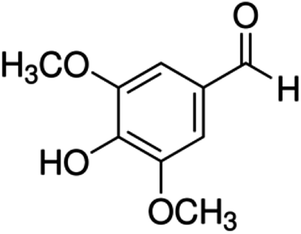
|
134-96-3 |
| 15 | Ethyl gallate | Phenolic acid |
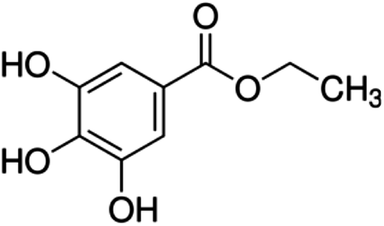
|
831-61-8 |
| 16 | Umbelliferone | Coumarin |
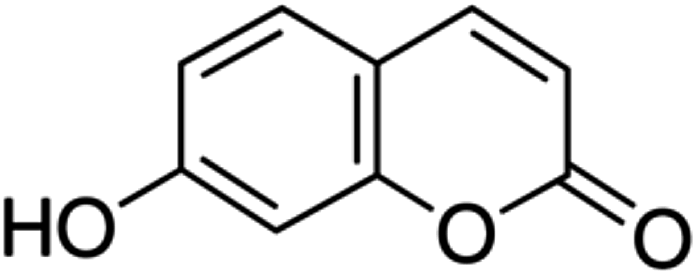
|
93-35-6 |
| 17 | p-Coumaric acid | Phenolic acid |
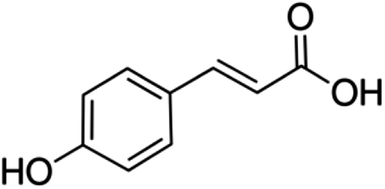
|
501-98-4 |
| 18 | Taxifolin | Flavone |
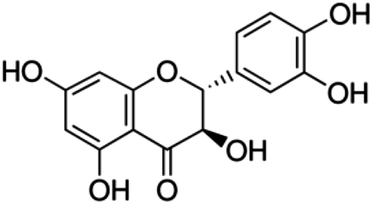
|
480-18-2 |
| 19 | Polydatin | Stilbene |

|
65914-17-2 |
| 20 | Ferulic acid | Phenolic acid |
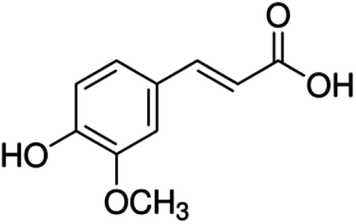
|
537-98-4 |
| 21 | Sinapic acid | Phenolic acid |
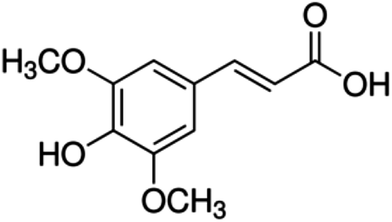
|
530-59-6 |
| 22 | Resveratrol | Stilbene |
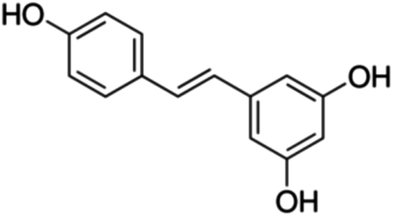
|
501-36-0 |
| 23 | Quercitrin hydrate | Flavone |
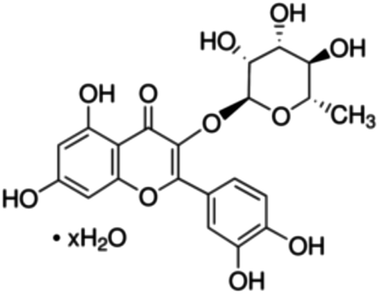
|
522-12-3 |
| 24 | Fisetin | Flavone |
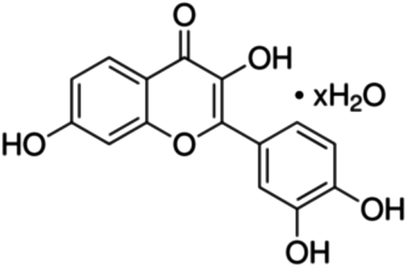
|
345909-34-4 |
| 25 | Quercetin dihydrate | Flavone |
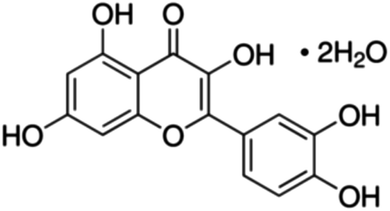
|
6151-25-3 |
| 26 | Kaempferol | Flavone |
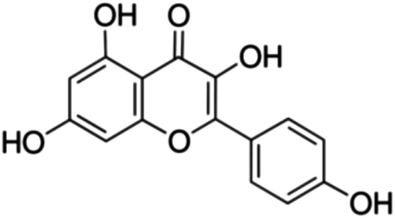
|
520-18-3 |
| 27 | Procyanidin A2 | Proanthocyanidins |
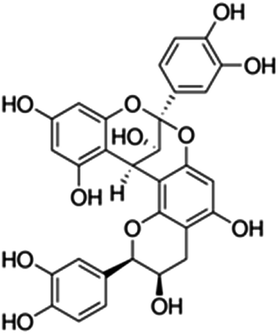
|
41743-41-3 |
| 28 | Procyanidin B2 | Proanthocyanidins |
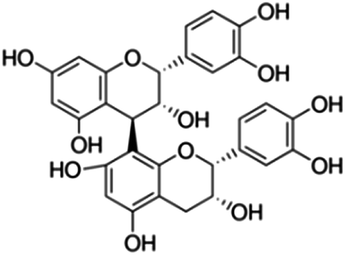
|
29106-49-8 |
| 29 | Procyanidin C1 | Proanthocyanidins |
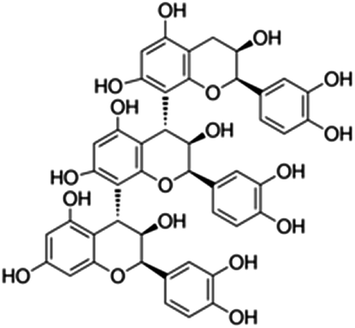
|
37064-30-5 |
LC-MS grade water and methanol, as well as formic acid (98–100%), toluene, chlorobenzene, anisole and acetone, were purchased from Sigma-Aldrich, and hydrochloric acid (98%) was provided by Merck (Seelze, Germany). Nitrogen (>99.995% pure) supplied by Abelló Linde (Barcelona, Spain) was used for the ionization sources, and high-purity argon (Ar1), purchased from Air Liquid (Madrid, Spain), was used as a collision-induced gas (CID gas) in the triple quadruple mass spectrometer.
Stock standard solutions of all polyphenols (∼1000 mg L−1) were prepared in methanol in amber glass vials. Intermediate working solutions were prepared weekly from these stock standard solutions by appropriate dilution with water. All stock solutions were stored at 4 °C for not more than 1 month.
Sample treatment
A total of 18 juice samples (9 based on cranberry and 9 based on grapes) from different brands were purchased from Barcelona markets. Additionally, a total of 14 raw extract materials and commercial cranberry-based pharmaceutical preparations presented as powder capsules, syrup, sachets and natural extracts were provided by Deiters S.L. Company (Barcelona, Spain).Prior to sample treatment, liquid samples (juices and cranberry pharmaceutical syrup) were freeze-dried to achieve fully lyophilized products with a texture similar to that of natural extracts and commercial pharmaceutical preparations (powdered samples). To this end, samples were kept for 24 h inside a lyophilizer (Telstar LyoQuest, Terrassa, Spain) with a gradient temperature ramp from −80 °C to room temperature, and then were kept for 6.5 h at 40 °C.
Sample treatment was carried out following a previously described method with some modifications.14,16 Briefly, 0.1 g of sample was dispersed in 10 mL of acetone/water/hydrochloric acid (70:29.9:0.1 v/v/v) and sonicated for 30 min. After that, the mixture was centrifuged for 15 min at 3500 rpm, and the supernatant extract was separated from the solid and stored at −4 °C until analysis. Before injection, extracts were filtered through 0.45 μm nylon filters (Whatman, Clifton, NJ, USA).
Apparatus
Chromatographic separation was performed on an ultrahigh pressure liquid chromatography (UHPLC) system (Open Accela system, Thermo Fisher Scientific, San José, CA, USA), equipped with a quaternary pump and a CTC autosampler. The performance of several chromatographic columns was tested in the present work (see characteristics of evaluated columns in Table S1 of the ESI†). Finally, a Syncronis C18 column and a Hypersil Gold C18 reversed-phase column (Thermo Fisher Scientific) were used for the proposed methods. Gradient separation was achieved using solvent A (0.1% formic acid aqueous solution) and solvent B (methanol).The UHPLC system was coupled to a TSQ Quantum Ultra AM (Thermo Fisher Scientific) triple quadrupole mass spectrometer, equipped with hyperbolic rods and with an Ion Max API source housing (Thermo Fisher Scientific) with ESI and APCI probes. When operating with heated-electrospray ionization (H-ESI II, Thermo Fisher Scientific) in negative mode, the electrospray voltage was −2.5 kV. For negative APCI, the discharge current was 10 μA. When operating with the APPI, the Ion Max source housing was mounted with a Syagen PhotoMate VUV light source (Krypton discharge lamp, 10.0 eV) (Syagen Technology Inc., Tustin, CA, USA), and the APCI probe was used as a nebulizer-desolvation device (no corona discharge was applied). For dopant-assisted APPI, 40 μL min−1 of acetone was post-column added by means of a Valco zero dead volume tee piece and using a chromatographic pump. For all API sources evaluated, both vaporizer temperature and ion transfer tube temperature were set at 350 °C. Nitrogen was employed as sheath gas, ion sweep gas, and auxiliary gas at a flow rate of 60, 0, and 20 arbitrary units (a.u.), respectively, for H-ESI, and 50, 0 and 5 a.u., respectively, for APCI and APPI. Full scan MS acquisition mode (m/z 50–1000) in Q1 (mass resolution of 0.7 m/z full width half maximum (FWHM)) with a scan time of 0.5 s was primarily used for characterization and evaluation. Selected reaction monitoring (SRM) acquisition mode (mass resolution of 0.7 m/z FWHM on both Q1 and Q3), with a scan width of 0.5 m/z and a scan time of 0.01 s, was used for quantitation purposes by monitoring two SRM transitions. Argon was used as collision gas at 1.0 mTorr, and the optimum collision energy (CE) for each transition monitored (quantifier and qualifier) is shown in Table S2 and S3 (ESI), for APPI and ESI, respectively, in the ESI.† Precursor and product ion assignments are also indicated in the tables. Instrument control and data acquisition were performed using Xcalibur v1.4 software (Thermo Fisher Scientific).
Data analysis
MATLAB (Version 6.5) was used for calculations. Principal component analysis (PCA) was from the PLS-Toolbox (Eigenvector Research Inc., Mason, WA, USA).35 A detailed description of this method is given elsewhere.36The data matrix to be treated consisted of peak area values of polyphenols in the different samples under study (see “Sample treatment”). The dimension of the matrix was 32 samples × 29 polyphenols. Since the concentration levels of some pharmaceutical samples were three orders of magnitude higher than those occurring in the juice samples, normalization pretreatment with respect to the overall polyphenolic peak areas was applied to provide similar weights to all the samples. The plot of scores showing the distribution of the samples on the principal components (PCs) revealed patterns that may be correlated with sample characteristics, such as the type of fruit in this case. The study of the distribution of variables from the loading plot provided information dealing with their correlations as well as dependencies of polyphenols on fruit product properties.
Results and discussion
Liquid chromatography
As previously commented in the Introduction section, one of the aims of this work is to develop a fast and reliable UHPLC-API-MS/MS method for the determination of polyphenolic profiles in the authentication and characterization of cranberry-based products. In order to reduce the total elution profile for this family of compounds, several reversed-phase LC columns of lower internal diameter (2.1 mm) were compared (see Table S1 in the ESI†), including sub-2 μm particle size columns of different lengths and particle sizes such as Syncronis C18 and Hypersil Gold C18, as well as porous-shell columns of different lengths such as Accucore C18 and Kinetex C18. Several applications dealing with gradient method transfers between conventional HPLC and UHPLC methods can be found in the literature.37–39 In the present work, the Thermo Scientific™ Method Transfer Tool was employed to calculate the final mobile phase flow rate and gradient elution program on each of the columns evaluated.40 The best polyphenolic profiles in terms of peak shapes, column efficiency and baseline noise were obtained with both sub-2 μm UHPLC columns in comparison to the porous-shell ones that for the same elution program always provided a very noisy chromatogram. So, both Syncronis C18 (100 × 2.1 mm, 1.7 μm) and Hypersil Gold C18 (50 × 2.1 mm, 1.9 μm) sub-2 μm UHPLC columns were selected for further studies. As an example, LC-ESI-MS/MS chromatograms obtained for the analysis of a mixture of the 29 targeted polyphenols are shown in Fig. 1. It can be seen that the performance of the transfer approach was quite limited and separations showed several full or partial coelutions between the analyzed polyphenols, especially for the shortest 50 mm column. However, baseline chromatographic separation is not mandatory because many of these coelutions could be selectively resolved by MS using the appropriate SRM transitions, taking into account that no ion suppression among the studied polyphenols was observed when ESI was used, as previously described.15 In general, all the columns studied allowed us to obtain the targeted polyphenolic profile in a much faster analysis time than the previously reported one (36 min) for the same group of compounds.15 This will represent an important reduction in the analysis time as well as in the consumption of mobile phase solvents in comparison to the methods previously proposed.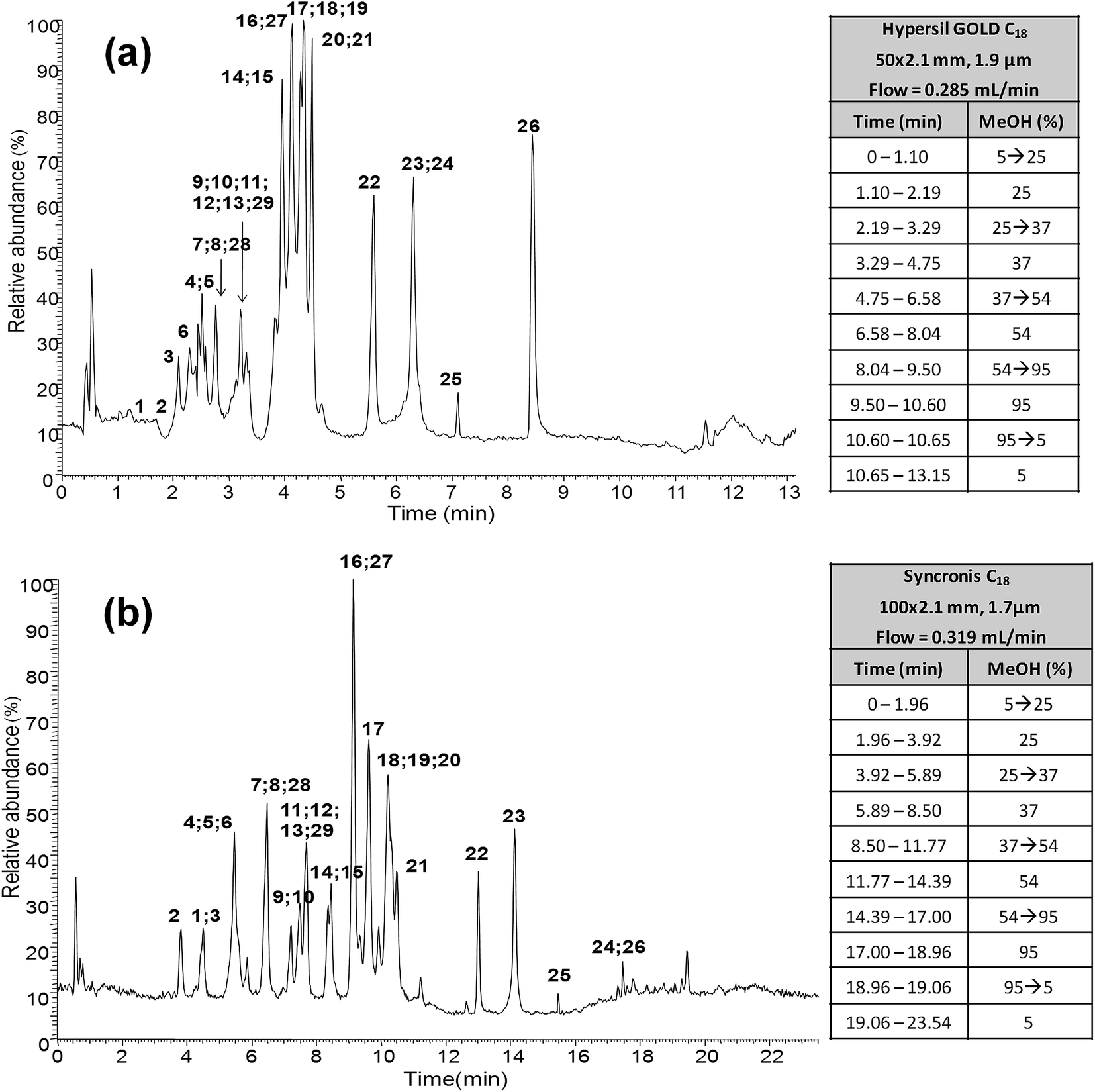 | ||
| Fig. 1 UHPLC-ESI-MS chromatograms of a mixture of the 29 analyzed polyphenols and gradient elution program by using (a) Hypersil Gold C18 (50 × 2.1 mm, 1.9 μm) and (b) Syncronis C18 (100 × 2.1 mm, 1.7 μm) reversed-phase columns. Peak identification as in Table 1. | ||
UHPLC-API-MS/MS conditions
Although ion suppression in ESI was not observed for this family of compounds, matrix effects were expected to be a handicap when dealing with the chromatographic polyphenolic profiles of real fruit-based and pharmaceutical preparation extracts. This was attributed to the fact that the important reduction on the total chromatographic elution program provoked multiple coelutions among polyphenols and matrix components. Indeed it is well known that other API sources such as APPI are typically more robust in the presence of matrix effects.41 For this reason, the performance of several API sources, such as H-ESI, APCI and APPI, was compared in order to reduce matrix effect influence on the polyphenolic characterization.The ionization of polyphenols under H-ESI, APCI and APPI conditions was thoroughly studied. In the case of APPI, several solvents such as toluene, anisole, acetone and chlorobenzene were assayed as dopants. First, full scan MS spectra (m/z 50–1000) of individual solutions of all polyphenols studied with the three API sources (and the four dopant solvents for APPI) in negative ionization mode were recorded. As an example, Fig. 2 shows the MS spectra of quercitrin with all the API sources evaluated. For H-ESI, all studied polyphenols showed the deprotonated [M − H]− ion (which was selected as the precursor ion) as the base peak of the MS spectra and, in general, neither adducts nor in-source fragmentation ions were observed at significant intensities, except for procyanidins, taxifolin, polydatin, gallic acid, (−)-epigallocatechingallate, quercitrin (Fig. 2) and quercetin that showed a small in-source fragmentation. Despite such an in-source fragmentation occurrence, the deprotonated ion continued to be the base peak signal in the MS spectra. In contrast, APCI and APPI showed MS spectra with higher in-source fragmentation for most of the analyzed polyphenols, one of the fragment ions being the base peak of the MS spectra. This fragmentation was higher for APCI than for APPI (see Fig. 2 for quercitrin). However, some of the base peak ions observed (for instance m/z 107 for quercitrin) were also common fragments in the MS spectra of other polyphenols and, thus, severe interferences may occur. Due to the high number of coelutions observed in the chromatographic separation, such common signals cannot be selected as precursor ions because of their poor selectivity. For this reason, and as a compromise, the deprotonated [M − H]− ion was also selected as the precursor ion for both APCI and APPI sources, with the exception of three polyphenols, catechin, chlorogenic acid and quercetin for which a fragment ion was selected as the precursor ion (see Table S1 in the ESI†). Regarding the different dopants used in APPI, no differences in the MS spectra of polyphenols were observed, apart from obtaining more noisy spectra in the case of anisole and chlorobenzene.
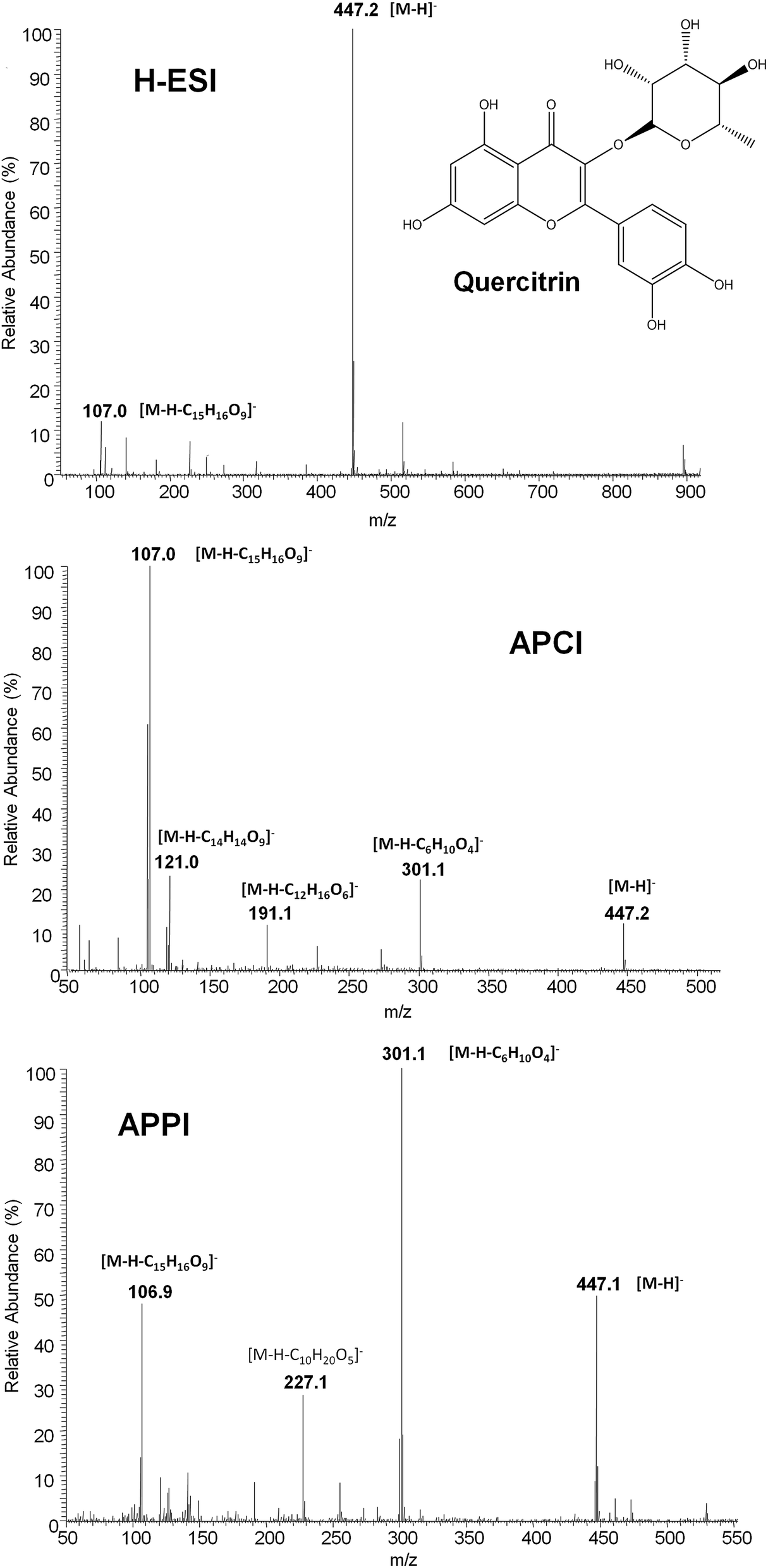 | ||
| Fig. 2 Full MS spectra of quercitrin obtained with H-ESI, APCI and APPI sources. | ||
The analyte responses obtained with dopant-assisted APPI by using different dopants were normalized for each compound to the highest signal obtained under the four conditions evaluated (see Fig. 3). As can be seen, for most of the polyphenols, the best results were obtained when acetone was used as the dopant. Differences in the dopant behavior were remarkable for some compounds such as gallic acid, (+)-catechin, chlorogenic acid, (−)-epigallocatechingallate, p-coumaric acid, resveratrol, quercitrin, fisetin, quercetin, kaempferol, procyanidin A2, and procyanidin B2. These results agree with the fact that acetone has been the selected dopant in other APPI studies of polyphenols reported in the literature.32,33 Thus, acetone was chosen as the optimum dopant for further APPI studies. Fig. 4 depicts the responses obtained when comparing H-ESI, APCI and acetone-assisted APPI normalized for each compound to the highest signal obtained under the three sources evaluated. As can be seen, H-ESI and APPI showed the highest signals, depending on the polyphenol. The worse results obtained with APCI were related to the fact that much higher in-source fragmentation was obtained and, as mentioned above, the high chromatographic coelution observed prevented the use of any of the fragment ions observed as the base peak in the MS spectra because of their poor selectivity. Hence, APCI was discarded as an efficient ionization source for our purposes. Both H-ESI and acetone-assisted APPI were selected as API sources appropriate to obtain the polyphenolic profiles for the authentication and characterization of cranberry-based products. Although APPI provides the best ionization efficiency for a limited number of polyphenols in comparison to H-ESI, it was selected for further studies because it will be less affected by matrix effects in comparison to H-ESI and probably better classification of the samples by principal components analysis will be achieved.
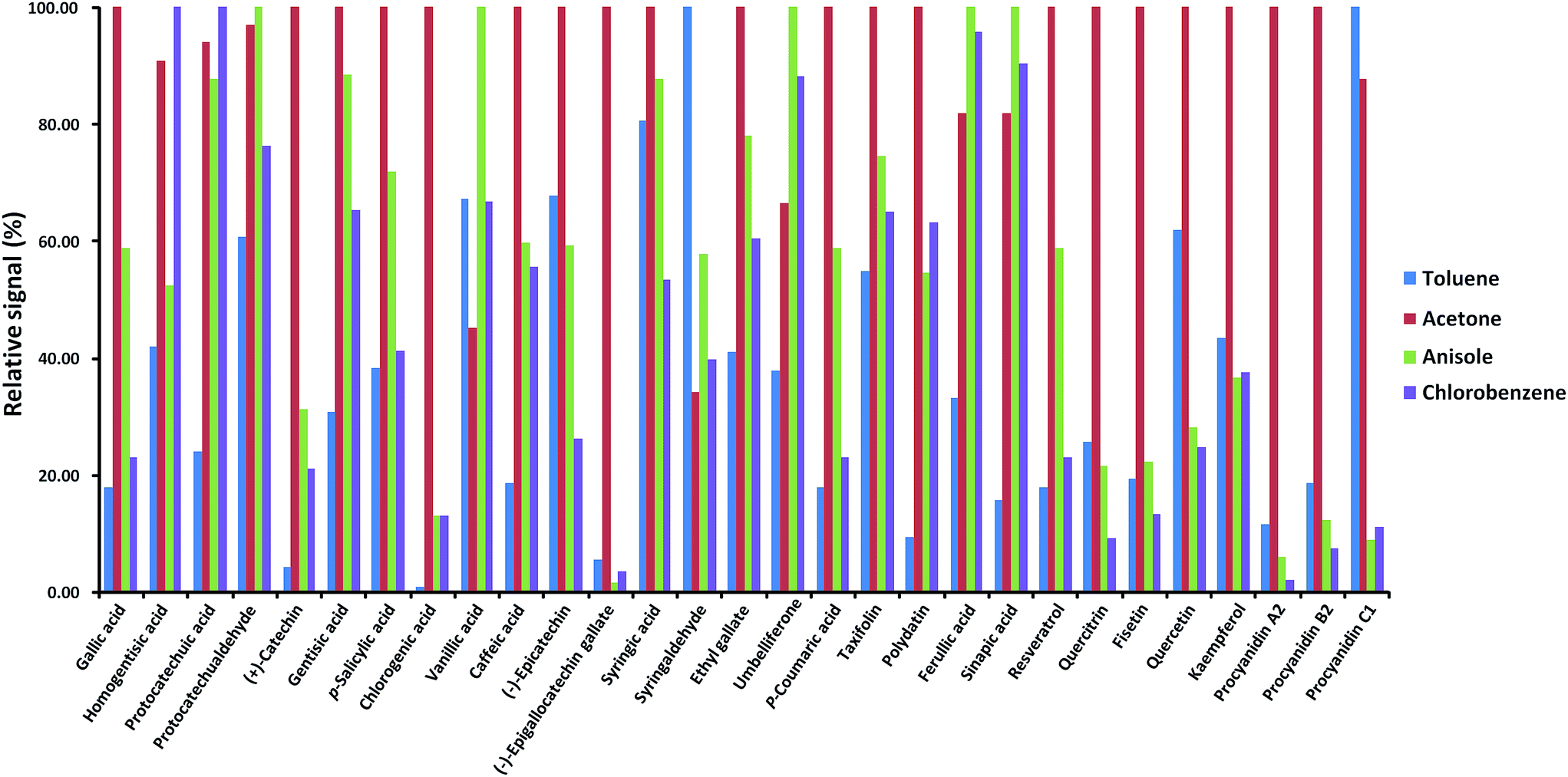 | ||
| Fig. 3 Normalized response of the signal of each polyphenol by dopant-assisted APPI when using different solvents as dopants. | ||
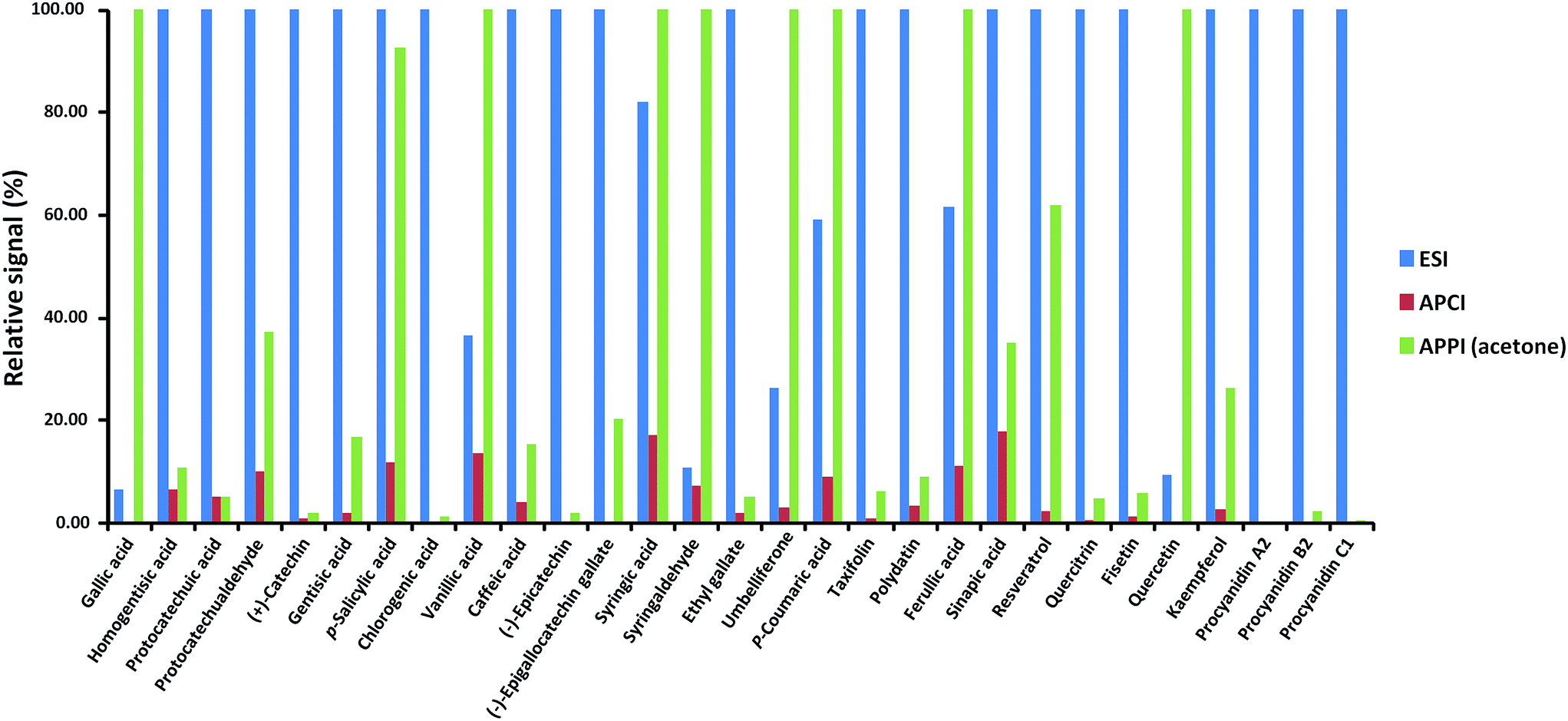 | ||
| Fig. 4 Normalized response of the signal of each polyphenol when using H-ESI, APCI and acetone-assisted APPI. | ||
The fragmentation of polyphenols in the triple quadrupole mass analyzer was also evaluated under tandem MS conditions using both H-ESI and APPI. For the correct product ion assignment, collision energy curves (5–80 eV) were assayed. After studying the product ion scan spectra of the compounds, the two most intense and characteristic transitions were selected for both quantitative and confirmation purposes. The assignments for the precursor ion and the two most intense product ions for each compound, which were selected as quantifier and qualifier SRM transitions, are given in Table S2 and S3 in the ESI.† Optimal collision energies for both quantifier and qualifier SRM transitions are given as well. In general, MS/MS spectra were similar for both H-ESI and APPI when [M − H]− was selected as the precursor ion, as previously described by Puigventós et al.15 with H-ESI, with the exception of three polyphenols, (+)-catechin, chlorogenic acid and quercetin that showed different fragment relative abundances between H-ESI and APPI, giving place to the selection of different quantifier and qualifier SRM transitions.
Instrumental quality parameters and matrix effect
Instrumental quality parameters of UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods using both Hypersil Gold and Syncronis C18 columns were calculated for the 29 polyphenolic studied compounds, and the figures of merit are given in Table 2. Limits of detection (LODs), based on a signal-to-noise ratio of 3:1, and limits of quantification (LOQs), based on a signal-to-noise ratio of 10:1, were calculated using standard solutions at low concentration levels. As can be seen in Table 2, for most of the studied compounds, APPI showed similar or worse LOD and LOQ values with both evaluated C18 columns in comparison to the figures achieved with ESI. These results were expected taking into account the ionization efficiency observed for ESI and APPI sources for the studied compounds (see Fig. 4), although it should be mentioned that for several polyphenols such as p-salicylic acid, vanillic acid, syringic acid, syringaldehyde, umbelliferone, p-coumaric acid, and ferulic acid similar or even better sensitivity was observed when APPI was used. Nevertheless, LOD values are always lower than 500 μg kg−1 and down to 0.5 μg kg−1 in the best of cases. Taking into account that most polyphenols and phenolic acids are usually expected at mg kg−1 in the fruit-based analyzed extracts, the sensitivity achieved with both UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS in either of the evaluated columns is enough for the detection and determination of this family of compounds in cranberry-based pharmaceuticals and fruit products.
| Peak | Compound | Hypersil Gold C18 column | Syncronis C18 column | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UHPLC-ESI-MS/MS | UHPLC-APPI-MS/MS | UHPLC-ESI-MS/MS | UHPLC-APPI-MS/MS | ||||||||||||||
| LODa | LOQa | Precisionb | Truenessc | LODa | LOQa | Precisionb | Truenessc | LODa | LOQa | Precisionb | Truenessc | LODa | LOQa | Precisionb | Truenessc | ||
| a μg kg−1. b % RSD (n = 5) at the LOQ value. c % Relative error (n = 5) at the LOQ value. | |||||||||||||||||
| 1 | Gallic acid | 5 | 17 | 5.5 | 6.5 | 100 | 330 | 3.6 | 3.3 | 50 | 165 | 5.1 | 3.6 | 50 | 165 | 7.2 | 6.3 |
| 2 | Homogentisic acid | 2 | 7 | 4.8 | 6.6 | 30 | 99 | 4.5 | 2.5 | 5 | 17 | 6.3 | 5.4 | 20 | 66 | 2.2 | 11.2 |
| 3 | Protocatechuic acid | 2 | 7 | 6.2 | 7.4 | 5 | 17 | 9.2 | 6.3 | 0.5 | 2 | 4.5 | 4.2 | 5 | 17 | 8.3 | 3.3 |
| 4 | Protocatechualdehyde | 1 | 3 | 5.8 | 5.6 | 3 | 10 | 5.8 | 8.2 | 5 | 17 | 9.7 | 8.2 | 10 | 33 | 4.1 | 4.4 |
| 5 | (+)-Catechin hydrate | 25 | 83 | 4.2 | 9.3 | 50 | 165 | 4.6 | 1.2 | 10 | 33 | 4.5 | 6.3 | 75 | 248 | 6.2 | 8.6 |
| 6 | Gentisic acid | 5 | 17 | 3.6 | 7.4 | 3 | 10 | 3.2 | 9.9 | 20 | 66 | 6.3 | 9.5 | 1 | 3 | 9.6 | 8.2 |
| 7 | p-Salicylic acid | 10 | 33 | 5.2 | 6.9 | 2 | 7 | 6.9 | 2.3 | 10 | 33 | 4.2 | 4.2 | 5 | 17 | 1.2 | 6.3 |
| 8 | Chlorogenic acid | 12 | 40 | 8.2 | 2.5 | 20 | 66 | 4.6 | 6.5 | 20 | 66 | 9.3 | 6.3 | 30 | 99 | 11.2 | 4.1 |
| 9 | Vanillic acid | 5 | 17 | 6.3 | 9.3 | 1 | 3 | 6.3 | 7.4 | 5 | 17 | 1.2 | 9.9 | 5 | 17 | 3.3 | 7.4 |
| 10 | Caffeic acid | 10 | 33 | 4.2 | 10.2 | 2 | 7 | 6.5 | 8.2 | 20 | 66 | 5.5 | 2.2 | 10 | 33 | 4.5 | 3.6 |
| 11 | (−)-Epicatechin | 22 | 73 | 3.5 | 4.5 | 50 | 165 | 8.0 | 6.3 | 10 | 33 | 3.6 | 4.4 | 70 | 231 | 2.2 | 8.4 |
| 12 | (−)-Epigallocatechin gallate | 1 | 3 | 9.3 | 7.6 | 250 | 825 | 9.2 | 6.3 | 100 | 330 | 4.4 | 4.6 | 500 | 1650 | 8.5 | 6.3 |
| 13 | Syringic acid | 0.75 | 2 | 4.2 | 3.3 | 0.5 | 2 | 1.9 | 5.2 | 10 | 33 | 9.3 | 3.3 | 2 | 7 | 3.3 | 7.2 |
| 14 | Syringaldehyde | 1.2 | 4 | 1.5 | 9.5 | 0.5 | 2 | 4.5 | 5.4 | 10 | 33 | 1.1 | 4.5 | 0.5 | 2 | 4.4 | 5.2 |
| 15 | Ethyl gallate | 125 | 412 | 3.5 | 2.5 | 5 | 17 | 6.3 | 1.6 | 10 | 33 | 6.3 | 5.2 | 10 | 33 | 7.1 | 3.9 |
| 16 | Umbelliferone | 0.75 | 2 | 4.5 | 8.2 | 0.5 | 2 | 11.5 | 8.2 | 2 | 7 | 4.4 | 5.8 | 0.5 | 2 | 6.5 | 6.1 |
| 17 | p-Coumaric acid | 1.2 | 4 | 7.8 | 4.6 | 1 | 3 | 2.6 | 4.5 | 5 | 17 | 6.3 | 3.3 | 10 | 33 | 9.3 | 4.0 |
| 18 | Taxifolin | 2 | 7 | 11.2 | 5.1 | 2 | 7 | 14.5 | 9.2 | 5 | 17 | 15.1 | 7.4 | 5 | 17 | 12.4 | 9.3 |
| 19 | Polydatin | 5 | 17 | 8.6 | 8.5 | 2 | 7 | 11.2 | 7.8 | 20 | 66 | 3.3 | 6.3 | 20 | 66 | 5.2 | 7.0 |
| 20 | Ferulic acid | 0.5 | 2 | 10.5 | 7.4 | 0.5 | 2 | 6.3 | 7.2 | 75 | 247 | 4.0 | 4.2 | 0.5 | 2 | 5.8 | 4.2 |
| 21 | Sinapic acid | 0.5 | 2 | 11.2 | 9.3 | 0.5 | 2 | 9.5 | 6.2 | 10 | 33 | 8.8 | 7.7 | 0.5 | 2 | 9.2 | 6.3 |
| 22 | Resveratrol | 6 | 20 | 5.6 | 2.8 | 10 | 33 | 4.5 | 8.2 | 10 | 33 | 4.4 | 6.3 | 10 | 33 | 4.2 | 4.1 |
| 23 | Quercitrin hydrate | 5 | 17 | 8.5 | 5.4 | 1 | 2 | 6.3 | 2.2 | 2 | 7 | 6.3 | 9.5 | 5 | 17 | 3.7 | 7.4 |
| 24 | Fisetin | 15 | 50 | 6.9 | 6.9 | 100 | 330 | 8.5 | 4.7 | 250 | 825 | 7.4 | 9.2 | 500 | 1650 | 8.2 | 9.6 |
| 25 | Quercetin dihydrate | 12 | 40 | 4.5 | 7.5 | 500 | 1650 | 7.4 | 3.6 | 100 | 330 | 4.1 | 1.4 | 500 | 1650 | 7.6 | 2.3 |
| 26 | Kaempferol | 5 | 17 | 6.3 | 4.6 | 500 | 1650 | 6.3 | 4.5 | 10 | 33 | 6.3 | 5.6 | 20 | 66 | 4.2 | 6.3 |
| 27 | Procyanidin A2 | 10 | 33 | 2.8 | 7.4 | 250 | 825 | 5.2 | 8.9 | 50 | 165 | 9.3 | 8.5 | 250 | 825 | 3.6 | 4.1 |
| 28 | Procyanidin B2 | 5 | 17 | 4.1 | 6.9 | 500 | 1650 | 4.4 | 9.3 | 50 | 165 | 4.5 | 6.3 | 250 | 825 | 4.2 | 3.3 |
| 29 | Procyanidin C1 | 1 | 3 | 6.3 | 7.1 | 500 | 1650 | 4.9 | 6.3 | 150 | 495 | 4.4 | 4.7 | 500 | 1650 | 7.1 | 7.4 |
Calibration curves based on the peak area at concentrations above the LOQ to 100 mg L−1 were established, and good linearity (r2 higher than 0.995) was observed for all compounds. Run-to-run precision for compound quantification was also calculated at the LOQ concentration level by performing five replicate determinations under optimal conditions. Relative standard deviation (%) values lower than 15.1% were observed. Trueness was also evaluated at the LOQ concentration level (n = 5) by comparing spiked with calculated concentrations using external calibration. Results were very acceptable, with relative errors (%) lower than 10.2%.
The results obtained showed that both UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods employing either of the two evaluated columns were satisfactory in terms of sensitivity, precision, and trueness for the determination of polyphenols at the expected concentration levels.
Matrix effects were also evaluated by comparing the external calibration slope of both UHPLC-API-MS/MS methods with that obtained by matrix-matched calibration. Due to the lack of real blank samples for all targeted polyphenols and phenolic acids, this study was carried out only with several polyphenols not detected (or at concentrations below LOD values) in some of the analysed samples. As an example, Fig. S1 in the ESI† shows the calibration plots obtained under external calibration and matrix-matched calibration (using a cranberry-based extract) for homogentistic acid and resveratrol (not present in the cranberry-based extract analyzed) when both UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods using the Hypersil GOLD C18 column were applied. As can be seen, a slight matrix effect by ion suppression was observed when ESI was employed (although lower than 20%), while APPI showed almost no matrix effect, as expected. A similar behaviour was observed for all the studied polyphenols in those extracts where they were not present. These results allow the proposal of external calibration as a suitable quantification method for polyphenols in the analyzed samples. However, it should be mentioned that the main objective of this work is not to quantify the concentration levels of polyphenols but to use polyphenolic signal profiles to perform sample classification by chemometric methods.
Principal component analysis
The characterization of cranberry and grape products, including berries, juices and pharmaceuticals, relied on polyphenolic chromatographic profiles resulting from UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods by employing both Syncronis C18 and Hypersil Gold C18 columns. In particular, peak areas of the quantifier SRM transition of each polyphenol corresponding to these four possibilities were used as analytical data to be treated chemometrically using PCA. As an example, Fig. 5 shows the UHPLC-ESI-MS/MS chromatogram of nine selected polyphenols found in a cranberry pill pharmaceutical sample. As indicated in the Data analysis section, results were interpreted to draw relevant patterns dealing with the characteristics of natural and processed products. Besides, the most significant polyphenols as the relevant markers of each class were also investigated.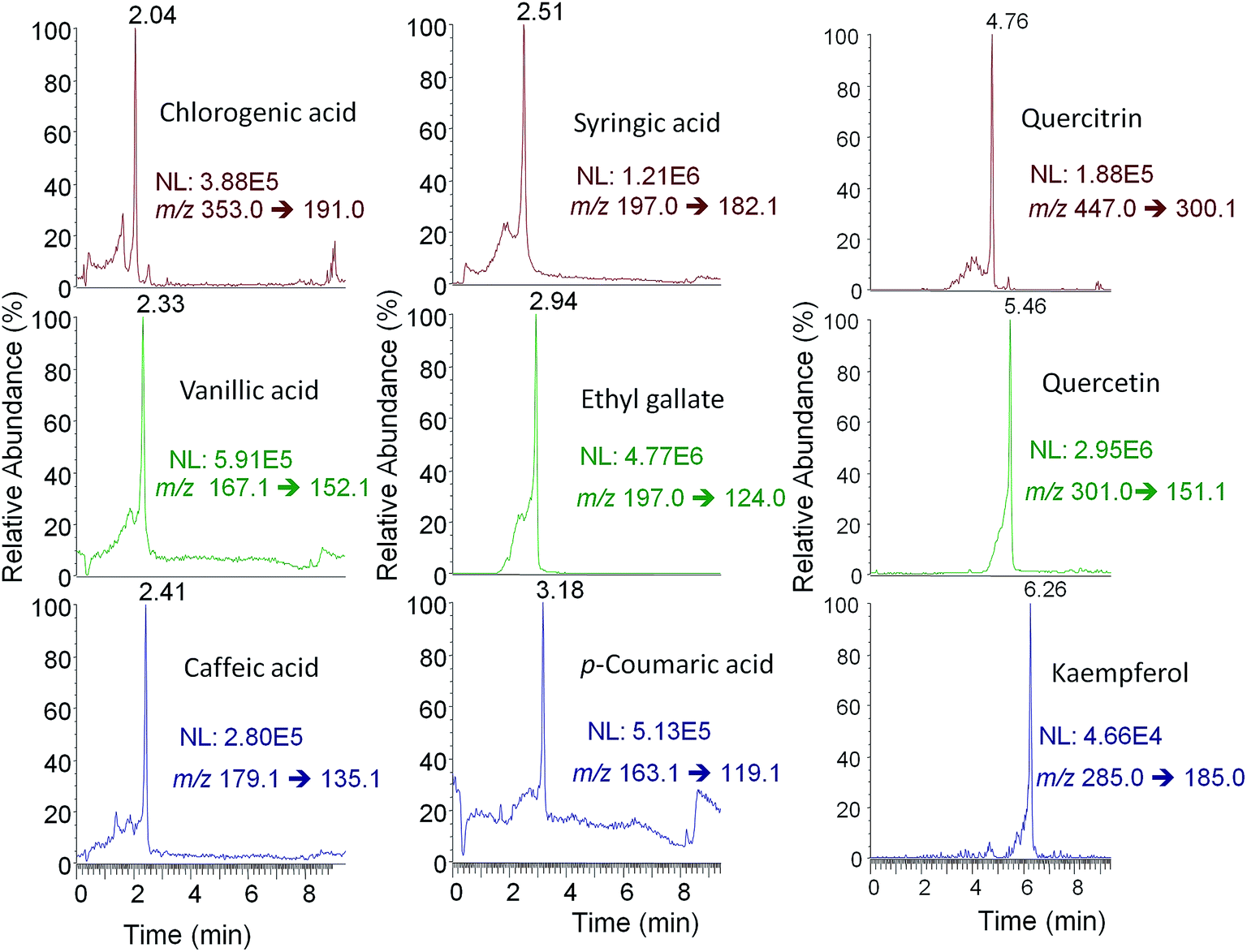 | ||
| Fig. 5 UHPLC-ESI-MS/MS chromatograms of 9 selected polyphenols found in a cranberry pill pharmaceutical sample. | ||
A first drawback to be faced was the much higher concentrations of polyphenols occurring in some pharmaceuticals as, indeed, such products were concentrated extracts of cranberries. Responses obtained for extracts and capsules were 2 or 3 orders of magnitude higher than those found in the juices. To try to reduce the influence of large values on the description models, data were pretreated. Data normalization and autoscaling were here applied to equalize the contribution of all kinds of samples in the model. After preprocessing, data were treated by PCA and the most noticeable findings are given as follows.
PCA results using normalized peak areas by UHPLC-ESI-MS/MS with Syncronis C18 (100 × 2.1 mm, 1.7 μm) and Hypersil Gold C18 (50 × 2.1 mm, 1.9 μm) columns are shown in Fig. 6. In the two cases, the plot of scores indicated that grape and cranberry samples were reasonably separated across PC1 although some cranberry-based samples appeared close to the grape cluster. The differentiation of samples into groups was more deficient in the case of the short analytical column (Hypersil Gold C18) from which the chromatographic resolution of components of the samples was limited and multiple overlapping among analytes and matrix components occurred. As pointed out above, the occurrence of coelutions hindered the quality of the chemometric results as responses were seriously affected by sample matrix effects. Conversely, results from UHPLC-APPI-MS/MS (Fig. 7) were more satisfactory and the discrimination of sample classes was excellent even when using the fast chromatographic method with the Hypersil Gold column. As depicted in Fig. 7b, sample clusters were perfectly delimited. The improvement of the sample map with respect to Fig. 6b was attributed to the more robust behavior of the APPI source in the presence of matrix effect issues. Grape and cranberry samples were better distinguished in the case of the long column, which allowed the analyte resolution to be improved chromatographically so the occurrence of matrix effects was minimized. Apart from fruit origin, differences within the cranberry group were observed. It can be seen that juices and berry samples appeared together in the bottom-left part of the scores plot. Besides, sachets, syrup and some capsules (e.g., C6, C8, etc.) were located in the same area, thus suggesting that polyphenolic contents were similar to those occurring in natural samples. The similarities in compositions were also in agreement with high correlation coefficients (r2 > 0.995). Conversely, another type of pharmaceuticals, located in the top-left, was noticeably different regarding composition. Such behavior was attributed to differences in polyphenol concentrations because of raw materials (e.g., berry variety) or manufacturing processes. From the point of view of authentication, PCA results suggested that no issues were found and all the samples were reasonably considered genuine.
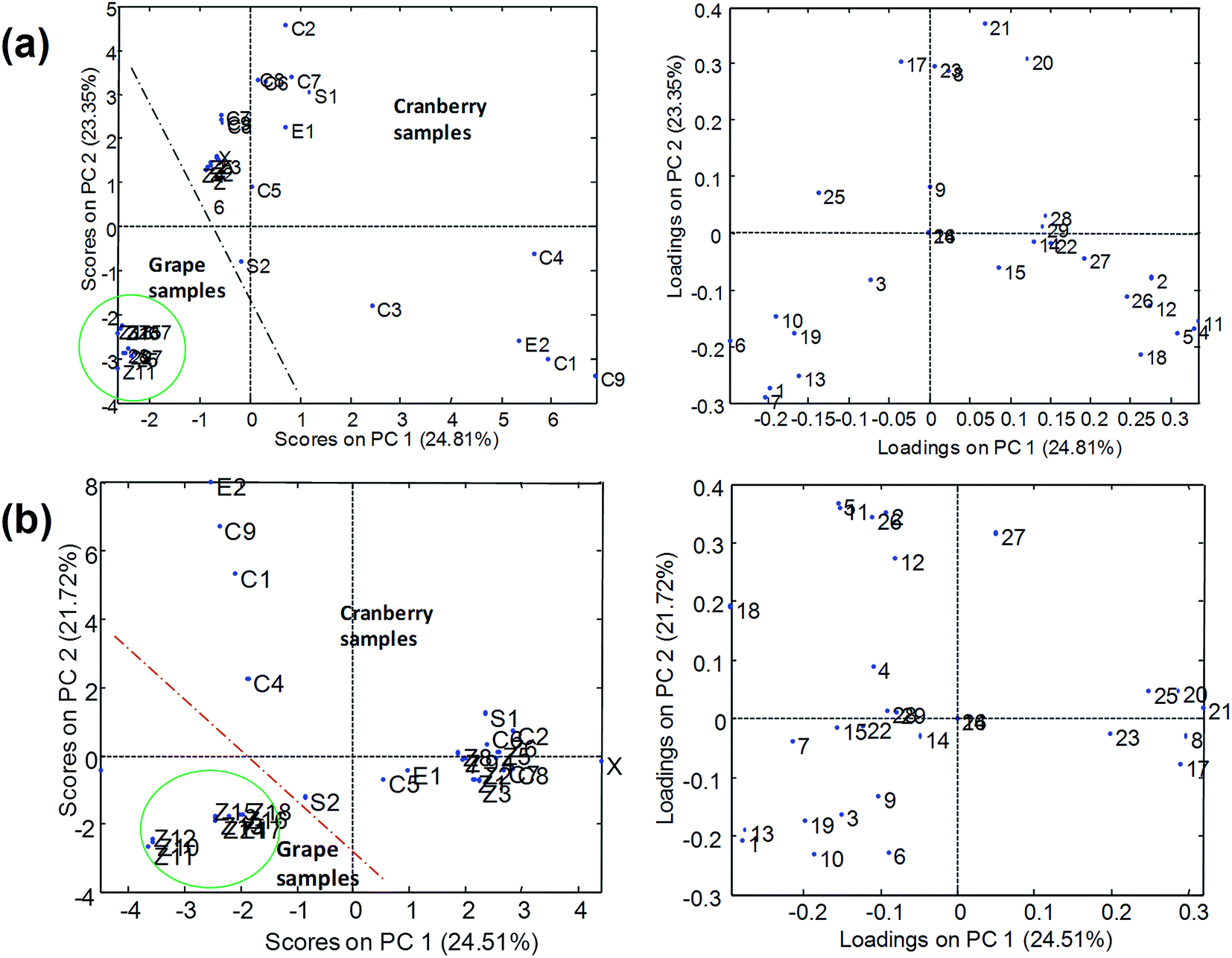 | ||
| Fig. 6 PCA results (scatter plots of scores and loadings of PC1 and PC2) using the normalized peak areas of the UHPLC-ESI-MS/MS polyphenolic profile obtained with (a) Syncronis C18 (100 × 2.1 mm, 1.7 μm) and (b) Hypersil Gold C18 (50 × 2.1 mm, 1.9 μm) reversed-phase columns as analytical data. Z, juices; C, capsules; S, sachets, E, extracts; X, syrup. The lines in the figure show the separation of grape and cranberry samples. | ||
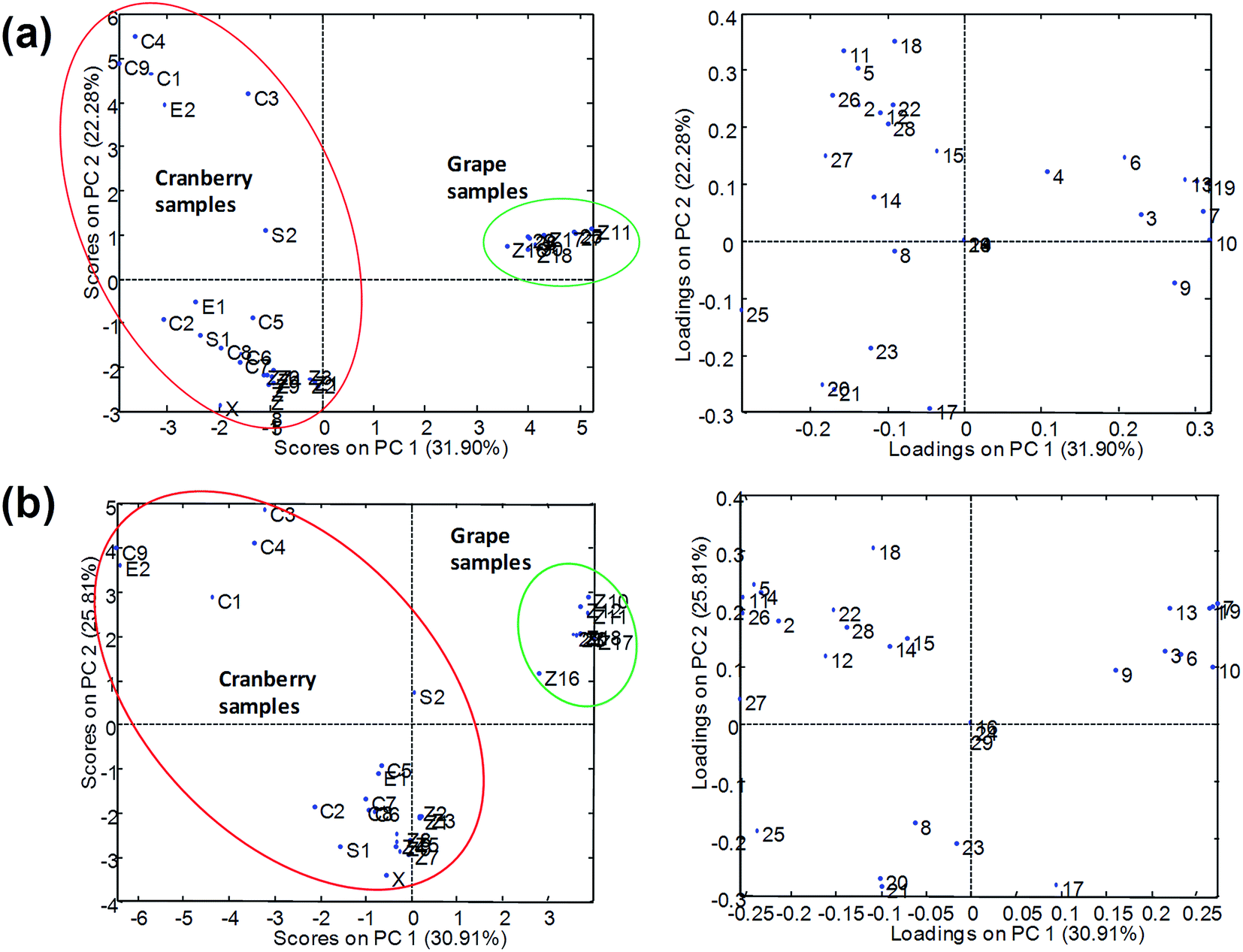 | ||
| Fig. 7 PCA results (scatter plots of scores and loadings of PC1 and PC2) using the normalized peak areas of the UHPLC-APPI-MS/MS polyphenolic profile obtained with (a) Syncronis C18 (100 × 2.1 mm, 1.7 μm) and (b) Hypersil Gold C18 (50 × 2.1 mm, 1.9 μm) reversed-phase columns as analytical data. Z, juices; C, capsules; S, sachets, E, extracts; X, syrup. | ||
From the plot of loadings it was deduced that syringic, salicylic, gallic and caffeic acids and polydatin were highly characteristic of grape products. These compounds were almost absent in cranberry juices so they were considered as representative grape markers. Gentisic and protocatechuic acids were also up expressed in grape-related samples. Regarding descriptors of cranberry products, ferulic, chlorogenic and sinapic acids were found to be quite specific compounds. Also, in general, quercetin and quercitrin were up expressed in cranberry juices with respect to the grape counterparts. As a summary of these comparative studies involving four different analytical methods, it was concluded that the APPI source provided the most satisfactory results as it was more robust in the presence of matrix interferences while ESI data were more sensitive to undergo variations due to the coelution of analytes and matrix components. Hence, when using ESI sources, reasonably good separations in the chromatographic domain will be required to minimize the disturbance of matrix components.
A complementary study was carried out based on combining data from the different methods as a way to enrich the datasets. Despite the shortcomings aforementioned regarding ESI results, it was realized that the sensitivity achieved for some compounds was superior to that of APPI. Indeed, the ESI approach may be of great interest in combination with a good chromatographic separation. In other words, those well separated compounds (without being affected by matrix component overlapping) could be successfully detected by ESI with no sensitivity issues. If so, data from ESI experiments could be considered as a complementary source of information to improve the description of the samples studied. Results from the study of such augmented data arrangements (considering both UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS polyphenolic profiles) by PCA are shown in Fig. S2 (ESI†). It was found that the sample distribution showed a clear separation among cranberry- and grape-related samples. Also, as above, cranberry samples appeared segregated in two groups, e.g., juices and pharmaceuticals with similar compositions (bottom left corner) and other pharmaceuticals with noticeably different compositions (top left corner). Sample groups were highly discriminated in the case of the long Syncronis column, but even in the case of the short Hypersil Gold column the separation was excellent. In the plot of loadings, the same class descriptors pointed out in the previous analyses of each individual dataset could be confirmed. As a result, although the characterization of cranberry and grape-based samples could be resolved successfully by fast chromatography with the APPI source, the combination of complementary information from multiple sources should not be underestimated as a way of dealing with more complex issues.
Conclusions
The results obtained in this work show that the developed UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods, using both Syncronis and Hypersil Gold C18 reversed-phase columns, can be proposed as fast and suitable methods for the determination of polyphenolic profiles in fruit-based products and pharmaceutical preparations. Both C18 reversed-phase columns studied allowed us to obtain the targeted polyphenolic profile in a much faster analysis time (13.5 min and 23.5 min for the Syncronis and Hypersil Gold columns, respectively) than the previously reported one (36 min) for the same group of compounds. This represents an important reduction in both the analysis time and the consumption of mobile phase solvents, being important advantages for high throughput analysis. However, multiple coelutions among polyphenols were observed due to the important reduction on the total chromatographic elution programs. Although ion suppression in ESI was not observed for this family of compounds, matrix effects were expected to be a handicap when dealing with the chromatographic polyphenolic profiles of real fruit-based and pharmaceutical preparation extracts. For that purpose, the performance of several API sources (H-ESI, APCI and APPI using toluene, acetone, anisole and chlorobenzene as dopants) was evaluated.In the comparison of API sources, APCI was not appropriate for this study because of its high in-source collision-induced dissociation fragmentation preventing the selection of selective enough SRM transitions. In contrast, H-ESI and acetone-dopant assisted APPI showed great performance, providing good sensitivity for most of the analyzed polyphenols and being able to propose two SRM transitions for the acquisition of polyphenolic profiles. Acceptable sensitivity and good run-to-run precision and trueness were obtained with both UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS methods proposed. No matrix effects were observed when APPI was employed as the ionization source, as expected.
The proposed UHPLC-API-MS/MS methods using both C18 reversed-phase columns were applied to the analysis of 32 grape-based and cranberry-based products and pharmaceutical preparations after a simple extraction procedure as a sample treatment consisting of acetone/water/hydrochloric acid extraction by sonication. PCA results using normalized peak areas by UHPLC-ESI-MS/MS and UHPLC-APPI-MS/MS showed that, in general, the differentiation of samples into groups was more deficient in the case of the short analytical column (Hypersil Gold C18) from which the chromatographic resolution of components of the samples was limited and multiple overlapping among analytes and matrix components occurred. Conversely, results obtained by UHPLC-APPI-MS/MS were more satisfactory and the discrimination of sample classes was excellent even when using the fast chromatographic method with the Hypersil Gold column, behavior attributed to the higher robustness of APPI source in the presence of matrix effects.
Thus, PCA of polyphenolic peak areas obtained by the UHPLC-APPI-MS/MS method using the Hypersil Gold C18 column can be proposed as a fast and reliable approach for the classification, characterization and the probable authentication of grape- and cranberry-based products according to the type of fruit.
Acknowledgements
The authors gratefully acknowledge the financial support received from the Spanish Ministry of Economy and Competitiveness under the projects CTQ2014-65324 and CTQ2015-63968-C2-1-P and from the Agency for Administration of University and Research Grants (Generalitat de Catalunya, Spain) under the projects 2014SGR-377 and 2014SGR-539. Authors wish to acknowledge Deiters S.L. Company for providing some cranberry-based raw material extracts and several commercial cranberry-based pharmaceutical preparations.References
- C. Manach, A. Scalbert, C. Morand, C. Remesy and L. Jimenez, Am. J. Clin. Nutr., 2004, 79, 727–747 CAS.
- H. El Gharras, Int. J. Food Sci. Technol., 2009, 44, 2512–2518 CrossRef CAS.
- S. Pastore, D. Lulli, A. Potapovich, P. Fidanza, V. Kostyuk, E. Dellambra, C. De Luca, R. Maurelli and L. Korkina, J. Dermatol. Sci., 2011, 63, 104–114 CAS.
- S. Pastore, A. Potapovich, D. Lulli, P. Fidanza, V. Kostyuk, C. De Luca, E. Mikhalchik and L. Korkina, Antioxid. Redox Signaling, 2012, 16, 314–328 CrossRef CAS PubMed.
- T. Pluemsamran, T. Onkoksoong and U. Panich, Photochem. Photobiol., 2012, 88, 961–968 CrossRef CAS PubMed.
- Y. Yin, W. Li, Y. O. Son, L. Sun, J. Lu, D. Kim, X. Wang, H. Yao, L. Wang, P. Pratheeshkumar, A. J. Hitron, J. Luo, N. Gao, X. Shi and Z. Zhang, Toxicol. Appl. Pharmacol., 2013, 269, 89–99 CrossRef CAS PubMed.
- V. Cheynier, Am. J. Clin. Nutr., 2005, 81, 223S–229S CAS.
- F. Sánchez-Patán, B. Bartolome, P. J. Martin-Alvarez, M. Anderson, A. Howell and M. Monagas, J. Agric. Food Chem., 2012, 60, 3396–3408 CrossRef PubMed.
- L. Y. Foo, Y. Lu, A. B. Howell and N. Vorsa, Phytochemistry, 2000, 54, 173–181 CrossRef CAS PubMed.
- A. B. Howell, J. D. Reed, C. G. Krueger, R. Winterbottom, D. G. Cunningham and M. Leahy, Phytochemistry, 2005, 66, 2281–2291 CrossRef CAS PubMed.
- L. Gu, M. A. Kelm, J. F. Hammerstone, G. Beecher, J. Holden, D. Haytowitz and R. L. Prior, J. Agric. Food Chem., 2003, 51, 7513–7521 CrossRef CAS PubMed.
- S. Guyot, Flavan-3-Ols and Proanthocyanidins, in Handbook of Analysis of Active Compounds in Functional Foods, ed. N. M. L. Nollet and F. Toldrá, CRC Press, Boca Raton, FL, USA, 2012 Search PubMed.
- L. Gu, M. A. Kelm, J. F. Hammerstone, G. Beecher, J. Holden, D. Haytowitz, S. Gebhardt and R. L. Prior, J. Nutr., 2004, 134, 613–617 CAS.
- M. Navarro, O. Núñez, J. Saurina, S. Hernández-Cassou and L. Puignou, J. Agric. Food Chem., 2014, 62, 1038–1046 CrossRef CAS PubMed.
- L. Puigventós, M. Navarro, E. Alechaga, O. Núñez, J. Saurina, S. Hernández-Cassou and L. Puignou, Anal. Bioanal. Chem., 2015, 407, 597–608 CrossRef PubMed.
- T. C. Wallace and M. M. Giusti, J. Food Sci., 2010, 75, C690–C696 CrossRef CAS PubMed.
- S. Rzeppa, C. Von Bargen, K. Bittner and H. Humpf, J. Agric. Food Chem., 2011, 59, 10594–10603 CrossRef CAS PubMed.
- R. Furuuchi, T. Yokoyama, Y. Watanabe and M. Hirayama, J. Agric. Food Chem., 2011, 59, 3738–3746 CrossRef CAS PubMed.
- M. P. Delgado de la Torre, C. Ferreiro-Vera, F. Priego-Capote and M. D. Luque de Castro, J. Agric. Food Chem., 2013, 61, 12539–12548 CrossRef CAS PubMed.
- A. I. Hamed, A. S. Al Ayed, J. Moldoch, S. Piacente, W. Oleszek and A. Stochmal, J. Mass Spectrom., 2014, 49, 306–315 CrossRef CAS PubMed.
- M. T. Engström, M. Palijarvi, C. Fryganas, J. H. Grabber, I. Mueller-Harvey and J. P. Salminen, J. Agric. Food Chem., 2014, 62, 3390–3399 CrossRef PubMed.
- R. M. Alonso-Salces, K. Ndjoko, E. F. Queiroz, J. R. Ioset, K. Hostettmann, L. A. Berrueta, B. Gallo and F. Vicente, J. Chromatogr. A, 2004, 1046, 89–100 CrossRef CAS PubMed.
- V. Jerkovic, D. Callemien and S. Collin, J. Agric. Food Chem., 2005, 53, 4202–4206 CrossRef CAS PubMed.
- V. Jerkovic and S. Collin, J. Agric. Food Chem., 2008, 56, 584–590 CrossRef CAS PubMed.
- M. Shrinivasan, V. Aparna, P. K. Puvvda and V. R. Kolte, J. Pharma Res., 2012, 5, 3486–3491 CAS.
- G. Jerz, J. A. Murillo-Velasquez, I. Skrjabin, R. Gok and P. Winterhalter, ACS Symp. Ser., 2012, 1098, 145–165 CrossRef CAS.
- A. Vallverdú-Queralt, O. Jauregui, A. Medina-Remon, C. Andres-Lacueva and R. M. Lamuela-Raventos, Rapid Commun. Mass Spectrom., 2010, 24, 2986–2992 CrossRef PubMed.
- I. I. Rockenbach, E. Jungfer, C. Ritter, B. Santiago-Schuebel, B. Thiele, R. Fett and R. Galensa, Food Res. Int., 2012, 48, 848–855 CrossRef CAS.
- I. Iswaldi, A. M. Gomez-Caravaca, D. Arraez-Roman, J. Uberos, M. Lardon, A. Segura-Carretero and A. Fernandez-Gutierrez, J. Pharm. Biomed. Anal., 2012, 58, 34–41 CrossRef CAS PubMed.
- J. Regueiro, C. Sanchez-Gonzalez, A. Vallverdu-Queralt, J. Simal-Gandara, R. Lamuela-Raventos and M. Izquierdo-Pulido, Food Chem., 2014, 152, 340–348 CrossRef CAS PubMed.
- D. B. Robb, T. R. Covey and A. P. Bruins, Anal. Chem., 2000, 72, 3653–3659 CrossRef CAS PubMed.
- J. B. Jean-Denis, R. Pezet and R. Tabacchi, J. Chromatogr. A, 2006, 1112, 263–268 CrossRef CAS PubMed.
- L. Riffault, C. Colas, E. Destandau, L. Pasquier, P. Andre and C. Elfakir, Phytochem. Anal., 2015, 26, 189–201 CrossRef CAS PubMed.
- J. P. Rauha, H. Vuorela and R. Kostiainen, J. Mass Spectrom., 2001, 36, 1269–1280 CrossRef CAS PubMed.
- B. Wise and N. B. Gallager, PLS_Toolbox for Use with MATLAB, Version 2.0, Eigenvector Research Inc, Mason, WA, 1992 Search PubMed.
- D. L. Massart, B. G. M. Vandeginste, L. M. C. Buydens, S. de Jong, P. J. Lewi and J. Smeyers-Verbeke, Handbook of Chemometrics and Qualimetrics, Elsevier, Amsterdam, 1997 Search PubMed.
- D. Guillarme, D. T. T. Nguyen, S. Rudaz and J. L. Veuthey, Eur. J. Pharm. Biopharm., 2007, 66, 475–482 CrossRef CAS PubMed.
- D. Guillarme, D. T. T. Nguyen, S. Rudaz and J. L. Veuthey, Eur. J. Pharm. Biopharm., 2008, 68, 430–440 CrossRef CAS PubMed.
- J. Schappler, J.-L. Veuthey and D. Guillarme, UHPLC Separations Using Sub-2μm Particle Size Columns, in Fast Liquid Chromatography Mass Spectrometry Methods in Food and Environmental Analysis, ed. O. Núñez, H. Gallart-Ayala, C. P. B. Martins and P. Lucci, Imperial College Press, London, UK, 2015, pp. 3–32 Search PubMed.
- H. Franz and S. Fabel, A Universal Tool for Method Transfer from HPLC to UHPLC, Technical Note 75, Thermo Fisher Scientific, Germering, Germany, 2016, http://www.dionex.com/en-us/webdocs/70287-TN75-RSLC-Method-Transfer-TN70828_E.pdf Search PubMed.
- H. B. Theron, M. J. van der Merwe, K. J. Swart and J. H. van der Westhuizen, Rapid Commun. Mass Spectrom., 2007, 21, 1680–1686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay00929h |
| This journal is © The Royal Society of Chemistry 2016 |