
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas-phase microsolvation of ubiquitin: investigation of crown ether complexation sites using ion mobility-mass spectrometry†
Melanie
Göth
ab,
Frederik
Lermyte‡
c,
Xiao Jakob
Schmitt‡
b,
Stephan
Warnke
b,
Gert
von Helden
 b,
Frank
Sobott
cde and
Kevin
Pagel
*ab
b,
Frank
Sobott
cde and
Kevin
Pagel
*ab
aDepartment of Biology, Chemistry, Pharmacy, Freie Universität Berlin, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
bDepartment of Molecular Physics, Fritz Haber Institute of the Max Planck Society, 14195 Berlin, Germany
cBiomolecular and Analytical Mass Spectrometry, Chemistry Department, University of Antwerp, 2020 Antwerp, Belgium
dAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
eSchool of Molecular and Cellular Biology, University of Leeds, Leeds, LS2 9JT, UK
First published on 26th July 2016
Abstract
In this study the gas-phase structure of ubiquitin and its lysine-to-arginine mutants was investigated using ion mobility-mass spectrometry (IM-MS) and electron transfer dissociation-mass spectrometry (ETD-MS). Crown ether molecules were attached to positive charge sites of the proteins and the resulting non-covalent complexes were analysed. Collision induced dissociation (CID) experiments revealed relative energy differences between the wild type and the mutant crown-ether complexes. ETD-MS experiments were performed to identify the crown ether binding sites. Although not all of the binding sites could be revealed, the data confirm that the first crown ether is able to bind to the N-terminus. IM-MS experiments show a more compact structure for specific charge states of wild type ubiquitin when crown ethers are attached. However, data on ubiquitin mutants reveal that only specific lysine residues contribute to the effect of charge microsolvation. A compaction is only observed for one of the investigated mutants, in which the lysine has no proximate interaction partner. On the other hand when the lysine residues are involved in salt bridges, attachment of crown ethers has little effect on the structure.
Introduction
Electrospray ionization-mass spectrometry (ESI-MS) has developed into a powerful and indispensable analytical tool for the structural investigation of proteins. It is still fiercely debated to what extent the protein structure is retained after ESI and transfer into the gas phase and there are a growing number of publications addressing this question.1–3 In this context it is generally accepted that the charge state presents a major determinant for the three-dimensional organization of a protein:4 species with a low amount of charge adopt compact, presumably native-like structures, whereas species in higher charge states adopt more extended structures.5–7 Intermediate charge states can show many different coexisting conformations, ranging from compact to more extended structures. Furthermore, it was proposed that after transfer into the gas phase, intramolecular charge self-solvation, i.e. the collapse of the charged side chains onto the backbone, can disrupt the intramolecular hydrogen network.1A powerful tool to obtain information about the molecular structure in the gas phase is the combination of ion mobility spectrometry and mass spectrometry (IM-MS).8–10 Its potential in the analysis of proteins and their assemblies has been shown for various examples.11–13 In IM-MS, ions are guided by a weak electric field through a cell, which is filled with an inert buffer gas such as helium or nitrogen. During this drift, ions with a compact shape undergo fewer collisions with the buffer gas and therefore traverse the drift cell faster than the ions with more extended conformations. As a result, the investigated molecules are not only separated according to their mass and charge as in a conventional MS experiment, but also according to their size and shape. In addition, the drift time (td) of a particular ion can be further converted into a rotationally averaged collision cross section (CCS)14 that can also be determined theoretically and compared universally.
Ubiquitin is a small (76 residue) protein that plays an important role in signal transduction and post-translational modification of other proteins.15 As a result, it has been studied extensively in the gas phase by various groups.3,6,16–23
Here, we investigate the influence of side chain to backbone interactions on the overall gas-phase structure of ubiquitin. To do so, we non-covalently attach crown ether molecules (18-crown-6, 264 Da; inset in Fig. 1A) to the protein. Generally, 18-crown-6 is known to bind strongly to protonated lysines in peptides and proteins via three hydrogen bonds.24–26
 | ||
| Fig. 1 (A) ESI mass spectrum of wild type (wt) ubiquitin in charge state 5+ with different numbers of 18-crown-6 (264 Da) attached. The inset structure shows in a simplified form how the crown ether (CE) binds noncovalently to protonated side chains. (B) Arrival time distributions (ATDs) corresponding to (A) with the same colour code for the CE complexes. With an increasing number of CEs attached, the overall structure becomes more compact, which is reflected by a decrease in the CCS. | ||
A representative mass spectrum of ubiquitin and its crown-ether (CE) complexes in charge state 5+ is shown in Fig. 1A. Up to six 18C6 adducts are observed under mild ESI conditions, each with a mass shift of m/z 52.8 (264/5). The corresponding arrival time distributions (ATDs) are shown in Fig. 1B with the drift times converted to collision cross sections (CCSs). The general effect of the CE-attachment on the structure of ubiquitin can be described as follows: for ions with a low number of charges (z = 4) and ions in high charge states (z ≥ 8) each additional CE leads to an increase in size and mass (Fig. S1†), which in turn is reflected in an increase of the corresponding drift times and CCSs. Intermediate charge states (5+ to 7+ for ubiquitin), however, yield a rather counterintuitive result: by the addition of CEs, the conformational heterogeneity is reduced and the gas-phase structure becomes more compact. In charge states 6+ and 7+ this is reflected with CCSs being significantly smaller than those of the bare ions. In general, three major conformations seem to exist across the charge states shown in Fig. S1,† with compact (∼1000 Å2), intermediate (∼1200 Å2), and extended (∼1500 Å2) structures. The population of the bare 5+ ions is rather heterogeneous, showing a mix of the compact (i.e. presumably native-like) and intermediate states, and attachment of multiple CEs leads to the disappearance of the intermediate species. As such, the compact form becomes dominant when three or more CEs are coordinated (Fig. 1B). The bare 6+ ion (Fig. S1†) occurs predominantly in the extended state and adopts the intermediate conformation after binding a single crown ether. Binding of five crown ethers eventually results in the appearance of the compact state, but it never becomes dominant. As such, and because the 5+ ion is the lowest and most native-like charge state for which the compaction phenomenon is observed via IM-MS, we will focus the rest of our analysis on this ion. In a previous study on cytochrome c-CE complexes we proposed that the crown ether molecules take over the role of the solvent and microsolvate the protonated lysine residues, such that the overall structure remains compact.27 In the present work, this effect was investigated further by analyzing the CE-binding of ubiquitin in comparison to ubiquitin mutants using CID-MS, ETD-MS and IM-MS. In particular, we focused on the following questions: (1) does microsolvation have the same effect when certain lysine residues are replaced by arginine and, in this context, do all lysines play an equal role in this effect? (2) Can we identify the binding sites of the crown ether? (3) Does binding to the charged N-terminus lead to a similar microsolvation effect?
Experimental
Sample preparation
Wild type ubiquitin (human) was purchased from Boston Biochem (Cambridge, USA) as a colourless powder and dissolved in water to a concentration of 100 μM. The mutants were obtained from Sigma Aldrich (Taufkirchen, Germany) as phosphate-buffered saline (PBS) solutions. Prior to use, all samples were buffer exchanged in 10 mM aqueous triethylammonium acetate solution (pH = 7, Fluka Analytics) twice for 2 hours and overnight using Slide-A-Lyzer dialysis tubes (MWCO = 2000 Da, Thermo Scientific). After dialysis the samples were diluted to 10 μM with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) solution of water:methanol. 18-Crown-6 was purchased from Sigma Aldrich (Taufkirchen, Germany) as a colourless powder and dissolved with water to a 10 mM stock solution. For the preparation of the protein-crown ether complexes 25 to 50 equivalents of 18C6 were added to a 10 μM protein solution.
:1 (v:v) solution of water:methanol. 18-Crown-6 was purchased from Sigma Aldrich (Taufkirchen, Germany) as a colourless powder and dissolved with water to a 10 mM stock solution. For the preparation of the protein-crown ether complexes 25 to 50 equivalents of 18C6 were added to a 10 μM protein solution.
Mass spectrometry
Results and discussion
Crown-ether microsolvation of ubiquitin and ubiquitin mutants
To study the impact of microsolvation of individual lysine (K) residues, wild type (wt) ubiquitin and ubiquitin lysine-to-arginine mutants are investigated. As arginine is positively charged under physiological conditions and therefore comparable to lysine, it is assumed that the mutation does not have major effects on electrostatic interactions and the protein structure (Fig. S6†). Although arginine also binds 18-crown-6 molecules, it has a considerably lower affinity (133 kJ mol−1) than lysine (150 kJ mol−1) and previous studies further suggested that binding to lysine is favoured over arginine when both residues are present.26 In Fig. 2A the amino acid sequence of the wild-type protein and its crystal structure are shown with the seven lysine residues highlighted in green. Fig. 2B shows a schematic overview of the mutant sequences in comparison to wt ubiquitin with arginines coloured in dark blue and the retained lysines in green. In the mutants investigated here, either all lysine residues (noK-mutant) or all except one lysine at a specific position are replaced by arginine (K6-, K11-, K27-mutant). We specifically focus on mutants with lysines retained close to the N-terminus, which enables us to perform ETD experiments on ubiquitin-CE complexes in which lysine-containing N-terminal fragments (possibly with a bound CE) are produced at high abundance. Inspection of the individual residues in the crystal structure (PDB accession code: 1UBQ) for a native-like structure shows that K11 and K27 are both involved in salt bridges, whereas K6 is not in close proximity to an acidic side chain.29 | ||
| Fig. 2 (A) Crystal structure (PDB: 1UBQ) and amino acid sequence of wt ubiquitin with its seven lysine residues highlighted in green. While K11 and K27 are involved in salt bridges, K6 has no interaction partner in close proximity. (B) Schematic amino acid sequence comparing wt to the mutants noK, K6, K11 and K27. Either all lysines (green) or all but one specific lysine are replaced by arginine (blue). | ||
Under soft ESI conditions up to six CE adducts are observed for wt ubiquitin and up to five for the mutants, with all spectra showing a similar distribution of the complexes (Fig. S2†). However, under slightly harsher conditions (increase of trap voltage from 2 V to 9 V), the distribution of the CE complexes changes significantly for the mutants, whereas the change is less obvious for the wt protein with a maximum intensity at the 2CE complex (Fig. 3A). The most intense shift between soft and harsh conditions is observed for the noK-mutant (B), where the bare protein ion and the 1CE complex show the highest intensity with an almost 1:1 ratio. The distributions of K6-, K11- and K27-mutant complexes (Fig. 3C, D and E) resemble each other with the 1CE complex as the most abundant species.
 | ||
| Fig. 3 Comparison of the relative peak intensities of the protein crown-ether (CE) complexes under harsh conditions (increase of trap voltage) at charge state 5+. | ||
Energy-resolved collision induced dissociation experiments
In order to better understand the CE-distributions in Fig. 3 and to analyze differences in CE-binding energies between the mutants, energy-resolved CID experiments were performed. To do so, either the 1CE or the 3CE complex was isolated as the precursor and subsequently fragmented via CID. The selection of the 3CE complex is based on the assumption that the third CE is neither attached to a lysine nor to the N-terminus in the case of the mutants. Representative MS/MS spectra, with the respective precursor ions shaded in grey, are shown in Fig. 4A and B for the noK-mutant at a collision voltage of 9 V. Spectra for the other investigated mutants are shown in Fig. S3.† Isolation and subsequent dissociation of the 1CE complex result in the formation of the bare protein ion; dissociation of the 3CE complex, on the other hand, leads to the formation of three different product ions at an increased collision voltage. To obtain a more quantitative picture for the different mutants, the relative intensity of the corresponding precursor ions was plotted versus the collision voltages for all samples. The resulting CID depletion curves are of a typical sigmoidal shape (Fig. 4C and D). In addition, the voltage that is necessary for a 50% depletion of the corresponding precursor ion is marked by a dashed line in both graphs. | ||
| Fig. 4 CID spectra of noK CE complexes with (A) 1CE and (B) 3CE as precursor ions (shaded in grey) at a trap collision voltage of 9 V. For noK the bare protein ion is already observed at low collision voltages. Depletion curves of (C) 1CE complexes and (D) 3CE complexes of all proteins. Collision voltages are plotted versus the relative abundance for the decrease of the corresponding CE complex. | ||
Comparing the graphs of the 1CE depletion amongst each other, three distinct trends become apparent: (1) the noK-species needs the lowest energy to dissociate. This was expected, as there are no lysine residues for the CE to coordinate to. (2) Slightly more energy is required for a 50% fragmentation of the K6-, K11- and K27-mutant complexes. (3) The highest energy has to be applied to dissociate the wt–1CE complex. These results show that a distinction between wt and the mutants and even between the two types of mutants is generally possible based on the relative dissociation energy. The resulting gap in the effective binding energy between wt and the other three mutants is somewhat unexpected, as the wt as well as the mutants offer similar binding sites to the CE. In the case of the wt the CE coordinates either to the N-terminus or to one of the seven lysine residues, which are believed to be in close spatial proximity in charge state 5+. Three considerations could explain the observed stability of the wt–1CE complex: (1) the CE-binding sites are statistically distributed. Thus, one particular site might be favored, but the CE can in principle be attached to other sites as well. These other sites would still be lysines in the case of the wt and likely arginines in the mutants. (2) The first CE is neither attached to K6, K11 or K27 nor to the N-terminus, but to one of the four remaining lysines that are not investigated in this study, and which somehow has a much greater affinity than the three we focused on. (3) The CE is not permanently bound to one specific site, but can “walk” between them when they are close enough to each other. This phenomenon was reported earlier30,31 and might offer an explanation for the increase in the effective binding strength of the wt.
Next, the 3CE complex of each variant was isolated, fragmented and its depletion plotted as described above (Fig. 4D). The collision voltage for a 50% fragmentation is roughly similar for all mutant species and in addition significantly lower than the energy necessary to dissociate the wt complex. In the case of the wt, the third CE is likely coordinated to a lysine, whereas the mutants presumably offer arginine as a binding site. Furthermore, a comparison of the wt depletion in Fig. 4C and D shows that the third CE is bound significantly weaker than the first CE. This might be attributed to the fact that the 1CE and 3CE complexes of the wt exhibit different structures as shown in Fig. 1. Alternatively, due to the two already occupied sites the third CE may also not be able to “walk” between different residues as freely as the first CE and is therefore not bound similarly strong. Nevertheless, the CID experiments and the resulting depletion graphs show that it is possible to differentiate between a coordination of the CE to arginine and lysine on the basis of the relative dissociation energy.
Electron transfer dissociation experiments
MS and CID experiments provide insights into the relative binding energies of the investigated protein–CE complexes and thus allow conclusions on the CE-binding sites. However, these experiments do not reveal the role of the N-terminus and to what extent it is involved in the CE-microsolvation.To answer this question, electron transfer dissociation (ETD) experiments were performed. This method provides the advantage of preserving non-covalent interactions while cleaving covalent bonds.32–34 Thus, fragments of different sizes can be formed with a CE still being attached to the corresponding lysine. It should therefore be possible to identify the binding sites from the ETD fragment mass spectra. To minimize loss and/or migration of the CE ligand in the gas phase, acceleration voltages were kept minimal in ETD experiments. While this limited the fragmentation efficiency and sequence coverage, fragmentation in the first 5 to 15 residues (depending on the precursor charge state) yields insight into CE binding and compaction in wt ubiquitin, as well as the K6, K11 and noK-mutants.
Protein–CE complexes with different numbers of CEs were selected as precursor ions in charge state 5+. Generally, a high sequence coverage and S/N ratio can be obtained in ETD for precursor ions with a high number of charges.35 The reason for this is twofold: first, the increased Coulomb attraction between the ETD reagent and precursor will result in an increased ion/ion reaction rate.36 Second, increased intramolecular electrostatic repulsion will reduce the degree to which the protein structure is stabilized by noncovalent interactions, facilitating fragment release after cleavage of the backbone N–Cα bond.37
In this case, sufficiently resolved fragment spectra were also obtained for the lowly charged 5+ ions investigated in this study and the resulting mass spectra of all complexes show a vast variety of fragments (Fig. S4†). In addition to shorter N-terminal fragments, charge-reduced species are also observed. Those species are formed either by proton or electron transfer, or gas-phase adduct formation with the ETD reagent as reported earlier.38,39,43Fig. 5 shows the lower m/z region of the ETD fragment spectra of the wt, K6, K11 and noK complexes for a 5+ precursor ion with two CEs. The colour code for the different number of CEs attached is the same as in Fig. 1. In general, a- and c-type fragments with no CE (yellow), one CE (red) and two CEs (blue) are observed. Despite the rather low fragmentation yield of the lowly charged precursor ions, a few intense N-terminal fragments (c1+, c2+ and c3+) carrying one CE are observed for all four proteins. As these fragments contain neither a lysine nor an arginine residue, they indicate binding of the CE to the protonated N-terminus. In addition, for the wt complex an a6+ fragment was identified with a single CE bound (i.e. either attached to the N-terminus or to K6). However, no further fragments with more than one CE were formed for wt 5+ ions. In charge state 6+, a higher fragmentation yield was achieved, where nearly all the fragments carried one or two CEs (Fig. S5†). These spectra show sequences with up to 17 residues and indicate that the second CE binds preferentially to K11, but can also bind to K6.
 | ||
| Fig. 5 ETD fragmentation spectra of the 5+ complex with two crown ethers attached. (A) Wild-type ubiquitin, (B) K6-mutant, (C) K11-mutant, and (D) noK-mutant. | ||
In turn, fragment spectra of the K6-mutant complex (Fig. 5B) show a slightly better sequence coverage for charge state 5+. In addition to clear N-terminal binding (c1+, c2+, c3+, c4+, and c5+), larger fragments (a6+, c6+, c7+, c8+, and c9+) are formed with one CE attached (i.e. either to the N-terminus or to K6). Furthermore, the occurrence of the fragments c92+, c102+ and c172+ with two CEs, suggests binding of the second CE to lysine at position six.
For the K11-mutant, ETD experiments were also performed under similar conditions (Fig. 5C). However, at charge state 5+ only fragments up to a length of nine amino acids with one or no CE attached are formed (Fig. 5C). These fragments suggest that the first CE is likely to bind to the N-terminus. Since only fragments up to c9 were observed, it is not possible to reveal further binding sites. However, based on known affinities and ETD of the K6-mutant, the second CE likely coordinates to K11. Given the limited sequence coverage observed under these soft conditions, ETD experiments were not performed with the K27-mutant.
ETD spectra of the noK-mutant complexes (Fig. 5D) show in addition to the intense N-terminal fragments with one CE also the fragments with two CEs attached (c62+, c72+ and c102+). These short sequences contain the N-terminus and one arginine residue at position 6 as possible coordination sites for the two CEs. Furthermore, a larger fragment is observed with c172+. In this case the single attached CE is able to bind to the N-terminus, R6 or R11.
From the ETD experiments, we conclude that N-terminal CE-binding likely occurs for all investigated species, as the corresponding fragments were among the most intense signals in the fragment spectra. These fragments indicate that it is the first CE that binds to the N-terminus. However, there are also fragments observed where it is not explicitly clear whether the first CE is coordinated to the N-terminus or to K6. In general, the low sequence coverage in charge state 5+ hinders a clear identification of every CE-binding site.
Ion mobility-mass spectrometry experiments
The major focus of this study was to test whether the effect of microsolvation can still be observed with one or several lysine residues being replaced by arginine and, thus, to reveal if all lysines contribute equally to this effect or if specific residues play a major role. To clarify this question, the protein–CE complexes were investigated further using ion mobility-mass spectrometry. The measured arrival time distributions (ATDs) for the proteins and their CE complexes were converted to contour plots, in which the colour intensity reflects the peak intensity of the raw data.The corresponding contour plot for wt ubiquitin is shown in Fig. 6A and reveals a similar trend for a microsolvation-induced compaction as discussed above. Experiments under the same conditions were performed with the mutant–CE complexes and the resulting ATDs are presented as contour plots in Fig. 6B. Raw IM-MS data of all possible lysine-to-arginine mutants (charge state 5+) are shown in Fig. S6.† Generally, a different behavior is observed for all mutants compared to the wt. The ATD of the bare noK mutant appears much narrower than that of the wt and reveals a compact conformation. Further compaction does not occur upon the addition of crown ethers. Similar behavior is observed for the K11- and K27-CE complexes (and K29-, K33, K48-, and K63-CE complexes, Fig. S6†). In all these cases, the drift time increases steadily with the growing mass of the complex, and shows a rather narrow and homogenous ATD, corresponding to a compact conformation. The K6-mutant, on the other hand, reveals the same conformational heterogeneity as the wt in the absence of CEs. More importantly, however, K6 shows a similar effect as the wt sequence when CE molecules are bound, and requires fewer ligands to undergo this effect. The second, more compact conformational family evolves with the first attached CE and dominates after addition of the second CE molecule. As a result, K6 requires at least two (compared to three for the wt) coordinated CEs to undergo a compaction of the structure. Therefore it is likely that both the N-terminus and K6 are involved in binding and that K6 plays a key role in extending the structure in ubiquitin. This assumption is supported by the ETD experiments, as both N-terminal and K6 CE-binding are observed with the first two CEs.
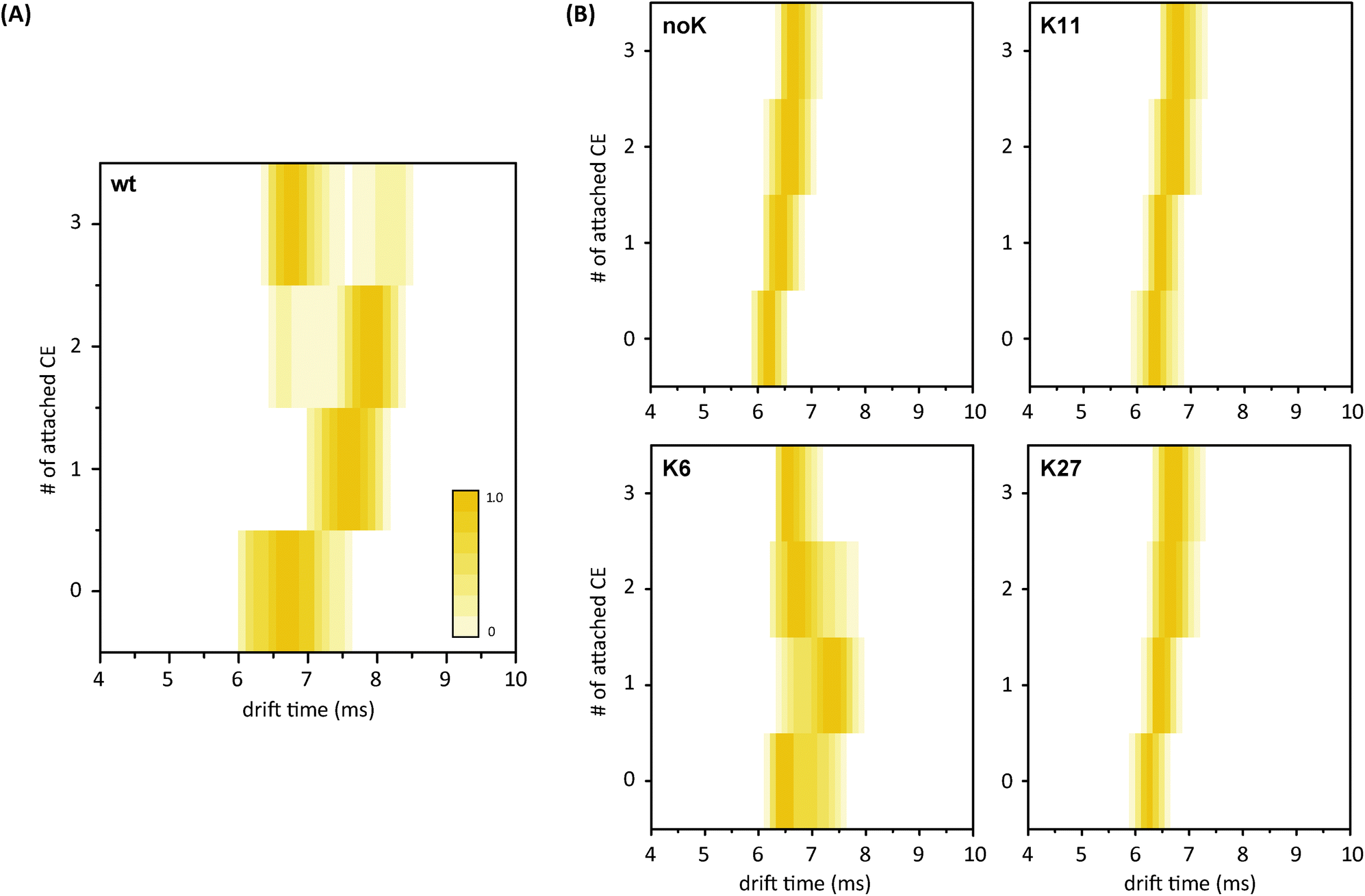 | ||
| Fig. 6 (A) Arrival time distributions (ATDs) for wt ubiquitin with 0 to 3 CEs (5+) presented as contour plots. Upon attachment of CE molecules, the gas-phase structure increases at first, but a second, more compact species evolves as well; this conformation dominates at higher numbers of crown ethers. (B) Contour plots for the ATDs of the mutant complexes. Only the K6-mutant shows the same conformational heterogeneity and compaction upon CE addition as the wt. The other mutant variants exhibit homogeneous and compact structures already without CE attachment. | ||
Comparison of the microenvironment of the particular lysine residues in the crystal structure of the protein (Fig. 2) reveals that K6 is the only one of the investigated residues that is not involved in an intramolecular salt bridge. As the next potential binding partner for K6 is approximately 5.4 Å away,29 the charged residue is more likely to self-solvate onto the protein backbone after transfer into the gas phase. This, in turn, leads to a disruption of structure-stabilizing intramolecular hydrogen bonds and, eventually, to the unfolding of the protein's compact conformation. Salt bridges, on the other hand, are likely to support the compact and native-like structure of the 5+ ubiquitin ions. Previous DFT calculations on the binding of 18-crown-6 to different possible conformations of protonated lysine revealed that in structures where lysine is involved in a salt bridge, the crown ether was likely to prefer binding to the corresponding N-terminus rather than the side chain in order to retain the stabilization by the salt bridge.26 Therefore, it is likely that lysine residues that take part in such structure-stabilizing interactions do not show a strong effect upon microsolvation with a CE. This agrees well with another study where CE complexes with ubiquitin lysine-to-asparagine mutants were analysed via selective noncovalent adduct protein probing mass spectrometry (SNAPP-MS).40–42 Here, the authors investigate the changing adduct distribution in mass spectra for mutants with only one lysine replaced in the sequence and reveal that intramolecular interactions, such as salt bridges and hydrogen bonds, can hamper the coordination of 18C6 to protonated lysine residues.29
Conclusions
In the present work we investigate the influence of a protein's microenvironment on its gas-phase structure by non-covalent attachment of crown ether molecules to ubiquitin and ubiquitin lysine-to-arginine mutants. We demonstrate that the structure of partially folded conformations of ubiquitin becomes more compact upon crown-ether attachment, which is in good agreement with our previous work on cytochrome c.27 Our results strongly indicate that crown ether molecules coordinate not only to protonated lysine and arginine residues, but also to the N-terminus. Applying harsher conditions in the MS and CID experiments showed that the single crown-ether complexes differ in relative dissociation energies, which enables the differentiation between a coordination of the crown ether to lysine or arginine. Furthermore, differences in the dissociation energies of the respective complexes are not significant between the mutants of the same type (K6, K11 and K27-mutant). ETD-MS experiments were performed to locate the crown-ether binding sites. Although a clear identification of all binding sites is not possible, the data reveal that the N-terminus is involved in the CE-microsolvation, and in some cases even by binding the first CE. When comparing the arrival time distributions of the different mutants, the major differences in the overall gas-phase structure are observed upon attachment of CEs: only with the K6-mutant a structural rearrangement similar to wild type ubiquitin is detected. All other mutants exhibit compact and homogeneous ATDs that increase with drift time without a noticeable structural change. In other words, the lysine residue on position 6 induces a structural extension and shows a compaction upon CE addition, whereas salt-bridged residues adopt a compact structure already without the CEs attached. We here assume that the formation and preservation of salt bridges to spatially adjacent residues are more favourable than backbone solvation. Altogether, our results strongly indicate that the effect of crown-ether microsolvation is dependent on the microenvironment of the specific residue.Acknowledgements
We thank Dr Kathrin Breuker for the inspiring discussions on this project. M. G. thanks Bayer HealthCare Pharmaceuticals for funding a PhD fellowship. F. L. thanks the Research Foundation – Flanders (FWO) for funding a PhD fellowship. The Synapt G2 instrument is funded by a grant from the Hercules Foundation – Flanders.Notes and references
- K. Breuker and F. W. McLafferty, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18145–18152 CrossRef CAS PubMed.
- S. Myung, E. R. Badman, Y. J. Lee and D. E. Clemmer, J. Phys. Chem. A, 2002, 106, 9976–9982 CrossRef CAS.
- E. Segev, T. Wyttenbach, M. T. Bowers and R. B. Gerber, Phys. Chem. Chem. Phys., 2008, 10, 3077–3082 RSC.
- Z. Hall and C. Robinson, J. Am. Soc. Mass Spectrom., 2012, 23, 1161–1168 CrossRef CAS PubMed.
- Y. Mao, M. A. Ratner and M. F. Jarrold, J. Phys. Chem. B, 1999, 103, 10017–10021 CrossRef CAS.
- H. Shi, N. A. Pierson, S. J. Valentine and D. E. Clemmer, J. Phys. Chem. B, 2012, 116, 3344–3352 CrossRef CAS PubMed.
- A. I. González Flórez, E. Mucha, D.-S. Ahn, S. Gewinner, W. Schöllkopf, K. Pagel and G. von Helden, Angew. Chem., Int. Ed., 2016, 55, 3295–3299 CrossRef PubMed.
- B. C. Bohrer, S. I. Merenbloom, S. L. Koeniger, A. E. Hilderbrand and D. E. Clemmer, Annu. Rev. Anal. Chem., 2008, 1, 293–327 CrossRef CAS PubMed.
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. R. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC.
- T. Wyttenbach and M. Bowers, in Modern Mass Spectrometry, ed. C. Schalley, Springer, Berlin, Heidelberg, 2003, vol. 225, ch. 6, pp. 207–232 Search PubMed.
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Science, 2005, 310, 1658–1661 CrossRef CAS PubMed.
- C. Bleiholder, N. F. Dupuis, T. Wyttenbach and M. T. Bowers, Nat. Chem., 2011, 3, 172–177 CrossRef CAS PubMed.
- K. Pagel, E. Natan, Z. Hall, A. R. Fersht and C. V. Robinson, Angew. Chem., Int. Ed., 2013, 52, 361–365 CrossRef CAS PubMed.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS PubMed.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425–479 CrossRef CAS PubMed.
- K. Breuker, H. Oh, D. M. Horn, B. A. Cerda and F. W. McLafferty, J. Am. Chem. Soc., 2002, 124, 6407–6420 CrossRef CAS PubMed.
- S. L. Koeniger, S. I. Merenbloom and D. E. Clemmer, J. Phys. Chem. B, 2006, 110, 7017–7021 CrossRef CAS PubMed.
- S. L. Koeniger and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2007, 18, 322–331 CrossRef CAS PubMed.
- T. Wyttenbach and M. T. Bowers, J. Phys. Chem. B, 2011, 115, 12266–12275 CrossRef CAS PubMed.
- O. S. Skinner, F. W. McLafferty and K. Breuker, J. Am. Soc. Mass Spectrom., 2012, 23, 1011–1014 CrossRef CAS PubMed.
- J. W. Lee, S. W. Heo, S. J. C. Lee, J. Y. Ko, H. Kim and H. I. Kim, J. Am. Soc. Mass Spectrom., 2013, 24, 21–29 CrossRef CAS PubMed.
- S. Warnke, C. Baldauf, M. T. Bowers, K. Pagel and G. von Helden, J. Am. Chem. Soc., 2014, 136, 10308–10314 CrossRef CAS PubMed.
- K. A. Servage, J. A. Silveira, K. L. Fort, D. E. Clemmer and D. H. Russell, J. Phys. Chem. Lett., 2015, 6, 4947–4951 CrossRef CAS PubMed.
- S. Maleknia and J. Brodbelt, J. Am. Chem. Soc., 1993, 115, 2837–2843 CrossRef CAS.
- C. C. Liou, H. F. Wu and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 1994, 5, 260–273 CrossRef CAS PubMed.
- R. R. Julian and J. L. Beauchamp, Int. J. Mass Spectrom., 2001, 210–211, 613–623 CrossRef.
- S. Warnke, G. von Helden and K. Pagel, J. Am. Chem. Soc., 2013, 135, 1177–1180 CrossRef CAS PubMed.
- P. R. Kemper, N. F. Dupuis and M. T. Bowers, Int. J. Mass Spectrom., 2009, 287, 46–57 CrossRef CAS.
- Z. Liu, S. Cheng, D. R. Gallie and R. R. Julian, Anal. Chem., 2008, 80, 3846–3852 CrossRef CAS PubMed.
- D. P. Weimann, H. D. F. Winkler, J. A. Falenski, B. Koksch and C. A. Schalley, Nat. Chem., 2009, 1, 573–577 CrossRef CAS PubMed.
- H. D. F. Winkler, D. P. Weimann, A. Springer and C. A. Schalley, Angew. Chem., Int. Ed., 2009, 48, 7246–7250 CrossRef CAS PubMed.
- S. N. Jackson, S. Dutta and A. S. Woods, J. Am. Soc. Mass Spectrom., 2011, 20, 176–179 CrossRef PubMed.
- L. Muller, S. N. Jackson and A. S. Woods, RSC Adv., 2014, 4, 42272–42277 RSC.
- F. Lermyte and F. Sobott, Proteomics, 2015, 15, 2813–2822 CrossRef CAS PubMed.
- D. M. Good, M. Wirtala, G. C. McAlister and J. J. Coon, Mol. Cell. Proteomics, 2007, 6, 1942–1951 CAS.
- S. A. McLuckey and J. L. Stephenson, Mass Spectrom. Rev., 1998, 17, 369–407 CrossRef CAS PubMed.
- S. J. Pitteri, P. A. Chrisman, J. M. Hogan and S. A. McLuckey, Anal. Chem., 2005, 77, 1831–1839 CrossRef CAS PubMed.
- S. J. Pitteri and S. A. McLuckey, Mass Spectrom. Rev., 2005, 24, 931–958 CrossRef CAS PubMed.
- F. Lermyte, J. P. Williams, J. M. Brown, E. M. Martin and F. Sobott, J. Am. Soc. Mass Spectrom., 2015, 26, 1068–1076 CrossRef CAS PubMed.
- T. Ly and R. R. Julian, J. Am. Soc. Mass Spectrom., 2006, 17, 1209–1215 CrossRef CAS PubMed.
- T. Ly and R. R. Julian, J. Am. Soc. Mass Spectrom., 2008, 19, 1663–1672 CrossRef CAS PubMed.
- B. N. Moore, O. Hamdy and R. R. Julian, Int. J. Mass Spectrom., 2012, 330–332, 220–225 CrossRef CAS PubMed.
- F. Lermyte, J. P. Williams, J. M. Brown, E. M. Martin and F. Sobott, J. Am. Soc. Mass Spectrom., 2015, 26, 1068–1076 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures. See DOI: 10.1039/c6an01377e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |