
Comparison of near-infrared diffuse reflectance (NIR) and attenuated-total-reflectance mid-infrared (ATR-IR) spectroscopic determination of the antioxidant capacity of Sambuci flos with classic wet chemical methods (assays)†
D.
Clara‡
a,
C. K.
Pezzei‡
a,
S. A.
Schönbichler
a,
M.
Popp
b,
J.
Krolitzek
b,
G. K.
Bonn
ac and
C. W.
Huck
*a
aInstitute of Analytical Chemistry and Radiochemistry, CCB – Center for Chemistry and Biomedicine, Leopold-Franzens University, Innrain 80/82, 6020 Innsbruck, Austria. E-mail: Christian.W.Huck@uibk.ac.at; Tel: +43 512 507 57304
bBionorica SE, Kerschensteinerstrasse 12-15, 92318 Neumarkt/Oberpfalz, Germany
cADSI – Austrian Drug Screening Institute, Innrain 66a, 6020 Innsbruck, Austria
First published on 26th August 2015
Abstract
Near-infrared diffuse reflectance (NIR) and attenuated-total-reflectance mid-infrared (ATR-IR) spectroscopy techniques in hyphenation with multivariate analysis were utilized to determine the antioxidant capacity of ground Sambuci flos samples. Folin–Ciocalteu (FC), ferric ion reducing antioxidant power (FRAP), cupric reducing antioxidant capacity (CUPRAC), 2,2-diphenyl-picrylhydrazyl (DPPH) and 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) were optimized and performed as reference methods. To remove systematic errors several spectral pretreatments like 1st and 2nd derivative Savitzky–Golay, standard normal variate (SNV) or multiplicative scatter correction (MSC) were applied. Cross-validations and test-set validations were performed for all assays. The quality parameters, standard error of prediction (SEP) and the ratio performance deviation (RPD), were calculated. An acceptable quality of the calibration can be confirmed for ATR-IR spectroscopy (e.g. for the CUPRAC assay: R2: 0.85, RPDcorr: 2.68, SECV: 0.13% GAE for cross-validation; R2: 0.81, RPDcorr: 2.20, SEP: 0.15% GAE for test-set validation). Surprisingly all models calculated for NIR spectroscopy were of poor quality and point to unpredictability of the antioxidative capacity. Further investigations of extracts by high performance liquid chromatography (HPLC) with a diode array detector (DAD) coupled to mass spectroscopy (MS) were performed to analyze the principal compounds. Thus, rutin and chlorogenic acid were confirmed to be the main components in the samples. This study demonstrates that ATR-IR spectroscopy is suitable to determine the antioxidative capacity in ground Sambuci flos samples and can be used for quality control.
1 Introduction
Natural reactive oxygen species (ROS) and reactive nitrogen species (RNS) can damage biomolecules. ROS describe free radicals that derive from oxygen, like superoxide anions (O2˙−) and hydroxyl radicals (˙OH) or reactive nonradical molecules such as singlet oxygen (1O2), hydrogen peroxide (H2O2) and hypochlorous acid.1–3 RNS describe radicals and reactive molecules with nitrogen in it: nitric oxide radicals (NO˙), nitrogen dioxide radicals (NO2˙), ONOO− and all products which form when NO˙ reacts with O2˙−, RO˙ and RO2˙.3 Normally, ROS and RNS are harmful or even important as second messengers. There is a delicate equilibrium between the radical production and scavenging. Environmental stresses such as drought, metal toxicity, salinity and UV-B radiation as well as pathogen attack increase the generation of radicals in plants. When the level of ROS transcends the defense mechanisms, the cell is “oxidatively stressed”.1 These radical molecules can modify and break DNA-strands and proteins causing cancer.3,4 They are also responsible for effecting some diseases including autoimmune, neurodegenerative, cardiovascular, pulmonary, renal, gastrointestinal and liver disorders. ROS are also involved in the ageing process.5Antioxidants can lead to inhibition of the aforementioned effects and can be useful as preventers regarding oxidative molecules. Fruits, vegetables and medicinal herbs are the most important sources of antioxidant compounds.5 Herbal medicine is still the most used medicine in comparison to synthetic drugs due to its better cultural acceptability and lower side effects.5 In this study, spectroscopic methods to determine the antioxidant power of ground Sambucus nigra flowers (Sambuci flos) were developed. Sambuci flos belongs to the lineage of Adoxaceae (prior Caprifoliaceae), to the subgenus European elderberry.6,7 The most important antioxidants in Sambuci flos are flavonoids (flavonol glycosides, up to 3%) and phenolic acids (ca. 3%).8
Sambuci flos has a diaphoretic effect for the treatment of cold, chills and fever.8 Of great importance are the antiviral activity9 and the antibacterial activity against hospital pathogens.10
The antioxidant capacity of plants, food and chemical compounds can be measured by various assays.11 The various chemical mechanisms of these assays can be divided into two basic groups, the HAT (hydrogen atom transfer) or the SET (single electron transfer) mechanism.11,12 The currently used assays Folin–Ciocalteu (FC), ferric ion reducing antioxidant power (FRAP), cupric reducing antioxidant capacity (CUPRAC), 2,2-diphenyl-picrylhydrazyl (DPPH) and 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) function via the SET mechanism and the last two assays (DPPH and ABTS) additionally exhibit the HAT mechanism.11 These assays are destructive, time and chemical consuming. Near-infrared (NIR) and attenuated-total-reflectance infrared (ATR-IR) spectroscopy techniques measure the interaction of light with the material in a nondestructive way.13 NIR and ATR-IR spectroscopies in hyphenation with partial least squares (PLS) regression have already proved in a case study their capability to replace the aforementioned assay methods.14
2 Materials and methods
2.1. Chemicals and Sambuci flos samples
Iron(III) chloride hexahydrate (FeCl3·6H2O, 97%), 2,2-diphenyl-1-picrylhydraziyl (DPPH, purity not stated), 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, ≥98%), copper(II) chloride dihydrate (CuCl2·2H2O, ≥99.0%), potassium persulfate (K2S2O8, 99.99%), Folin–Ciocalteu's phenol reagent, sodium carbonate (Na2CO3, ≥99.8%), gallic acid ≥97.5%, neocuproine ≥99.0% and sodium acetate ≥99.5% were obtained from Sigma-Aldrich (St. Louis, USA). 2,4,6-Tris(2-pyridyl)-s-triazine (TPTZ, ≥98%) and ammonium acetate ≥98.0% were purchased from MERCK (Darmstadt, Germany). Ethanol absolute 99.9%, hydrochloric acid and acetic acid (analytical grade) were received from VWR (Radnor, USA). 19 dried Sambuci flos samples were provided by Bionorica SE (Neumarkt, Germany) and 49 samples were bought from local pharmacies. For HPLC-MS measurements, bidistilled water and acetonitrile (ACN) both of ultra LC-MS grade were obtained from ROTH (Karlsruhe, Germany).2.2. Sample preparation and extraction
68 ground Sambuci flos samples were milled in a Retsch ZM 200 rotormill (Retsch, Haan, Germany) to a particle size lower than 0.5 mm. 100 mg of all samples were weighed by using a METTLER TOLEDO Ax 205 DR/M scale (Mettler Toledo, Greifensee, Switzerland) and extracted with 5 ml ethanol/water (50/50, v/v) for 15 minutes in an ultrasonic bath Elma/TRANSSONIC T 820/H (Elma, Singen, Germany) five times each. All portions were combined and adjusted to 25 ml. The extracts were centrifuged at 3500 rpm for 30 minutes and the supernatant was aliquoted and stocked at −80 °C until the day of measurement (Fig. 1). Before performing the assays, the aliquots were quickly unfrozen and centrifuged for 15 minutes, where the aliquots were acclimated to room-temperature (22 °C). All assays were measured in triplicate.2.3. Folin–Ciocalteu
Folin and Ciocalteu15 developed this method and it was improved by Singleton et al.16 for the measurement of phenols in plants. The enhanced method of Singleton et al. was performed with small modifications. 100 μl sample extract, 1500 μl of H2O followed by 100 μl Folin–Ciocalteu's phenol reagent were put together. After five minutes, 1300 μl Na2CO3 solution (471.75 mM) was added and mixed. After incubation at 60 °C for 30 minutes, the mixture was cooled down for ten minutes. The absorbance was determined at 750 nm.2.4. FRAP
This assay was first developed by Benzie and Strain17 to measure the reducing power of plasma.11 Later it was adapted to measure the antioxidant power of botanicals.18 The assay was performed with slight modifications. First, the FRAP reagent was prepared by mixing together 5 ml of TPTZ solution (10 mM TPTZ in 458 mM HCl), 5 ml FeCl3 solution (12 mM FeCl3·6H2O) and 50 ml acetate buffer (1870 mg sodium acetate anhydrous and 16 ml of acetic acid adjusted to 1000 ml with H2O). 300 μl sample extract was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.5 with ethanol/H2O (50/50, v/v). 300 μl of diluted sample extract, 1600 μl H2O and 1100 μl FRAP reagent were mixed together. After incubation at 60 °C for 45 minutes and cooling for five minutes the absorbance was measured at 593 nm.
:9.5 with ethanol/H2O (50/50, v/v). 300 μl of diluted sample extract, 1600 μl H2O and 1100 μl FRAP reagent were mixed together. After incubation at 60 °C for 45 minutes and cooling for five minutes the absorbance was measured at 593 nm.
2.5. CUPRAC
Apak, Güçlü et al. described the possibility of measuring the antioxidant potential by reducing a copper(II)-complex to a copper(I)-complex.19,20 This assay was performed with minor revisions. Ammonium acetate buffer (1 M), copper(II) chloride solution (10 mM CuCl2·2H2O), and neocuproine solution (7.5 mM in ethanol absolute) were mixed in the ratio 1:1:1 (v/v/v). This mixture (CUPRAC reagent) was allowed to rest at least for one hour. 50 μl sample extract, 775 μl H2O and 2250 μl CUPRAC reagent were mixed and incubated at 60 °C for 20 minutes. The absorbance was detected at 450 nm after the samples were cooled down for five minutes.
2.6. DPPH
According to Brand-Williams et al.21 stable radicals of DPPH can be used to measure the antioxidant activity. 50 μl sample extract, 950 μl ethanol/H2O (50/50, v/v) and 2000 ml DPPH solution (238 nM DPPH in ethanol absolute) were mixed. After incubation at 60 °C for 15 minutes, the samples were cooled down for five minutes and were measured at 517 nm.2.7. ABTS
ABTS is an improvement of the TEAC (trolox equivalent antioxidative capacity) assay.22 In the novel method ABTS reacts with potassium persulfate.23 In this study this assay was adapted. The concentrate of the ABTS reagent (19.33 mg ABTS and 3.31 mg K2S2O8 solved in 10 ml H2O) was allowed to react for at least 16 h at 4 °C. 50 μl sample extract, 2350 μl ethanol/H2O (50/50, v/v) and 600 μl of diluted ABTS reagent (8.75 ml of concentrate diluted to 50 ml with ethanol absolute) were mixed. The mixture reacted at room temperature for 10 minutes and was measured afterwards at 734 nm.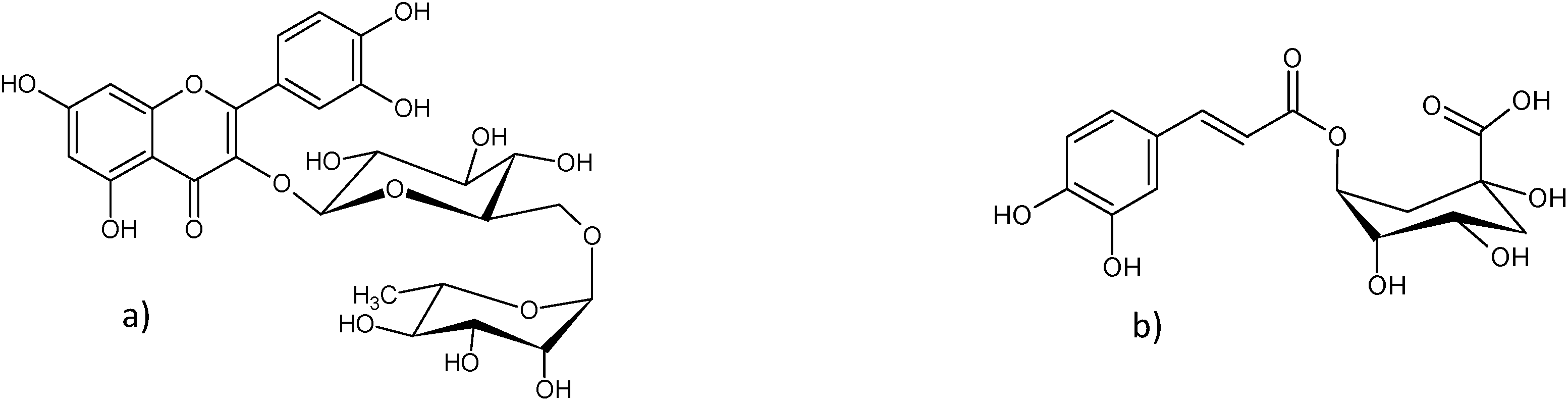 | ||
| Fig. 1 Main compounds of Sambuci flos extracts: (a) rutin and (b) chlorogenic acid. | ||
2.8. UV spectroscopy and measurement of the assays
Rotilabo 4.5 ml polystyrol cuvettes (Carl Roth, Karlsruhe, Germany) were used applying a Perkin Elmer 3051 Lambda 2 UV-VIS spectrometer (Perkin Elmer, Waltham, USA) to perform all measurements.All measurements were performed in triplicate against a blank which contained the same amount of H2O instead of the sample in FC, FRAP and CUPRAC and the same amount of ethanol/H2O (50/50, v/v) instead of the sample in DPPH and ABTS.
All calibrations were performed using gallic acid as a reference compound in the ranges of 35–350 mg l−1, 2.3–18.4 mg l−1, 30–300 mg l−1, 14–140 mg l−1 and 12–120 mg l−1 for FC, FRAP, CUPRAC, DPPH and ABTS, respectively. The antioxidative capacities were calculated as % GAE (gallic acid equivalent) to enable a comparison. The measurements of one assay lasted three weeks on average. Ten replicate measurements at high, middle and low concentration levels were performed and the relative standard deviation (RSD) was calculated for each level to provide the various precisions.
2.9. ATR spectroscopy
To collect the ATR-IR spectra, a Perkin Elmer Spectrum 100 FT-IR spectrometer (Perkin Elmer, Massachusetts, USA) equipped with a DicompTM crystal composed of a diamond ATR-IR with a ZnSe focusing element was utilized. All ATR-IR spectra were collected using Spectrum version 6.3.1.0134 software (Perkin Elmer, Massachusetts, USA). Every sample was split into five subsamples and each subsample was recorded with 50 scans. The recorded wavenumber ranged from 4000 cm−1 to 650 cm−1 and the resolution was set to 4 cm−1 with a datapoint interval of 1 cm−1. All measurements were done at 22 °C within twelve days.2.10. NIR spectroscopy
A Büchi NIRFlex N-500 FT-NIR spectrometer (Büchi, Flawil, Switzerland) in combination with the Büchi solids device equipped with a rotating cylinder was used to collect the NIR spectra. The internal and external references were recorded using a Spectralon® reflector. The samples were measured in diffuse reflectance using the NIR Ware 1.4.3010 software (Büchi, Flawil, Switzerland) in the range of 10000 cm−1 to 4000 cm−1. The absolute wavelength accuracy was ±2 cm−1 and the relative reproducibility was 0.2 cm−1. Every ground sample was measured in triplicate with 64 scans and the resolution was set to 8 cm−1. All measurements were performed at 22 °C within three days.
2.11. Multivariate analysis and statistical analysis
Between the different assays, the Pearson correlations were calculated using Excel (Microsoft, Redmond, USA).For spectral pretreatments and multivariate data analysis like the principal component analysis (PCA) with NIPALS (nonlinear iterative partial least squares) algorithm, partial least square regression (PLS) and all statistical analyses, The Unscrambler 10.2 chemometric software (Camo, Oslo, Norway) was used.
The spectra were transformed log1/R and the sample average of the sample measurements (three for NIR and five for ATR-IR) was determined. The ATR-IR spectra were first normalized between one and zero then averaged and subsequently treated with 1st or 2nd derivative Savitzky–Golay.24 The NIR spectra were pretreated with 1st or 2nd derivative Savitzky–Golay,24 standard normal variate (SNV)25 and multiplicative scatter correction (MSC).26 Wavenumber regions with low information content or higher noise were removed. For all derivative calculations the optimal polynomial order and window size for smoothing were evaluated. For each assay a PLS regression model was calculated. To get the significant wavelength regions for every model, the uncertainty test function of The Unscrambler 10.2 software was executed. For both spectroscopic methods test-set and cross-validation models were performed. For the test-set validation, two thirds of the samples representing the reference data (calibration set) and the other one third that generates the test set (validation set) were chosen.
To evaluate each PLS calibration method, the quality parameters, standard error of prediction (SEP), standard error of cross-validation (SECV), R2 and the ratio performance to deviation (RPD), were computed and compared. The SEP is the standard deviation of differences between reference data and the results in the validation set.27 The squared coefficient of determination (R2) describes the squared quotient of the covariance of the reference data and the predicted data, divided by the product of their standard variations.28 The RPD is defined as the ratio of the standard deviation of the reference data to the SEP or SECV.28 The best PLS and the optimum number of PCs were selected according to the SECV/SEC or SEP/SEC ratio near to one, the highest R2 and the highest RPD value. SEC is the standard error of calibration.
2.12. HPLC-DAD-MS
The measurements were performed with a Shimadzu (Shimadzu, Tokyo, Japan) HPLC-DAD/ESI-MS. The HPLC system was composed of two Shimadzu LC-30AD pumps, a SIL-30AC auto-injector, a CTO-20AC column oven, a SPD-M20A diode array detector and an electrospray ionization single quadrupole liquid chromatograph mass spectrometer LC/MS (LCMS2020) with a DGU-20A5R online degasser unit. The system control and data evaluation were achieved by using the manufacturer's LabSolutions LCMS software packages. For this analysis a precolumn Security Guard ULTRA cartridge for C18 UHPLC with 2.1 mm internal diameter (Thermo Scientific, Berlin) and a Phenomenex Kinetex PFP 4.6 mm × 150 mm, 2.6 μm, 100 Å (Phenomenex, Aschaffenburg, Berlin, Germany) column were employed.Solvent A was water with 0.5% formic acid and B acetonitrile. A gradient program was executed in the following steps (min/% B): 0/5, 15/16, 30/25, 40/35, 41/95 and 47/95. The flow rate was 1 ml min−1 and the injection volume was 10 μl. The detection wavelength for rutin was 260 nm and for chlorogenic acid 320 nm. The mass spectrometry measurements were executed using ESI in negative-ion mode. The scan range was 200–1000 m/z and the scan speed was 830 u s−1. The interface voltage was 0.95 kV. The temperature of the desolvation line (DL) was 250 °C and that of the heat block was 200 °C. Drying gas flow was set at 15 l min−1 and nebulizing gas flow was 1.5 l min−1. The unfrozen extracts of samples 15 and 29 were centrifuged once again at 3500 rpm for 30 minutes at 22 °C and 200 μl were filled in a HPLC vial inlet. Both samples were measured by HPLC-DAD-MS.
The results of FC, FRAP and CUPRAC assays showed high Pearson correlations among each which are displayed in Table 1.
| FC | FRAP | CUPRAC | DPPH | ABTS | |
|---|---|---|---|---|---|
| FC | 1.00 | 0.94 | 0.95 | 0.85 | 0.88 |
| FRAP | 1.00 | 0.94 | 0.86 | 0.84 | |
| CUPRAC | 1.00 | 0.86 | 0.85 | ||
| DPPH | 1.00 | 0.84 | |||
| ABTS | 1.00 |
3 Results
3.1. ATR spectroscopy
The average ATR-IR spectrum of all Sambuci flos samples which were used to develop the models is shown in Fig. 2. Marked vibrational bands were assigned according to Kellner et al.:29 3282 cm−1 (O–H stretching of water, carbohydrates and flavonoids), 2924 cm−1 (C–H asymmetric stretching), 2853 cm−1 (C–H symmetric stretching), 1732 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, non-conjugated), 1606 cm−1 (CC stretching and CO stretching, conjugated) and 1027 cm−1 (C–O, C–C stretch of carbohydrates and flavonoids).
O stretching, non-conjugated), 1606 cm−1 (CC stretching and CO stretching, conjugated) and 1027 cm−1 (C–O, C–C stretch of carbohydrates and flavonoids).
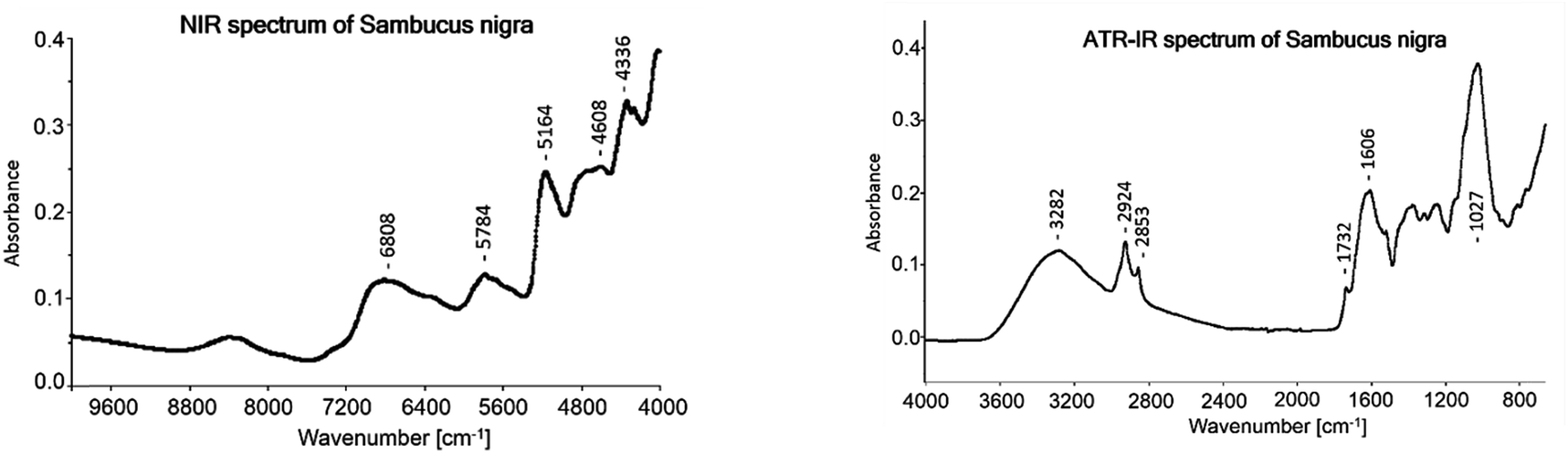 | ||
| Fig. 2 Averaged NIR and ATR-IR spectrum of 68 Sambuci flos samples. | ||
For the PLS methods (test-set and cross-validation) of all assays the 2nd derivative Savitzky–Golay with a polynomial order of three and a window size of 23 (see Fig. 5) showed the best results. To compare the PLS models the cross-validation results were used because all samples are included in the test set. In the test-set validation the validation samples vary which complicates a direct comparison. The cross-validation gave the best results for the CUPRAC assay and its regression coefficient plot of the model is displayed in Fig. 3. Most prominent regression coefficients appeared at 1187 cm−1, 1123 cm−1 and 986 cm−1 and are assigned to aromatic CH in plane bending vibrations. The band at 814 cm−1 was assigned to C–O–C symmetric stretching vibrations of saturated heterocycles like sugars and to aromatic CH out of plane bending. The band at 1726 cm−1 was assigned to carbonyl stretching. Band assignments were realized according to Pretsch, Bühlmann and Badertscher.30
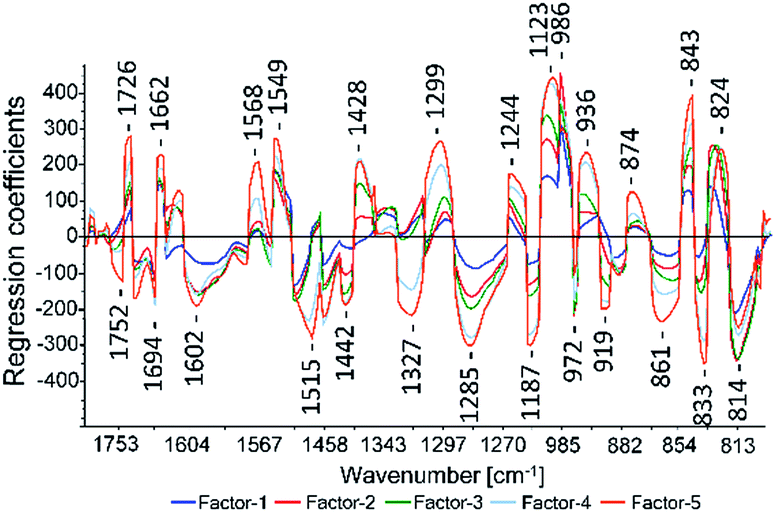 | ||
| Fig. 3 CUPRAC regression coefficient plot calculated with ATR-IR spectra. | ||
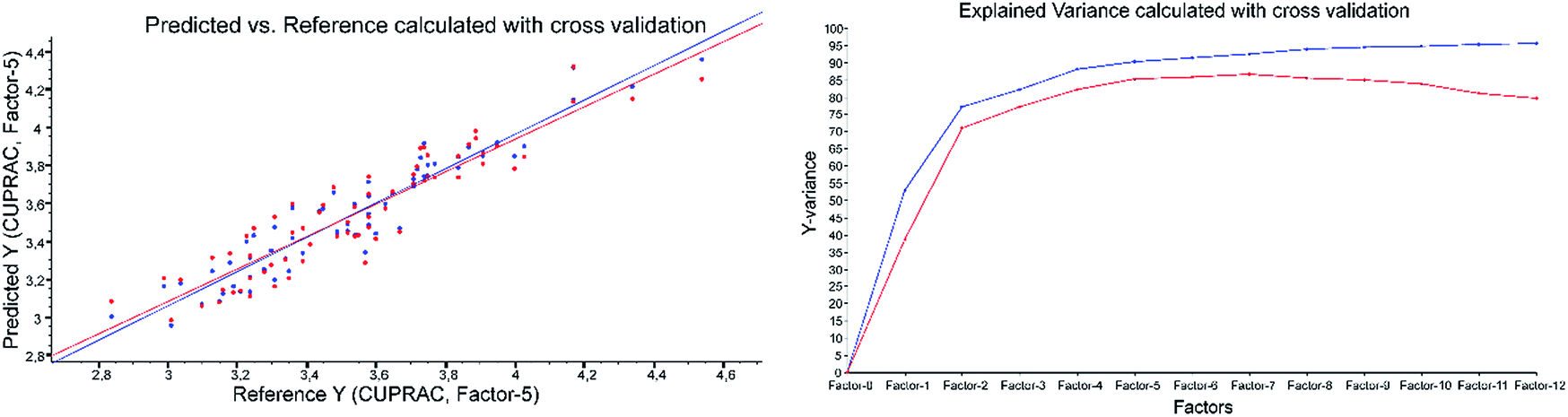 | ||
| Fig. 4 CUPRAC predicted vs. reference plot and explained variance plot calculated with ATR-IR spectra applying cross-validation. | ||
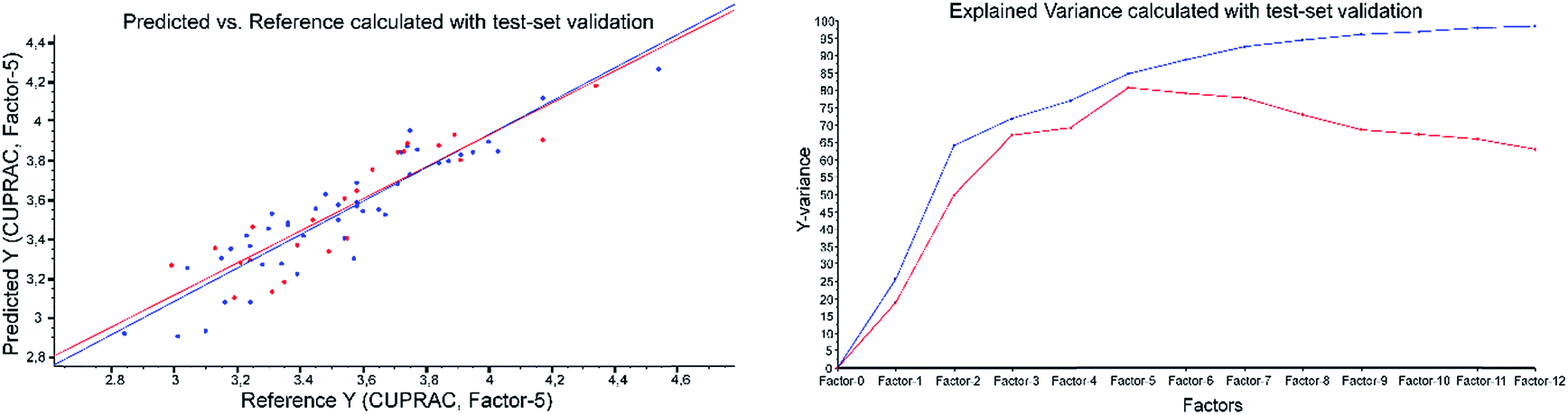 | ||
| Fig. 5 CUPRAC predicted vs. reference plot and explained variance plot calculated with ATR-IR spectra applying test-set validation. | ||
The quality parameters of the PLS models are listed in Table 3.
Example of the ATR-results with CUPRAC as the reference method: both validations were performed applying 2nd derivative Savitzky–Golay, polynomial order of three and a window size of 23 (Fig. 4 and Fig. 5).
3.2. NIR spectroscopy
The average NIR spectrum of the 68 samples is displayed in Fig. 2. The following main bands were assigned according to Workman and Weyer:31 6808 cm1 (O–H 1st overtone of phenols and water), 5784 cm−1 (C–H 1st stretching overtone of CH2), 5164 cm−1 (combination of O–H stretching and O–H deformation), 4728 cm1 to 4608 cm−1 (combination of C–H stretching and deformation and combination of O–H stretching and C–O deformation of carbohydrates), 4336 cm−1 (combination of C–H stretching and C–H deformation) and 4260 cm−1 (combination of C–H stretching and C–H deformation). In Table 4 just the cross-validation results are shown and the test-set validation gave miserable results.4 Discussion
As proven in previous publications, spectroscopic methods can be used to replace destructive and time consuming color assays14 and HPLC analysis as well.33,34 In this study, ATR-IR and NIR in hyphenation with multivariate data analysis were performed to develop and to compare fast methods to determine the anti-oxidant capacity of ground Sambuci flos samples.Looking at the different assays executed in this study, the Folin–Ciocalteu (FC) assay showed the best precision and accuracy among the used wet chemical methods as shown in Table 2. Another advantage of this assay was its simple preparation procedure.
| Inter-day precision of different concentrations | Intra-day precision of different concentrations | Accuracy | |||||
|---|---|---|---|---|---|---|---|
| Low | Middle | High | Low | Middle | High | ||
| FC | 0.72 | 0.73 | 0.60 | 0.51 | 0.41 | 0.38 | 99.27 |
| FRAP | 1.48 | 1.70 | 1.25 | 0.66 | 1.15 | 0.66 | 97.40 |
| CUPRAC | 1.37 | 1.12 | 0.96 | 0.59 | 0.65 | 0.38 | 98.68 |
| DPPH | 2.53 | 1.46 | 1.11 | 1.56 | 0.94 | 0.82 | 86.61 |
| ABTS | 3.01 | 2.72 | 2.48 | 2.06 | 1.74 | 1.40 | 95.90 |
However, the results of the ATR-IR PLS models did not show the best RPD and R2 values for the FC assay. The various models resulted in the following order with decreasing RPD and R2 values: CUPRAC > ABTS > FC > DPPH > FRAP. All RPD values of the test-set validation were a bit lower than the corresponding values in cross-validation, because the cross-validation includes all samples for validation, compared with the test-set validation the results of which depend on the selected validation samples (see Table 3).
| R 2 | SECV | SEC | SECV/SEC | LV | RPD | RPDcorr | RPDPrec | RBE | Range | PrecMean | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATR-IR cross-validation | |||||||||||
| FC | 0.84 | 0.15 | 0.11 | 1.38 | 6 | 2.54 | 2.62 | 10.16 | 3 | 4.1–6.0 | 0.68 |
| FRAP | 0.82 | 0.09 | 0.07 | 1.23 | 5 | 2.29 | 2.47 | 6.11 | 7 | 1.6–2.6 | 1.48 |
| CUPRAC | 0.85 | 0.13 | 0.10 | 1.25 | 5 | 2.56 | 2.68 | 8.72 | 4 | 2.8–4.6 | 1.15 |
| DPPH | 0.83 | 0.07 | 0.05 | 1.27 | 5 | 2.43 | 2.56 | 7.74 | 5 | 0.9–1.8 | 1.70 |
| ABTS | 0.84 | 0.04 | 0.04 | 1.24 | 5 | 2.47 | 3.85 | 3.23 | 36 | 0.9–1.5 | 2.73 |
| R 2 | SEP | SEC | SEP/SEC | LV | RPD | RPDcorr | RPDPrec | RBE | Range | PrecMean | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATR-IR test-set validation | |||||||||||
| FC | 0.84 | 0.17 | 0.14 | 1.25 | 6 | 2.27 | 2.32 | 10.25 | 2 | 4.1–6.1 | 0.68 |
| FRAP | 0.80 | 0.09 | 0.08 | 1.16 | 5 | 2.35 | 2.53 | 6.27 | 7 | 1.6–2.6 | 1.48 |
| CUPRAC | 0.81 | 0.15 | 0.13 | 1.14 | 5 | 2.12 | 2.20 | 8.29 | 3 | 2.8–4.6 | 1.15 |
| DPPH | 0.84 | 0.07 | 0.06 | 1.17 | 5 | 2.39 | 2.51 | 8.04 | 5 | 0.9–1.9 | 1.70 |
| ABTS | 0.80 | 0.05 | 0.04 | 1.04 | 5 | 2.24 | 3.42 | 2.97 | 34 | 0.9–1.5 | 2.73 |
The best results for the ATR-IR models were achieved with the 2nd derivative Savitzky–Golay. A polynomial order of three and a window size of 23 were used to perform the PLS models. Performing the FC and CUPRAC PLS models no sample was removed from the sample set. Compared to the other PLS models two, three and four samples were removed from the sample set of FRAP, DPPH and ABTS, respectively. Except for the PLS model of the FRAP assay, all developed modes showed a RPD value higher than 2.5 in cross-validation. In test-set validation only ABTS reached a good RDP value about 2.66. According to Williams et al. RPD values over 2.5 are acceptable.28 In both validation methods a R2 value higher than 0.8 was achieved. Five LVs were needed for almost all PLS models except for FC (LV = 6). As displayed in Fig. 3 wavelengths between 800 cm−1 and 1800 cm−1 were important to perform the models. The bands over 2800 cm−1 (O–H stretching of water, carbohydrates and flavonoids and C–H symmetric and asymmetric stretching) were not important to perform the models.
The NIR models performed really poor and the cross-validation results are depicted in Table 4. The number of LVs needed for all PLS models varied between five and seven. Only for the FC PLS model acceptable values for R2 and RPD were obtained, but the explained variance plot showed that the calculated model was of low quality because the first two LVs resulted in explained variances lower than 8%. A reason for the worse prediction power of NIR could be that the antioxidant power is carried mainly by two single antioxidative compounds (rutin and chlorogenic acid) in the plant material which belongs to two different structural classes. Former studies revealed a higher predictive power for the determination of single compounds with ATR-IR in plant matrices,33–36 which is advantageous in this application. In ATR-IR, fundamental vibrations are present exhibiting less overlap than the combination and overtone bands of NIR. This makes NIR more sensitive to matrix variations,35 which are usually severe in natural products. Also in earlier publications ATR-IR spectroscopy revealed better results than NIR spectroscopy for the quantification of single compounds in plant materials.33,34 Comparing the standard errors obtained from the studies of Navarro et al.,33 Schönbichler et al.,34 Krähmer et al.35 and Baranska et al.,36 which dealt with quantification of single compounds in plant materials, the NIR exhibited values with a factor 1.1–2.7 times higher than the ATR-IR in the cross-validation results. Even if there is evidence pointing to the advantages of ATR-IR in single compound prediction, it has not been reported until now that ATR-IR gives good results for an application and the NIR fails completely in the same application. The exact reason for this difference in performance of the two spectroscopic methods in this study could not be found. On top of that, our previous work comparing ATR-IR and NIR determining the antioxidative capacity of primulae flos powder revealed better results for NIR.14 The good results for NIR in the case of primulae flos can be explained by the contribution of mainly one class of substances to the antioxidative capacity. Here the broad bands of NIR seem to have an advantage for predicting a sum parameter of those substances. In the case of Sambuci flos mainly two substances contribute to the antioxidative capacity, and they belong to different structural classes. The authors assume that for NIR either the interference of chlorogenic acid with rutin or the interference of other matrix compounds causes problems in the PLS regression models. Probably ATR-IR can distinguish better between those two substances and matrix components.
| R 2 | SECV/SEC | LV | RPD | RPDcorr | RBE | Cumulative explained variance for the first LVs in % | |||
|---|---|---|---|---|---|---|---|---|---|
| 1st LV | 2nd LV | 3rd LV | |||||||
| FC | 0.88 | 1.30 | 7 | 2.93 | 3.06 | 4 | 2 | 5 | 60 |
| FRAP | 0.77 | 1.25 | 7 | 2.10 | 2.24 | 6 | 3 | 5 | 17 |
| CUPRAC | 0.76 | 1.33 | 7 | 2.09 | 2.15 | 3 | 2 | 7 | 30 |
| DPPH | 0.78 | 1.33 | 6 | 2.02 | 2.09 | 3 | 5 | 2 | 25 |
| ABTS | 0.79 | 1.20 | 5 | 1.92 | 2.34 | 18 | 5 | 15 | 40 |
5 Conclusion
This study demonstrates that ATR-IR spectroscopy in hyphenation with multivariate analysis can replace the currently used wet chemical methods (Folin–Ciocalteu, FRAP, CUPRAC, DPPH and ABTS) to determine the antioxidant capacity of Sambuci flos samples. NIR spectroscopy in combination with multivariate analysis showed results of poor quality. Employing HPLC-DAD-MS measurements the main compounds of two ethanol–water extracts were successfully analyzed and identified and the results correspond to literature data.32Acknowledgements
Special thanks are given to Bionorica SE (Prof. Popp, Dr Krolitzek; Neumarkt/Oberpfalz, Germany), the Ministry for Science and Research and the Ministry for Health, Family and Youth (Vienna, Austria) (Novel analytical tools for quality control of immunomodulatory, anti-inflammatory and neuroprotective agents in Traditional Chinese Medicine).References
- P. Sharma, A. B. Jha, R. S. Dubey and M. Pessarakli, Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions, Journal of Botany, 2012, 2012, 1–26 CrossRef
.
- B. Halliwell and O. I. Aruoma, DNA damage by oxygen - derived species, FEBS Lett., 1991, 281, 9–19 CrossRef CAS PubMed
- H. Wiseman and B. Halliwell, Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer, Biochem. J., 1996, 313, 17–29 CrossRef CAS PubMed
- S. Loft and H. E. Poulsen, Cancer risk and oxidative DNA damage in man, J. Mol. Med., 1996, 74, 297–312 CrossRef CAS PubMed
- S. Sen, R. Chakraborty, C. Sridhar, Y. S. R. Reddy and B. De, Free radicals, antioxidants, diseases and phytomedicines: current status and future prospect, Int. J. Pharm. Sci. Rev. Res., 2010, 3, 91–100 CAS
- G. Kite, S. Larsson, N. Veitch and E. A. Porter, Acyl spermidines in inflorescence extracts of elder (Sambucus nigra L., Adoxaceae) and elderflower drinks, J. Agric. Food Chem., 2013, 61, 3501–3508 CrossRef CAS PubMed
-
D. Charlebois, Elderberry as a Medicinal Plant. Issues new Crop. new uses, 2007, pp. 284–292 Search PubMed
-
World Health Organization, WHO Monographs on Selected Medicinal Plants, 2002, vol. 2. p. 1 Search PubMed
- L. C. Chiang, W. Chiang, M. Y. Chang, L. T. Ng and C. C. Lin, Antiviral activity of Plantago major extracts and related compounds in vitro, Antiviral Res., 2002, 55, 53–62 CrossRef CAS PubMed
- C. Hearst and G. McCollum, Antibacterial activity of elder (Sambucus nigra L.) flower or berry against hospital pathogens, J. Med. Plants Res., 2010, 4, 1805–1809 Search PubMed
- R. L. Prior, X. Wu and K. Schaich, Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements, J. Agric. Food Chem., 2005, 53, 4290–4302 CrossRef CAS PubMed
- D. Huang, B. Ou and R. L. Prior, The chemistry behind antioxidant capacity assays, J. Agric. Food Chem., 2005, 53, 1841–1856 CrossRef CAS PubMed
-
D. A. Skoog and J. J. Leary, Instrumentelle Analytik: Grundlagen - Geräte – Anwendungen, Springer, 1996 Search PubMed
- S. A. Schönbichler,
et al. Comparison of NIR and ATR-IR spectroscopy for the determination of the antioxidant capacity of Primulae flos cum calycibus, Anal. Methods, 2014, 6, 6343–6351 RSC
- O. Folin and V. Ciocalteu, On tyrosine and tryptophane determinations in proteins, J. Biol. Chem., 1927, 73, 627–650 CAS
- V. L. Singleton, R. Orthofer and R. M. Lamuela-RAventós, Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent, Methods Enzymol., 1999, 299, 152–178 CAS
- F. F. Benzie and J. J. Strain, The ferric reducing ability of plasma (FRAP) as a measure of ‘antioxidant power’: the FRAP assay, Anal. Biochem., 1996, 239, 70–76 CrossRef PubMed
- B. Ou, D. Huang, M. Hampsch-Woodill and J. Flanagan, a & Deemer, E. K. Analysis of antioxidant activities of common vegetables employing oxygen radical absorbance capacity (ORAC) and ferric reducing antioxidant power (FRAP) assays: a comparative study, J. Agric. Food Chem., 2002, 50, 3122–3128 CrossRef CAS PubMed
- R. Apak, K. Güçlü, M. Özyürek and S. E. Çelik, Mechanism of antioxidant capacity assays and the CUPRAC (cupric ion reducing antioxidant capacity) assay, Microchim. Acta, 2007, 160, 413–419 CrossRef
- R. Apak, K. Güçlü, M. Ozyürek and S. E. Karademir, Novel total antioxidant capacity index for dietary polyphenols and vitamins C and E, using their cupric ion reducing capability in the presence of neocuproine: CUPRAC method, J. Agric. Food Chem., 2004, 52, 7970–7981 CrossRef CAS PubMed
- W. Brand-Williams, M. E. Cuvelier and C. Berset, Use of a free radical method to evaluate antioxidant activity, LWT--Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS
- N. J. Miller, C. Rice-Evans, M. J. Davies, V. Gopinathan and A. Milner, A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates, Clin. Sci., 1993, 84, 407–412 CrossRef CAS PubMed
- R. Re, N. Pellegrini and A. Proteggente, Antioxidant activity applying an improved ABTS radical cation decolorization assay, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed
- A. Savitzky and M. J. E. Golay, Smoothing and differentiation of data by simplified least squares procedures, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS
- R. J. Barnes, M. S. Dhanoa and S. J. Lister, Standard normal variate transformation and de-trending of near-infrared diffuse reflectance spectra, Appl. Spectrosc., 1989, 43, 772–777 CrossRef CAS
- P. Geladi, D. MacDougall and H. Martens, Linearization and scatter-correction for near-infrared reflectance spectra of meat, Appl. Spectrosc., 1985, 39, 491–500 CrossRef
-
W. Kessler, Multivariate Datenanalyse: für die Pharma-, Bio- und Prozessanalytik, Wiley-VCH, 2006 Search PubMed
- P. C. Williams and D. Sobering, Comparison of commercial near infrared transmittance and reflectance instruments for analysis of whole grains and seeds, J. Near Infrared Spectrosc., 1993, 1, 25–32 CrossRef CAS
-
R. Kellner, J.-M. Mermet, M. Otto and E. Al, Analytical Chemistry: a Modern Approach to Analytical Science, Wiley-VCH, Weinheim, 2004 Search PubMed
-
E. Pretsch, P. Bühlmann and M. Badertscher, Structure Determination of Organic Compounds: Tables of Spectral Data, Springer, 2009 Search PubMed
-
J. Workman Jr and L. Weyer, Practical Guide to Interpretive Near-infrared Spectroscopy, CRC Press, 2007 Search PubMed
- M. Scopel, L. A. Mentz and A. T. Henriques, Comparative analysis of Sambucus nigra and Sambucus australis flowers: development and validation of an HPLC method for raw material quantification and preliminary stability study, Planta Med., 2010, 76, 1026–1031 CrossRef CAS PubMed
- M. Navarro Escamilla,
et al. Rapid determination of baicalin and total baicalein content in Scutellariae radix by ATR-IR and NIR spectroscopy, Talanta, 2013, 114, 304–310 CrossRef CAS PubMed
- S. A. Schönbichler,
et al. Simultaneous quantification of verbenalin and verbascoside in Verbena officinalis by ATR-IR and NIR spectroscopy, J. Pharm. Biomed. Anal., 2013, 84, 97–102 CrossRef PubMed
- A. Krähmer,
et al. Characterization and quantification of secondary metabolite profiles in leaves of red and white clover species by NIR and ATR-IR spectroscopy, Vib. Spectrosc., 2013, 68, 96–103 CrossRef
- M. Baranska, W. Schütze and H. Schulz, Determination of lycopene and β-carotene content in tomato fruits and related products: Comparison of FT-raman, ATR-IR, and NIR spectroscopy, Anal. Chem., 2006, 78, 8456–8461 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay01314c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |