
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of microcystin-LR in surface water by a magnetic bead-based colorimetric immunoassay using antibody-conjugated gold nanoparticles†
A.-C.
Neumann
,
X.
Wang
,
R.
Niessner
and
D.
Knopp
*
Institute of Hydrochemistry and Chemical Balneology, Chair for Analytical Chemistry, Technical University Munich, Marchioninistrasse 17, Munich 81377, Germany. E-mail: dietmar.knopp@ch.tum.de; Fax: +49-89-2180-78255; Tel: +49-89-2180-78252
First published on 10th September 2015
Abstract
Herein we describe the development of a homogeneous competitive colorimetric immunoassay using antigen-functionalized magnetic beads (MBs) and antibody-immobilized gold nanoparticles (AuNPs) combined with the established gold staining method for the determination of microcystin-leucine-arginine (MC-LR) in surface water. Solid phase extraction proved to be the most beneficial sample preparation method in order to reduce matrix effects which might lead to agglomeration of the AuNPs and failing of the assay. The developed method could detect MC-LR in a linear range from 0.51–11.68 μg L−1 with a limit of detection (LOD) of 0.38 μg L−1. The recovery rates of the toxin in spiked water samples were in the range from 88.8% to 117.5%. This proposed immunoassay constitutes a simple and cost-effective method for the detection of sanitary relevant concentrations around 1 μg L−1 of MC-LR in fresh waters without any expensive or challenging equipment.
Introduction
Eutrophication of lakes and rivers caused by an enrichment of nutrients can lead to cyanobacterial water blooms. Cyanobacteria, also called blue-green algae, produce a wide variety of toxins, for example neurotoxins like anatoxin and saxitoxin and hepatotoxins such as nodularin and microcystins (MCs). These secondary metabolites are potent inhibitors of intracellular protein phosphatase 1 (PP1) and 2A both in vivo and in vitro.1 Furthermore, MCs belong to the okadaic acid class of tumor promotors.2,3 A chronic exposure to trace amounts of MCs in drinking water can lead to primary liver cancer. Microcystin-leucine-arginine (MC-LR) is the most toxic MC congener (Fig. 1). This toxin is a cyclic heptapeptide including a unique amino acid, so called ADDA [(2S,3S,8S,9S)-3-amino-9-methoxy-2,6,8-trimethyl-10-phenyldeca-4E,6E-di-enoic acid].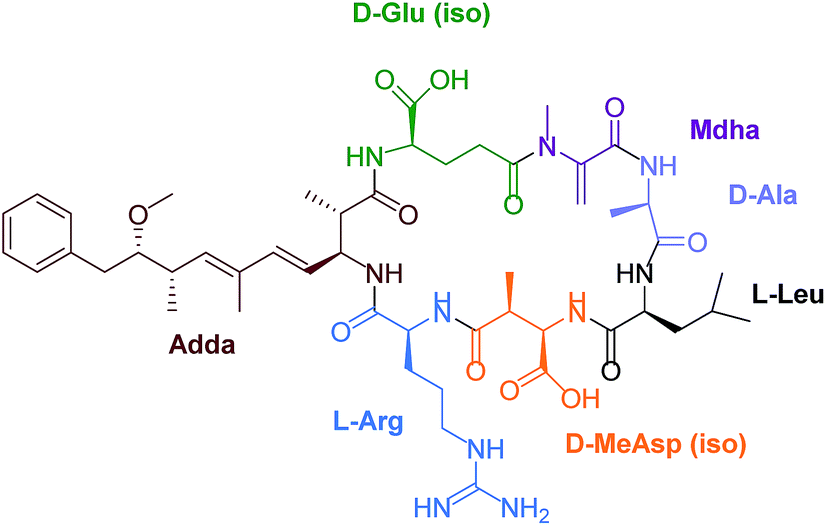 | ||
| Fig. 1 Chemical structure of MC-LR. | ||
Considering the current knowledge of MC toxicity the World Health Organisation (WHO) recommends that the amount in water of free plus cell bound MC-LR should be limited to 1 μg L−1.4 Germany aspires this proposal. To date, the most widespread methods for the identification and quantification of MCs in water samples are based on HPLC coupled to different detection methods like mass spectrometry (MS),5,6 fluorescence (FL)7 and ultraviolet (UV) spectroscopy.8–10 However, these methods are time-consuming and require sophisticated instrumentation and trained personnel.
Consequently, the development of rapid and cost-effective methods is needed. In the past, an enzyme-linked immunosorbent assay (ELISA) based on the interaction between antigen and antibody for the detection of MC-LR was established in our institute and yielded an IC50 value of 0.06 μg L−1 using the monoclonal antibody MC10E7.11 Several attempts have been made to shorten the assay time of immunoassays by utilizing bio-functionalized nanomaterials. Gold nanoparticles (AuNPs), for example, are used for rapid colorimetric assays because they own a lot of advantages like facile preparation, unique catalytic property and simplicity in modification.12–15 In combination with magnetic beads (MBs) which deserve good biocompatibility and a fast separation from the reaction mixture by an external magnet, it is possible to generate fast and simple assays where no expensive equipment is needed.16,17 For instance, Reverté et al. developed magnetic particle-based enzyme assays and immunoassays for MCs where the monoclonal antibody was coupled to protein G-coated MBs.18 In another study, Yu et al. developed a competitive fluorescence immunoassay for sensitive and rapid detection of MCs using analyte-coated MBs and antibody-conjugated quantum dots (QDs).16 The signal could be directly read out by a fluorescence spectrophotometer, which eliminated the signal development steps of traditional ELISA.19 Very recently, a competitive colorimetric immunoassay for the detection of the mycotoxin aflatoxin B1 was developed by our group based on homogeneous gold staining.20 Taking advantages of the catalytic amplification effect of immunogold colloids as well as rapid separation using MBs, the established method is fast and highly sensitive. Depending on the complexity of the sample, dilution is required to eliminate interfering matrix effects. In this work, solid phase extraction (SPE) was combined with the competitive colorimetric immunoassay for the determination of low concentrations of MC-LR in lake and river water.
Experimental
Materials and reagents
Sephadex G-25 columns were purchased from GE Healthcare Europe GmbH (Freiburg, Sweden). Chloroauric acid (HAuCl4), sodium citrate, bovine serum albumin (BSA, fraction V, ∼99%), ferrous sulfate heptahydrate (FeSO4·7H2O), ammonium hydroxide (28% in water), polyoxyethylene (20) sorbitan monolaurate (Tween-20), sodium iodoacetate, potassium hydrogen phosphate (K2HPO4), potassium dihydrogen phosphate (KH2PO4), 5,5′-dithiobis(2-nitrobenzoic acid) (Ellman's reagent), sodium azide (NaN3), potassium dihydrogen citrate, potassium sorbate, ethylenediaminetetraacetic acid (EDTA), dimethyl sulfoxide (DMSO), sodium thiosulfate (Na2S2O3), methanol (MeOH), ethanol (EtOH), ascorbic acid and sulphuric acid (H2SO4) were purchased from Sigma-Aldrich (Taufkirchen, Germany). Glutaraldehyde (25% in water), potassium carbonate (K2CO3), sodium dihydrogen phosphate (NaH2PO4·H2O), disodium hydrogen phosphate (Na2HPO4·2H2O), sodium hydrogen carbonate (NaHCO3) and hydroxylammonium chloride (NH4OH·HCl) were obtained from Merck Millipore (Darmstadt, Germany). Iron(III) chloride hexahydrate (FeCl3·6H2O), 3-aminopropyl-triethoxysilane (APTES, ∼99%), were purchased from Fluka (Buchs, Switzerland). Disposable cuvettes (cat. no. 759015), polyethylene glycol 8000 (PEG-8000), sodium hydroxide (NaOH), nitric acid (HNO3), hydrochloric acid (HCl), cetyltrimethylammonium bromide (CTAB) and sodium chloride (NaCl) were purchased from Carl Roth GmbH (Karlsruhe, Germany). N-Succinimidyl-S-acetylthio propionate (SATP) was obtained from Thermo Fisher Scientific (Braunschweig, Germany). Microcystin-LR was purchased from Enzo Life Science (Lörrach, Germany). The mouse monoclonal antibody against MC-LR (anti-MC-LR, clone MC10E7, affinity purified) was from our group. Ultrapure water was produced using reverse osmosis with UV treatment (Milli-RO 5 Plus, Milli-Q185 Plus, Millipore, Eschborn, Germany). The absorption spectra were measured on UV/Vis spectrometer Specord 250 Plus (Analytik Jena, Jena, Germany). For the freeze-drying of the MC-LR-BSA conjugate an Alpha 1-4 LSC (Christ, Osterode/Harz, Germany) was used. For LC-MS measurements an autosampler surveyor AS and a Surveyor MS PumpPlus were purchased from Thermo Finnigan (Bremen, Germany). The Orbitrap-based Exactive™ Benchtop Mass Spectrometer equipped with an electrospray source (ESI), a Hypersil GOLD column (100 mm × 2.1 mm, 1.9 μm particle size) for the LC separation were obtained from Thermo Fisher Scientific (Bremen, Germany). The column oven Hotdog 5090 was purchased from ProLab (Reinach, Switzerland).Synthesis of the MC-LR-BSA conjugate
MC-LR was coupled to bovine serum albumin (BSA) via a Michael addition. Starting the reaction, 1.64 mg of BSA (2.5 × 10−5 mmol, 1 eq.) was added to 84 μL NaHCO3 buffer (0.1 M in water, pH 8.0) and 9 μL of a SATP solution (9.5 mg SATP in 0.5 mL DMSO, 3.9 × 10−2 mmol, 1549 eq.) and stirred at ambient temperature for 1.5 h. A volume of 0.87 μL of a freshly prepared NH4OH·HCl solution (0.86 M in water) and 2.5 μL of NaOH solution (5 M in water) were added, followed by a purification step with a Sephadex-G25 column. The added sample was eluted with 1.5 mL of phosphate buffer (PBS, 1 mM KH2PO4, 70 mM K2HPO4, 145 mM NaCl in water, pH 7.4) and dropwise collected in a low-binding microtiter plate. To each drop 3 μL of Ellman's solution (10 mM in PBS) were added and fractions containing free thiol groups identified by yellow colour formation. These fractions were combined, flushed with nitrogen and 200 μL of a 1 mg mL−1 MC-LR solution (2.0 × 10−4 mmol, 8 eq.) were added to the reaction vessel followed by addition of 11 μL NaOH solution (5 M in water). The mixture was stirred in the dark for 12 h at ambient temperature. To block free thiol groups, 400 μg sodium iodoacetate (1.9 × 10−3 mmol, 1300 eq.) were added and the solution was further stirred for 15 min. For the purification of the MC-LR-BSA conjugate, the solution was dialysed against water for 4 days. After a lyophilisation step, the conjugate was redissolved in PBS containing 0.05% NaN3 to a final concentration of 1 mg mL−1. The solution was stored at 4 °C until utilisation.The molecular weight of the conjugate was determined by MALDI-TOF mass spectrometry and yielded a mass for MC-LR-BSA of 70.5 kDa, which denotes a coupling rate of 3 MC-LR molecules per mole of BSA.
Synthesis of MC-LR-BSA coated magnetic beads (MC-LR-BSA-MBs)
In general, when working with MBs/AuNPs the phosphate buffer was prepared without addition of NaCl and with sodium salts. The synthesis was performed by a modification of a procedure published by Wang et al.21 Briefly, 1.35 g of FeCl3·6H2O (5 mmol, 2.08 eq.) and 0.67 g of FeSO4·7H2O (2.4 mmol, 1 eq.) were added to 25 mL of nitrogen saturated deionized water. The resulting suspension was heated at 80 °C for 10 min under reflux conditions followed by the addition of 2 mL of ammonia (28% in water) and stirring for 30 min at 80 °C. Using an external magnet, the obtained particles were washed three times with pure water and twice with EtOH. The particles were dried at 120 °C for approximately 1 h. Then 60 mg of the resulted Fe3O4 particles were dispersed in 10 mL EtOH under sonication for 15 min followed by the addition of 100 μL deionized water and 200 μL APTES (0.96 mmol, 3.8 eq.). This suspension was shaken for 5 h at 200 rpm and ambient temperature followed by a magnetic separation and 5 washing steps with phosphate buffer (2.65 mM NaH2PO4 and 47.35 mM Na2HPO4 in water, pH 8.0) to get rid of any physically adsorbed APTES. The amino-functionalized particles were dispersed in 4 mL of deionized water.To one millilitre of washed NH2-MBs (∼15 mg mL−1) 0.5 mL glutaraldehyde (5.6 mmol, 25% in water) were added and the suspension was shaken for 1 h at 100 rpm and ambient temperature. The activated particles were separated by an external magnet, washed thoroughly and finally resuspended in 1 mL of phosphate buffer (∼15 mg mL−1, 2.65 mM NaH2PO4 and 47.35 mM Na2HPO4 in water, pH 8.0). Fifty microliters of the MC-LR-BSA conjugate solution (7.1 × 10−2 nmol) were added and the suspension was shaken overnight at 200 rpm. To block free binding sites on the surface of the MBs, 100 μL of a 50 mg mL−1 BSA solution (in phosphate buffer, 3.8 mM NaH2PO4 and 16.2 mM Na2HPO4 in water, pH 8.0) were added and the mixture shaken at 200 rpm for another 1 h. The obtained MC-LR-BSA-MBs (∼3.3 μm) were washed five times with deionized water, dispersed in 5 mL water and stored at 4 °C when not in use (∼3 mg mL−1). These particles are stable for approximately 3 months.
Synthesis of antibody-conjugated AuNPs (Ab-AuNPs)
The synthesis of the AuNPs was performed according to Frens et al.22 with minor modification. In brief, all used glass vessels were cleaned with aqua regia (HNO3/HCl, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) and deionized water to eliminate possible nucleation. In order to prepare AuNPs with a size of 35 nm a protocol which was recently published by our group was used.21 Fifty millilitres of a 0.25 mM HAuCl4 solution were heated up followed by adding 1.7 mL of a sodium citrate solution (1% in water). The AuNPs were heated up for further 15 min and cooled down to ambient temperature and stored at 4 °C when not in use.
:3, v/v) and deionized water to eliminate possible nucleation. In order to prepare AuNPs with a size of 35 nm a protocol which was recently published by our group was used.21 Fifty millilitres of a 0.25 mM HAuCl4 solution were heated up followed by adding 1.7 mL of a sodium citrate solution (1% in water). The AuNPs were heated up for further 15 min and cooled down to ambient temperature and stored at 4 °C when not in use.
For the coupling of antibody MC10E7, 5 mL of the obtained AuNPs solution was adjusted to pH 8–9 by adding 100 μL of a K2CO3 solution (0.1 M in water). Fifty microliters of MC10E7 solution (1.1 mg mL−1 in PBS, 0.05% NaN3) were added and the solution was stirred for 1 h at ambient temperature. To block free binding sites on the AuNPs, 570 μL of a BSA solution (5% in 50 mM phosphate buffer, pH 7.4) was added. After an incubation time of 1 h at 200 rpm and ambient temperature, 635 μL of a Tween-20 solution were added (1 wt% in water) and again stirred for 1 h. To remove surplus chemicals the solution was centrifuged at 9500g at 4 °C for 12 min. The resulting supernatant was removed and the nanoparticles were resuspended in washing solution (phosphate buffer, 0.95 mM NaH2PO4 and 4.05 mM Na2HPO4 in water, pH 7.4, 0.1% PEG-8000). After another centrifugation step, the Ab-AuNPs were dispersed in a total volume of 4 mL of washing solution and stored at 4 °C.
Preparation of the gold growth solution
Twenty-five microliters of a 0.1 M HAuCl4 solution were added to 10 mL of a 0.1 M CTAB and the mixture was heated at 50 °C in a water bath. When all the yellow precipitates were dissolved, 100 μL of a 0.1 M ascorbic acid solution were added. Until the solution has ambient temperature, it was ready for use.General protocol of the competitive colorimetric immunoassay
Protocol for one sample: the needed amount of MC-LR-BSA-MBs (40 μL) was washed three times with deionized water, by discharging the supernatant (magnetic separation), and finally resuspended in 40 μL deionized water. The MC-LR-BSA-MBs suspension was added to 100 μL of the laid MC-LR sample followed by adding 100 μL of the Ab-AuNPs solution. The suspension was shaken for 1 h at 20 °C and a speed of 500 rpm. The pink supernatant was separated from antibody bound MC-LR-BSA-MBs by an external magnet. Contemporary the growth reaction of the AuNPs-Ab-MC-LR particles was carried out. This was performed by adding 1 mL of a freshly prepared growth solution to a cuvette (PS) followed by adding 100 μL of the supernatant. The reaction occurred for exactly 10 min and was stopped with addition of 100 μL of a Na2S2O3 solution (10 mM in water). The absorbance was measured on a UV-Vis spectrophotometer. All measurements were performed at least in triplicate. For evaluation of the results, a sigmoidal calibration curve was set up using Rodbard's four parameter function and the absorbance was plotted against the log concentration of MC-LR standards.Water sampling
The surface water samples were collected from two lakes near Munich and two rivers. The pH values were determined, being 8.3 for lake Starnbergersee (Percha), 8.3 for lake Ammersee (Eching), 8.0 for river Isar (Munich) and 8.1 for river Windach (Windach). All water samples were filtered on a glass microfiber filter (Whatman Cat. no. 1822 047, Dassel, Germany) to remove particles larger than 1.2 μm and were stored at 4 °C.Solid phase extraction (SPE)
For the enrichment of MC-LR and simultaneous elimination of matrix constituents (e.g. natural organic matter, Ca2+ and Mg2+ ions) potentially interfering with the AuNPs an SPE was performed for rapid sample preparation of toxin spiked fresh water samples. Aliquots of 50 mL were spiked with MC-LR to obtain a final toxin concentration of 1.0 μg L−1. Prior to the water sample addition, the OASIS HLB SPE cartridge (Waters GmbH, Eschborn, Germany) was conditioned with 5 mL of MeOH and 10 mL of deionized water. The cartridges were then attached to a vacuum manifold apparatus, washed with 7 mL of 5% (v/v) aqueous MeOH and dried under low pressure for 2 min to eliminate residual wash solvent. The MC-LR was eluted by adding 5 mL of 100% MeOH and the collected eluate was dried under a gentle N2 stream at ambient temperature. Reconstruction of the residues was performed by adding 1 mL of 50% (v/v) aqueous MeOH. The extracts were stored at −20 °C until determination.HPLC-MS measurements
For the quantification of MC-LR in the extracts a liquid chromatography system coupled to mass spectrometry detection was performed. The column was at 30 °C, the flow rate was 0.2 μL min−1 and the injection volume was 10 μL. A gradient solvent program was used where MeOH and water (both containing 0.1% NH4OH) were used as mobile phase. The initial composition of 25% MeOH was increased to 70% in 12 min. After 1 min of constant composition, the MeOH was linearly decreased to 25% in 1 min and held for 1 min.The ESI was operated in negative ionization mode. It was operated at resolution of 50000 (2 Hz) and the maximum injection time was 50.0 ms. During measurements the nebulization gas was set at 40 L h−1 and the auxiliary gas set at 2 L h−1. Spray voltage was set at 2.6 kV, capillary temperature to 375 °C, capillary voltage to −60.00 V, tube lens voltage to 190.00 V and the skimmer voltage to −22.00 V. MC-LR was identified by its retention time of 8.5 ± 0.3 min and its exact mass [M − H]− = 993.54149. An accuracy of ± 5 ppm was acceptable. The calibration was done in triplicate by measuring standards in the range from 1 to 200 μg L−1 in 50% aqueous MeOH.
Results and discussion
The principle of the colorimetric immunoassay based on MC-LR-BSA-MBs and Ab-AuNPs is illustrated in Fig. 2. The MC-LR-BSA-MBs compete with MC-LR in solution for the limited amount of Ab-AuNPs binding sites. After the magnetic separation of the immune complex, the supernatant containing unbound Ab-AuNPs-MC-LR particles, was directly submitted for the gold growth reaction. The final absorbance mainly depends on the amount of the added AuNPs, which directly correlates with the MC-LR concentration in solution.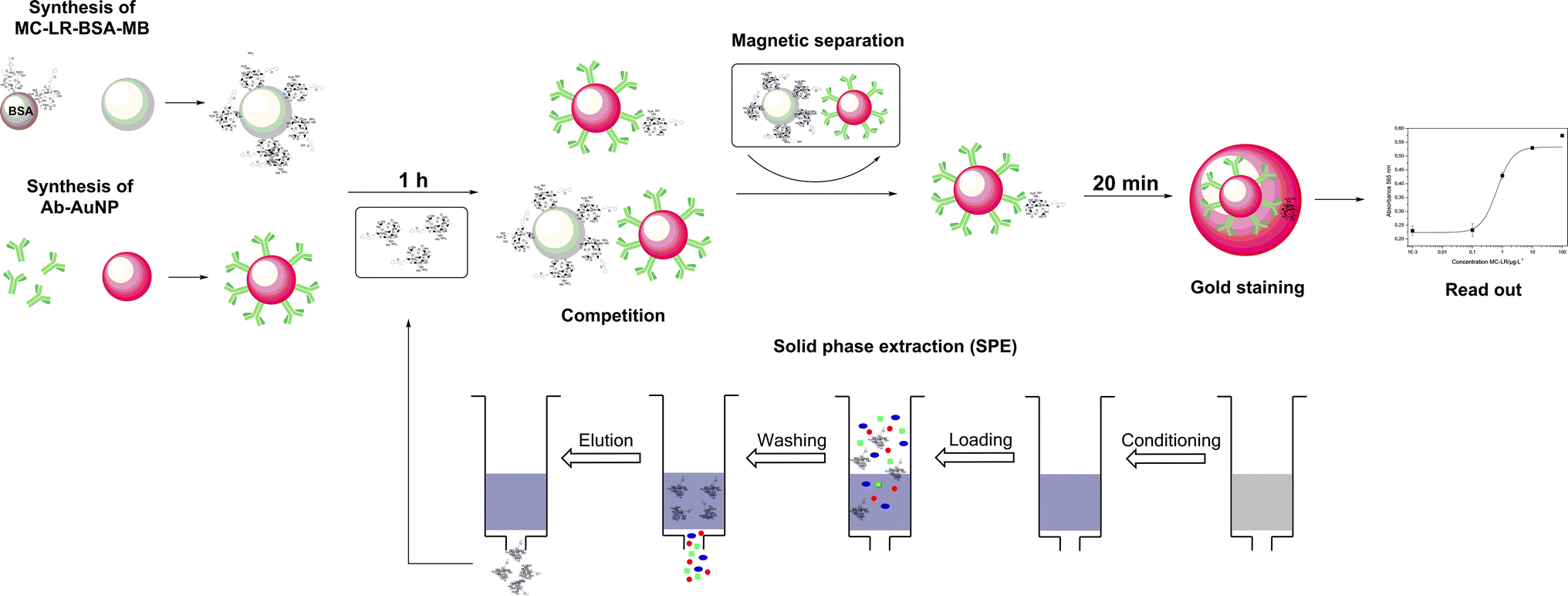 | ||
| Fig. 2 Scheme for the gold staining for amplified optical detection of MC-LR in solution. | ||
The mechanism of this enlargement of the AuNPs was recently published by our group and it was demonstrated that the sensitivity can be improved significantly.20
Optimization of the sample pre-treatment
Usually, any sample preparation should be as easy and rapid as possible. First experiments were performed using distilled water as solvent for preparing the MC-LR standards. After the competitive step and magnetic separation the expected absorbance change between 0 and 100 μg L−1 MC-LR was not denoted. The reason for this result is that a certain salt content is needed to keep certain ionic strength.23 When the pH is > 7, both MC-LR-BSA-MBs and Ab-AuNPs are negatively charged. Electrostatic repulsions occur between these two kinds of particles. So a certain ionic strength was required to reduce the electrostatic interactions. Based on our previous study, phosphate buffer revealed optimal for the immunoreaction, because it also adjusted the pH value of the reaction mixture.19,24An experiment was conducted where different PBS concentrations were tested. As seen in Fig. 3, with the increase of salt content from 0 mM to 50 mM PBS the absorbance difference between samples which contained 0 and 100 μg L−1 MC-LR increased up to ∼50 mM PBS. Higher PBS concentrations lead to the adverse effect, i.e., the absorbance difference dropped because some antibodies might be exfoliated from the gold surface resulting in a decrease of assay sensitivity. Using 50 mM PBS (9.5 mM NaH2PO4 and 40.5 mM Na2HPO4, pH 7.4) the highest absorbance difference was obtained. In fresh water samples, depending on the origin, a certain ionic strength is existing mainly by the presence of bivalent ions like Ca2+ and Mg2+. Indeed, first determinations of surface water samples spiked at 1 μg L−1 MC-LR, revealed unsatisfactory results. The concentration of the major bivalent ions in fresh water samples was determined by flame photometry (Ca2+) and atomic absorption spectroscopy (Mg2+). The results gave rise to the speculation on an interaction of the particles with cations and natural organic matter present in the surface water (Table 1).
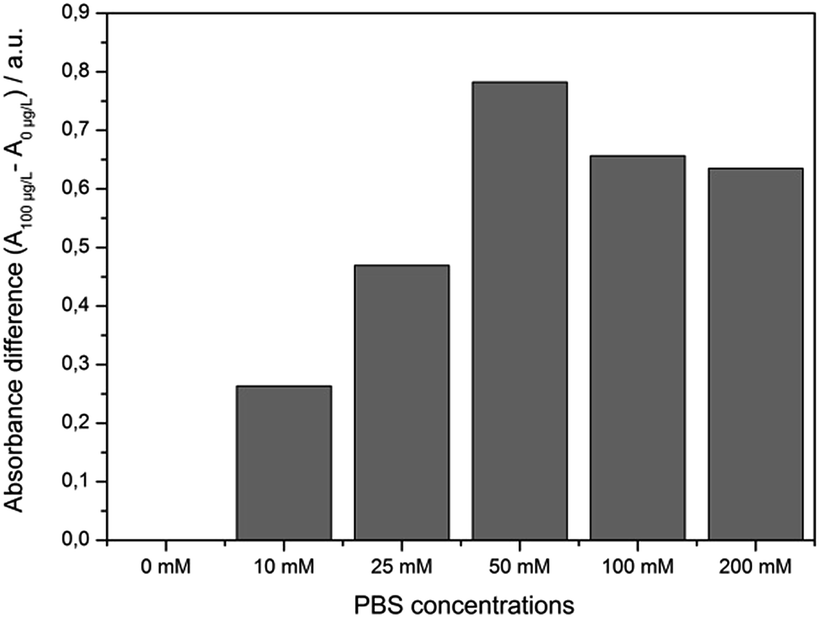 | ||
| Fig. 3 Correlation between the salt content and the absorbance difference after the competition step of the immunoassay between samples containing 0 and 100 μg L−1 MC-LR (m = 1, n = 6). | ||
| Sample | Ca2+ [mg L−1] | Mg2+ [mg L−1] |
|---|---|---|
| Starnbergersee | 39.8 ± 0.2 | 15.7 ± 0.1 |
| Ammersee | 54.4 ± 0.2 | 17.7 ± 0.1 |
| Isar | 54.8 ± 0.1 | 15.8 ± 0.0 |
| Windach | 93.9 ± 0.2 | 25.7 ± 0.1 |
According to the Hofmeister series which is a classification of ions in order of their ability to salt out or salt in proteins, cations like Ca2+ and Mg2+ can interact with peptide backbones by affecting the carbonyl oxygen or the amide moiety. By destroying the secondary and tertiary structure of the protein, the antibodies lose their binding properties.25 For example, de la Cruz et al. observed similar effects of Ca2+ at a concentration of 250 mg L−1, using other MC-LR immunoassays.26 Therefore, to get rid of bivalent ions a common chelating reagent was used in further sample measurements. The water samples were spiked at 1.0 μg L−1 of MC-LR and EDTA was added to a final concentration of 10 mM for complexation of bivalent ions. After the incubation step and the magnetic separation, however, no AuNPs were observed in the supernatant, i.e., no red colour was visible to the naked eye. As a conclusion, the bivalent ions tested at relevant concentrations in surface water did not interfere with the assay. Based on this finding, we supposed natural organic matter like fulvic and humic acids could bind to single bio-functionalized AuNPs and/or aggregates (the latter formed, e.g., because of the mineralization of the samples), and then were removed after non-specific adsorption to MBs. The determined dissolved organic carbon content in the water samples (DOC; determined with TOC Analyzer Multi N/C 2000, Analytik Jena) and the conductivity values (σ, measured with conductivity meter WTW Cond 3110, Weilheim, Germany) are summarized in Table 2. The DOC and mineralization was quite high in all samples. Consequently, a solid phase extraction (SPE) step was tested for sample preparation in order to eliminate these problems.
| Sample | DOC [mg L−1] | σ [μS cm−1] |
|---|---|---|
| Starnbergersee | 4.63 ± 0.2 | 317.0 ± 0.0 |
| Ammersee | 5.07 ± 0.1 | 484.0 ± 0.0 |
| Isar | 2.53 ± 0.1 | 378.0 ± 0.0 |
| Windach | 7.57 ± 0.1 | 590.0 ± 0.0 |
Solid phase extraction
The extraction efficiency of SPE in general depends on several operational parameters, like the extraction flow rate, the column material and the complexity of the samples. According to the literature, C18 material is commonly used for the enrichment and pure MeOH as eluent of microcystins.27 In this study, after filtration, each water sample was spiked at 1 μg L−1 of MC-LR and a preconditioned SPE-column was loaded with 50 mL of sample. In the followed washing step with 7 mL of MeOH–water (5:95, v/v) unbound chemicals were flushed through the column. To make sure that all MC-LR was eluted, 7 mL of pure MeOH were flushed very slowly through the column (1 drop per 2 seconds). After careful evaporation and resuspension of the residue in 1 mL of 50% (v/v) aqueous MeOH they were stored at −20 °C until use. The recoveries of were determined by HPLC-MS and were between 77.4% and 89.9% with relative standard deviations between 0.3% and 1%. In order to obtain concentrations that are in the linear range of the assay, the extracts were diluted 60 times in phosphate buffer (9.5 mM NaH2PO4 and 40.5 mM Na2HPO4, pH 7.4). To determine the influence of different amounts of MeOH in the samples, resulting from the dilution of the extract on the sensitivity of the assay, a set of different concentrations of MeOH in phosphate buffer (0, 5, 10 and 15%) was used to prepare blanks and standards containing 100 μg L−1 of MC-LR.
All these experiments were conducted with the same batch of functionalized MBs, AuNPs and gold growth solution. The obtained results are summarized in Fig. 4. The standard deviation for the values of the blank was 8.2% and for 100 μg L−1 MC-LR standard 5.5%. So, the content of MeOH in the range of 1% to 15% had no estimable effect on assay performance.
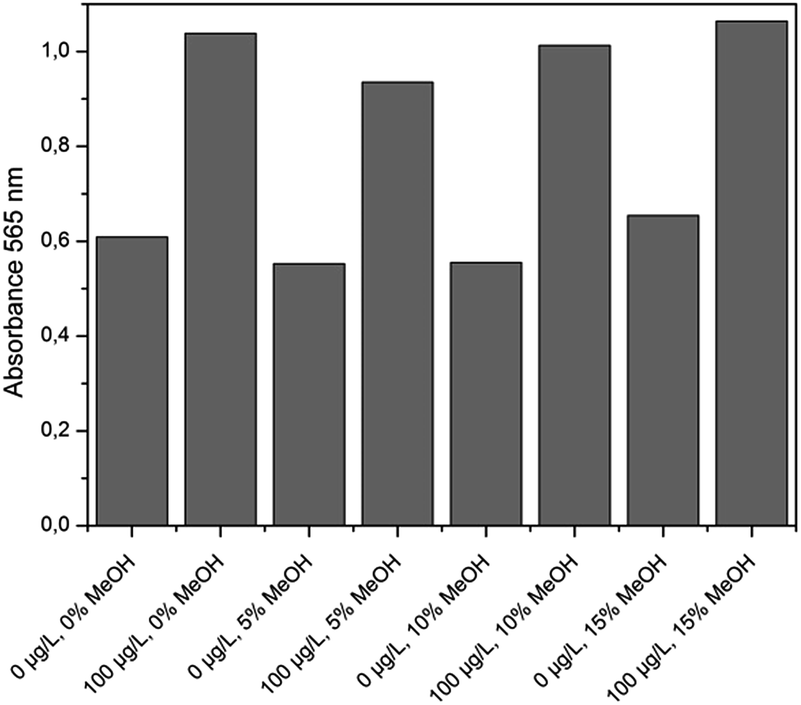 | ||
| Fig. 4 Correlation between the MeOH content in the sample and the absorbance at 565 nm after the incubation step (1 hour) and gold staining (m = 1, n = 8). | ||
Fresh water sample measurement
The optimized assay conditions were disposed by preparing MC-LR standards in the range from 0.001 to 100 μg L−1 in 50 mM phosphate buffer, pH 7.4. Fig. 5 shows the absorption spectra of the supernatants after the competition step and gold staining for 10 min (Fig. S1† represents the corresponding photographic image). With the increase of MC-LR concentration, the absorbance at 565 nm increased correspondingly and the maximum absorption was slightly blue-shifted which can be described by the smaller enlargement of Ab-AuNPs at higher immunogold concentration.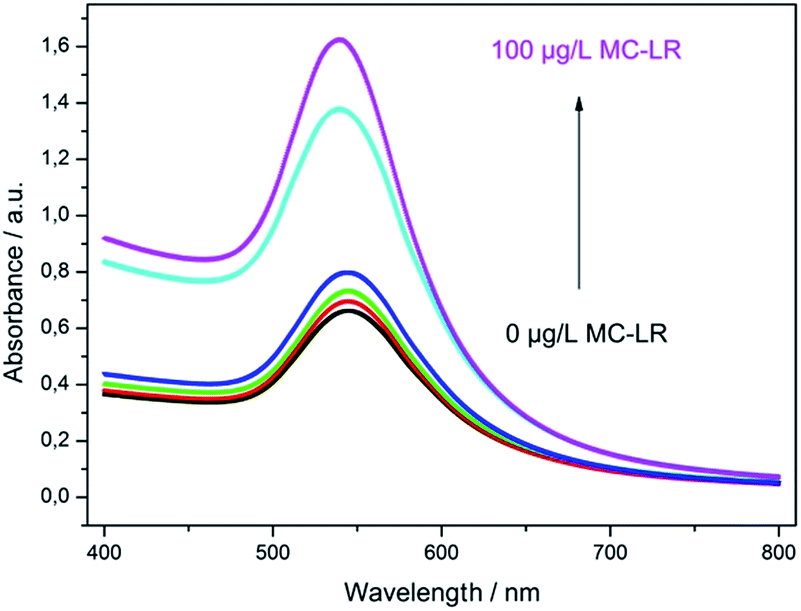 | ||
| Fig. 5 UV-Vis absorption spectra of supernatants after competition step and gold staining reaction for 10 min (m = 1, n = 6). Black: 0, red: 0.01, green: 0.1, blue: 1, cyan: 10, magenta: 100 μg L−1 MC-LR. | ||
In Fig. 6, the normalized absorbance values at 565 nm for each MC-LR concentration were plotted against the log concentration of MC-LR, yielding a sigmoidal curve which was set up using Rodbard's four parameter function. B/B0 was calculated for all samples by subtracting the average blank value from the sample absorbance measurement and divided by the difference between blank and maximum absorbance, then multiplied by 100 to obtain B/B0 in %. The error bars (±1 s) were calculated based on three-fold determinations.
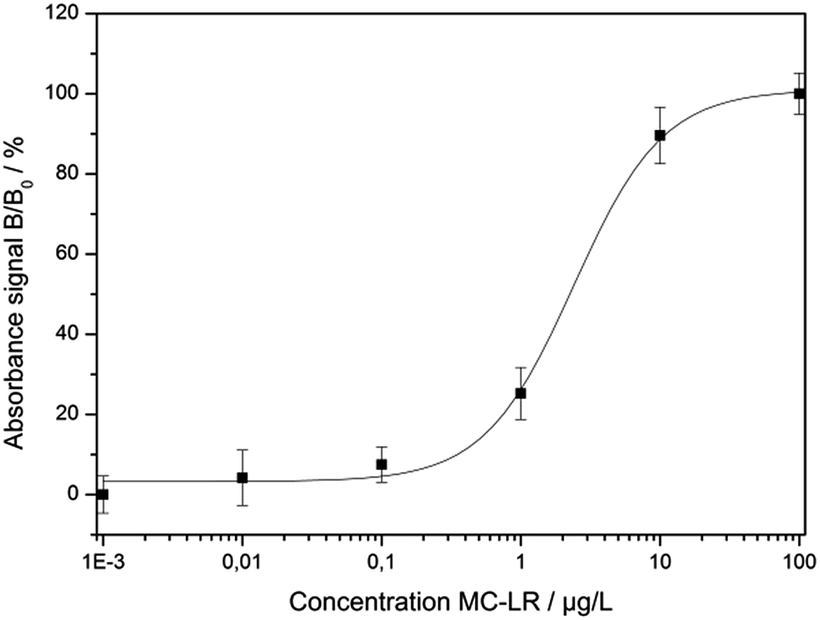 | ||
| Fig. 6 Calibration curve resulting from the normalised absorption values at 565 nm (R2 = 0.998, m = 3, n = 6). | ||
The quantification range set between 10% and 90% of the maximum absorbance was 0.51–11.63 μg L−1. The limit of detection (LOD) was calculated to be 0.38 μg L−1 based on a S/N ratio of 3. The relative standard deviations for the different concentrations were between 2.5% and 6.6%. The IC50 value was determined at 2.43 ± 0.75 μg L−1. For testing the colorimetric immunoassay's applicability to fresh water analysis, different MC-LR spiked surface water samples (Starnbergersee, Ammersee, Isar and Windach) were measured including SPE sample preparation. The results are summarized in Table 3.
With acceptable recoveries in the range of 88.8% to 117.5% the established immunoassay represents a relatively fast method with a total assay time of approximately 3 h. The proposed method is sufficiently sensitive and covers a relatively broad linear working range. Furthermore, no expensive or challenging equipment is needed.
Conclusions
This manuscript reports a method for the detection of MC-LR in surface waters based on a homogenous colorimetric immunoassay with toxin coated MBs and MC10E7 antibody-conjugated AuNPs. While real water samples, because of interfering matrix constituents, could not be analysed directly, determination of the toxin was possible after sample preparation, i.e., filtration, solid phase extraction and dilution with phosphate buffer. With the colorimetric assay it was possible to obtain an LOD of 0.38 μg L−1 and recovery rates for MC-LR spiked water samples in the range of 88.8% to 117.5%. This novel approach provides an alternative assay for efficient and above all low cost determination of MC-LR in fresh water samples.Acknowledgements
The financial support by the Deutsche Forschungsgemeinschaft (DFG) of project KN348/16-1 (Depletion of algal toxin-contaminated water using selective biofilters based on plant-produced antibodies (plantibodies)) is gratefully acknowledged. We thank Jessica Löprich for technical assistance.Notes and references
- C. MacKintosh, K. A. Beattie, S. Klumpp, P. Cohen and G. A. Codd, FEBS Lett., 1990, 264, 187–192 CrossRef CAS PubMed.
- H. Fujiki and M. Suganuma, Anti-Cancer Agents Med. Chem., 2011, 11, 4–18 CrossRef CAS PubMed.
- I. R. Falconer, Environ. Toxicol. Water Qual., 1991, 6, 177–184 CrossRef.
- WHO, Guidlines for Drinking Water Quality, Addendum to Vol 2, Health Criteria and Other Supporting Information, Geneva, 2nd edn, 1998 Search PubMed.
- H. Sirén, M. Jussila, H. Liu, S. Peltoniemi, K. Sivonen and M.-L. Riekkola, J. Chromatogr. A, 1999, 839, 203–215 CrossRef.
- W. Li, J. Duan, C. Niu, N. Qiang and D. Mulcahy, J. Chromatogr. Sci., 2011, 49, 665–670 CAS.
- K.-I. Harada, M. Oshikata, T. Shimada, A. Nagata, N. Ishikawa, M. Suzuki, F. Kondo, M. Shimizu and S. Yamada, Nat. Toxins, 1997, 5, 201–207 CrossRef CAS PubMed.
- F. M. de Andrade, A. N. de Macedo and E. M. Vieira, J. Liq. Chromatogr. Relat. Technol., 2012, 37, 1310–1319 CrossRef.
- P. I. Benke, M. C. S. Vinay Kumar, D. Pan and S. Swarup, Analyst, 2015, 140, 1198–1206 RSC.
- T. Hayama, K. Katoh, T. Aoki, M. Itoyama, K. Todoroki, H. Yoshida, M. Yamaguchi and H. Nohta, Anal. Chim. Acta, 2012, 755, 93–99 CrossRef CAS PubMed.
- A. Zeck, A. Eikenberg, M. G. Weller and R. Niessner, Anal. Chim. Acta, 2001, 441, 1–13 CrossRef CAS.
- H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849–2866 RSC.
- W. Li, J. Li, W. Qiang, J. Xu and D. Xu, Analyst, 2013, 138, 760–766 RSC.
- Y. Li, H. Schluesener and S. Xu, Gold Bull., 2010, 43, 29–41 CrossRef CAS.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028–2045 RSC.
- H. D. Hill and C. A. Mirkin, Nat. Protoc., 2006, 1, 324–336 CrossRef CAS PubMed.
- A. del Campo, T. Sen, J.-P. Lellouche and I. J. Bruce, J. Magn. Magn. Mater., 2005, 293, 33–40 CrossRef CAS.
- L. Reverté, D. Garibo, C. Flores, J. Diogène, J. Caixach and M. Campàs, Environ. Sci. Technol., 2012, 47, 471–478 CrossRef PubMed.
- H.-W. Yu, A. Jang, L. H. Kim, S.-J. Kim and I. S. Kim, Environ. Sci. Technol., 2011, 45, 7804–7811 CrossRef CAS PubMed.
- X. Wang, R. Niessner and D. Knopp, Analyst, 2015, 140, 1453–1458 RSC.
- X. Wang, R. Niessner and D. Knopp, Sensors, 2014, 14, 21535–21548 CrossRef CAS PubMed.
- G. Frens, Nature-Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- C. Burns, W. U. Spendel, S. Puckett and G. E. Pacey, Talanta, 2006, 69, 873–876 CrossRef CAS PubMed.
- K. Zabetakis, W. Ghann, S. Kumar and M.-C. Daniel, Gold Bull., 2012, 45, 203–211 CrossRef CAS.
- P. Jungwirth and P. S. Cremer, Nat. Chem., 2014, 6, 261–263 CrossRef CAS PubMed.
- A. A. d. l. Cruz, T. J. Lynche and D. D. Dionysiou, J. Environ. Prot. Ecol., 2012, 3, 1275–1285 CrossRef.
- L. Cong, B. Huang, Q. Chen, B. Lu, J. Zhang and Y. Ren, Anal. Chim. Acta, 2006, 569, 157–168 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay02164b |
| This journal is © The Royal Society of Chemistry 2016 |