
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid phase microextraction–comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry: a new tool for determining PAHs in airport runoff water samples†
Anna Maria
Sulej-Suchomska
 *ab,
Żaneta
Polkowska
a,
Tomasz
Chmiel
a,
Tomasz Marcin
Dymerski
a,
Zenon Józef
Kokot
b and
Jacek
Namieśnik
a
*ab,
Żaneta
Polkowska
a,
Tomasz
Chmiel
a,
Tomasz Marcin
Dymerski
a,
Zenon Józef
Kokot
b and
Jacek
Namieśnik
a
aGdańsk University of Technology, Faculty of Chemistry, Department of Analytical Chemistry, 11/12 G. Narutowicza St., 80-233 Gdańsk, Poland
bPoznan University of Medical Sciences, Faculty of Pharmacy, Department of Inorganic and Analytical Chemistry, 6, Grunwaldzka Str., 60-780 Poznań, Poland. E-mail: sulejsuchomska@ump.edu.pl; Fax: +48 61 854 66 09; Tel: +48 61 854 66 16
First published on 3rd May 2016
Abstract
A fundamental aspect of airport operations is the pollution caused by airport runoff waters. Polycyclic aromatic hydrocarbons (PAHs) are one of the most important groups of xenobiotics which are commonly found in runoff water originating from airports. Only very limited data on the analysis of airport runoff water have been published until now. Therefore, a reliable and accurate analytical method based on headspace solid-phase microextraction (HS-SPME) coupled with comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry (GC × GC-TOF-MS) for simultaneous determination of 16 PAHs in airport runoff water was developed. The optimization of the HS-SPME procedure resulted in the following extraction conditions: extraction time of 45 min, temperature of 70 °C, salt addition of 3.0 g, and desorption time of 10 min at 270 °C. The recovery values obtained using this method (63–108%) mostly fell within the acceptable range for the analytical procedures. This indicates that HS-SPME is a suitable and efficient tool for the extraction of PAHs from airport wastewater, the latter being characterized by a very complex matrix composition. In addition, the developed procedure exhibited satisfactory selectivity, accuracy and a low MDL (0.22–2.20 ng L−1). It should be emphasized that the presented procedure is new with respect to the determination of toxic, mutagenic and carcinogenic analytes in the original environmental samples, elaboration of the detailed metrological characteristics, and the diversity of places from which runoff water samples are collected. The validated analytical protocol was successfully applied to determine the aforementioned organic pollutants in real samples collected from different international airports. Regardless of the airport location, chrysene, phenanthrene and pyrene were the most abundant PAH compounds detected in all analyzed samples (1.8–26.3 μg L−1). The presented methodology can be used for tracking the environmental fate of PAHs and assessing the impact of airports on the environment.
1. Introduction
Runoff water is one of the most important problems related to airport operations. Such water forms as a result of the airport platform (i.e. runways, aircraft cleaning and de-icing area, taxiways, apron areas, transfer stations, and repair shops) being washed by precipitation or atmospheric deposition.1–6 Polycyclic aromatic hydrocarbons (PAHs) are one of the most important groups of xenobiotics found in runoff water from airports.7–9 PAH compounds released at airports mainly originate from the combustion of jet fuel and fuel and lubricant spills during aircraft refueling operations, fuel transport and aircraft repairs. These compounds also form during sharp turns of an aircraft, especially at high speeds, as a result of abrasion of both aircraft tires and the asphalt surface of the airport platform.10–13 To date, few data have been published on the analysis of airport runoff waters. Nevertheless, interest in such data is definitely on the increase, as they are the information source about the potentially adverse effect of airport activities on the environment.1,2,5 Regardless of where and the extent to which runoff water samples are investigated, this is a very great analytical challenge. The most important problems to be solved and the obstacles to be overcome include low or even very low contents of a large number of contaminants; very considerable variability in the concentration levels of particular contaminants in runoff samples from different airports; difficulties in standardizing the measurements; possible co-occurrence of sample constituents with very similar physicochemical properties; and the lack of standard techniques for sample collection.3,4,12 The conclusion from the above is that determining the content of xenobiotics in runoff water is an exceedingly complex problem, and yet an indispensable one in view of the increasing threat to the environment from the activities of many airports.14The development of appropriate techniques for analyte isolation and enrichment is necessary in order to obtain reliable information about the content of analytes in airport wastewater. The use of various extraction techniques for the preparation of samples from the original material, such as airport runoff water, may have a significant effect on the final determination of analytes. So far, mostly the procedures based on liquid–liquid extraction (LLE) and solid-phase extraction (SPE) have been used to determine PAHs in different types of runoff water samples.15–18 Both the aforementioned extraction techniques are generally effective, but have some drawbacks, e.g. they require a large volume of the aqueous sample for isolating the analytes (up to few liters). LLE is time-consuming and labor intensive, and it uses large volumes of high-purity solvents. In the case of SPE, a relatively small amount of solvent is required to rinse the sorption media, however, the sorbent bed and/or sorbent pores can be blocked by the particles of the suspension present in the sample.18–20 It is always advisable to develop new techniques that have an advantage over the old ones, and ensure successful application of the new approach. Therefore, green sample preparation techniques such as solid-phase microextraction (SPME) have been introduced to extract a broad spectrum of volatile and semi-volatile organic compounds, including PAHs, from different types of environmental samples.19,21–23 SPME does not have any of the aforementioned limitations because it is a solvent-free technique that combines sampling, sample clean-up and preconcentration into a single step.19,22,24 SPME is currently recognized as the most useful preparation technique for determining the PAH levels in complex matrices such as oily wastewaters, natural stream water or sludge.23,25,26
The selection of a suitable separation technique is the next stage of an appropriate analytical procedure used for the detection, identification and quantitative determination of analytes, often found at trace or even ultratrace levels, in samples characterized by their complex matrix composition. Chromatographic techniques play an increasingly important role in this area. PAHs are commonly analyzed by gas chromatography coupled with mass spectrometry (GC-MS) and flame ionization detection (GC-FID) as well as high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS), spectrofluorometric detection or UV-diode array detection (HPLC-UV-DAD).27,28 The analysis of runoff water or any other samples with a complex matrix composition by means of a single chromatographic technique, e.g. one-dimensional GC, may be insufficient. This is due to the fact that the investigated compounds can be indistinguishable from the baseline noise or hidden in larger peaks of co-eluted sample components. In our study, this crucial problem was resolved by employing comprehensive two-dimensional gas chromatography (GC × GC), which has been shown to be a powerful high-resolution technique well suited for the separation of target compounds present in highly complex sample matrices, such as runoff water.22,29 The benefits of multidimensional systems applied in this kind of research study include highly improved sensitivity, large separation power, and high selectivity.22,30 A detailed description of the working principles and references to the application of GC × GC can be found in a number of articles.31–35 The GC × GC chromatograms are characterized by very narrow peaks which require the use of a fast scanning detector.22,32 The time-of-flight mass spectrometry (TOF-MS) detector has been commonly used to allow successful identification of numerous compounds.22,32,36,37
The purpose of this study was to develop, optimize and validate the procedure for determining polycyclic aromatic hydrocarbons in the samples of airport runoff water based on headspace SPME (HS-SPME) coupled with GC × GC-TOF-MS. To the best of our knowledge, only very limited data have been published on the analysis of airport runoff water. The developed and validated analytical procedure was successfully applied to determine hazardous PAHs in the original samples of runoff water. The samples were collected from three international airports located in different geographical regions and characterized by different levels of activity (number of flights/passengers per year).
2. Experimental
2.1 Chemicals and materials
The reagents, standards, reference materials and consumables used in the analytical testing of collected runoff water are presented in the ESI, Table S1.†2.2 SPME conditions
SPME fiber coated with a 100 μm-thick film of polydimethylsiloxane (PDMS) was used.38,39 HS-SPME extractions were performed in 20 mL amber glass vials sealed with stainless steel screw caps equipped with a PTFE/silicone-coated septum. The headspace-to-sample ratio was 2. Appropriate amounts of a mixture of 16 polycyclic aromatic hydrocarbons (at a concentration level of 2000 μg mL−1) and isotopically labelled standards (naphthalene-d8, benzo(a)anthracene-d12) were added to the model sample for optimization purposes. For the analysis of real samples and the recovery calculation, only deuterated PAH standards were used to spike the runoff water samples.The fibre was conditioned at the injection port at 320 °C for 60 min, according to the manufacturer's specifications. Prior to extraction, the sample solution in the vial was maintained at the target temperature for 15 min under continuous agitation (700 rpm). This incubation step was intended to promote the transfer of volatile compounds from the sample solution to the headspace. All the solutions were stirred at a constant rate (700 rpm) throughout the extraction period. Following the extraction, the fibre was withdrawn into the needle, removed from the sample vial and immediately transferred into the GC injector port for thermal desorption. In order to avoid sample carryover, SPME fiber was post-baked at 270 °C for 5 min with the injector in the splitless mode. Moreover, the fiber and chromatographic blanks were run periodically during the analysis to confirm the cleanliness of the GC × GC system. Each extraction was performed in triplicate. All HS-SPME steps were carried out automatically by using a Gerstel MultiPurpose Sampler (MPS) using the Gerstel MAESTRO software.
2.3 GC × GC-TOF-MS analysis
On the basis of literature data and conclusions resulting from the authors' prior experience, an appropriate set of chromatographic columns was selected together with basic operating parameters of the GC × GC system. Information about technical specifications and conditions of procedures carried out by means of two-dimensional gas chromatography–time-of-flight mass spectrometry is summarized in the ESI, Table S2.†2.4 Data acquisition and processing
Chromatographic data were processed and consecutively visualised on 2D and 3D chromatograms using LECO ChromaTOF™ software version 4.44 (LECO Corporation, St. Joseph, MI, USA). After data acquisition, the analytical results were submitted to a data processing module where the individual peaks were automatically detected on the basis of a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 signal to noise ratio. Each peak area was automatically acquired, and the compound identification proceeded via examination and comparison with the mass spectra available in the National Institute of Standards and Technology (NIST) library as well as the retention times, authentic standards and elution order described in the scientific literature. The amount of each compound was calculated using the most abundant ions in the mass spectrum of the compound (ESI, Table S3†). Data files were collected and stored on a Pegasus 4D instrument.
:1 signal to noise ratio. Each peak area was automatically acquired, and the compound identification proceeded via examination and comparison with the mass spectra available in the National Institute of Standards and Technology (NIST) library as well as the retention times, authentic standards and elution order described in the scientific literature. The amount of each compound was calculated using the most abundant ions in the mass spectrum of the compound (ESI, Table S3†). Data files were collected and stored on a Pegasus 4D instrument.
A schematic representation of the analytical procedure used to determine PAHs is shown in Fig. 1.
 | ||
| Fig. 1 Diagram of the analytical procedure for determining PAHs by using SPME and the GC × GC-TOF-MS system. | ||
2.5 Airport wastewater samples
In the period 2011–2013, stormwater was sampled at three international airports, i.e. two airports located in Poland and one in Great Britain. The samples were collected from surface depressions near drain inlets and airport drainage systems. The sample collection sites were selected in the vicinity of airport areas where most of the technical servicing is performed, e.g. aircraft refueling, cargo aircraft loading and unloading, spraying aircrafts with de-icing substances, and parking and servicing of technical support vehicles. Thus, these were the places which produce the largest amount of contaminants delivered with runoff water into drainage ditches and then to other elements of the environment. The description of sampling sites at the investigated airport areas is presented in Table 1.| Airport/sample number | Location of sampling site | ||
|---|---|---|---|
| International Large Airport PL | International Small Airport PL | International Large Airport UK | |
| 1 | Influent of a river | Vicinity of an airport terminal | De-icing area (1) |
| 2 | Effluent of a river | De-icing area | A river in the vicinity of the airport |
| 3 | Municipal water catchment area | Machinery stock, parking places | De-icing area (2) |
| 4 | CARGO water catchment area | Runway | De-icing area (3) |
| 5 | Airport ramp | Parking places | De-icing area (4) |
| 6 | Car park | The periphery of an airport | A road near the airport |
| 7 | De-icing area | Car park | — |
| 8 | Airport ramp | — | — |
The samples of runoff water were mostly collected manually during continuous atmospheric precipitation lasting for at least 5 h. The samples were collected in 1000 mL bottles made of dark glass by using a syringe (100 mL) with Teflon tubing. Prior to use, the syringes and tubing were rinsed with ultrapure water and sampled runoff water.
Due to safety regulations, assembly and locating of any devices at the airport platform is not possible. These conditions result from the necessity of maintaining the regular schedule of work related to the proper airport functioning and more stringent procedures introduced recently in connection to increased air traffic while maintaining the high service standard and the safety level of air operations.40
After each sample collection, the samples of airport runoff water were transported to the laboratory and stored at 4 °C until further analysis.41
3. Results and discussion
3.1 Optimization of SPME
Based on the published literature and conclusions resulting from the authors' prior experience, solid-phase microextraction in a headspace mode was used to determine PAH analytes.19,23,42 HS-SPME has been proved to be a useful tool for the effective isolation of PAHs from water samples, especially when the sample preheating step and analyte sampling at higher temperatures are applied.23,42–44 This type of SPME technique allows for the isolation and enrichment of PAH analytes from runoff water samples which are characterized by a very complex and sometimes variable composition of the matrix. Fibre is exposed to the sample headspace, which prevents the degradation of the fiber's stationary phase that can be caused by the action of non-volatile contaminants present in the sample matrix; this considerably reduces the operating costs.23,44 Based on the published literature and conclusions resulting from our previous experience, a suitable type of extraction fibre was selected. The investigated PAHs belong to the semi-polar class of chemicals with high octanol–water partition coefficients and low water solubility. It has been demonstrated that PDMS coating with higher film thickness is characterized by good sorption of PAHs.19,21,39 In order to optimize the parameters of HS-SPME extraction, an environmental sample resembling the complex composition of the airport wastewater matrix (a sample of runoff water collected from a runway) and spiked with an appropriate amount of the PAH standard mixture, including isotopically labeled standards, was used as a model. During the fibre exposure to the headspace above the sample, all analyzed samples were stirred at a constant, maximum agitation level to improve the analyte extraction and reduce the time of extraction. The following parameters were optimized: extraction temperature, extraction time, desorption time, and the sample ionic strength. The temperature of the system and extraction time are two basic parameters that allow for controlling the efficiency of analyte extraction via SPME because SPME directly depends on these factors. An increase in the extraction temperature results in increased transport of analytes from the solution to the solution headspace, however, too high temperature may cause desorption of analytes from the extraction fiber. Increased temperature may lead to lowered values of partition coefficients, which will result in smaller amounts of analytes retained on the SPME fiber. The extraction temperature is also selected based on the composition of the investigated medium, volatility of extracted analytes, and the affinity of analytes for the sample matrix components. Heating of the investigated sample is most frequently used to increase the vapor pressure of volatile analytes above that of the analyzed solution. This is due to the fact that the time needed to attain equilibrium is usually shorter for the analytes sampled from the headspace than for those sampled directly from the solution, which is connected to faster diffusion of analytes in the gaseous phase.24 The addition of salting out agents may also improve the extraction efficiency and sensitivity of analytical procedures.24 The addition of a small amount of salting out agents, i.e. NaCl, KCl, and Na2SO4, to the analyzed sample increases the solution's ionic strength, resulting in lowered solubility of organic components and increased values of partition coefficients. This results in an increased concentration of organic components in the headspace, and consequently in the higher amount of analytes retained in the extraction medium.42,45 The optimized parameters of the HS-SPME procedure are presented in Fig. 2. | ||
| Fig. 2 Effects of the extraction temperature (A), extraction time (B), ionic strength (C), and desorption time (D) on the efficiency of the HS-SPME procedure for the determination of PAHs. | ||
In the present study, five different temperature levels were examined, i.e. 50, 60, 70, 80 and 90 °C. Preliminary experiments on the effect of extraction time on the SPME efficiency were carried out for 30 and 60 min at different temperatures in order to evaluate the possible relationships between the extraction time and temperature. This approach allowed for performing the optimization of extraction temperature using the most appropriate extraction time (data not included). A longer extraction time has been accepted as sufficient for the extraction of PAHs with relatively low and medium molecular weights (LMW; MMW), and as fully suitable for the effective extraction of high-molecular-weight (HMW) PAHs. HS-SPME is very sensitive to temperature variations and therefore it is crucial to ensure conditions which minimize even minor differences between consecutive analyses. Consequently, in order to maintain constant analytical conditions and reduce possible variations between chromatographic runs, each analysis was preceded by 15 min incubation at 70 °C. PAHs show a wide range of volatility, and the release of these with lower volatility to the headspace during extraction is limited. In general, Henry's constants and diffusion coefficients of PAHs increase with increasing temperature, and consequently the vapor pressure and the concentration of the analytes in the headspace increase.42 On the other hand, since the adsorption of analytes is an exothermic process, the partition coefficients decrease with increasing temperature. Because of that the extraction capacity is a compromise between these two factors. Fig. 2a shows the effect of extraction temperature ranging between 50 and 90 °C for selected PAH compounds, which displayed different extraction behaviors. The low-ring PAHs reached equilibrium at a temperature of 50 °C. As shown in the figure, an increase in temperature had a negative effect on the extraction of the most volatile PAHs, especially naphthalene. However, the extraction efficiency of MMW- and HMW-PAHs characterized by high diffusion coefficients increases at temperatures above 50 °C. Similar observations have also been reported by others.19,42,46Fig. 2a illustrates that the highest extraction yield of 5- and 6-ring PAHs occurred at a temperature of 90 °C. Thus, an extraction temperature of 70 °C was selected as the optimal SPME parameter for efficient extraction of all investigated PAHs.
The extraction time is the next crucial parameter in the SPME process. Since HS-SPME is an equilibrium extraction technique, the time required for reaching equilibrium determines the maximum amount of the target analyte that can be extracted by the fibre, which affects the sensitivity of the technique.19,24 Therefore, it is important to determine the time required for reaching the equilibrium state.42 In this work, the sorption time profiles were studied by measuring the peak area as a function of exposure time. The fibre was exposed to the headspace of model samples during five different extraction periods. Fig. 2b illustrates the equilibrium time profiles of selected PAHs. The extraction times longer than 70 min were not applied in order to avoid the excessive time of routine analysis and reduce the interval between consecutive GC × GC runs to a minimum. Based on the obtained results, it can be concluded that, in general, LMW- and MMW-PAHs reached the highest extraction yield after 45–50 min. On the other hand, PAHs characterized by high molecular mass reached equilibrium after 50 min extraction. The equilibrium times for the analyzed PAH compounds increased with increasing molecular mass, which is in agreement with the previous studies on porewater samples from a sediment core,25 rainwater and stormwater samples.46 This finding mainly results from the fact that HMW PAHs are characterized by low diffusion coefficients, and consequently slower mass transfer. Therefore, a longer time is required to reach equilibrium and provide sufficient SPME yield. Taking into account the obtained results and a suitable extraction period to ensure continuous running time of the GC × GC system, an extraction time of 45 min was selected as the optimal parameter for isolating PAHs via HS-SPME.
As mentioned before, enhanced ionic strength due to the addition of salt has been reported to increase the extraction efficiency; this phenomenon is known as the “salting-out” effect. In the present work, potassium chloride was used for adjusting the ionic strength. Different amounts of KCl (1.0 g, 2.0 g, and 3.0 g) were added to the model solution in order to determine its optimal ionic strength. Higher doses of salt were not tested because of the possible saturation of the sample solution. The results of the extraction of selected PAHs from samples containing different amounts of added salt are shown in Fig. 2c. The obtained data indicate that the addition of KCl had a strong positive effect on the extraction efficiency, resulting in increased sensitivity of the analytical method. All of the investigated PAHs reached the highest extraction yield when 3.0 g of KCl was added to the model sample. Naphthalene was an exception because a slightly higher extraction efficiency was observed for the addition of 2.0 g of salt. Other authors also reported that the values of partition coefficients of PAHs increased with increasing ionic strength of sample solution, resulting in higher SPME yields.45,46 Therefore, the addition of 3.0 g of salt has been chosen as the optimal extraction parameter for determining PAHs in runoff water samples by the proposed method.
Suitable desorption time and temperature are important parameters that ensure a complete desorption of analytes from the fibre and prevent the occurrence of the memory effect or carryover. In the present study, desorption times were optimized by inserting the fibre into the injection port for a period of 5–15 min (Fig. 2d). The desorption of analytes was carried out at a temperature of 270 °C. Temperatures above 270 °C are not recommended due to the thermal instability of the septum, which may cause a noticeable blank signal as well as gas leak. Optimal results were obtained for a desorption time of 10 min (Fig. 2d). Moreover, in order to eliminate any sample carryover, the post-baking of SPME fibre for additional 5 min was applied. Also, the blank sample runs for the fibre were performed between two consecutive sample analyses to check the background level of the GC × GC system. Under these conditions, no sample carryover was observed.
As described above, the following HS-SPME conditions were applied: an extraction time of 45 min, an extraction temperature of 70 °C, a desorption temperature of 270 °C, a desorption time of 10 min, addition of 3.0 g KCl, and sample agitation at 700 rpm. A 2D chromatogram of the model sample extracted via HS-SPME under the optimized conditions is shown in Fig. S1 (see ESI†).
3.2 GC × GC-TOF-MS analysis
Gas chromatography is commonly used for the separation and determination of PAH compounds. Although capillary columns provide higher resolution, GC is often ineffective for separating a large number of compounds that are usually present in environmental samples. The peak capacity of GC × GC is much higher in comparison to that of one-dimensional GC, resulting in a significantly improved separation of individual analytes, and, what is even more important is their separation from interfering matrix components. Additionally, the main advantage of the trapping and refocusing processes occurring during modulation is a 3–10-fold increase in the signal-to-noise ratio (S/N) compared to 1D-GC, which results in enhanced sensitivity of the two-dimensional approach. Furthermore, the identification of compounds as well as their quantification is more reliable when using GC × GC due to two retention times for each analyte and well-ordered bands of compound groups (chromatographic fingerprint).9,22,34,37 Referring to our previous studies12,13,18 and the aforementioned usefulness of the GC × GC-TOF-MS technique in the analysis of highly complex samples, this particular technique has been chosen for determining PAHs in airport runoff water. The parameter optimization of the applied technique is described below.GC oven temperature program is an important parameter that affects the extent of the analyte resolution. Prior to the application of the GC × GC system, different temperature programs for 1D GC separation were evaluated with respect to their effect on the behavior of PAHs. The best 1D separation was achieved with an initial temperature of 50 °C held for 0.2 min; then ramped at 10 °C min−1 to 200 °C; ramped again at 5 °C min−1 to a final temperature of 300 °C; and held for 3 min. After several attempts, the temperature program of the secondary column was optimized as follows: an initial temperature of 80 °C held for 0.2 min; then ramped at 10 °C min−1 to 230 °C; ramped again at 5 °C min−1 to 330 °C; and held for 5 min. A further effort to reduce the run time by increasing the ramping rate disturbed the separation of analytes, and thus has been abandoned. We also attempted to optimize different flow programs but this approach did not offer any meaningful improvement in the resolution of the above co-eluting compounds.
Modulation is a key step in GC × GC separation, which consists of three tasks: trapping, focusing and releasing small fractions of the effluent from the first column to the second one. The duration of a single complete cycle of these modulation events is called the modulation period (PM). The optimization of PM is important as it ensures the appropriate peak shape, high sensitivity and separation through cryofocusing. Therefore, the modulation periods of 2, 3, 4, 5 and 6 s with the hot pulse duration set at 20% of PM were investigated. In order to select the optimal PM that provides comprehensive separation of individual analytes as well as their separation from other volatile components of the sample matrix, the airport runoff water sample spiked with a standard mixture of PAHs was used as a model in all tests. In general, it was observed that with increasing PM the peak height increased, while the peak area was not significantly affected. This finding indicated the enhancement of the S/N ratio, and thus a higher sensitivity of the method. Fig. 3 shows the experimental verification of the effect of PM on the GC × GC separation of selected PAHs. For example, phenanthrene (Ph) and anthracene (An) that had the same retention time on the 2D column were well separated on the 1D column. The separation of these two analytes is well preserved using a PM ≤ 5 s (three modulations per 1D peak; PM = 5 s), as the two compounds yielded two separate spots (Fig. 3d). However, in some cases, a longer modulation period led to deterioration or a complete loss of the separation achieved on the 1D column, e.g. similar to Ph and An, chrysene (Chry) co-eluted with benzo(a)anthracene (BaA) at PM = 6 s. On the other hand, shortening the modulation period to less than 5 s caused the wraparound effect. For short modulation periods of 2, 3 or even 4 s, this effect was observed when the 2D retention of an analyte became longer than PM, which caused some or all of these analytes to elute during the successive modulation cycle(s). For example, fluoranthene (Flt) and pyrene (Py) appeared as partially wraparound peaks in the total ion current (TIC) chromatograms obtained for the PM value of 2 and 4 s (Fig. 3a and c). The same problematic effect was also reported for fluorene (Flu) for PM = 3 s. The wraparound effect disturbs the structure of 2D chromatograms and may lead to co-elution of analytes eluting in the following modulation cycle(s). For these reasons, the PM value of 5 s was chosen as the optimized modulation period in this study.
 | ||
| Fig. 3 GC × GC contour plots (TIC mode) of airport runoff water samples spiked with PAH standards obtained at different modulation periods: (a) 2 s; (b) 3 s; (c) 4 s; and (d) 5 s. | ||
3.3 Quality assurance/quality control (QA/QC)
After parameter optimization, the procedure for determining petroleum-derived compounds in the samples of airport runoff water was validated to ensure the appropriate level of quality control and quality assurance of measurements. The measuring system was calibrated by using a standard solution (Naph, Acy, Ace, Flu, Ph, An, Flt, Py, Chry, BbF, BkF, BaP, BaA, InPy, DBahA, and BghiP). For this purpose, two dilution series of standard solutions were prepared from the stock solution, resulting in the two concentration ranges of standard compounds, i.e. 0.01–10 μg L−1 and 10–100 μg L−1. These solutions were prepared immediately prior to each series of measurements. Three independent measurements were carried out for each calibration point. The determined calibration curves were used to calculate the concentration of analytes in the extracts of runoff water samples. For each analyte, the linearity of calibration curves was assessed by plotting the peak area against the concentration of the respective standard, and expressed by the coefficient of determination (R2). The limit of detection (LOD) was determined based on three replicate measurements. The LOD values were calculated using the equation LOD = 3.3SD/b, where b is the slope of the calibration curve and SD is the standard deviation. The limit of quantification (LOQ) was determined as three times the LOD. The above results were used to determine the method detection limits (MDLs) and method quantification limits (MQLs) of the developed analytical procedure, taking into account all its steps, i.e. from sampling to statistical processing of the sets of measurements. The MDL was defined as the lowest concentration of a substance that can be detected with a specified probability using a given analytical methodology. On the other hand, the MQL was the lowest concentration of a substance that can be quantified with a certain accuracy, precision and uncertainty using a given analytical methodology. The precision of the developed procedure was expressed as the coefficient of variation calculated for five replicates (n = 5) according to the equation CV = SD/![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) × 100%, where SD is the standard deviation of the concentration of analytes and is the average concentration of the analytes. The determination of intermediate precision included both intra-day and inter-day assays. The intra-day precision study comprised five independent runs of each sample at three concentration levels in a single batch during one day. The inter-day precision was evaluated based on the analysis of quality control samples (five replicates per sample) at three concentration levels on separate days within a five-day period. An acceptable limit of ≤15% was applied to all QC samples. The accuracy was tested via the recovery studies of runoff water samples spiked with known amounts of PAHs at low, medium and high concentration levels. The analyzed samples were spiked with 0.2, 2.0 and 20 μg L−1 of PAHs (levels of PAHs in spiked samples were selected based on the expected concentration of analytes in real samples) and additionally with isotopically labeled standards at the same concentration levels. The measurements were performed in triplicate for each spiked sample. As mentioned earlier, the recovery values were estimated by the standard addition method. A known amount of analyte(s) and isotopically labeled standards was added to one sample, while another sample was only spiked with isotopically labeled standards at the same concentration level. After completing the analysis with the sample without the added analytes (x) and the sample containing added analytes (x + si), the recovery was calculated using the equation % recovery = [(x + si) − (x)/s] × 100, where si is the amount of the determined analyte. The sensitivity of the procedure was expressed as the slope of the calibration curve for each of the analytes. The range of linearity, calibration curve parameters, MDL, MQL and repeatability are shown in Table 2. The values of coefficient of determination (R2) ranged from 0.9721 to 0.9987, and from 0.9776 to 0.9994 for the analyte concentration levels of 0.01–10 μg L−1 and 10–100 μg L−1, respectively. Additionally, linearity was tested by using the Visual Linearity Test and Mandel's Fitting Test with a probability P = 0.99. Mandel's Fitting Tests were passed in all cases. The values of intra-day precision for the samples of airport runoff water ranged from 5.5 to 13.0%. However, in the inter-day studies, the repeatability was reduced and CV values were found to be higher than 15% in some cases. Similar numerical data on reproducibility obtained by using the SPME technique have also been published by others.19,21,47 The recoveries for runoff water samples obtained by GC × GC-TOF-MS ranged from 63 to 108%. These values meet the requirements of the analytical procedures wherein the recovery should range from 70–120% depending on matrix complexity.48 The sources of uncertainty associated with the determination of PAH compounds in real samples are presented graphically using the Ishikawa diagram (Fig. 4). Taking into account the sources of uncertainty associated with the determination of PAHs, the values of combined standard uncertainty of the results obtained for real samples were calculated.
× 100%, where SD is the standard deviation of the concentration of analytes and is the average concentration of the analytes. The determination of intermediate precision included both intra-day and inter-day assays. The intra-day precision study comprised five independent runs of each sample at three concentration levels in a single batch during one day. The inter-day precision was evaluated based on the analysis of quality control samples (five replicates per sample) at three concentration levels on separate days within a five-day period. An acceptable limit of ≤15% was applied to all QC samples. The accuracy was tested via the recovery studies of runoff water samples spiked with known amounts of PAHs at low, medium and high concentration levels. The analyzed samples were spiked with 0.2, 2.0 and 20 μg L−1 of PAHs (levels of PAHs in spiked samples were selected based on the expected concentration of analytes in real samples) and additionally with isotopically labeled standards at the same concentration levels. The measurements were performed in triplicate for each spiked sample. As mentioned earlier, the recovery values were estimated by the standard addition method. A known amount of analyte(s) and isotopically labeled standards was added to one sample, while another sample was only spiked with isotopically labeled standards at the same concentration level. After completing the analysis with the sample without the added analytes (x) and the sample containing added analytes (x + si), the recovery was calculated using the equation % recovery = [(x + si) − (x)/s] × 100, where si is the amount of the determined analyte. The sensitivity of the procedure was expressed as the slope of the calibration curve for each of the analytes. The range of linearity, calibration curve parameters, MDL, MQL and repeatability are shown in Table 2. The values of coefficient of determination (R2) ranged from 0.9721 to 0.9987, and from 0.9776 to 0.9994 for the analyte concentration levels of 0.01–10 μg L−1 and 10–100 μg L−1, respectively. Additionally, linearity was tested by using the Visual Linearity Test and Mandel's Fitting Test with a probability P = 0.99. Mandel's Fitting Tests were passed in all cases. The values of intra-day precision for the samples of airport runoff water ranged from 5.5 to 13.0%. However, in the inter-day studies, the repeatability was reduced and CV values were found to be higher than 15% in some cases. Similar numerical data on reproducibility obtained by using the SPME technique have also been published by others.19,21,47 The recoveries for runoff water samples obtained by GC × GC-TOF-MS ranged from 63 to 108%. These values meet the requirements of the analytical procedures wherein the recovery should range from 70–120% depending on matrix complexity.48 The sources of uncertainty associated with the determination of PAH compounds in real samples are presented graphically using the Ishikawa diagram (Fig. 4). Taking into account the sources of uncertainty associated with the determination of PAHs, the values of combined standard uncertainty of the results obtained for real samples were calculated.
| Analytes | Concentration range used for calibration [μg L−1] | Coefficients of the calibration curve (y = ax + b) | Coefficient of determination, R2 | MDL | MQL | Intra-day precision [%] | Inter-day precision [%] | Recovery [%] | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | ||||||||||||
| PAHs | I | II | a | b | a | b | I | II | [ng L−1] | ||||
| Naphthalene | 0.01–10 | 10–100 | 5774412 |
130797 |
5920862 |
−9549526 |
0.9979 | 0.9987 | 0.23 | 0.70 | 5.5–7.1 | 3.2–29 | 77–97 |
| Acenaphthylene | 12380813 |
1412444 |
11939174 |
4184651 |
0.9954 | 0.9970 | 0.34 | 1.03 | 5.7–8.9 | 2.3–30 | 74–96 | ||
| Acenaphthene | 7491619 |
1406948 |
7541935 |
−7308033 |
0.9873 | 0.9968 | 0.58 | 1.73 | 5.6–9.4 | 23–28 | 75–95 | ||
| Fluorene | 10957828 |
1379730 |
10689221 |
−4738740 |
0.9915 | 0.9974 | 0.47 | 1.42 | 5.9–7.6 | 5.9–30 | 70–103 | ||
| Phenanthrene | 3994454 |
9015966 |
3752905 |
39079632 |
0.9869 | 0.9899 | 2.2 | 6.50 | 6.0–9.9 | 12–30 | 79–108 | ||
| Anthracene | 27972896 |
−795103 |
27560297 |
1685462 |
0.9885 | 0.9929 | 0.55 | 1.65 | 6.1–10.4 | 11–31 | 71–90 | ||
| Fluoranthene | 13885150 |
5136377 |
14324786 |
−17284631 |
0.9987 | 0.9994 | 1.63 | 4.89 | 7.8–11.3 | 12–25 | 69–87 | ||
| Pyrene | 8741436 |
−283600 |
9340469 |
−48801048 |
0.9979 | 0.9980 | 2.08 | 6.25 | 6.9–12.0 | 2.0–26 | 70–81 | ||
| Benz[a]anthracene | 6331871 |
−161647 |
6677654 |
−8315466 |
0.9980 | 0.9985 | 0.23 | 0.68 | 5.8–12.9 | 4.0–21 | 65–87 | ||
| Chrysene | 2361801 |
339560 |
2496327 |
−1883088 |
0.9900 | 0.9948 | 0.51 | 1.53 | 5.6–12.5 | 1.3–28 | 63–79 | ||
| Benzo[b]fluoranthene | 49585 |
0 | 53546 |
−155738 |
0.9721 | 0.9875 | 1.85 | 5.55 | 6.0–11.6 | 5.6–12 | 64–77 | ||
| Benzo[k]fluoranthene | 1222702 |
117776 |
1244465 |
−377049 |
0.9981 | 0.9975 | 0.22 | 0.67 | 5.7–13.1 | 1.4–23 | 64–79 | ||
| Benzo[a]pyrene | 1186529 |
−153344 |
1135200 |
300546 |
0.9817 | 0.9841 | 1.67 | 5.01 | 6.9–12.6 | 13–30 | 67–78 | ||
| Indeno[1,2,3-c,d]pyrene | 83005 |
13762 |
85907 |
0 | 0.9812 | 0.9847 | 0.70 | 2.11 | 11.7–13 | 16–28 | 64–70 | ||
| Dibenz[a,h]anthracene | 11486 |
1450 | 13841 |
−76560 |
0.9812 | 0.9857 | 0.71 | 2.12 | 12.0–12.9 | 15–27 | 63–67 | ||
| Benzo[g,h,i]perylene | 11760 |
1402 | 13588 |
−52378 |
0.9793 | 0.9776 | 0.74 | 2.22 | 12.8–13.5 | 19–26 | 64–71 | ||
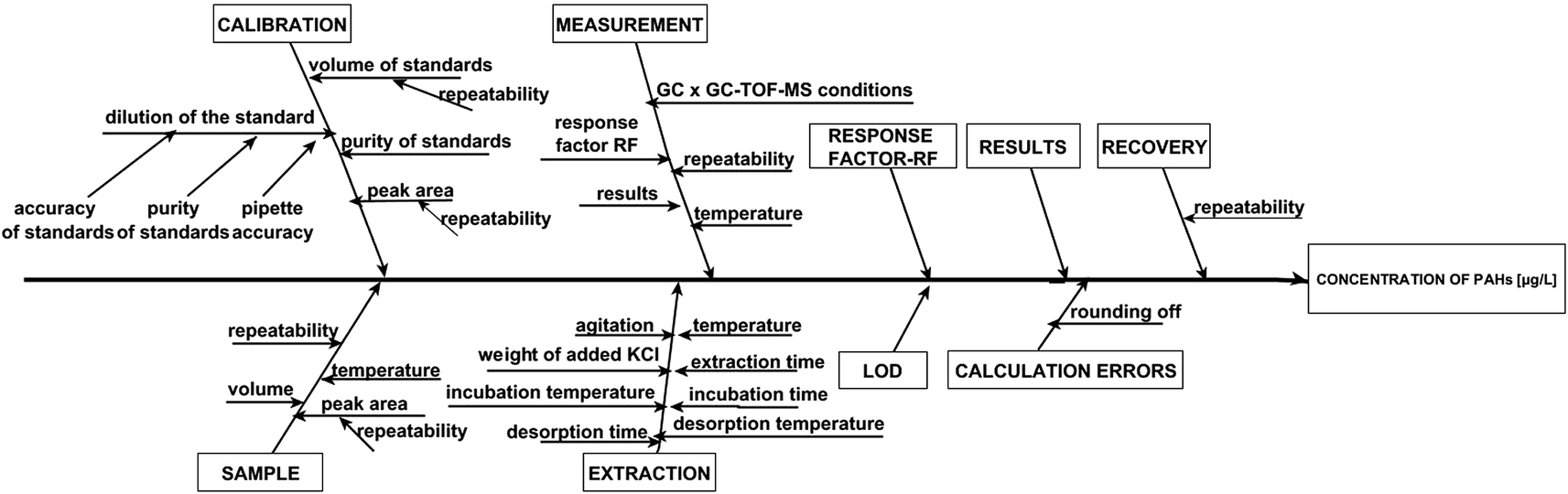 | ||
| Fig. 4 The Ishikawa diagram presenting the influence of uncertainty of individual parameters in the analytical process on the value of combined standard uncertainty for the determination of PAH analytes in airport runoff water. | ||
The obtained results indicate that the MDL values of the HS-SPME/GC × GC-TOF-MS procedure elaborated in the present study are generally lower than those reported for the SPE/GC-MS (1–57 ng L−1),18 LLE/GC-MS (1600–7800 ng L−1),49 and SPME/GC-MS (1–29 ng L−1)50 methods previously applied to determine 16 PAHs in water/wastewater samples. The above-mentioned studies also show that the efficiency of the developed analytical procedure is quite similar to that of the SPE/GC-MS method (recovery of 71–86%), and slightly lower than that of LLE/GC-MS (81–106%).50 The precision experiments demonstrated that the repeatability of the proposed methodology is slightly lower than that of the SPE/GC-MS procedure (CV = 1.5–5.2%), and similar to that of the LLE/GC-MS procedure (CV = 6–12%) applied in our previous study.18 The obtained results indicate the suitability of the HS-SPME technique for efficient extraction of PAHs from airport runoff water. Moreover, the MQL values of the proposed analytical procedure are significantly lower than the corresponding values obtained by applying SPE/GC-MS and LLE/GC-MS procedures to determine PAHs in airport runoff waters.18,49,50 This finding confirms the higher sensitivity of the methodology applied in the present study, especially the two-dimensional gas chromatography in comparison to its one-dimensional counterpart. Furthermore, the developed method, being very selective, solves the problem of coelution and interferences that is associated with the analysis of PAHs in airport runoff water samples. As a result of the study, the quantitation of acenaphthylene, acenaphthene, fluoranthene, pyrene, phenanthrene, anthracene, benzo[b]fluoranthene and benzo[k]fluoranthene has been improved in comparison to the outcome of SPE/GC-MS and LLE/GC-MS procedures.18 Also, it should be emphasized that the elaborated HS-SPME procedure is a solvent-free extraction technique as opposed to LLE and SPE; the latter two also require the sample concentration step prior to GC analysis. The advantages and limitations of the procedure for PAH determination in the samples of airport stormwater are presented in Table 3.
| Advantages | Disadvantages |
|---|---|
| Suitable for highly contaminated samples | Qualified staff is necessary for handling the apparatus |
| A Small amount of sample is required | Time-consuming |
| Relatively high recovery | Low repeatability in some case |
| Low MDL | |
| Good accuracy | |
| Good selectivity | |
| Good resolution |
3.4 Application to real samples
An elaborated and validated analytical procedure based on HS-SPME/GC × GC-TOF-MS was applied to determine the contents of PAH analytes in runoff water collected from three different airports. The samples of stormwater were collected in the period from autumn 2012 to winter 2013 (75 samples from Small Airport PL, 90 samples from Large Airport PL, and 27 samples from Large Airport UK). Fig. 5 presents the concentration levels of individual PAHs determined in the samples collected from the specific areas of three airports. The airports, located in different geographical regions, are characterized by various levels of the activity as expressed by the number of passengers and the number of flights. Generally, the contents of 16 PAHs in the samples collected from two Polish international airports were significantly higher (i.e. 217 ± 15 μg L−1 and 187 ± 14 μg L−1 in the samples from Large Airport PL (31 Jan 2013) and Small Airport PL (8 Dec 2012), respectively) than those determined in the samples from the UK international airport. The highest concentration of phenanthrene was measured in the sample collected from the airport ramp at the Large Airport PL (26.3 ± 1.8 μg L−1). Regardless of the airport location, chrysene, phenanthrene and pyrene were the most abundant PAH compounds detected in all the analyzed samples (concentration range 1.8–26.3 μg L−1). Fig. 6 and S2 (ESI†) show exemplary chromatograms of the sample of runoff water collected from the aircraft de-icing areas at the International Large Airport PL, and the sample of river outflow from the airport area. It is noticeable that PAH analytes were well separated from interfering compounds present in runoff water. This can be seen in the case of acenaphthylene and acenaphthene as well as fluoranthene and pyrene detected in the sample from the area of aircraft de-icing (Fig. 6a). Similarly, the peaks of phenanthrene and anthracene were fully separated from other substances present in the sample, as shown in Fig. 6b. Based on our previous studies, it can be stated that the aforementioned analytes had not been well separated when the GC-MS system was used. We conclude that co-elution of analytes and interferences present in the sample matrix may occur if one-dimensional gas chromatography is applied. These exemplary data confirm that the use of comprehensive two-dimensional gas chromatography is necessary to separate target analytes from interfering compounds in both the first and second dimensions. Therefore, GC × GC with a high peak capacity and separation efficiency is recommended as a tool for analyzing environmental samples characterized by complex matrix composition, such as runoff waters. | ||
| Fig. 5 The content of PAHs in runoff water collected from different airports (International Large PL, Small Airport PL, and International Large Airport UK). | ||
 | ||
| Fig. 6 GC × GC extracted chromatograms of the samples of runoff water collected on 14 Jan 2013 from (a) the aircraft de-icing area, and (b) the river outflow at the International Large Airport PL. | ||
4. Conclusion
As a result of the study, a procedure based on green extraction techniques, i.e. SPME and GC × GC-TOF-MS, for determining one of the most toxic xenobiotics, namely, PAH compounds, in the samples of airport runoff water was developed, optimized and validated. Until now, a very limited number of procedures were available that ensure a reliable data about the concentration levels of PAHs in new types of environmental samples, such as the airport runoff water samples, which are characterized by a very complex and sometimes variable matrix composition, and the possibility of interference associated with the presence of components displaying similar physico-chemical properties. The obtained experimental data show that the SPME technique is a powerful method for extracting PAHs from the airport runoff water samples. The SPME–GC × GC-TOF-MS procedure presented here allowed for selective, sensitive, precise and accurate identification and quantification of PAHs in the samples of airport stormwater. The developed and validated analytical protocol has been successfully applied to determine these organic pollutants in real samples collected from three international airports. It should be emphasized that the established procedure is new in relation to the determination of toxic, mutagenic and carcinogenic analytes in the original type of environmental sample, e.g. airport runoff waters; elaboration of the detailed metrological characteristics; and the diversity of places from which samples were collected. Owing to the development and application of appropriate analytical methods, it is possible to obtain new information on the content of different groups of xenobiotics in airport runoff water, and expand knowledge about the processes of transport as well as chemical, photochemical and biological transformations of airport runoff water streams. The presented methodology can be used as a tool for tracking the environmental fate of PAH compounds and assessing the impact of airports on the environment.Acknowledgements
The authors would like to acknowledge the Polish Ministry of Science and Higher Education for its role in financing the presented research via the grant awarded within the framework of the Project no. 0528/IP3/2011/71.References
- L. Luther, Environmental Impacts of Airport Operations, Maintenance, and Expansion, Report 0704-0188, Congressional Research Service, Washington, 2007 Search PubMed.
- S. R. Corsi, S. W. Geis, G. Bowman, G. Failey and T. D. Rutter, Environ. Sci. Technol., 2009, 43, 40–46 CrossRef CAS PubMed.
- S. Barash, J. Covington and C. Tamulonis, Preliminary Data Summary Airport Deicing Operations (Revised) Report EPA-821-R-00–016, United States Environmental Protection Agency, Washington, 2000 Search PubMed.
- A. M. Sulej, Ż. Polkowska and J. Namieśnik, Crit. Rev. Environ. Sci. Technol., 2012, 42, 1691–1734 CrossRef CAS.
- A. M. Sulej, Ż. Polkowska and J. Namieśnik, Crit. Rev. Anal. Chem., 2011, 41, 190–213 CrossRef CAS.
- A. M. Sulej, Ż. Polkowska and J. Namieśnik, Pol. J. Environ. Stud., 2012, 21, 725–739 CAS.
- M. Rawa-Adkonis, L. Wolska and J. Namieśnik, Crit. Rev. Anal. Chem., 2006, 36, 63–72 CrossRef CAS.
- I. Iavicoli, M. Chiarotti, A. Bergmaschi, R. Marsili and G. Carelli, J. Chromatogr. A, 2007, 1150, 226–235 CrossRef CAS PubMed.
- T. Ieda, N. Ochiai, T. Miyawaki, T. Ohura and Y. Horii, J. Chromatogr. A, 2011, 1218, 3224–3232 CrossRef CAS PubMed.
- EPA, Survey of New Findings in Scientific Literature Related to Atmospheric Deposition to the Great Waters: Polycyclic Aromatic Hydrocarbons (PAH), United States Environmental Protection Agency, 2007 Search PubMed.
- B. J. Mahler, P. C. V. Metre and J. T. Wilson, Concentrations of Polycyclic Aromatic Hydrocarbons (PAHs) and Major and Trace Elements in Simulated Rainfall Runoff From Parking Lots, United States Department of the Interior United States Geological Survey, Austin, 2004 Search PubMed.
- A. M. Sulej, Ż. Polkowska, L. Wolska, M. Cieszynska and J. Namieśnik, Environ. Sci.: Processes Impacts, 2014, 16, 1083–1093 CAS.
- A. M. Sulej, Ż. Polkowska and J. Namieśnik, Sensors, 2011, 11, 11901–11920 CrossRef CAS PubMed.
- M. Switzenbaum, S. Veltman, M. D. Wagoner and T. Schoenberg, Chemosphere, 2001, 43, 1051–1062 CrossRef CAS PubMed.
- W. Zhang, S. Zhang, D. Yue, C. Wan, Y. Ye and X. Wang, Environ. Toxicol. Chem., 2008, 27, 31–37 CAS.
- K. Lamprea and V. Ruban, Environ. Technol., 2011, 32, 1141–1149 CrossRef CAS PubMed.
- M. S. Garcia-Falcon, C. Perez-Lamera and J. Simal-Gandara, Anal. Chim. Acta, 2004, 508, 177–183 CrossRef CAS.
- A. M. Sulej, Ż. Polkowska, A. Astel and J. Namieśnik, Talanta, 2013, 117, 158–167 CrossRef CAS PubMed.
- R. D. S. Chang and Y. Sun, J. Chromatogr. A, 2000, 879, 177–188 CrossRef.
- M. Rawa-Adkonis, L. Wolska, A. Przyjazny and L. Wolska, Anal. Lett., 2006, 39, 2317–2331 CrossRef CAS.
- D. Cam, S. Gagni, L. Meldolesi and G. Galletti, J. Chromatogr. Sci., 2000, 38, 55–60 CAS.
- G. Purcaro, P. Morrison, S. Moret, L. S. Conte and P. J. Marriott, J. Chromatogr. A, 2007, 1161, 284–291 CrossRef CAS PubMed.
- R. B. Gomes, R. Nogueira, J. M. Oliveira, J. Peixoto and A. G. Brito, Environ. Sci. Pollut. Res., 2009, 16, 671–678 CrossRef CAS PubMed.
- E. P. Barros, N. Moreira, G. E. Pereira, S. G. F. Leite, C. M. Rezende and P. G. Pinho, Talanta, 2012, 101, 177–186 CrossRef PubMed.
- A. King, J. Readman and J. Zhou, Anal. Chim. Acta, 2004, 523, 259–267 CrossRef CAS.
- A. King, J. Readman and J. Zhou, Environ. Geochem. Health, 2003, 25, 69–75 CrossRef CAS PubMed.
- F. Busetti, A. Heitz, M. Cuomo, S. Badoer and P. Traverso, J. Chromatogr. A, 2006, 1102, 104–115 CrossRef CAS PubMed.
- C. Maldonaldo, J. Bayona and L. Bodineau, Environ. Sci. Technol., 1999, 33, 2693–2702 CrossRef.
- B. M. F. Avila, R. Pereira, A. O. Gomes and D. A. Azevedo, J. Chromatogr. A, 2011, 1218, 3208–3216 CrossRef CAS PubMed.
- T. Ieda, N. Ochiai, T. Miyawaki, T. Ohura and Y. Horii, J. Chromatogr. A, 2011, 1218, 3224–3232 CrossRef CAS PubMed.
- X. Di, R. A. Shellie, P. J. Marriot and C. W. Huie, J. Sep. Sci., 2004, 27, 451–458 CrossRef CAS PubMed.
- L. N. Williamson and M. G. Bartlett, Biomed. Chromatogr., 2007, 21, 664–669 CrossRef CAS PubMed.
- F. J. Santos and M. T. Galceran, TrAC, Trends Anal. Chem., 2002, 21, 9–10 CrossRef.
- P. Marriott and R. Shellie, TrAC, Trends Anal. Chem., 2002, 21, 9–10 CrossRef.
- M. Adahchour, J. Beens, R. J. J. Vreuls and U. A. T. Brinkman, TrAC, Trends Anal. Chem., 2006, 25, 438–454 CrossRef CAS.
- K. Banerjee, S. H. Patil, S. Dasgupta, D. P. Oulkar, S. B. Patil, R. Savant and P. Adsule, J. Chromatogr. A, 2008, 1190, 350–357 CrossRef CAS PubMed.
- E. Skoczyńska, P. Korytar and J. Boer, Environ. Sci. Technol., 2008, 42, 6611–6618 CrossRef.
- A. J. King, J. W. Readman and J. L. Zhou, Environ. Geochem. Health, 2003, 25, 67–75 Search PubMed.
- A. J. King, J. W. Readman and J. L. Zhou, Anal. Chim. Acta, 2004, 523, 259–267 CrossRef CAS.
- A. M. Sulej-Suchomska, Ż. Polkowska, Z. J. Kokot, M. d. l. Guardia and J. Namieśnika, Microchem. J., 2016, 126, 466–473 CrossRef CAS.
- M. Korhonen, A. Kiviranta and R. Ketola, Toxicol. Environ. Chem., 1998, 66, 37–45 CrossRef CAS.
- S. Maghsoudi and E. Noroozian, Chromatographia, 2012, 75, 15–16 Search PubMed.
- P. Hsieh, J. Jen, C. Lee and K. Chang, Environ. Eng. Sci., 2015, 32, 301–309 CrossRef CAS.
- Z. Zhang and J. Pawliszyn, Anal. Chem., 1993, 65, 1843 CrossRef CAS.
- F. F. Lei, J. Huang, X. N. Zhang, X. J. Liu and X. J. Li, Chromatographia, 2011, 74, 99–107 CAS.
- E. Rianawati and R. Balasubramanian, Phys. Chem. Earth, 2009, 34, 857–865 CrossRef.
- E. Coelho, C. Fereira and C. M. M. Almeida, J. Braz. Chem. Soc., 2008, 19, 1087–1097 Search PubMed.
- T. Chmiel, D. Abogado and W. Wardencki, Anal. Bioanal. Chem., 2014, 406, 4965–4986 CrossRef CAS PubMed.
- EPA, Methods for organic chemical analysis of multicipal and industrial wastewater, EPA, 2012 Search PubMed.
- S. Ozcan, A. Tor and M. Aydin, Anal. Chim. Acta, 2010, 665, 193–1999 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay00401f |
| This journal is © The Royal Society of Chemistry 2016 |