
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Portable Sequentially Shifted Excitation Raman spectroscopy as an innovative tool for in situ chemical interrogation of painted surfaces
Claudia
Conti
*a,
Alessandra
Botteon
a,
Moira
Bertasa
ab,
Chiara
Colombo
a,
Marco
Realini
a and
Diego
Sali
*c
aInstitute for the Conservation and Valorization of Cultural Heritage (ICVBC), National Research Council, Via Cozzi 53, 20125, Milano, Italy. E-mail: c.conti@icvbc.cnr.it
bDepartment of Chemistry, University of Turin, Via Pietro Giuria 7, 10125, Torino, Italy
cBruker Italia S.r.l. Unipersonale, V. le V. Lancetti 43, 20158, Milano, Italy. E-mail: diego.sali@bruker.com
First published on 8th June 2016
Abstract
We present the first validation and application of portable Sequentially Shifted Excitation (SSE) Raman spectroscopy for the survey of painted layers in art. The method enables the acquisition of shifted Raman spectra and the recovery of the spectral data through the application of a suitable reconstruction algorithm. The technique has a great potentiality in art where commonly a strong fluorescence obscures the Raman signal of the target, especially when conventional portable Raman spectrometers are used for in situ analyses. Firstly, the analytical capability of portable SSE Raman spectroscopy is critically discussed using reference materials and laboratory specimens, comparing its results with other conventional high performance laboratory instruments (benchtop FT-Raman and dispersive Raman spectrometers with an external fiber optic probe); secondly, it is applied directly in situ to study the complex polychromy of Italian prestigious terracotta sculptures of the 16th century. Portable SSE Raman spectroscopy represents a new investigation modality in art, expanding the portfolio of non-invasive, chemically specific analytical tools.
Introduction
Non-invasive analysis of painted surfaces is an analytical challenge commonly present in a wide range of disciplines. The chemical information is crucially important, for example, in the conservation field to develop a deeper understanding of the artist's technique, artwork history and to supply the background for the suitable conservation procedures. The key goal in all these applications is to avoid the sampling which is either highly undesirable or not permissible due to the uniqueness and high cultural value of the object analysed. Portable Raman spectroscopy is one of the advanced analytical techniques currently used for in situ non-invasive analyses, together with portable reflectance FT-IR and handheld XRF spectrometers.1–4 The quick and relatively straightforward molecular recognition of inorganic and organic compounds provides high potentiality to Raman spectroscopy. However, conventional portable Raman spectrometers suffer from critical limitations in the analysis of art samples due to the following main reasons:(a) low Raman signal sensitivity; (b) short spectral range availability; (c) high impact of fluorescence, which often completely overwhelms the Raman signal, especially the less intense peaks; (d) interferences from ambient light and environmental conditions in general, hard to control. Moreover, in the presence of scaffolding, positioning and stability are seriously compromised.
Hence the use of portable Raman spectrometers for in situ art applications is challenging and in many cases the acquired information is limited just to the identification of high Raman scattering cross section compounds. Noticeably different is the laboratory approach: when the samples are available for laboratory analysis, fluorescence can be typically overcome using a near IR 1064 nm excitation wavelength, since the fluorescence effect decreases with the increasing of the excitation laser wavelength (later called λ0). All the conventional benchtop spectrometers equipped with a 1064 nm excitation source use FT technology,5 rather than the dispersive one, due to the need to have the maximum possible sensitivity in that spectral range (namely an absolute range approximately between 6000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1) without the use of really high power lasers. It is worth noting that the standard multiplex detectors as the silicon based CCDs (mainly used in commercial dispersive Raman spectrometers), have a poor efficiency in the spectral range that is Raman shifted relative to 1064 nm, and to detect this radiation with enough sensitivity it is necessary to use different kinds of semiconductors such as InGaAs or germanium.
000 cm−1) without the use of really high power lasers. It is worth noting that the standard multiplex detectors as the silicon based CCDs (mainly used in commercial dispersive Raman spectrometers), have a poor efficiency in the spectral range that is Raman shifted relative to 1064 nm, and to detect this radiation with enough sensitivity it is necessary to use different kinds of semiconductors such as InGaAs or germanium.
In other situations, when the sensitivity is the main issue and fluorescence can be kept under control, the conventional dispersive Raman technology should be applied (since the Raman sensitivity is inversely proportional to λ04), selecting a suitable laser wavelength (typically 785 nm and 532 nm).6
With the availability of the mentioned benchtop instruments it is often possible to achieve straightforward Raman information to be used for identification.
At present, no FT-Raman portable spectrometers equipped with a 1064 nm laser are available on the market due to technical issues, but only dispersive systems, where the low sensitivity is a really big issue. To increase the sensitivity, high power lasers are typically used for the commercial portable Raman spectrometers (up to 500 mW). However, the application of such a laser power at that wavelength is often impossible, due to the thermal effects and transferred heating which could lead to damaging or burning of the surface.
An alternative and relatively recent method to mitigate the fluorescence with benchtop instruments is Shifted Excitation Raman Difference Spectroscopy (SERDS). In simplistic terms, this method is based on the changing of the excitation laser wavelength during Raman spectral acquisition;7,8 the location of Raman intensities in spectral space changes with the excitation wavelength, while unwanted spectral intensities corresponding to fluorescence, stray light, fixed pattern detector noise, etc., remain unchanged in spectral space. This difference allows extracting the Raman spectrum separated from the fluorescence spectrum. A second similar method is Subtracted Shifted Raman Spectroscopy (SSRS), where the shifted spectrum was obtained by moving the spectrometer grating from its initially calibrated position to the shifted value by monitoring the position through the Raman lines of a standard compound (i.e. polystyrene).8–10
Despite the good results achieved with these methods (also used for the analysis of organic dyes, colorants and lakes8,9) there is significant difficulty in reconstructing the Raman spectra from the derivative data. In addition, the instrumentation can be large and expensive, thus limiting some of the main advantages of dispersive Raman spectroscopy.
After that, a new promising approach was proposed, Sequentially Shifted Excitation (SSE), where the diode lasers operate at different temperatures, providing slightly shifted wavelengths; a suitable algorithm allows extracting the spectral data in true Raman space.11–13 This technical solution has already been extensively tested for typical pharmaceutical applications (raw material identification, liquid or powders) and nowadays it is well accepted as a dedicated analyzer.11,14
In the present study, the SSE concept is tested in a new generation of portable Raman spectrometers and for the first time it is applied to the non-invasive in situ analysis of an artwork painted surface; the aim is to explore its performance outside the pharmaceutical field, where samples are typically neither powders nor liquids as in artworks. First, its analytical capability is critically discussed using laboratory specimens and comparing its performance with other conventional high performance laboratory instruments (benchtop FT-Raman and dispersive Raman spectrometers with an external fiber optic probe). Secondly, it is used directly in situ to study the complex polychromy of Italian prestigious terracotta sculptures of the 16th century.
Experimental
Analytical methods
The average specified spectral resolution is about 11 cm−1 and the applied laser power is always less than 100 mW for both lasers. When used directly for in situ analysis, BRAVO is adapted on a standard tripod (less than 1 mm of working distance). The spectra were acquired with acquisition time ranging from 500 ms to 2 s and accumulations ranging from 5 to 300. For all the measurements, pSSE-RS has been used with external notebook and OPUS™ software (Version 7.7), in order to manually select the appropriate integration time and number of coadditions.
The method validation was carried out comparing BRAVO data with those of FT and dispersive high performance benchtop spectrometers:
Spectra acquired with pSSE-RS have been used as generated by the spectrometer, without any additional manipulations to the data processing steps necessary to convert the shifted excitation spectra into a resultant spectrum (described in the mentioned patent). FOP-RS and FT-RS data have been baseline corrected using an additional concave rubberband correction via OPUS™ algorithm, with an appropriate number of iterations.
Materials
This comparative study was also used for a critical and accurate interpretation of the pSSE-RS performance when used directly in situ. The following representative test samples were selected for validation purposes:
1. chocolate and madder lake pigments were used to investigate the pSSE-RS potentiality in the case of fluorescent samples. Chocolate is commonly used to demonstrate the FT-RS capability on fluorescence suppression. The identification of madder lakes (or organic dyes in general) represents one of the most analytical challenges in conservation science due to its low concentration in painted layers and high fluorescence, even using NIR excitation sources; commonly, the Raman spectrum of madder lakes is recovered by surface enhancement Raman spectroscopy.15 Here we tested a commercial milk chocolate and a madder lake purchased by Kremer (natural red 9).
2. Beeswax and polystyrene, two good CH scattering samples, were used to evaluate the general performance at the CH stretching spectral range (i.e. around 2900 cm−1), an unusual range for portable and handheld Raman spectrometers.
3. The same polystyrene sample was used to calculate the average sensitivity and the signal to noise ratio: the polystyrene sample is commonly adopted as the reference sample from each laboratory Raman spectrometers (FT and dispersive one) to measure and specify the throughput or signal to noise ratio.
4. A red modern pigment in a mixture with an acrylic binder (Naphthol Crimson Red 292 Liquitex Heavy Body PR170) was used as the test sample to estimate wavenumber accuracy and stability on the whole available spectral range; a comparative study among the band positions of the spectra acquired with pSSE-RS, FOP-RS and FT-RS was carried out.
Here we report a selection of the most significant measurements (see Fig. 1) carried out in three representative sculptures: Nicodemus (N), Joseph of Arimathea (GA) and Woman with child (DB).
 | ||
| Fig. 1 Selected measurement points and their location in Nicodemus (N), Joseph of Arimathea (GA) and Woman with child (DB) painted sculptures (from left to right). | ||
Results and discussion
Validation
1. The fluorescence mitigation of pSSE-RS was checked using a commercial milk chocolate (solid) and a madder lake (powder). The standard dispersive Raman data (FOP-RS) show a really intense fluorescence background. The spectra obtained removing the baseline with a suitable OPUS™ algorithm do not provide any characteristic bands of the chocolate or madder lake; it is possible to observe only broad artifacts with a low contrast. FT-RS and pSSE-RS data show a reasonable spectrum for both samples, where all the peaks are well reproducible with good contrast, allowing an ideal molecular identification (Fig. 2).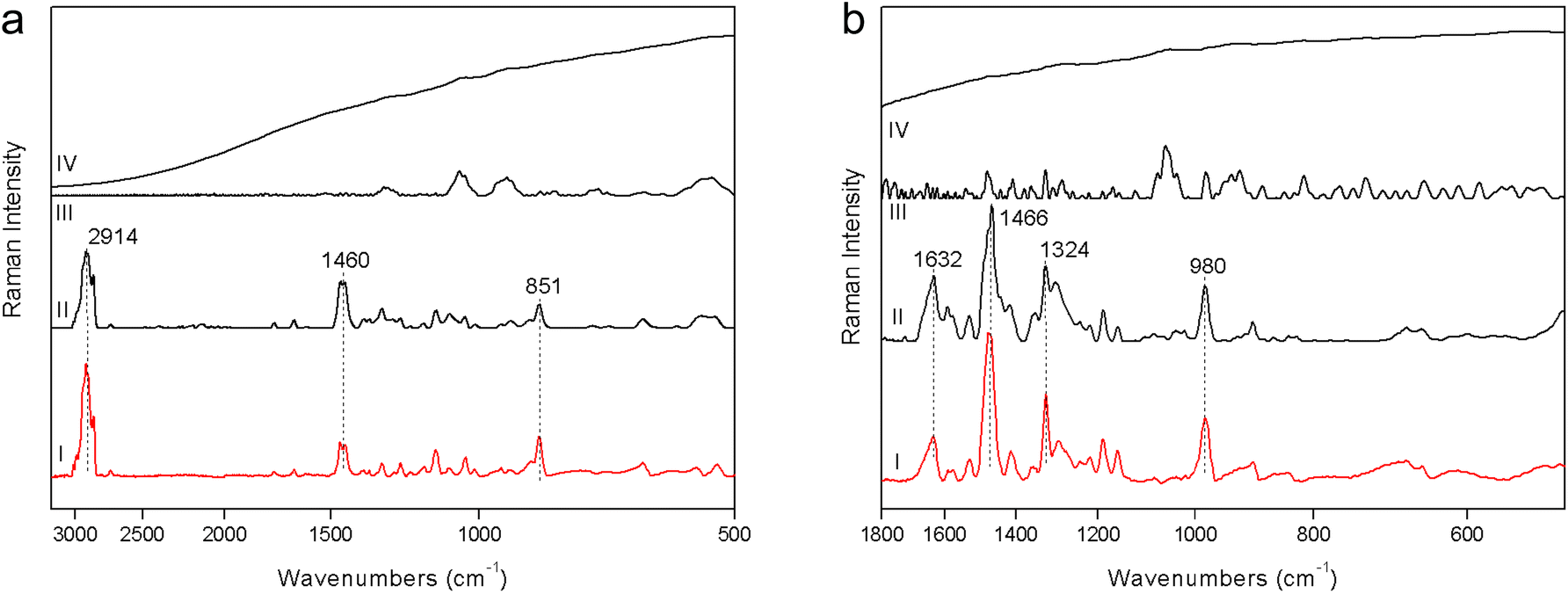 | ||
| Fig. 2 Raman spectra acquired on a commercial milk chocolate (a) and on a madder lake (b) with pSSE-RS (I), FT-RS (II), FOP-RS baseline corrected (III) and FOP-RS raw (IV). Spectra are plotted on a logarithmic scale. | ||
2. Beeswax and polystyrene were used to evaluate the performance at the CH stretching spectral range; data comparison is carried out mainly with the FT-RS data, due to its best sensitivity in that spectral range (provided by the germanium diode detector and its optical components) with respect to the dispersive one. However, the spectra acquired with FOP-RS are also reported in Fig. 3. The comparison shows really good agreement between data in terms of band contrast, peak position and band shape.
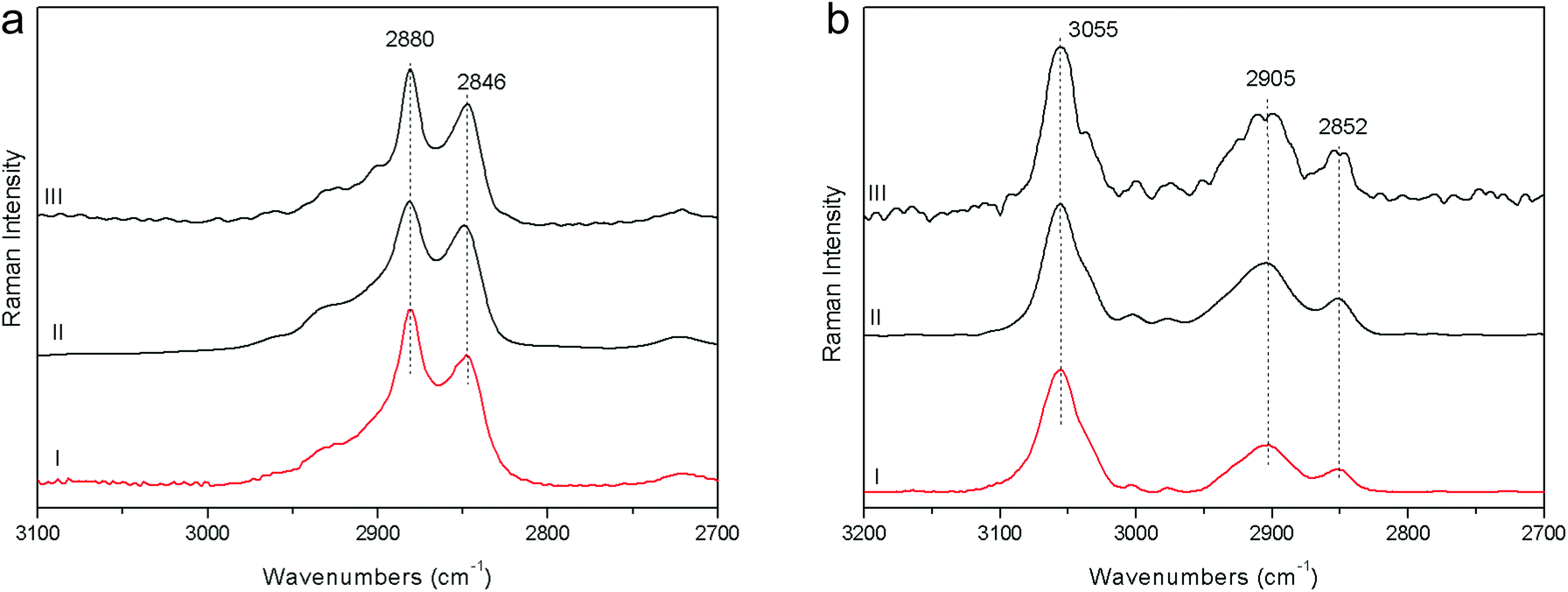 | ||
| Fig. 3 Raman spectra acquired on beeswax (a) and polystyrene (b) with pSSE-RS (I), FT-RS (II), FOP-RS (III). | ||
3. To compare pSSE-RS sensitivity with the laboratory benchtop instruments, it is essential to define a method to measure the signal to noise ratio (SNR) and to identify a suitable sample to do it. Typically, for dispersive instruments, the SNR is estimated measuring the throughput, i.e. the quantity of the signal read by the CCD, using a suitable standard sample (always necessary to generate a signal). The throughput is a measure of the absolute Raman signal, thus it is proportional to the CCD counts. Different instruments use different methods to integrate the signal (i.e. the number of coadditions can be normalized using the average or just the sum), therefore the throughput measure is not commonly standardized, especially if FT-RS is involved in the comparison. In the presence of a reference sample, it is reasonable to consider a signal (i.e. the intensity of a specified band), to measure the noise in a certain spectral range and finally to calculate their ratio. Applying this procedure to the spectra acquired with each technique, it is possible to obtain a sensible value functional to their comparison.
The SNR measurement (peak–peak) of polystyrene has been carried out taking the intensity value of the most significant band (at about 1003 cm−1), normalizing the spectrum intensity with an offset of this value and finally measuring the SNR (peak–peak) in the 1800–1700 cm−1 spectral range with a suitable OPUS™ function (Fig. 4a). This is the standard procedure to define the SNR specification for a FT-Raman spectrometer. The obtained SNR values reveal that pSSE-RS has good intrinsic sensitivity, showing a SNR value of the same order of magnitude with respect to FOP-RS and FT-RS ones (about 200:1 as the average value).
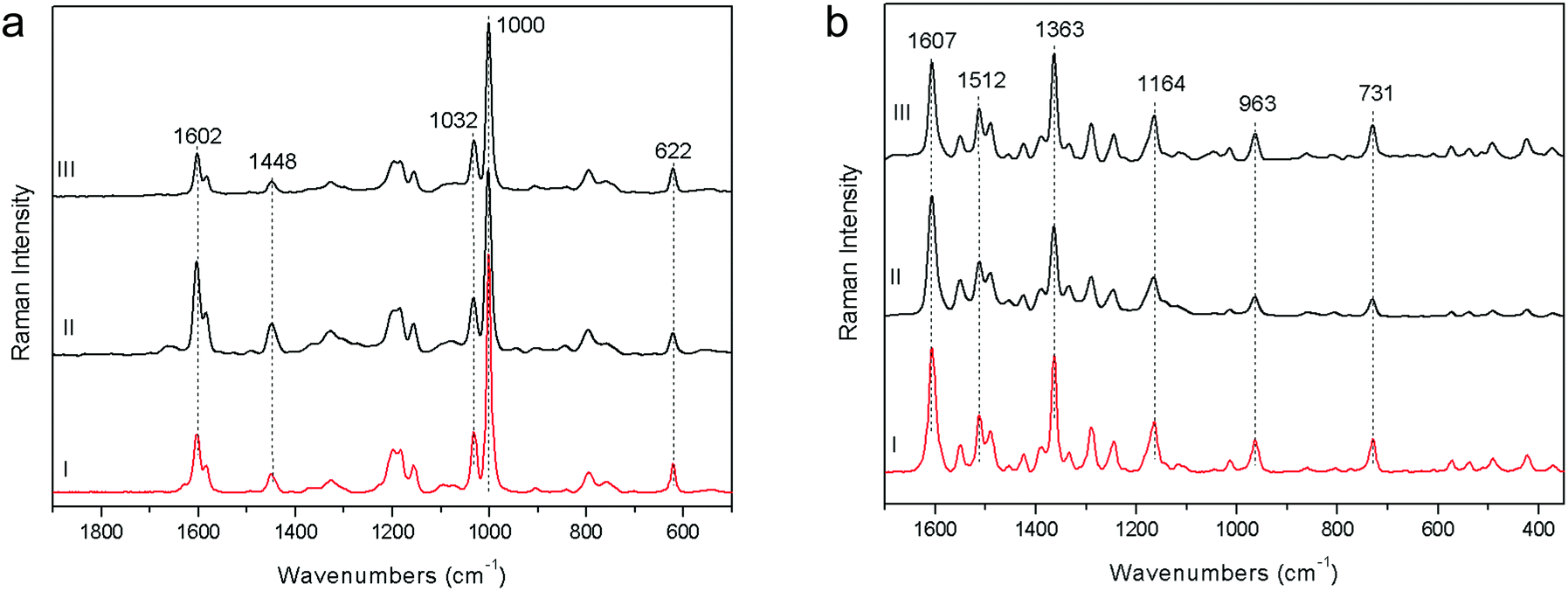 | ||
| Fig. 4 Raman spectra acquired on polystyrene (a) and red colorant PR170 (b) with pSSE-RS (I), FT-RS (II), FOP-RS (III). | ||
4. Generally speaking, the highest wavenumber accuracy (and also the highest spectral resolution) is given by the FT-Raman system (where an interferometer is used to have frequencies dispersion), mainly due to the use of a HeNe laser as internal sampling reference6 (the spectral resolution of such a FT system is mainly dependent on the maximum excursion of the interferometer moving mirror). The results obtained on the red colorant PR170 (Fig. 4b), analysed at low spectral resolution (8–11 cm−1) for a more confident comparison among the instruments, are really excellent: considering the bands with good contrast, the average of the relative difference in their positions is approximately 0.4 cm−1. Moreover, monitoring the wavenumber accuracy over several days, it is possible to state that no band shifts take place, due to the intrinsic wavenumber calibration system. The conclusion is that the wavenumber stability is good enough for this kind of application, in particular for a molecular identification library search, where the absolute peak position is really critical.
Case study
The demonstration of the analytical capability of pSSE-RS on sculptures was carried out on different painted areas; here we present the most significant results of the main colours. The identification of the compounds has been obtained with the help of literature data as well as comparing the Raman spectra with the extensive pigment database acquired with pSSE-RS on standard references.The pigment mixtures used in Nicodemus's red jacket (N4) and in Joseph of Arimathea's red/orange mantle (GA25) were clearly distinguished by pSSE-RS. The N4 spectrum (Fig. 5a) shows the presence of the most characteristic signature of cinnabar [α-mercury(II) sulfide], lead white (basic lead carbonate) and barium white (barium sulphate): despite the cutting of the spectral range at low wavenumbers of this portable instrument, cinnabar is well detectable due to the strong band at 344 cm−1, while the most intense peak at 253 cm−1 is not visible. Lead white is one of the most common white pigments used as an extender and it is identified by its intense ν(CO32−) band at 1050 cm−1; barium white is inferred by the band at 989 cm−1 and it has been used as an extender since the early 19th century,17 thus it gives an indication of post quem this area was re-painted. Joseph of Arimathea's mantle (Fig. 5a) has an orange hue that was obtained mixing chrome yellow [lead(II) chromate] to all the other pigments previously found in Nicodemus's red jacket. Chrome yellow was identified through its most intense band at 842 cm−1; the lighter hue of the mantle was also due to the higher amount of lead white.
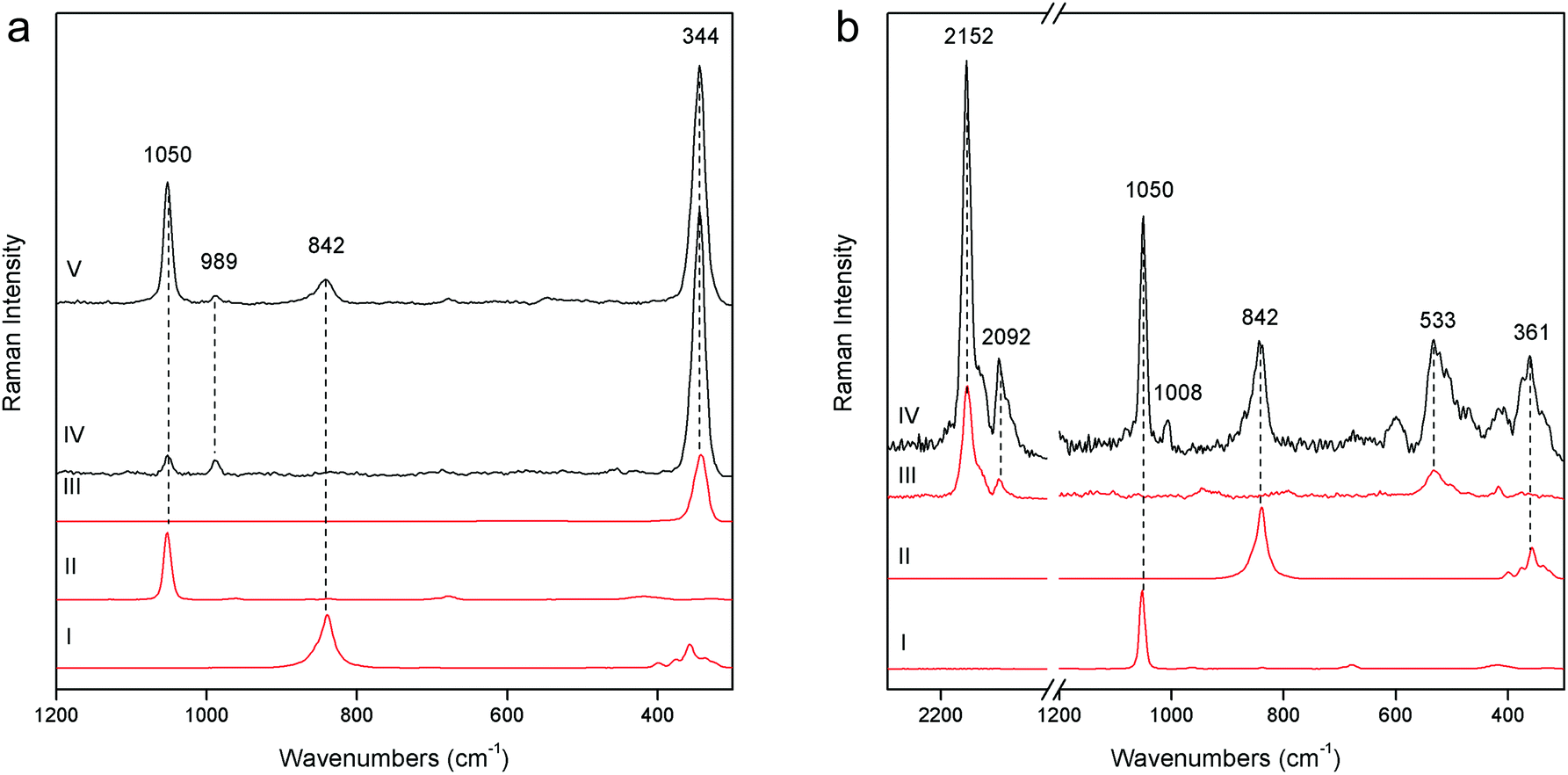 | ||
| Fig. 5 (a) pSSE Raman spectra acquired on red/orange areas: Joseph of Arimathea's red/orange mantle (GA25 – V), Nicodemus's red jacket (N4 – IV), reference spectra of cinnabar (III), lead white (II) and chrome yellow (I). (b) Raman spectra acquired on the Nicodemus's green mantle (N25 – IV) and reference spectra of (I) lead white, (II) chrome yellow, (III) Prussian blue. Reference spectra are acquired with pSSE-RS. | ||
The pSSE Raman spectrum acquired on the Nicodemus's green mantle (N25) exhibits intense bands unequivocally assigned to Prussian blue [iron(III) hexacyanoferrate(II)], chrome yellow and lead white (Fig. 5b). Prussian blue shows the most intense band at 2152 cm−1 and other two diagnostic bands at 2092 and 533 cm−1; in this area chrome yellow is clearly distinguishable by two very intense peaks at 842 and 361 cm−1, and lead white was used as an extender. The mixture of Prussian blue and chrome yellow is one of the most important commercial green pigments (chrome green);17 due to its high homogeneity usually the components cannot be distinguished microscopically. Prussian blue and chrome yellow have been introduced in the early 18th and 19th centuries,17 respectively. No date for the introduction of chrome green can be given; it must have been available and in use shortly after the introduction of chrome yellow in the first quarter of the 19th century. Thus, it gives an indication of post quem this area was re-painted.
More challenging is the recognition of the pigments used for the Woman with child's green mantle (DB21); the inability of pSSE-RS to provide satisfactory results can be due to the excitation line in NIR which is strongly absorbed by green pigments, besides the high fluorescence background; in fact, the previous micro-destructive measurements performed on small fragments with a benchtop Raman spectrometer equipped with a 514 nm excitation line showed the presence of emerald green (copper acetoarsenite).16 However, pSSE-RS allows achieving interesting information on the stratigraphy of the Woman with child's mantle, discovering a completely different palette in the layers under the surface (Fig. 6a). Microscopical observations performed in a hole caused by a surface fracture show the presence of particles underneath with different colours (blue, red, orange and white – Fig. 6b); even though the spectra acquired with pSSE-RS are rather complex, the presence of several pigments can be inferred: orpiment (arsenic(III) sulphide) with characteristic bands at low wavenumbers (309, 355 and 384 cm−1), red lead [dilead(II) lead(IV) oxide] with very sharp peaks at 546 and 391 cm−1 and azurite [basic copper(II) carbonate] are clearly identifiable through the Raman band at 1094 cm−1, while its most characteristic peak around 400 cm−1 is covered by the more intense red lead; finally, lead white occurs as an extender. To verify the interpretation proposed for this complex mixture of pigments and specially to shed light into the overlapping Raman peaks in a red lead–azurite mixture, a mock-up specimen has been prepared in laboratory mixing together an appropriate amount of red lead and azurite powders to simulate the peak intensity ratio observed in the Raman spectrum acquired in the real artwork. As shown in Fig. 6a the Raman signature of the artificial mixture compares well with the real one. Although the order of these pigments in the stratigraphy remains obscure, pSSE-RS provides the complete identification of the crystal pigments observed under the green surface which can supply the background for the re-investigation of the sculpture history; just to cite an example, the absence of barium white (barium sulphate) in this sublayer area could lead to the discrimination between the more internal and ancient mixture (where lead white is used as a single extender) and the more external and recent layer (where barium white is in a mixture with lead white, as deduced by almost all pSSE-RS measurements acquired in the sculpture surfaces).
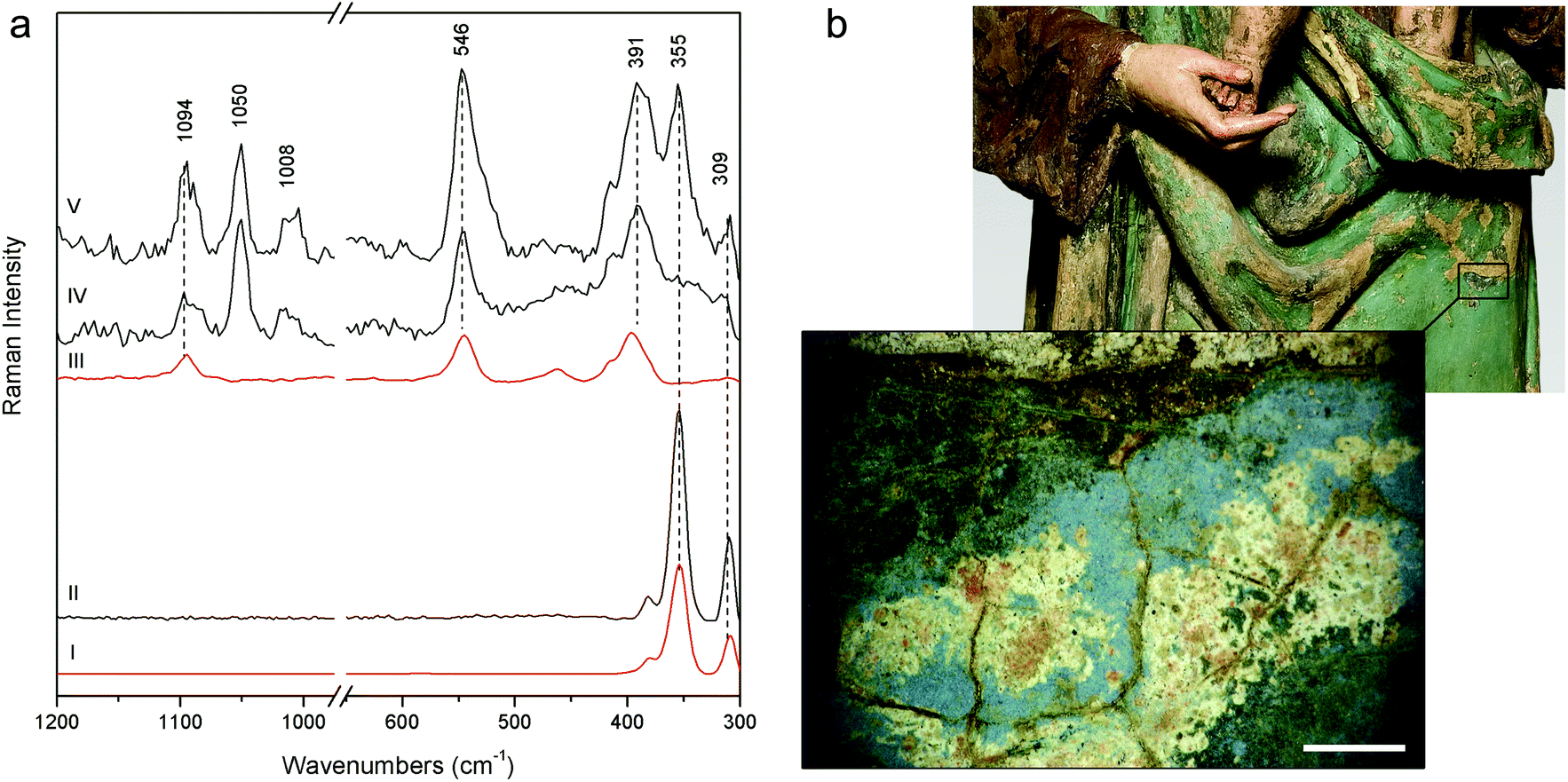 | ||
| Fig. 6 (a) pSSE Raman spectra acquired on the Woman with child's green mantle (DB21) sublayer in three different locations (V, IV, II), reference spectra of orpiment (I) and of a red lead and azurite mixture (III). Reference spectra are acquired with pSSE-RS. (b) Image acquired with a portable optical microscope in the green mantle sublayer (black rectangle in a detail of Woman with child's mantle); scale bar 1 mm. | ||
Joseph of Arimathea's shirt consists of yellow and blue portions; pSSE-RS carried out on the yellow one (GA21) allows distinguishing the complete Raman signatures of chrome yellow including the less intense bands at lower wavenumbers (326, 341, 361, and 379 cm−1); moreover, together with the most typical yellow chrome, the strong shoulder at 828 cm−1 suggests the presence of the chrome yellow–orange pigment which includes lead oxide in the chemical composition.18 Again, barium and lead white occur as extenders (Fig. 7a).
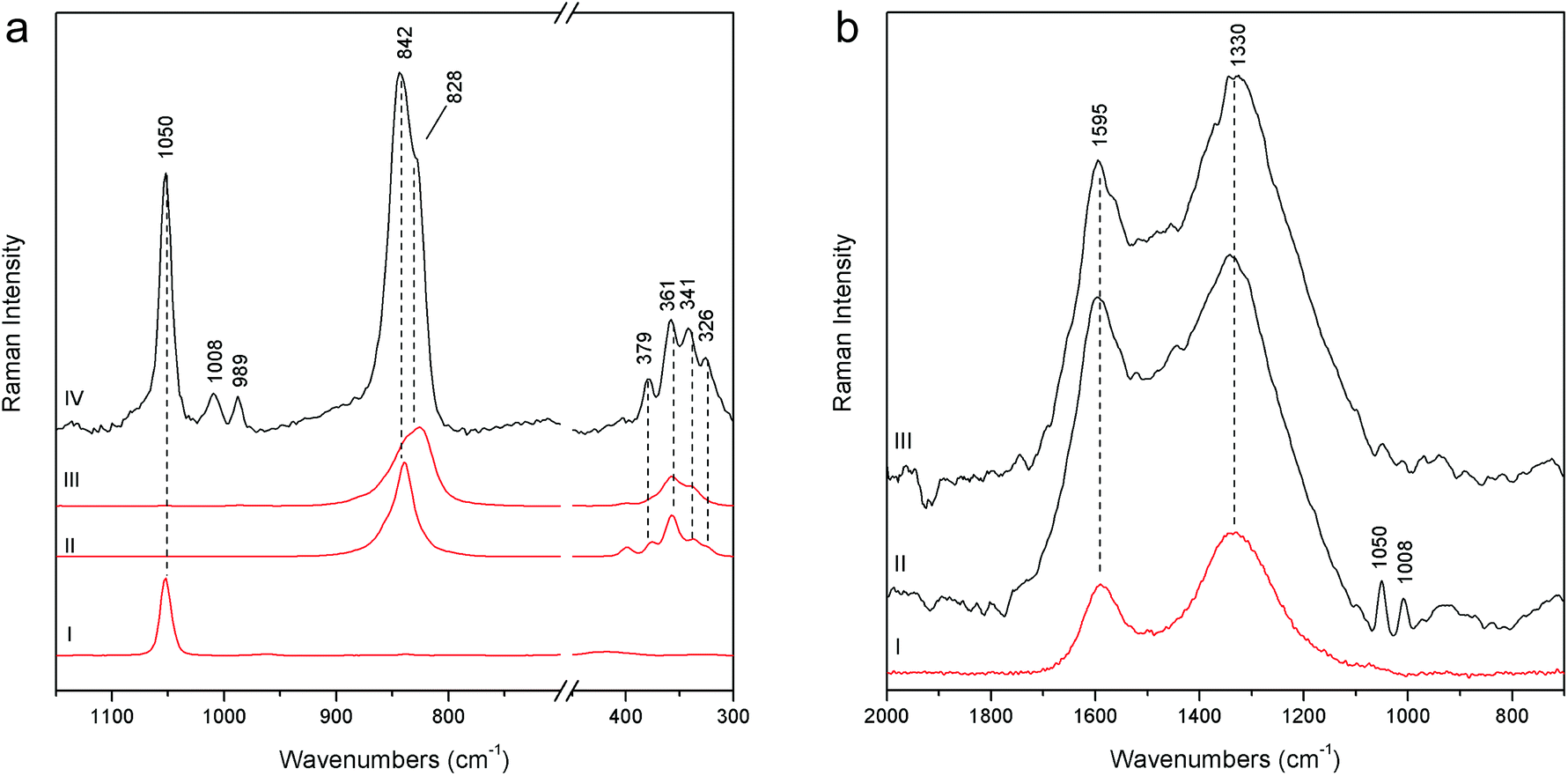 | ||
| Fig. 7 (a) pSSE Raman spectrum acquired on the yellow area of the Joseph of Arimathea's shirt (GA21 – IV), reference spectra of chrome yellow-orange (III), chrome yellow (II) and lead white (I). (b) Raman spectra acquired on the Joseph of Arimathea's mantle lapel (GA24 – III), Nicodemus's hat (N10 – II), and reference spectra of vine black (I). Reference spectra are acquired with pSSE-RS. | ||
Particularly meaningful is the Raman spectrum (Fig. 8a) acquired on the blue areas of the Joseph of Arimathea's shirt (GA20): ultramarine blue (lazurite) is detected through its most intense and characteristic band at 548 cm−1, assigned to the symmetric stretching ν mode of the S3− ions in a sodium alumino-silicate matrix.18 In line with expectations, barium and lead white are present as extenders and the typical bands ascribable to organic compounds dramatically arise in the range 1200–1500 cm−1 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and around 3000 cm−1 (νC–H). Five most intense bands have to be considered at 1297, 1354, 1440, 2860 and 2910 cm−1; although a minor band of lead white at 1365 cm−1 could give a contribution to the band at 1354 cm−1, on the basis of these Raman features the presence of linseed oil and/or egg white can be inferred, even though the unequivocal distinction between these two kinds of binders is not straightforward due to the similarity in their Raman band position.19,20 However, it is well known that in the presence of lead white, linseed oil is the most common binder for the preparation of the renaissance painted terracotta sculptures,21,22 as also confirmed during the previous diagnostic campaign performed on the same sculptures where linseed oil was detected on the external painted layers using benchtop Fourier Transform Infrared (FT-IR) spectroscopy. A further check has been carried out preparing a second mock-up specimen in laboratory mixing together an appropriate amount of linseed oil and lead white. After drying and accelerated decay with the UV camera, the specimen has been analysed with pSSE-RS and the spectrum compares well with those acquired in situ (Fig. 8b).
C) and around 3000 cm−1 (νC–H). Five most intense bands have to be considered at 1297, 1354, 1440, 2860 and 2910 cm−1; although a minor band of lead white at 1365 cm−1 could give a contribution to the band at 1354 cm−1, on the basis of these Raman features the presence of linseed oil and/or egg white can be inferred, even though the unequivocal distinction between these two kinds of binders is not straightforward due to the similarity in their Raman band position.19,20 However, it is well known that in the presence of lead white, linseed oil is the most common binder for the preparation of the renaissance painted terracotta sculptures,21,22 as also confirmed during the previous diagnostic campaign performed on the same sculptures where linseed oil was detected on the external painted layers using benchtop Fourier Transform Infrared (FT-IR) spectroscopy. A further check has been carried out preparing a second mock-up specimen in laboratory mixing together an appropriate amount of linseed oil and lead white. After drying and accelerated decay with the UV camera, the specimen has been analysed with pSSE-RS and the spectrum compares well with those acquired in situ (Fig. 8b).
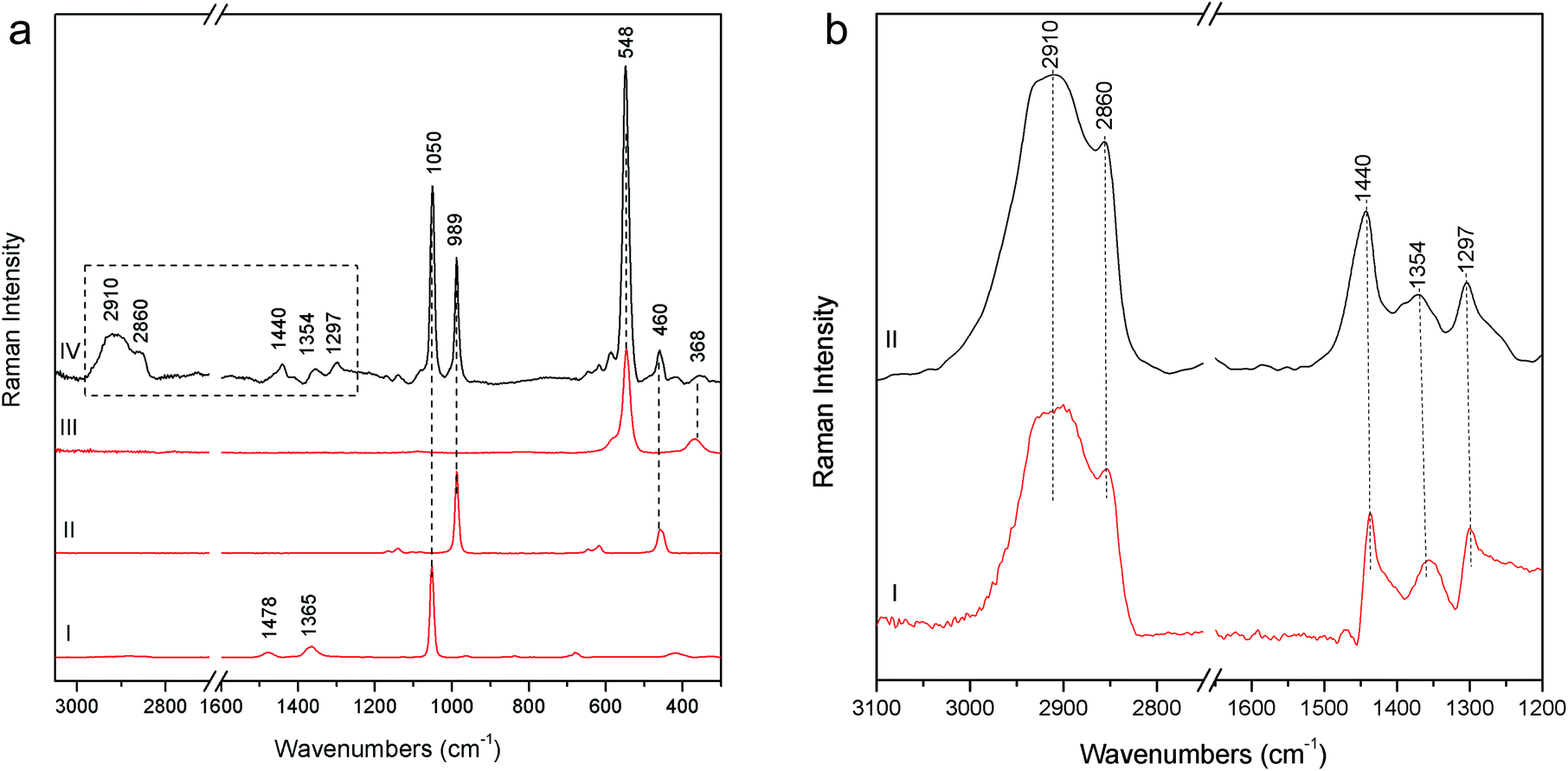 | ||
| Fig. 8 (a) pSSE Raman spectrum acquired on the blue area of the Joseph of Arimathea's shirt (GA20 – IV), reference spectra of ultramarine blue (III), barium white (II) and lead white (I). (b) Zoom on a detail (the dotted rectangle of a figure) of GA20 (II) and the pSSE reference spectrum acquired on linseed oil and lead white mock-up aged specimen (I). Reference spectra are acquired with pSSE-RS. | ||
It is worth pointing out that the detection by Raman spectroscopy of the binder fraction of painted layers is challenging with benchtop instruments, and even more with portable devices, as the scarce number of literature papers on this subject proves. This can be due to the critically low amount of binder residuals and to the low Raman sensitivity in the organic spectral regions, especially using NIR laser excitation wavelength.
The measurements carried out on the Joseph of Arimathea's mantle lapel (GA24) highlight that black carbon is the pigment employed for the black areas, as the two broad bands at approximately 1330 and 1595 cm−1 unambiguously prove (Fig. 7b). The same pigment mixed with cinnabar and lead white was used for the Nicodemus's hat (N10).
Each Raman spectra shows the presence of a relatively small amount of gypsum and/or anhydrite at 1008 and 1018 cm−1, respectively. The investigation of the composition of an area restored during the most recent conservation work allowed for ascribing the presence of calcium sulphate to the material used for the reintegration of surface gaps, mainly composed of calcium sulphates and cellulose.
Conclusions
The analytical capability of a portable sequentially shifted excitation instrument has been proved also in complex matrices as those encountered in the art field. The validation process enabled the assessment of its ability to analyse fluorescent samples in a short measurement time, to evaluate its wavenumber accuracy and stability on the whole available spectral range, its sensitivity, even at the CH stretching spectral range, and the signal to noise ratio. The method is conceptually demonstrated on a range of reference materials and its performance is benchmarked against two conventional benchtop Raman instruments indicating a largely undiminished benchtop performance.A first overall view of pSSE-RS potentiality for in situ non-invasive analysis of artworks has been given. The chemical makeup of the complex palette has been almost completely revealed, even in the binder fraction and with considerably better findings as regards the last non-invasive diagnostic campaign; pigments located within surface fractures and related to previous paintings have been identified, paving the way to a new exploration of artwork history; moreover, compounds of the previous restoration work have been detected.
The natural limitations of pSSE-RS are the positioning, rather complicated in a non-flat surface like in the case of sculptures, the low lateral resolution, critical in such heterogeneous surfaces, the unlikely detection of some pigments due to the short spectral range in the low wavenumbers (i.e. ochre) or to the NIR excitation laser wavelength (i.e. green pigments).
A portable SSE instrument paves the way for the establishment of Raman spectroscopy as an effective analytical tool applicable to conservation science expanding the portfolio of non-invasive, chemically specific analytical tools.
Acknowledgements
We would like to thank Fabio Bevilacqua, restorer of C.R.C. Restauri and Mari Mapelli, restorer of the MIBACT Regional Secretariat for Lombardy, for their support in the conservation site; Tommaso Poli for the specimens availability.References
- D. Lauwers, A. G. Hutado, V. Tanevska, L. Moens, D. Bersani and P. Vandenabeele, Spectrochim. Acta, Part A, 2014, 118, 294–230 CrossRef CAS PubMed.
- G. Barone, D. Bersani, J. Jehlička, P. P. Lottici, P. Mazzoleni, S. Raneri, P. Vandenabeele, C. Di Giacomo and G. Larinà, J. Raman Spectrosc., 2015, 46, 989–995 CrossRef CAS.
- F. Daniel, A. Mounier, J. Aramendia, L. Gómez, K. Castro, S. Fdez-Ortiz de Vallejuelo and M. Schlicht, J. Raman Spectrosc., 2016, 47, 162–167 CrossRef CAS.
- G. Simsek, P. Colomban, F. Casadio, L. Bellot-Gurlet, G. Zelleke, K. T. Faber, V. Milande and L. Tilliard, J. Am. Ceram. Soc., 2015, 98, 3006–3013 CrossRef CAS.
- P. R. Griffiths and J. A. De Haseth, Fourier Transform Infrared Spectrometry, Wiley, Hoboken, New Jersey, 2nd edn, 2007 Search PubMed.
- J. M. Chalmers and P. R. Griffiths, Handbook of Vibrational Spectroscopy, Wiley, Hoboken, New Jersey, 2001 Search PubMed.
- P. Matousek, M. Towrie and A. W. Parker, Appl. Spectrosc., 2005, 59, 848–851 CrossRef CAS PubMed.
- I. Osticioli, A. Zoppi and E. M. Castellucci, J. Raman Spectrosc., 2006, 37, 974–980 CrossRef CAS.
- F. Rosi, M. Paolantoni, C. Clementi, B. Doherty, C. Miliani, B. G. Brunetti and A. Sgamellotti, J. Raman Spectrosc., 2010, 41, 452–458 CAS.
- S. E. J. Bell, E. S. O. Bourguignon and A. Dennis, Analyst, 1998, 123, 1729–1734 RSC.
- J. Kauffman, J. D. Rodriguez and L. F. Buhse, Am. Pharm. Rev., 2011, 14, 34–40 CAS.
- J. B. Cooper, S. Marshall, R. Jones, M. Abdelkader and K. L. Wise, Appl. Opt., 2014, 53, 3333–3340 CrossRef CAS PubMed.
- J. B. Cooper, M. Abdelkader and K. L. Wise, Appl. Spectrosc., 2013, 67, 973–984 CrossRef CAS PubMed.
- S. Assi, Eur. Pharm. Rev., 2015, 20, 2–8 Search PubMed.
- F. Casadio, M. Leona, J. R. Lombardi and R. Van Duyne, Acc. Chem. Res., 2010, 43, 782–791 CrossRef CAS PubMed.
- C. Colombo, F. Bevilacqua, L. Brambilla, C. Conti, M. Realini, J. Striova and G. Zerbi, Anal. Bioanal. Chem., 2011, 401, 757–765 CrossRef CAS PubMed.
- J. Gettens and L. Stout, Painting materials, Dover Publications, New York, 2011 Search PubMed.
- I. M. Bell, J. H. R. Clark and P. J. Gibbs, Spectrochim. Acta, Part A, 1997, 53, 2159–2179 CrossRef.
- L. Burgio and R. J. H. Clark, Spectrochim. Acta, Part A, 2001, 57, 1491–1521 CrossRef CAS.
- P. Vandenabeele, B. Wehling, L. Moens, H. Edwards, M. De Reu and G. Van Hooydonk, Anal. Chim. Acta, 2000, 407, 261–274 CrossRef CAS.
- P. Bensi , in La scultura in terracotta, Ed. Centro Di della Edifici srl, Firenze, 1996, Alla vita della terracotta era necessario il colore: appunti sulla policomia della statuaria fittile, 34–46.
- G. Vasari, Le vite dè più eccellenti architetti, pittori, et scultori italiani, da Cimabue insino a’ tempi nostri, Einaudi, Torino, 1986 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |